Amplified plasmonic detection of DNA hybridization using doxorubicin-capped gold particles
Jolanda
Spadavecchia
*a,
Ramesh
Perumal
a,
Alexandre
Barras
b,
Joel
Lyskawa
c,
Patrice
Woisel
c,
William
Laure
c,
Claire-Marie
Pradier
a,
Rabah
Boukherroub
a and
Sabine
Szunerits
*a
aLaboratoire de Réactivité de Surfaces, UMR CNRS 7197, Université Pierre & Marie Curie – Paris VI, Site d'Ivry – Le Raphaël, 94200 Ivry-sur-Seine, France. E-mail: jolanda.spadavecchia@upmc.fr; sabine.szunerits@iri.univ-lille1.fr; Fax: +33 (0)1 44 27 60 33; Fax: +33 (0)3 62 53 17 01; Tel: +33 (0)1 44 27 55 12 Tel: +33 (0)3 62 53 17 25
bInstitut de Recherche Interdisciplinaire (IRI, USR 3078 CNRS), Université Lille 1, Parc de la Haute Borne, 50 Avenue de Halley, BP 70478, 59658 Villeneuve d'Ascq, France
cUniversité Lille 1, Unité des Matériaux Et Transformations (UMET, UMR 8207 CNRS), Ingénierie des Systèmes polymères (ISP) Team, F-59655 Villeneuve d'Ascq Cedex, France
First published on 25th October 2013
Abstract
We show in this article that doxorubicin-modified gold nanoparticles (Au NP-DOX) can be used for the post-amplification of the wavelength shift of localized surface plasmon resonance (LSPR) signals after DNA hybridization events. We take advantage of the intercalation properties of DOX with guanine-rich oligonucleotides and the plasmon coupling between surface-linked gold nanostructures and Au NP-DOX in solution to detect in a sensitive manner DNA hybridisation events. Post-treatment of double-stranded DNA with Au NP-DOX resulted in a detection limit of ≈600 pM, several times lower than that without post-incubation (LOD ≈ 40 nM).
1. Introduction
DNA hybridization assays are widely employed in molecular biology and for forensic tests as they provide enormous potential for rapid analysis of nucleic acid samples.1 While traditionally DNA assays are based on fluorescence read out by labeling the target DNA with a fluorescent molecule,2 advances in DNA biosensors have been realized through the use of sensitive label-free detection schemes based on electrochemical and plasmonic read out. Indeed, as the concentration of the target DNA is extremely low, the sensitivity of DNA biosensors has to be very high in order to detect DNA directly. This request can be achieved by the use of controlled and efficient surface DNA probe immobilization strategies allowing maximization of the hybridization efficiency of the biosensor and thus lowering the detection limit.3,4 In addition, different research groups have utilized signal amplification strategies to increase the sensitivity of the DNA biosensor.5–7 In an attempt to enhance the sensitivity of localized surface plasmon resonance (LSPR) interfaces for the detection of hybridization events, metal nanostructure enhanced fluorescence has been proposed.7 Fluorescently labeled DNA oligomers interacted with covalently immobilized DNA on thin amorphous silicon–carbon layers capping the gold metal nanostructures. Through optimization of the coating thickness and by working close to resonance conditions for plasmon and fluorophore excitation, the hybridization and thus the detection of very dilute (5 fM) oligomers was easily attained. More recently we have proposed the use of plasmonic coupling between surface linked gold nanostructures and gold nanorod (Au NR) or gold nanostar (Au NS) modified DNA molecules in solution as a DNA hybridization amplification strategy, which allowed a decrease of the limit of detection from 40 nM (without amplification) to 200 pM (with amplification).8This article widens the concept of enhancing the sensitivity for DNA hybridization assays with plasmonic read out by post-labeling the formed double stranded DNA with doxorubicin-conjugated gold nanoparticles. Indeed, gold nanoparticles (Au NPs) are excellent candidates for biological assays as a wide range of molecules can be tethered onto the Au NP surface by means of thiol groups and others.9–11 In this work, doxorubicin (DOX) modified Au NPs capped with dicarboxylic acid functionalized polyethylene glycol are used for this purpose (Fig. 1). DOX is an approved chemotherapeutic agent that binds to duplex DNA via sequence specific intercalation, e.g. 5′-CG/CG, with the daunosamine sugar lying in the minor groove, extending to an additional DNA base pair adjacent to the intercalation site.12 The binding constant of DOX with double stranded DNA is as high as 106 M−1. We take advantage of the sequence specificity of Au NPs modified with DOX to post-amplify the plasmonic read out of the hybridization signal. LSPR sensors, employing noble metal nanostructures, have lately attracted considerable attention as a new class of plasmonic nanosensors.13,14 The position of the nanoparticle extinction band maximum (λmax) is located in the visible range and is highly dependent on the local refractive index of the nanoparticles. This enables biomolecular binding events such as DNA hybridization to be followed in real-time by monitoring the temporal variation of the LSPR signal.7,15–17 In the case of oligonucleotide-based LSPR interfaces, sensitivity loss can arise as LSPR sensors are functionalized with long DNA surface ligands.18 In this article we investigate if DOX-labeled gold nanoparticles rather than DNA-modified particles8 would allow for sensitive detection of DNA hybridization. One of the advantages of this approach would be that DOX-Au nanostructures can be easily synthesized and are highly stable over time.
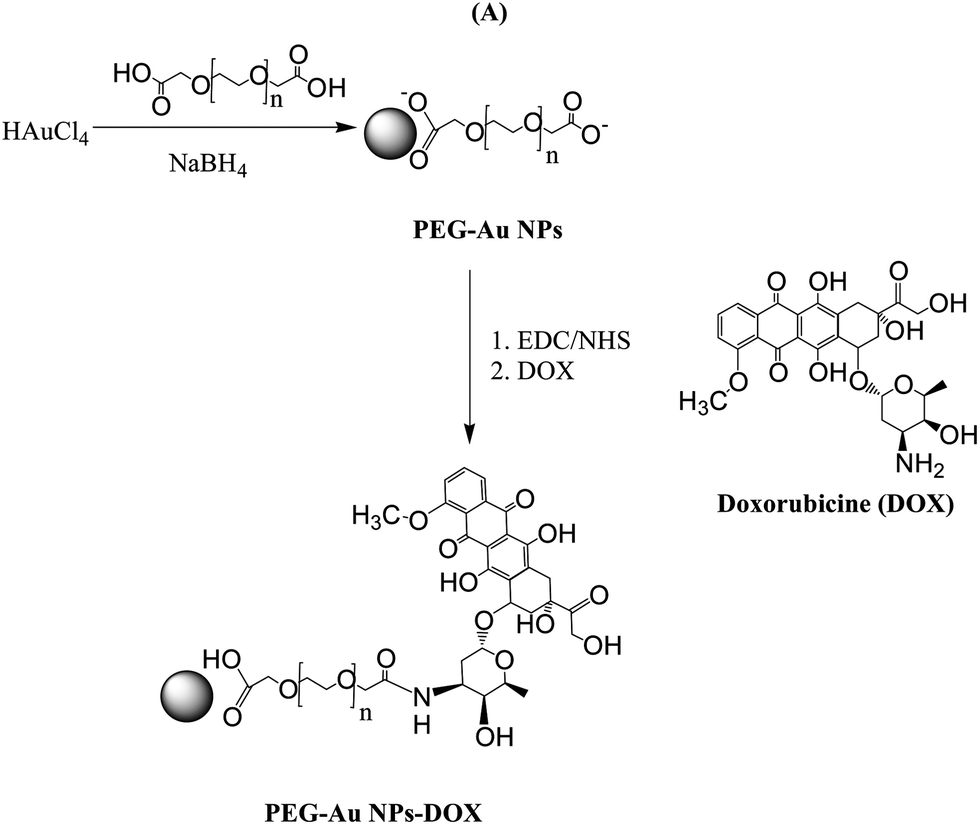 | ||
| Fig. 1 Preparation of PEG-capped gold nanoparticles (PEG-Au NPs) by reduction of chloroauric acid (HAuCl4) with sodium borohydride (NaBH4) in the presence of PEG-diacid and subsequent covalent modification of the free carboxylic acid of the PEG-Au NPs with the NH2-functions of DOX using EDC/NHS. | ||
2. Experimental
2.1. Materials
All chemicals were of reagent grade or higher and were used as received unless otherwise specified. Tetrachloroauric acid (HAuCl4), sodium borohydride (NaBH4), N-hydroxysuccinimide (NHS), 1-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), ethanol (Normapur 99%), 1,4-phenylenediisothiocyanate (PDC), polyethylene glycol 600 diacid (PEG-diacid), sodium hydroxide (NaOH), sodium dodecyl sulphate (SDS), salmon sperm DNA, formamide, phosphate buffered saline (PBS, 0.1 M, pH 7.4), acetone, acetonitrile (CH3CN), methanol (CH3OH), doxorubicin hydrochloride (98%), and ethanol (C2H5OH) were purchased from Sigma Aldrich. Saline sodium citrate (SSC) buffer was obtained from Fluka.The 28mer oligonucleotides were purchased from Eurogentec and have the following sequences.
DNA on the LSPR interface (probe 1): 5′-HS-TTT-TGG-GAT-GGT-TGA-GGG-TGC-CTC-TGG-C-3′
Complementary DNA in solution (target 1): 5′-GCC-AGA-GGC-ACC-CTC-AAC-ACT-CCC-A-3′
Noncomplementary DNA probe in solution (target 2): AAG-CGA-TCG-ATA-GTC-CAG-AAG-AAT-A.
Stock solutions of 5 μM were prepared in PBS buffer (100 mM, pH 7.4) containing 0.01% sodium dodecyl sulphate (SDS).
The hybridization buffer was a solution of NaCl (0.5 M), phosphate buffered solution (0.01 M), and ethylenediaminetetraacetic acid (0.01 M, pH 5.5).
2.2. Formation of DNA-modified LSPR interfaces
2.3. Synthesis of doxorubicin-capped gold nanoparticles (DOX-Au NPs)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 26 min (three times) and then the supernatant was discarded and the residue was redispersed in an equivalent amount of buffer solution (PBS, pH: 7). This was repeated twice principally to remove excess of dicarboxylic PEG. Stock solutions were stored at 25 °C and characterized using UV-Vis spectroscopy and transmission electron microscopy (TEM).
000 rpm for 26 min (three times) and then the supernatant was discarded and the residue was redispersed in an equivalent amount of buffer solution (PBS, pH: 7). This was repeated twice principally to remove excess of dicarboxylic PEG. Stock solutions were stored at 25 °C and characterized using UV-Vis spectroscopy and transmission electron microscopy (TEM).
2.4. DNA hybridization
For end-point measurement, the surface was exposed to the DNA target at 37 °C during 1 h in a hybridization chamber. The hybridization solution was made of 2× SSC, 0.1% SDS, 0.1% salmon sperm DNA, 35% formamide and the target oligonucleotide at a concentration in the range of 20 pM to 100 nM. After hybridization, the sample was subjected to three successive wash steps of 2 min each in the following solutions: 2× SSC, 0.1% SDS, then 1× SSC, 0.1% SDS and finally 0.1× SSC, all at pH 7. The sample was finally dried under a stream of argon before LSPR measurement.Amplification with Au NP-DOX, DOX, and Au NPs was performed by coating the hybridized LSPR interface with 200 μL of the respective aqueous solution for 30 min at room temperature. The sample was then washed intensively with water before LSPR measurements were performed.
2.5. Instrumentation
000 rpm. The upper part of the colorless solution was removed and the solution fraction was re-dispersed in 1 mL buffer solution (PBS, pH 7). 2 μL of this re-dispersed particle suspension was placed on a carbon-coated copper grip and dried at room temperature.
3. Results and discussion
3.1. Synthesis of doxorubicin modified PEG-Au NPs
Morphological analyses of the resulting PEG-Au NPs by transmission electron microscopy (TEM) reveal their regular spherical shape, smooth surface and homogeneous size distribution without agglomeration with an average particle diameter of 7.5 nm and a standard deviation of 2 nm (Fig. 2A). A typical PMIRRAS spectrum of washed and dried PEG-Au NPs, deposited on a planar gold surface, is shown in Fig. 3. The presence of a strong stretching band at 1115 cm−1 due to C–O–C of the PEG polymer together with the C–H bending modes at 1461 cm−1 and the carboxylic acid group (COOH) at 1713 cm−1 suggests the stabilization of the Au NPs with PEG molecules. The presence of the bands at 1713 cm−1 is evidence of the presence of PEG units on Au NPs, in particular “free” acid functions that will be possibly used for the covalent linkage of biomolecules. The band at 1544 cm−1 is attributed to the vibration band of carboxylate anions (COO−) formed on the particles.25 The surface charge of the PEG-Au NPs was determined by zeta-potential analysis and was found to be −16.8 ± 2 mV, suggesting in addition the presence of free negatively charged carboxylic acid functions.
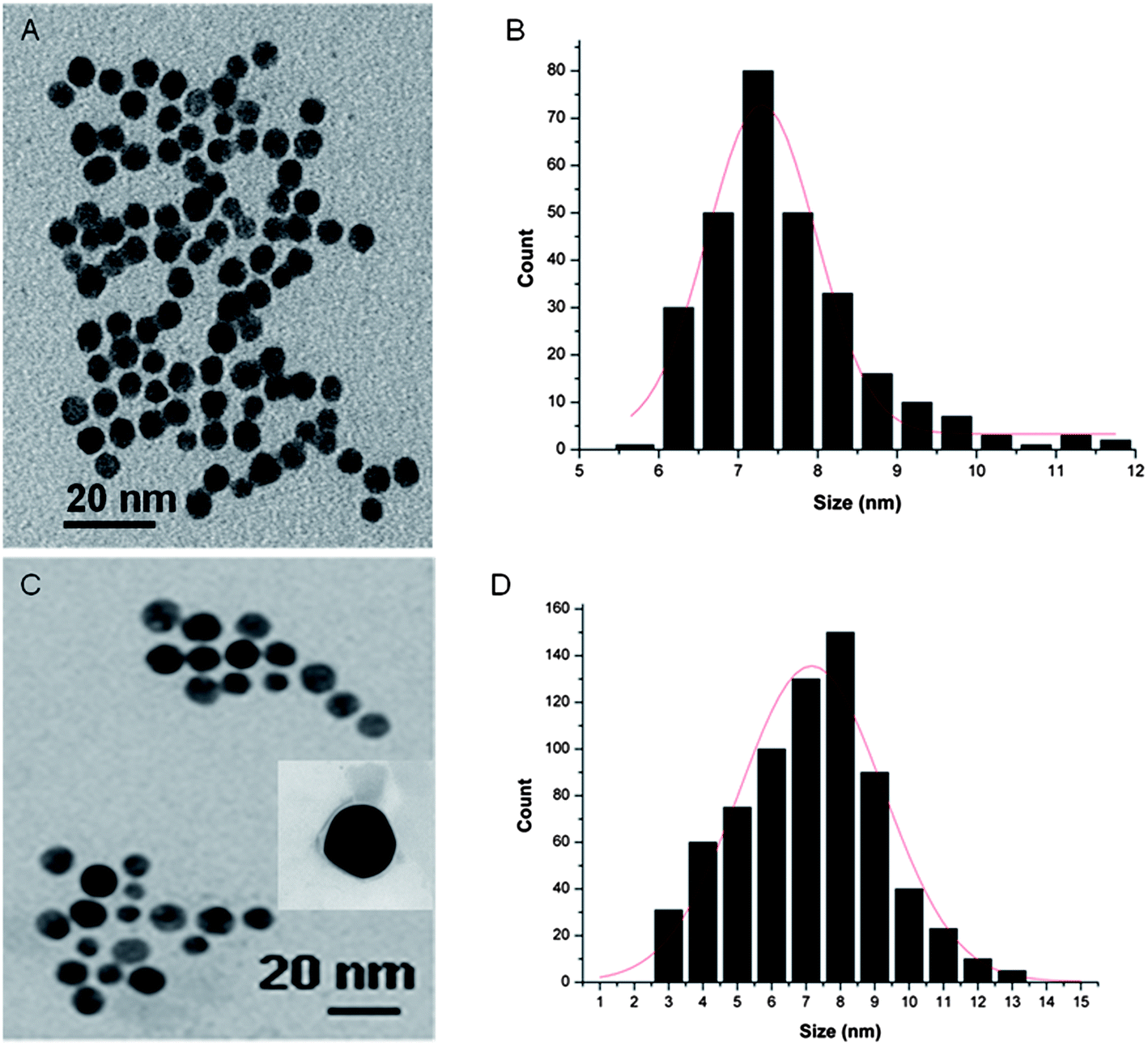 | ||
| Fig. 2 TEM images and particle size distribution of PEG-Au NPs (A and B) and PEG-Au NP-DOX (C and D). | ||
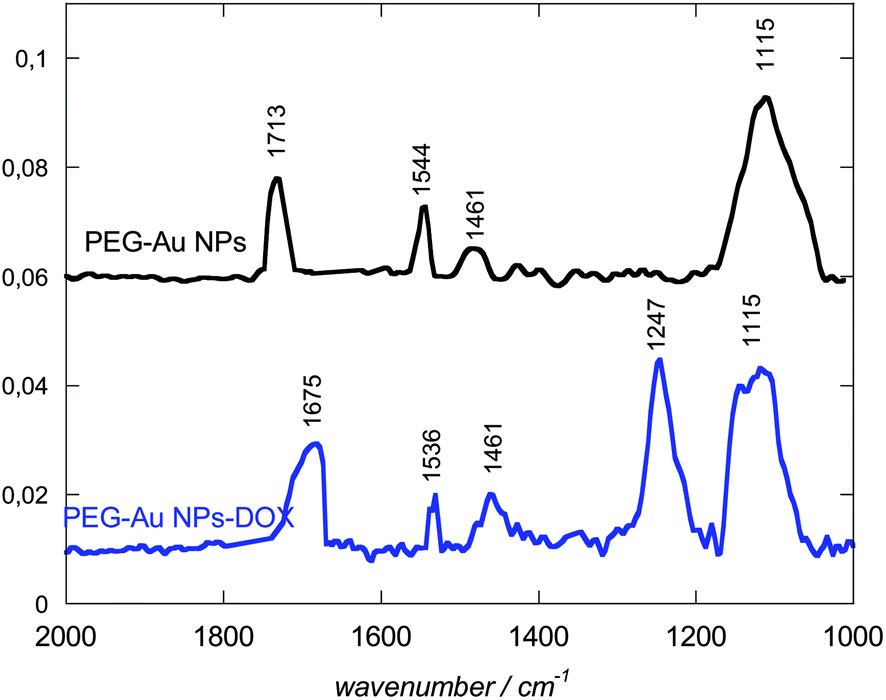 | ||
| Fig. 3 PM-IRRAS of PEG-Au NPs before (black) and after covalent conjugation with DOX (blue). | ||
The extinction spectra of a colloidal solution of PEG-Au NPs before and after DOX linking are displayed in Fig. 4. Gold nanoparticles are known to absorb at a specific wavelength of ≈520 nm, where the position as well as the absorption intensity depends on the morphology of the particles such as particle size, shape, and particle density.14 Both, PEG-capped and DOX conjugated gold particles exhibit strong absorption bands around this wavelength, being centred at 512 nm in the case of PEG-Au NPs and 528 nm for PEG-Au NP-DOX particles. The significant red shift in the LSPR signal with peak broadening is a consequence of the surface modification and is in accordance with other reports on DOX modified gold nanostructures.27–29 The UV/Vis spectra remain unaltered after storage for more than three months at room temperature suggesting the formation of stable particle suspensions.
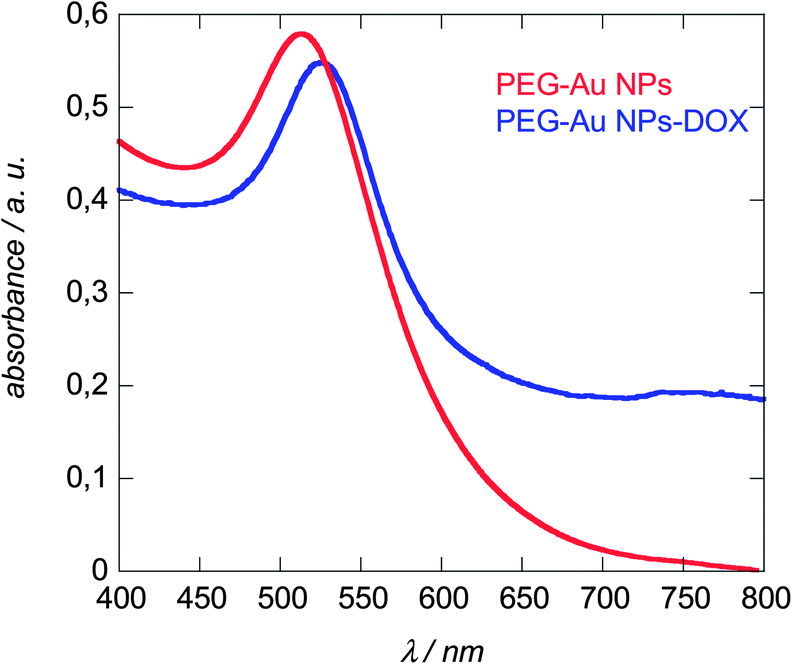 | ||
| Fig. 4 LSPR spectra of Au NPs before (red) and after linking DOX (blue) recorded in water. | ||
3.2. Anchoring DNA onto LSPR interfaces
To demonstrate the ability of DOX capped Au NPs to amplify DNA hybridization signals, DNA-modified LSPR interfaces were prepared as reported previously.8 A multilayered LSPR interface was used in this case (Fig. 5A). This interface was obtained by thermal evaporation of thin (2–4 nm) gold films on glass and post-annealing at elevated temperature.14,16 The thermal treatment results in the formation of individual spheroidal to ellipsoidal shaped particles, which are however mechanically rather frail.16 Post-coating the particles with a nanometer thick dielectric film such as 10 nm SiOx overcomes this poor adhesion of the metal islands to the substrate, resulting in stable LSPR interfaces which can be further used for sensing.30 Oligonucleotides were covalently linked to such LSR interfaces using the surface modification route as outlined in Fig. 5A. We opted for an attachment chemistry based on maleimide-modified dopamine as such dopamine derivatives prevent self-oxidation and polymerization due to the absence of free amine groups and results consequently mostly in the formation of surface monolayers.8 Immobilization of thiolated oligonucleotides (probe 1) was achieved through reaction of the maleimide function with the thiol groups of the DNA molecules (Fig. 5A). The covalent linking of the thiolated DNA stand is unambiguously confirmed by the presence of the S2p band in the XPS survey spectrum (Fig. 5B). The high resolution XPS of the S2p band shows contributions at 164.2 eV (S2p3/2) and 165.4 eV (S2p1/2), characteristic of C–S bonds and small peaks at 167.1 (S2p3/2) and 168.2 eV (S2p1/2) from some oxidized sulphur. A detailed characterization of the surface composition after modification of the LSPR interface with maleimide-terminated dopamine is reported in ref. 8. Fig. 5C displays the UV transmission spectra of the glass/Au NI/SiOx interface before and after DNA immobilization. Covalent linking of DNA results in a red shift of the plasmonic signal of the interface from 565 nm (glass/Au NI/SiOx) to 576 nm as previously reported.16,30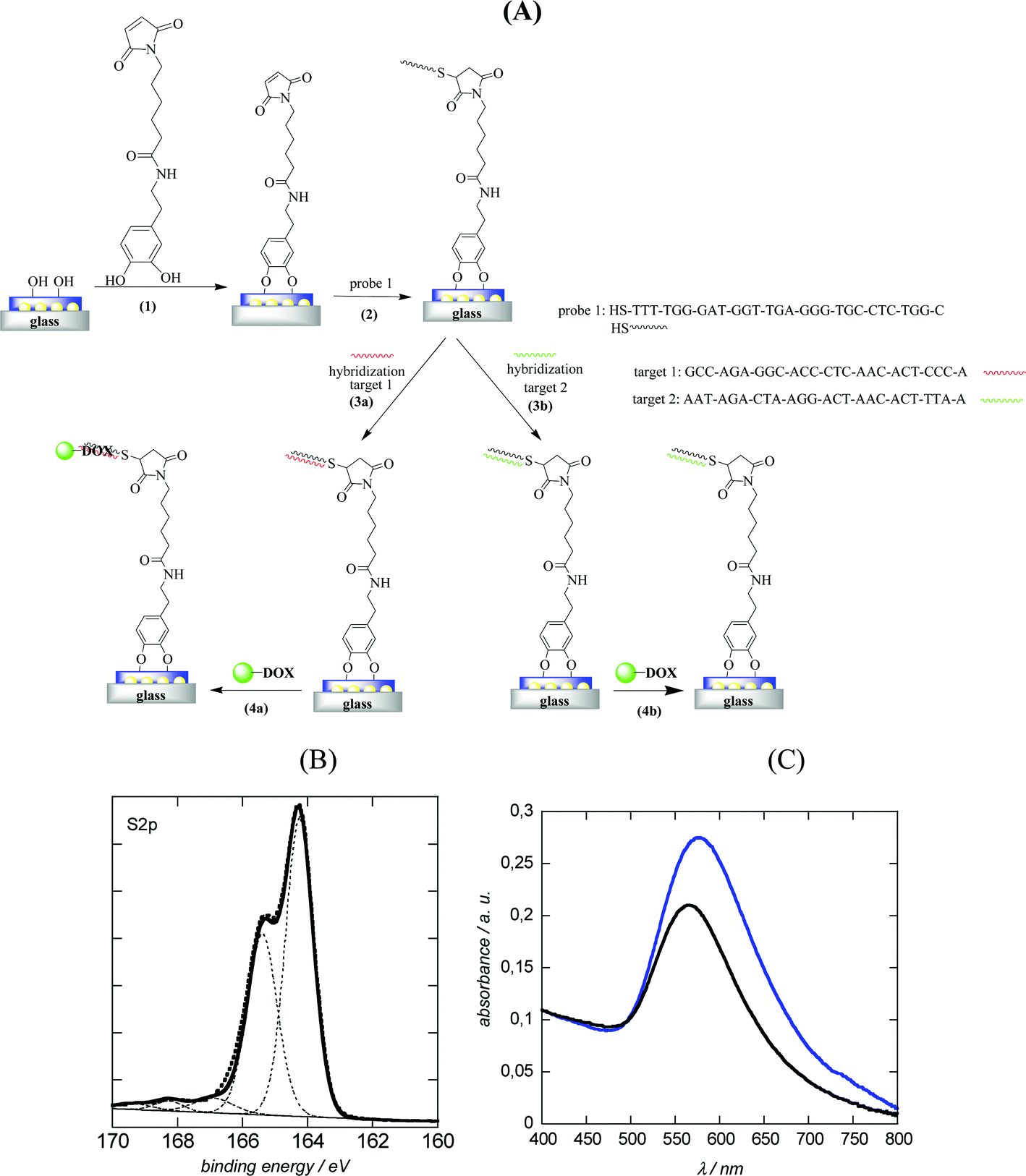 | ||
| Fig. 5 (A) Schematic illustration of the immobilization of DNA probe 1 on glass/Au NI/SiOx interfaces followed by amplification with PEG-Au NP-DOX. (1) Linking of maleimide-dopamine, (2) reaction of maleimide function with thiolated DNA molecules (probe 1), (3a) hybridization with complementary target 1, (3b) hybridization with non-complementary target 2; (4) amplification by post-incubation for 1 h with PEG-Au NP-DOX; (B) high resolution XPS spectrum of S2p of glass/Au NI/SiOx modified with thiolated DNA (probe 1); (C) LSPR spectra before (black) and after modification with probe 1 (blue). | ||
3.3. Hybridization experiments
To demonstrate the ability of the DNA-modified LSPR interface to bind specifically to complementary DNA, the LSPR interface was incubated for 1 h at 37 °C with 100 nM complementary DNA (target 1) as well as with a non-complementary one (target 2). Following the hybridization, the extinction spectrum of the LSPR sensor was again recorded and showed a red shift in λmax of 6 nm for the complementary DNA (Fig. 6A), while in the case of non-complementary DNA a negligible red shift of 1 nm was seen. The hybridized interfaces were post-treated with DOX-loaded Au NPs and a 24 nm blue shift of the plasmonic signal (when compared to λmax of the double-stranded LSPR interface) was observed (Fig. 6A). The blue shift was rather surprising as other reported hybridization events recorded by LSPR showed strongly red shifted signals.8,16 One possible reason for the blue shift of the LSPR signal might be the optical properties of DOX itself.29,31,32 Indeed DOX is a fluorescent molecule with an emission wavelength at 560 nm when excited at 470 nm. The absorption spectrum of an aqueous solution of free DOX in Fig. 6C confirms the presence of strong absorbance bands between 474 and 536 nm, the region where the LSPR interface and the PEG-Au NPs show their plasmon bands. In a control experiment, LSPR interfaces hybridized with complementary DNA were post-treated in a similar way with free DOX and PEG-Au NPs. As seen from Fig. 6B no significant change in the position of the LSPR band was observed by post-incubation in a PEG-Au NP solution. A blue shift of 6 nm and an increase in the absorption intensity was however seen in the case of free DOX. This blue shift is much smaller than that in the case of post-treatment with PEG-Au NP-DOX, but indicates that it is indeed the presence of DOX which seems to be mainly responsible for the significant blue shift observed in the experiment (Fig. 6A).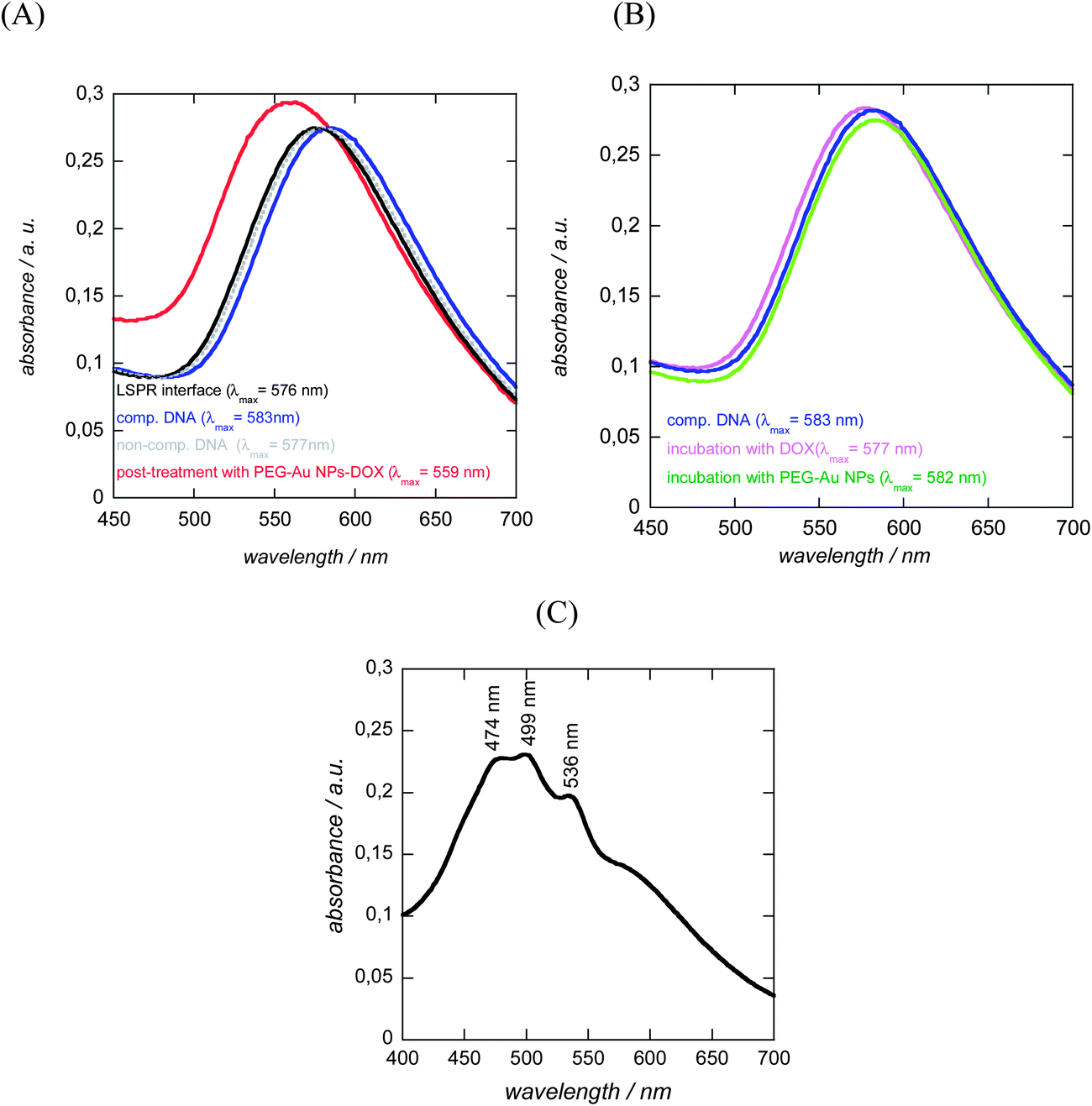 | ||
| Fig. 6 LSPR spectra of glass/Au NI/SiOx/DNA interfaces: (A) initial interface (black); and after incubation with 100 nM complementary DNA (target 1, blue), non-complementary DNA (target, grey) and after post-incubation with PEG-Au NP-DOX (red); (B) control experiments: post-incubation with free DOX (10 μg mL−1, violet) and PEG-Au NPs (green) of glass/Au NI/SiOx/DNA interfaces hybridized with complementary DNA (blue); (C) UV/Vis spectrum of an aqueous solution of DOX. | ||
To determine the dynamic range of the signal amplification, the LSPR shift upon hybridization with different concentrations of complementary DNA and post-incubation with PEG-Au NP-DOX was recorded. As seen from Fig. 7, a linear relationship between the change in the LSPR position (Δλmax) and concentration of complementary DNA is obtained. The detection limit was determined to be ≈600 pM from five blank noise signals (95% confidence level), being considerably lower than those without post-incubation, where the detection limit was reported to be ≈40 nM.8 The LOD is however higher than that recently reported using DNA-labeled gold nanostars and nanorods with LOD = 400 pM. This is most likely due to the fact that hybridization results in an overall significant red shift of the surface plasmon band, due to plasmon coupling effects between the surface-linked gold nanostructures and the different DNA-modified Au NPs in solution.
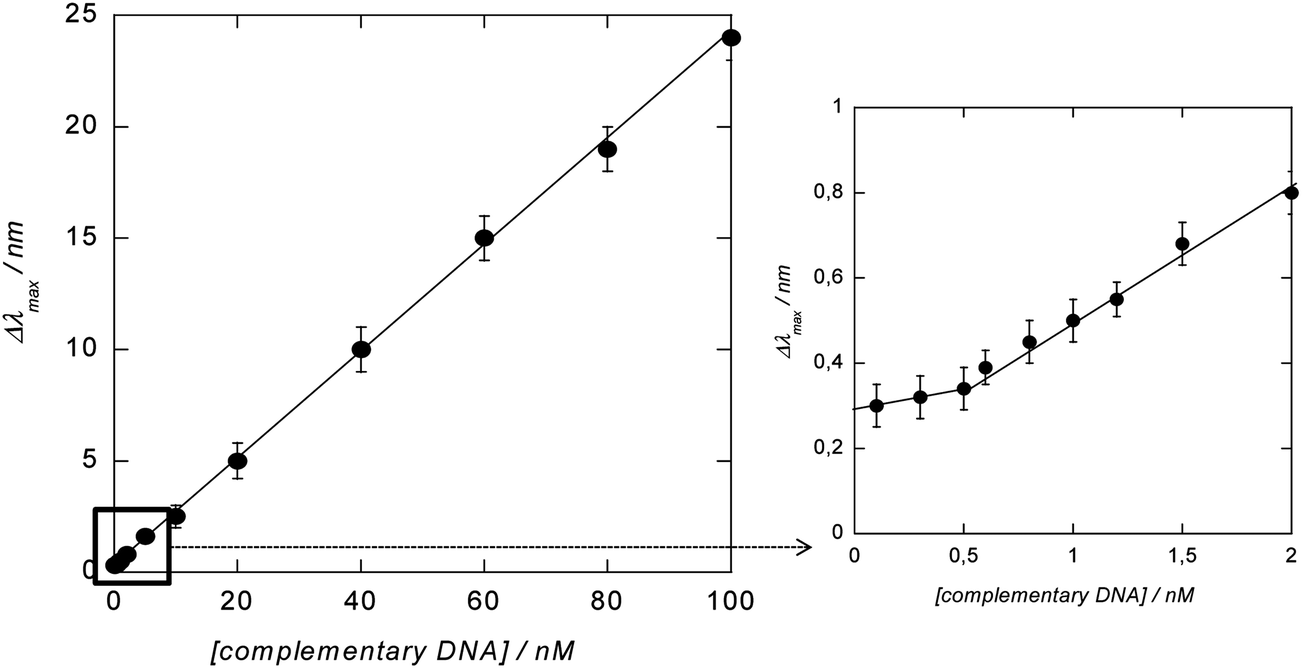 | ||
| Fig. 7 Change (Δλmax) in the position of the LSPR signal recorded on glass/Au NI/SiOx/DNA interfaces after hybridization with different concentrations of complementary DNA for 1 h at 37 °C and post-incubated with PEG-Au NP-DOX for 30 min. | ||
4. Conclusion
We demonstrated that doxorubicin-modified gold nanoparticles are efficient for the amplification of the LSPR-based wavelength shift associated with DNA hybridization events. The post-incubation of hybridized LSPR interfaces with PEG-Au NP-DOX particles allows a decrease of the detection limit to ≈600 pM. The stability of the Au NP-DOX over a long time when compared to Au NPs modified with DNA makes the concept new and an interesting alternative for LSPR based DNA sensing in an easy manner.Acknowledgements
The EU-FEDER and Interreg IV, project ‘‘Plasmobio’’, the Centre National de la Recherche Scientifique (CNRS), the Institut Universitaire de France (IUF) and the Nord-Pas-de Calais region are gratefully acknowledged for financial support.References
- J. Wang, Nucleic Acids Res., 2000, 28, 3011 CrossRef CAS PubMed.
- A. C. Pease, D. Solas, E. J. Sullivan, M. Cronn, C. P. Holmes and S. P. A. Fodor, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5022–5026 CrossRef CAS.
- P. Guedon, T. Livache, F. Martin, F. Lesbre, A. Roget, G. Bidan and Y. Levy, Anal. Chem., 2000, 72, 6003 CrossRef CAS.
- D. Meziane, A. Barras, A. Kromka, J. Houdkova, R. Boukherroub and S. Szunerits, Anal. Chem., 2012, 84, 194 CrossRef CAS PubMed.
- B. P. Ting, J. Zhang, Z. Gao and J. Y. Ying, Biosens. Bioelectron., 2009, 25, 282 CrossRef CAS PubMed.
- W. P. Hall, S. N. Ngatia and R. P. Van Dyne, J. Phys. Chem. C, 2011, 115, 1410 CAS.
- L. Touahir, E. Galopin, R. Boukherroub, A. C. Gouget-Laemmel, J.-N. Chazalviel, F. Ozanam and S. Szunerits, Biosens. Bioelectron., 2010, 25, 2579 CrossRef CAS PubMed.
- J. Spadavecchia, A. Barras, J. Lyskawa, P. Woisel, L. William, C.-M. Pradier, R. Boukherroub and S. Szunerits, Anal. Chem., 2013, 85, 3288 CrossRef CAS PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. Boeneman Gemmill, B. J. Casey, E. Oh, M. H. Stewaft and I. L. Medintz, Chem. Rev., 2013, 113, 1904 CrossRef CAS PubMed.
- H. Jang, S.-R. Ryoo, K. Kostarelos, S. W. Han and D.-H. Min, Biomaterials, 2013, 34, 3503 CrossRef CAS PubMed.
- Z. Xiao, C. Ji, J. Shi, E. M. Pridgen, J. Frider, J. Wu and O. Farokhzad, Angew. Chem., Int. Ed., 2012, 51, 11853 CrossRef CAS PubMed.
- J. B. Chaires, J. E. Herrera and M. J. Waring, Biochemistry, 1990, 29, 6145 CrossRef CAS.
- K. M. Mayer and J. H. H. Hafner, Chem. Rev., 2011, 111, 3828 CrossRef CAS PubMed.
- S. Szunerits and R. Boukherroub, Chem. Commun., 2012, 48, 8999 RSC.
- E. Galopin, L. Touahir, J. Niedziółka-Jönsson, R. Boukherroub, A.-C. Gouget-Laemmel, J.-N. Chazalviel, F. Ozanam and S. Szunerits, Biosens. Bioelectron., 2010, 25, 1199 CrossRef CAS PubMed.
- S. Szunerits, M. R. Das and R. Boukherroub, J. Phys. Chem. C, 2008, 12, 8239 Search PubMed.
- L. Touahir, R. Boukherroub, A. C. Gouget-Laemmel, J.-N. Chazalviel, J. Peretti, F. Ozanam and S. Szunerits, Analyst, 2011, 136, 1859 RSC.
- A. J. Haes, S. Zou, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. B, 2004, 108, 109 CrossRef CAS.
- M. Mazur, A. Barras, V. Kuncser, A. Galatanu, V. Zaitzev, P. Woisel, J. Lyskawa, W. Laure, A. Siriwardena, R. Boukherroub and S. Szunerits, Nanoscale, 2013, 5, 2692–2702 RSC.
- C. Li, D. Li, G. Wan, J. Xu and W. Hou, Nanoscale Res. Lett., 2011, 6, 440 CrossRef PubMed.
- J. Manson, D. Kumar, B. J. Meenan and D. Dixon, Gold Bull., 2011, 44, 99–105 CrossRef CAS PubMed.
- H. J. Parab, J.-H. Huang, T.-C. Lai, Y.-H. Jan, R.-S. Liu, J.-L. Wang, M. Hsiao, C.-H. Chen, Y.-K. Hwu, D. P. Tsai, S.-Y. Chuang and J.-H. S. Pang, Nanotechnology, 2011, 22, 395706 CrossRef PubMed.
- C. Li, D. Li, G. Wan, J. Xu and W. Hou, Nanoscale Res. Lett., 2011, 6, 440 CrossRef PubMed.
- K. G. Neoh and E. T. Kang, Polym. Chem., 2011, 2, 747 RSC.
- N. Maalouli, A. C. Gouget-Laemmel, B. Pinchemel, M. Bouazaoui, J.-N. Chazalviel, F. Ozanam, Y. Yang, P. Burkjard, R. Boukherroub and S. Szunerits, Langmuir, 2011, 27, 5498 CrossRef CAS PubMed.
- G. Das, A. Nicastri, L. Coluccio, F. Gentile, P. Candeloro, G. Cojoc, C. Liberale, F. De Angelis and E. Di Fabrizio, Microsc. Res. Tech., 2011, 5, 991 Search PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- M. Prabaharan, J. J. Grail, S. Pilla, D. A. Steebern and S. Gong, Biomaterials, 2009, 30, 6065 CrossRef CAS PubMed.
- S. Aryal, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, J. Mater. Chem., 2009, 19, 7879 RSC.
- S. Szunerits and R. Boukherroub, Langmuir, 2006, 22, 1660 CrossRef CAS PubMed.
- Y. Wang, H.-Z. Jia, K. Han, R.-X. Zhuo and X.-Z. Zhang, J. Mater. Chem. B, 2013, 1, 3344 RSC.
- F. Wang, Y.-C. Wang, S. Dou, M.-H. Xiong, T.-M. Sun and J. Wang, ACS Nano, 2011, 5, 3679 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |