Aptasensor based on the synergistic contributions of chitosan–gold nanoparticles, graphene–gold nanoparticles and multi-walled carbon nanotubes-cobalt phthalocyanine nanocomposites for kanamycin detection
Xia
Sun
b,
Falan
Li
b,
Guanghui
Shen
b,
Jiadong
Huang
*ac and
Xiangyou
Wang
*b
aCollege of Medicine and Life Sciences, University of Jinan, Jinan250022, P.R. China. E-mail: chm_huangjd@ujn.edu.cn; Fax: +86 531 82769122; Tel: +86 531 89736122
bSchool of Agriculture and Food Engineering, Shandong University of Technology, Zibo 255049, P.R. China. E-mail: wxy@sdut.edu.cn
cKey Laboratory of Chemical Sensing & Analysis in Universities of Shandong, School of Chemistry and Chemical Engineering, University of Jinan, Jinan 250022, P.R. China
First published on 14th October 2013
Abstract
An electrochemical aptasensor was developed for the detection of kanamycin based on the synergistic contributions of chitosan–gold nanoparticles (CS–AuNPs), graphene–gold nanoparticles (GR–AuNPs) and multi-walled carbon nanotubes–cobalt phthalocyanine (MWCNTs–CoPc) nanocomposites. The aptasensor was prepared by sequentially dripping CS–AuNPs, GR–AuNPs and MWCNTs–CoPc nanocomposites onto a gold electrode (GE) surface. During the above process, these nanomaterials showed a remarkable synergistic effect towards the aptasensor. CS–AuNPs, GR–AuNPs and MWCNTs–CoPc as the nanocomposites mediator improved electron relay during the entire electron transfer process and the aptasensor response speed. The electrochemical properties of the modified processes were characterized by cyclic voltammetry (CV). The morphologies of the nanocomposites were characterized by scanning electron microscopy (SEM). The experimental conditions such as the concentration of the aptamer, the time, temperature and the pH were optimized. Based on the synergistic contributions of CS–AuNPs, GR–AuNPs and MWCNTs–CoPc nanocomposites, the proposed aptasensor displayed high sensitivity, high specificity, a low detection limit (5.8 × 10−9 M) (S/N = 3) and excellent stability. It was successfully applied to the detection of kanamycin in real milk spiked samples.
1. Introduction
Kanamycin, an aminoglycoside antibiotic, is used to treat a wide variety of infections. In susceptible bacteria, the role of kanamycin is to bind the 30S subunit of the bacterial ribosome and inhibit protein synthesis.1 When it is abused in humans through the intake of food or medicinal over-prescription, it will cause serious side effects, including toxicity to the kidneys, loss of hearing and allergic reactions to the drug.2 Therefore, it is critical to develop sufficiently sensitive methods to detect kanamycin residues for food safety and clinical diagnosis. Recently, many analytical methods, such as capillary electrophoresis,3 surface plasmon resonance,4 square-wave cathodic adsorptive stripping voltammetry,5 immunoassay,6 HPLC,7 the microbiological multi-residue system8 and immunological methods,9 have been reported for the detection of kanamycin. However, most of the above-mentioned methods are often tedious, time-consuming and require a great amount of solvents, reagents and expensive apparatus. Thus, a low-cost, lower reagent consumption, highly sensitive and selective technique for kanamycin detection is very desirable.Aptamers are RNA or DNA molecules with specific 3-D structures. They can be selected through an in vitro selection process called systematic evolution of ligands by exponential enrichment (SELEX), and they are capable of recognizing and binding a variety of targets ranging from small molecules to organisms. Owing to their unique advantages of chemical recognition, accuracy, ease of modification, in vitro synthesis, high purity and chemically stability, aptamers have been used as a strong competitor to antibodies in analysis applications.10 Moreover, due to their relative ease of tailored binding affinity, modification, isolation and storage,11 aptamers exhibit several unprecedented advantages as compared with antibodies. Various aptamer-based strategies and technologies for detections have been developed, such as surface plasmon resonance (SPR),12 quartz crystal microbalance (QCM),13 electrochemical detection,14 colorimetry,15 fluorescence,16 chemiluminescence,17 dry-reagent strip18 and so on.
In addition, an enzyme is used as an electroactive label to introduce further signal amplification based on the enzymatic catalytic reaction. The commonly used enzymes that serve as labels in bioassays include horseradish peroxidase (HRP),19 glucose dehydrogenase20 and alkaline phosphatase.21 Among these enzyme labels, HRP is the most commonly used label since it is the smallest and most stable enzyme.22 Using HRP as the enzyme label, an electrochemical aptasensor for thrombin detection based on an enzymatic reaction has been developed.19 Recently, an RNA aptasensor labeled with methylene blue has been reported for the detection of kanamycin and some other aminoglycoside antibiotics in blood serum.23
To enhance the sensitivity of the electrochemical sensors, composites of nanomaterials and conducting polymers have also been used.24 As a counterpart of graphite with well-separated 2D aromatic sheets, graphene possesses a high electron mobility under ambient conditions25 and a large specific surface area with low manufacturing cost.26 Recently, graphene-based composite materials have received increasing attention due to the synergistic contribution of two or more functional components and their potential applications.27 In addition, as excellent acceptors, gold nanoparticles (AuNPs) can allow ultra-efficient adsorption of proteins as a result of their exceptional properties, such as large surface area, superior mechanical properties, excellent conductivity and large thermal conductivity.28 Many investigations have demonstrated that graphene–gold nanoparticle (GR–AuNP) nanocomposites have remarkable electrochemical properties. Our group has also reported that GR–AuNPs can improve the electrochemical response and effective surface area of the electrode.29 Moreover, some biocompatible materials such as chitosan (CS), gold nanoparticles (AuNPs) and ionic liquid have also been introduced into the modified layers to improve the stability of the enzyme.30 CS is a typical biological macromolecule, a polysaccharide copolymer of glucosamine and N-acetylglucosamine. The solubility of CS in slightly acidic solution allows the co-processing of CS with other biomolecules because its primary amines can be chemically cross-linked with other reagents. The –NH2 ligand of CS has been used to stabilize the AuNPs in the preparation process. The application of the CS–AuNPs nanocomposites has been reported and they can provide a suitable micro-environment for enzyme immobilization.31 AuNPs also assemble through electrostatic interactions (as polyion complexes) on CS with a solution of citrate-stabilized gold nanoparticles (negatively charged),32 and the nanoparticles can act as tiny conduction centers to facilitate electron transfer.33 Among the various types of nanomaterials, multi-walled carbon nanotubes (MWCNTs) possess a relatively well-characterized behavior in terms of their electron transport mechanism34,35 and surface binding phenomenon.36 Recently, the immobilization of metal phthalocyanine (MPc) at the surface of MWCNTs has been achieved by means of non-covalent adsorption.37 Moreover, MWCNTs and cobalt phthalocyanine (CoPc) (MWCNTs–CoPc) composites have been widely used to modify electrodes and the enhancement effect of the current response is significant.38
In this work, considering benefits of the CS–AuNPs, GR–AuNPs and MWCNTs–CoPc nanocomposites, we integrated them into an aptasensor to exploit the synergy contributions on the improvement of aptasensor characteristics. Kanamycin as a small molecule and an aptamer modified with different groups (Apt1: 5′-amino-TGGGGGTTGAGGCTAAGCCGAC-3′; Apt2: 5′-biotin-TGGGGGTTGAGGCTAAGCCGAC-3′)39 were used to recognize kanamycin by formation of a sandwich structure. The aptamers (AptI: 5′–NH2–(CH2)6-GGTTGGTGTGGTTGG-3′; AptII: 5′–NH2–(CH2)6-GGTTGGTGTGGTTGG-3′) have been reported to recognise different sites of thrombin.40,41 Here, the sequences of AptI and AptII were the same, and they can combine different parts of one thrombin to form a sandwich structure. Aptamers with a sandwich structure (AptA: 5′-biotin-AAAAAAAAAATACTCAGGGCACTGCAAGCAATTGTGGTCCCAATGGGCTGAGTAT-3′; AptB: 5′-amino-AAAAAAAAAATACTCAGGGCACTGCAAGCAATTGTGGTCCCAATGGGCTGAGTAT-3′) were reported to recognise different sites of the platelet-derived growth factor B-chain homodimer (PDGF-BB).42 The sequences here for AptA and AptB were also the same but modified with different groups. Mutational experiments on the kanamycin aptamer revealed that kanamycin binds to its loop region (GG sequence).39 The loop region (GG sequence) of Apt1 (5′-amino-TGGGGGTTGAGGCTAAGCCGAC-3′) and Apt2 (5′-biotin-TGGGGGTTGAGGCTAAGCCGAC-3′) have a high affinity for kanamycin. Kanamycin can bind to the loop region (GG sequence) of the aptamer. Therefore, we suspected that the same two aptamers can combine different parts of one kanamycin (Fig. 1(b) and 1(c)), and the experiments showed that our guess is correct.
 | ||
| Fig. 1 (a) Predicted secondary structure of the kanamycin aptamer; (b) the molecular structure of kanamycin; (c) sandwich structure model. | ||
To the best of our knowledge, such an aptasensor has not been reported. The proposed aptasensor has the advantages of high sensitivity and low background current. This electrochemical aptasensor is simple, rapid, sensitive and highly specific, and it was developed for the sensitive detection of kanamycin in real milk samples.
2. Experimental
2.1. Reagents and chemicals
NaH2PO4·2H2O and Na2HPO4·12H2O were purchased from Beijing Chemical Technology Co., Ltd. (Beijing, China). Graphene (GR), kanamycin sulfate, neomycin sulfate, chlortetracycline hydrochloride and gentamicin sulfate were all purchased from Jingchun Co., Ltd. (Shanghai, China). Chitosan (CS), DNA oligonucleotides modified with biotin and amino groups (Apt1: 5′-amino-TGGGGGTTGAGGCTAAGCCGAC-3′; Apt2: 5′-biotin-TGGGGGTTGAGGCTAAGCCGAC-3′) were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). The aptamer sequences specific for kanamycin, were as identified by Song et al.39via the SELEX process from a library (1 × 1015 to 1 × 1016 molecules, 100 mL) of single-stranded DNA randomized at 40 contiguous positions. Chloroauric acid (HAuCl4) and ethanol were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Cobalt phthalocyanine (CoPc) was obtained from Alfa Aesar (Ward Hill, MA). Multi-walled carbon nanotubes (MWCNTs) were purchased from Sigma-Aldrich (St. Louis, USA). All other chemicals were of analytical reagent grade. All the solutions were prepared with ultrapure water which was purified with a Milli-Q purification system (Branstead, USA).2.2. Apparatus
Electrochemical measurements were carried out using a CHI660C workstation (China). A three-electrode configuration was employed, consisting of a gold electrode (GE, ∅ = 2 mm) serving as the working electrode, a saturated calomel electrode (SCE) and a platinum wire serving as the reference and counter electrodes, respectively. The solution pH values were measured using an FE 20 Mettler-Toledo pH meter (Switzerland). Ultrasonication was performed using a KQ-100E ultrasonic cleaner (Kunshan, China). Scanning electron micrographs (SEM) were studied using a JSM-6360LV SEM (Japan). All electrochemical experiments were carried out at room temperature (RT).2.3. Preparation of CS–AuNPs nanocomposites
The CS was prepared according to ref. 43. The CS–AuNPs nanocomposites were prepared as follows: all glassware used in the preparation was thoroughly cleaned in aqua regia (3 parts HCl, 1 part HNO3), rinsed in triply ultrapure water and oven-dried prior to use. 20 mL of 1% CS (w/v) acetic acid solution was added into the stirring 200 μL of 1% HAuCl4 (w/v) aqueous solution. After stirring for about 60 min, 100 μL of KBH4 (0.4 mol L−1) was added slowly. The solution quickly turned a ruby-red color, indicating the formation of the AuNPs. After continuing with vigorous stirring of the solution for 1 h, the as-prepared nanocomposites were obtained. The resulting solution of colloidal particles was stored in a brown bottle and kept at 4 °C. The resulting nanocomposites were used for all the characterizations and experiments.2.4. Preparation of GR–AuNPs nanocomposites
The synthesis of GR–AuNPs nanomaterials consisted of the following three steps according to a previous method.44 Firstly, graphene oxide was prepared from natural flake graphite power by the modified Hummers’ method.45 1 g graphite powder was added to a big beaker containing 0.5 g sodium nitrate and 23 mL of 98% H2SO4, then the mixture were stirred for 30 min in an ice–salt bath at 0 °C. 3 g KMnO4 was slowly added into the solution at 20 °C. After being stirred for 2 h, the solution temperature was raised gradually to 35 °C in a water bath and maintained at this temperature for more than 30 min. Then 46 mL ultrapure water was added to the solution and heated to 98 °C for 15 min. After adding 140 mL ultrapure water to dilute the solution, 30% H2O2 was added into the mixture until the color of the suspension changed to brilliant yellow. The reaction mixture was filtered and washed with HCl aqueous solution and ultrapure water. Finally, the obtained solid was dried at 50 °C for 24 h and we obtained graphite oxide. Secondly, polyvinylpyrrolidone-protected graphene (PVP-protected GR) was prepared. It was prepared as follows: the prepared graphite oxide (5 mg) was then dispersed into 20 mL of ultrapure water ultrasonication for 2 h. After this the dispersed solution was sonicated until it became translucent with no visible particulate matter. Then 80 mg of polyvinylpyrrolidone (PVP) was added to the resulting dispersion with continuous stirring for 12 h. The above mixture was mixed with 7 μL of hydrazine solution (50 wt% in water) and 80 μL of ammonia solution (25 wt% in water), respectively. Then the mixture was stirred for 1 h at a temperature of 95 °C and we obtained a black dispersion. After this, it was centrifuged for 10 min and dissolved in 2.5 mL of ultrapure water. Thirdly, GR–AuNPs were synthesized by the NaBH4 reduction method: 250 μL of HAuCl4 (1 wt%) solution was added into 2.5 mL of PVP-protected GR obtained above and the mixture was adequately stirred at RT for 0.5 h. Afterwards, 12.5 μL of NaBH4 solution (0.2 mol L−1) was added into the mixture solution under stirring. After continuous stirring for 1 h at RT, the color of the solution was dark brown and we obtained GR–AuNPs.2.5. Preparation of MWCNTs–CoPc nanocomposites
The MWCNTs were purified by adopting the conditions described previously in the literature.46 MWCNTs were chemically shortened and functionalized with carboxylic acid groups on their tips. 50 mg of MWCNTs was suspended in a 40 mL mixture of concentrated sulfuric acid and nitric acid (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). After ultrasonic agitation for 4 h, the mixture was washed with ultrapure water, with centrifugation (10000 rpm) until the resulting solution was neutral. The MWCNTs–CoPc nanocomposites were formed by mixing CoPc (2 mg) in ultrapure water (1 mL) with a 2 mg mL−1 MWCNTs solution, followed by ultrasonication for 30 min.47 Then, the MWCNTs–CoPc nanocomposites were obtained.
:1, v/v). After ultrasonic agitation for 4 h, the mixture was washed with ultrapure water, with centrifugation (10000 rpm) until the resulting solution was neutral. The MWCNTs–CoPc nanocomposites were formed by mixing CoPc (2 mg) in ultrapure water (1 mL) with a 2 mg mL−1 MWCNTs solution, followed by ultrasonication for 30 min.47 Then, the MWCNTs–CoPc nanocomposites were obtained.
2.6. The fabrication of the aptasensor
Prior to modification, the gold electrodes were polished with 0.3 μm alumina powder and sonicated in ultrapure water. Then, the electrodes were cleaned in piranha solution (v/v, 7:3 H2SO4/H2O2; warning: piranha solution reacts violently with organic solvents) for 20 min, and washed with ultrapure water. Next, the electrodes were electrochemically cleaned in 0.5 M H2SO4 with potential scanning between −0.5 V and 1.5 V for 5 min, followed by washing with ultrapure water and drying under nitrogen. After being dried at RT, 6 μL of the CS–AuNPs nanocomposites film was pipetted onto the surface of the GE. Then the modified electrode (CS–AuNPs/GE) was dried at RT, before 6 μL of the GR–AuNPs nanocomposites film was cast onto the pre-treated substrate and dried over 2 h at RT. Finally, 6 μL of the MWCNTs–CoPc was added dropwise onto the above-modified electrode. Then the MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE was obtained. The aptamer (Apt1) was assembled on a modified GE surface, while the other aptamer (Apt2) was modified with biotin at the 5′-end, which could be further labeled with streptavidin–horseradish peroxidase (SA–HRP). Following that, 6 μL of the aminated kanamycin Apt1 was added dropwise onto the MWCNTs–CoPc/GR–AuNPs/CS–AuNPs-modified electrode. The electrode was incubated with 0.5% BSA at RT for 0.5 h in order to block possible remaining active sites and avoid non-specific adsorption. Finally, 6 μL of the prepared Kana–SA–HRP–Apt2 mixture was added dropwise onto the modified electrode surface and incubated for 1 h. The resulting aptasensor was stored in 0.1 M PBS (pH 7.5) at 4 °C when not in use. After every adsorption step, the modified electrode was thoroughly rinsed with ultrapure water and dried with nitrogen. The stepwise preparation of the aptasensor was shown in Scheme 1.
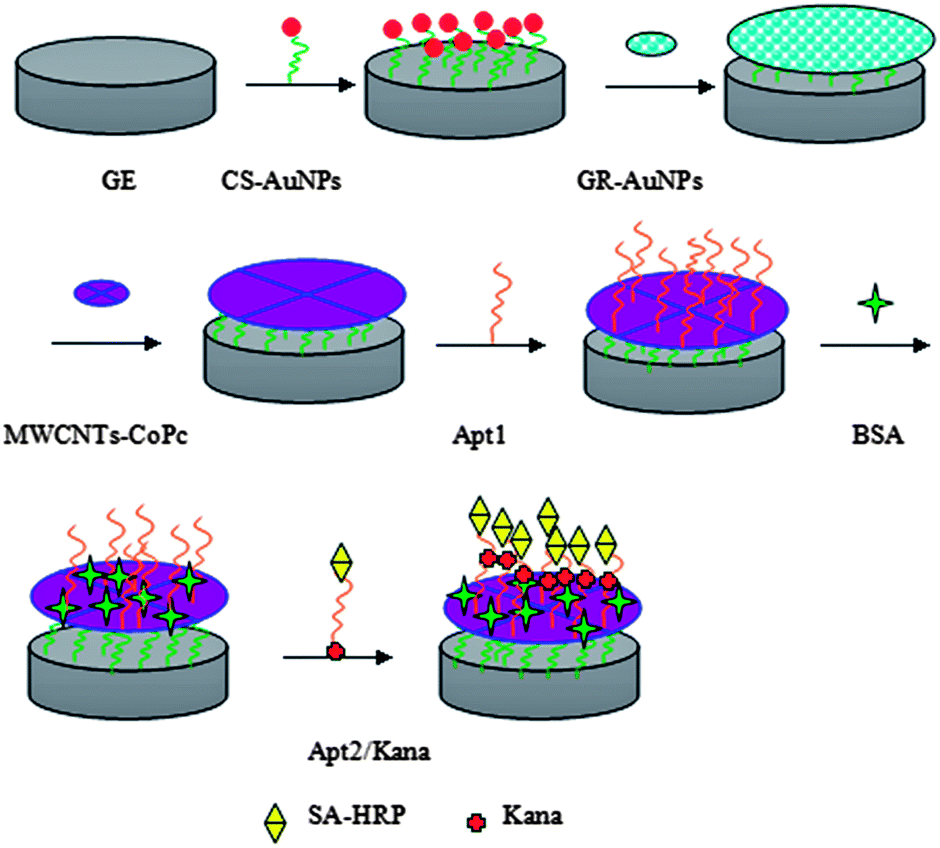 | ||
| Scheme 1 Stepwise preparation of the aptasensor. | ||
2.7. Electrochemical measurements
Electrochemical measurements were carried out in a conventional electrochemical cell at 37 °C. Cyclic voltammetry (CV) was performed in 0.01 M PBS (pH 7.4) containing 0.1 M KCl from −0.2 to 0.6 V at a scan rate of 100 mV s−1. The electrochemical differential pulse voltammetry (DPV) measurements were carried out under the following conditions: The voltage was scanned from −0.05 V to 0.4 V with a pulse height of 50 mV, the step height and the frequency were kept as 4 mV and 15 Hz, respectively. DPV measurements of the modified GE were carried out in PBS containing 1 mM hydroquinone (HQ) and 1 mM H2O2. Upon binding with kanamycin, the HRP-labeled aptamer–kanamycin complex on the electrode would increase the reduction current of HQ in the presence of H2O2. The sensitivity and the specificity of the proposed electrochemical aptasensor were investigated by DPV.3. Results and discussion
3.1. SEM characterization of modified electrodes
The morphologies and microstructures of the as-prepared different films were studied by SEM observation (Fig. 2). When the first CS–AuNPs layer was assembled, gold nanoclusters were deposited onto the multilayer film, and many different diameter clusters were observed (Fig. 2(a)). Fig. 2(b) presents the SEM image of the GR–AuNPs nanocomposites. The SEM view of the GR–AuNPs clearly illustrates a number of bright dots positioned over the entire surface, particularly at the folded edges and wrinkle sites, suggesting that the AuNPs were indeed linked to the surface of the GR sheets. As shown in Fig. 2(c), the SEM image revealed that the MWCNTs were well distributed on the surface of the GE with the formation of small bundles (tubes). Fig. 2(d) presents the SEM image of the GR–AuNPs nanocomposites immobilized on the CS–AuNPs/GE, which is familiar to the SEM view of the GR–AuNPs. As shown in Fig. 2(e), the SEM image reveals that this view iss similar to the SEM of MWCNTs–CoPc, when the MWCNTs–CoPc was immobilized on the GR–AuNPs/CS–AuNPs/GE. Therefore it could be concluded that these nanocomposites had been successfully dispersed on the surface of the GE to form nanocomposite films. | ||
| Fig. 2 SEM images of (a) CS–AuNPs/GE; (b) GR–AuNPs/GE; (c) MWCNTs–CoPc/GE; (d) GR–AuNPs/CS–AuNPs/GE; (e) MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE. | ||
3.2. Fabrication mechanism of the sensor
In this paper, CV was used in the study to investigate the processes of modification of the electrodes. The cyclic voltammograms of differently modified electrodes are presented in Fig. 3A. It can be seen that the bare GE has an obvious redox peak (Fig. 3A(a)). After the CS–AuNPs (Fig. 3 A(b)) are immobilized on the bare GE, a larger current response was exhibited. The reason for this might be that AuNPs can effectively increase the surface area and the current response of the electrode. With the addition of 6 μL GR–AuNPs on the CS–AuNPs/GE, the GR–AuNPs/CS–AuNPs/GE exhibited a much higher current (Fig. 3A(c)) response due to the synergistic effects of the good electrical conductivity of the GR and AuNPs. After MWCNTs–CoPc (Fig. 3A(d)) was immobilized on the GR–AuNPs/CS–AuNPs/GE, a larger current response was exhibited. It was found that the sensitivity of the aptasensor was more 6-fold better than that for the CS–AuNPs (Fig. 3B), GR–AuNPs and MWCNTs–CoPc used alone (after the CS–AuNPs was immobilized alone on the bare GE, the current only increased 12.5 μA. After the GR–AuNPs was immobilized alone on the bare GE, the current only increased 18.3 μA. When the MWCNTs–CoPc was immobilized alone on the bare GE, the current only increased 8.1 μA. After the MWCNTs–CoPc composite was immobilized on the GR–AuNPs/CS–AuNPs/GE, the current increased 231.3 μA.). This could be ascribed to the improvement in conductivity between the graphene nanosheets in the GR–AuNPs due to the introduction of the CS–AuNPs nanocomposites. Fast charge transfer was obtained from MWCNTs–CoPc to the graphene sheets and CS–AuNPs due to the unique electrochemical properties of the MWCNTs–CoPc. When Apt1 (Fig. 3A(e)) and BSA (Fig. 3A(f)) were modified on the MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE surface, the peak current decreased in sequence, which suggested the aptamer and BSA severely reduced the effective area and active sites for electron transfer, and blocked non-specific sites. Upon self-assembly of Kana–SA–HRP–Apt2 on the BSA/Apt1/MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE the voltammetric characteristics of HQ at the Kana–SA–HRP–Apt2/BSA/Apt1/MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE changed dramatically (Fig. 3A(g)). This result demonstrated that SA–HRP was successfully labelled to the aptamer complex and maintained its electrocatalytic activity to H2O2 since the increased reduction current at Kana–SA–HRP–Apt2/BSA/Apt1 was due to the catalytic effect of HRP. HQ serves as an effective mediator of shuttling electrons between the electrode surface and the redox center of HRP. As shown in the following mechanism, H2O2 in the solution is firstly reduced by the immobilized HRP(Red). Then the HRP can be regenerated with the aid of the mediator, while the mediator HQ can be oxidized to BQ in the enzymatic reaction, leading to the disappearance of the HQ oxidation peak. Finally, the BQ can be reduced back to HQ by receiving electrons from the electrode:| H2O2 + HRP(Red) → HRP(Ox) + H2O |
| HRP(Ox) + HQ → BQ + HRP(Red) |
| BQ + 2H+ + 2e−→ HQ |
 | ||
| Fig. 3 (A) Cyclic voltammograms of: (a) the bare GE; (b) CS–AuNPs/GE; (c) GR–AuNPs/CS–AuNPs/GE; (d) MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE; (e) Apt1/MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE; (f) BSA/Apt1/MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE; and (g) Kana–SA–HRP–Apt2/BSA/Apt1/MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE. (B) Cyclic voltammograms of: (a) the bare GE; (b) CS–AuNPs/GE; (c) GR–AuNPs/GE; (d) MWCNTs–CoPc/GE; and (e) MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE. | ||
3.3. Optimization of the analytical parameters
The experimental parameters were optimized in terms of aptamer concentration, incubation time, temperature and pH. The optimum concentration of the capturing Apt1 was determined by drop coating with different concentrations (0.5, 1.0, 2.0, 5.0, 10.0, 15.0, 20.0 μM) of the aptamer onto the activated surface of the MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE. The aptamer-modified probe was treated with 10 μM kanamycin in 0.1 M PBS (pH 7.4). Then, DPVs were recorded for different concentrations of the aptamer-modified sensors. The effect of aptamer concentration on the response is shown in Fig. 4(a). The current response increased with increasing aptamer concentration. However, there was no significant increase in the response that was observed for an aptamer concentration of above 5.0 μM, due to the saturation of the active sites for immobilizing the aptamer. Thus, an aptamer concentration of 5.0 μM was used for sensor fabrication.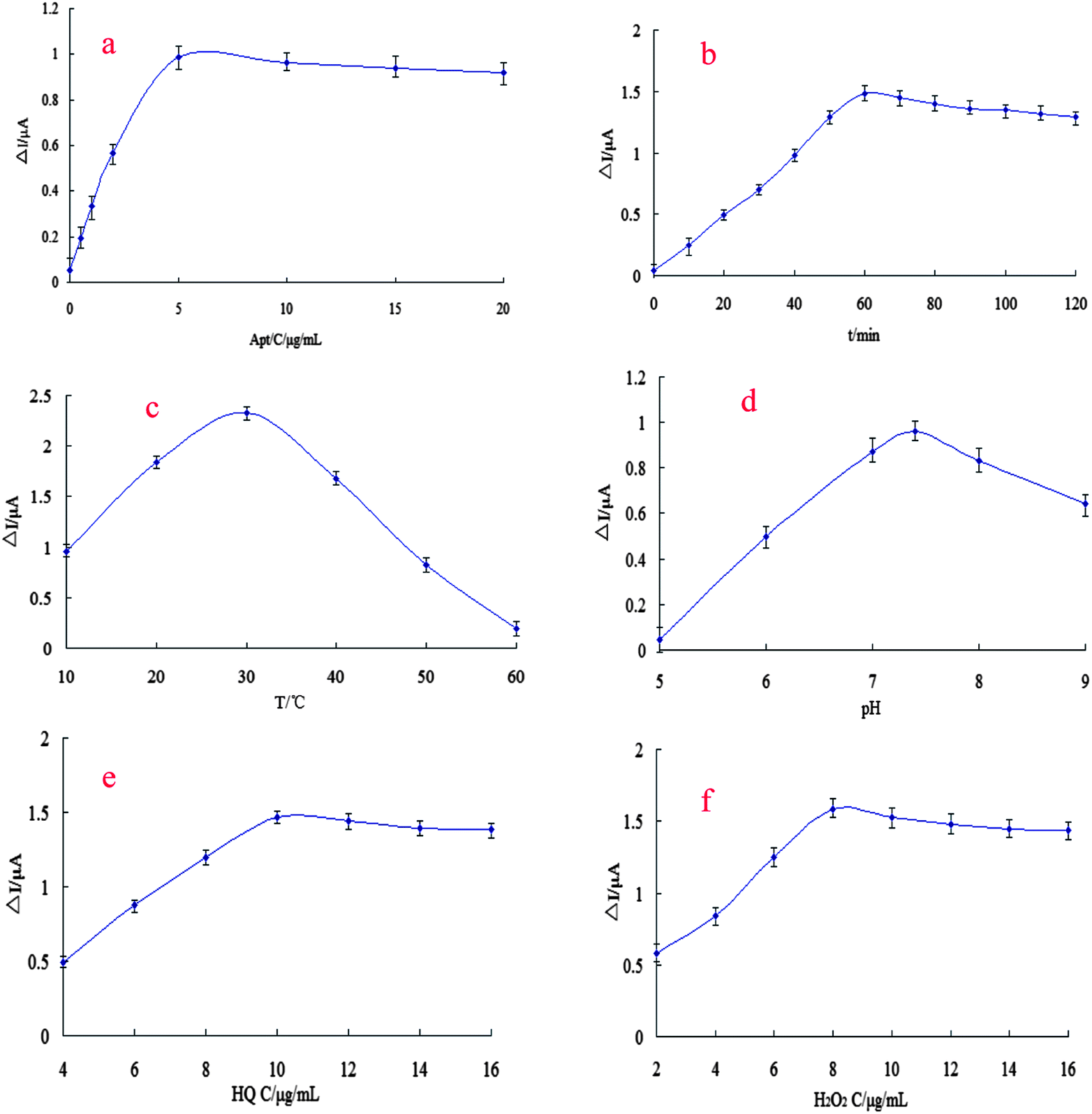 | ||
| Fig. 4 The optimization of experimental parameters. The influence of: (a) aptamer concentration, (b) the incubation time, (c) the working temperature, (d) the working pH, (e) HQ concentration, (f) H2O2 concentration. | ||
To investigate the effect of incubation time with respect to kanamycin uptake, the Apt/MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE was immersed in 0.1 M PBS (pH 7.4) containing 10 μM kanamycin for different time intervals (0–120 min) and DPVs were recorded for the captured kanamycin. The relationship between the sensor response and the incubation time is shown in Fig. 4(b). The current response increased with the increasing incubation time and reached a maximum at 60 min. However, there was no significant increase in the peak current observed after 60 min due to the saturation of the active sites for kanamycin binding. Thus, 60 min was selected as the adequate incubation time for the Apt/MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE to interact with kanamycin.
Further, the effect of incubation temperature was investigated for temperatures ranging from 10 to 60 °C. The signal responses of the assay to the different incubation temperatures are shown in Fig. 4(c). The peak current increased significantly as the temperature increased from 10 to 30 °C and then suddenly decreased when over 30 °C. The decrease of the sensor response over 30 °C might be due to the partial denaturation of the aptamer structure at the elevated temperatures. Taking the stability of the aptamer into consideration, 30 °C was selected as the optimal temperature.
In addition, the effect of pH on the sensor response was examined by recording DPVs for the kanamycin-captured electrode in 0.1 M PBS. The current increased as the medium pH was increased from 5.0 to 9.0 and then decreased when the pH value was higher than 7.4 (Fig. 4(d)). The maximum current was observed at a pH of 7.4. Therefore, all subsequent experiments were performed in 0.1 M PBS at pH 7.4.
Detection parameters, such as the H2O2 concentration and HQ concentration could also affect the detection sensitivity of the assay. A series of optimizations for these parameters was performed to improve the sensitivity of the aptasensor. As shown in Fig. 4(e), the peak current of the proposed aptasensor increased until 10 mM HQ. The current responses reached a saturation value for HQ at over 10 mM. As shown in Fig. 3(f), the current response increased with the increase of H2O2 concentration and leveled off to a saturation value at 8 mM of H2O2 when the concentration of HQ was 10 mM. Therefore, pH 7.4, 10 mM HQ and 8 mM H2O2 were chosen as the optimized experimental conditions of the detection system.
3.4. Calibration curve
The calibration plots for kanamycin detection with the prepared aptasensor under the optimal experimental conditions are shown in Fig. 5. A gradual increase in current was observed with increasing kanamycin concentration and the corresponding calibration curve exhibited good linearity. The changes of the oxidation peak current response (ΔI) of the aptasensor were found to be proportional to the kanamycin concentration in the linear range from 10.0 to 150.0 ng mL−1, with a detection limit of 5.8 nM (S/N = 3). The linear slope was 0.5428 and the correlation coefficient was 0.9987. Compared with other reported colorimetric methods, the proposed aptasensor exhibited a higher sensitivity and a lower detection limit (Table 1).39,48 The results of this comparison are shown in Table 1.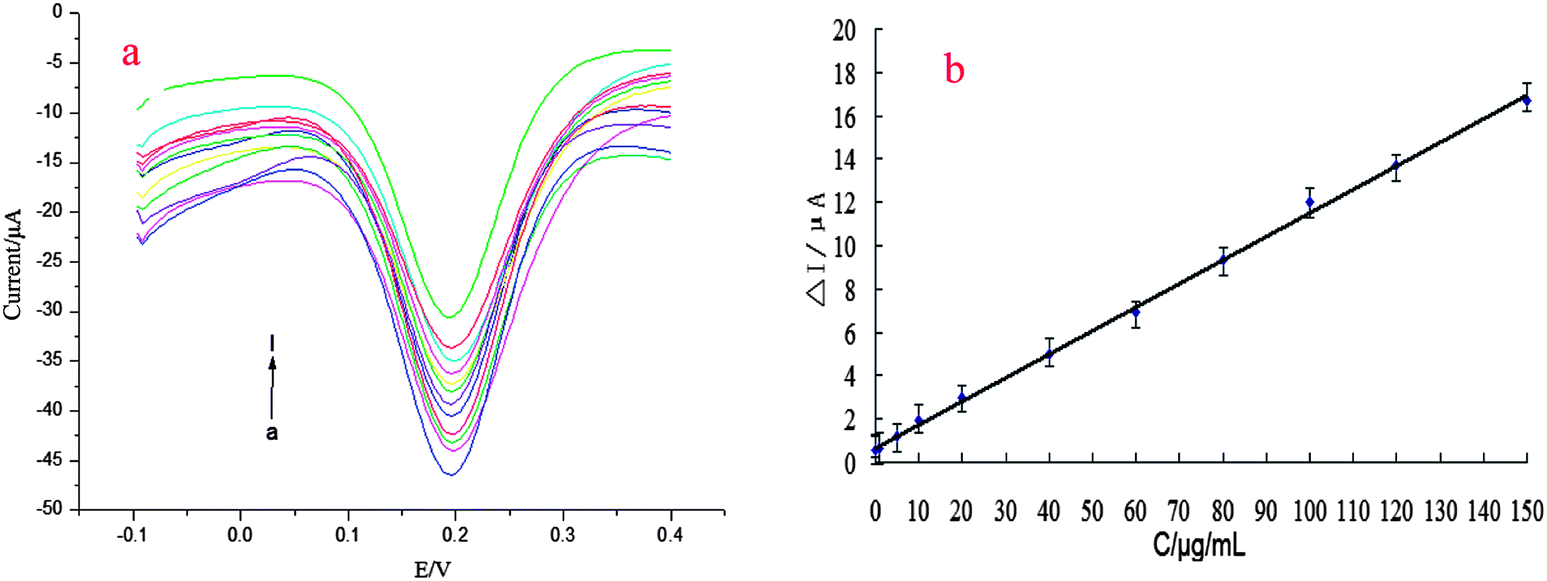 | ||
| Fig. 5 (a,b) Calibration curve of the aptasensor. | ||
| Method of detection | Limit of detection/M | Linear range/M | Ref. |
|---|---|---|---|
| Fluorescence detection | 8.95 × 10−9 to 2.56 × 10−8 | 2.7 × 10−8 to 6.7 × 10−5 | Yu et al., 200949 |
| ELISA | 4.15 × 10−9 | — | Watanabe et al., 199948 |
| ELISA | 2.1 × 10−8 | — | Loomans et al., 20036 |
| Colorimetric detection | 2.5 × 10−8 | — | Song et al., 201139 |
| Aptasensor detection | 9.4 ± 0.4 × 10−9 | 5 × 10−8 to 9 × 10−6 | Zhu et al., 201247 |
| Aptasensor detection | 5.8 × 10−9 | 1 × 10−8 to 1.5 × 10−7 | This work |
3.5. Selectivity, reproducibility, repeatability and stability of the aptasensor
Selectivity is an important property of the aptasensor. It was evaluated by measuring the DPV response. We introduced five kanamycin analogues such as oxytetracycline (QTC), neomycin sulfate (NEO), chloromycetin (CHI), chlortetracycline (CTC) and gentamicin sulfate (GM) to investigate the selectivity performance of the sensor. The Apt/MWCNTs–CoPc/GR–AuNPs/CS–AuNPs/GE was incubated in 0.1 M PBS (pH 7.4) containing 10.0 mM kanamycin and 100 mM interfering antibiotics for 60 min. After washing the electrode with PBS, further experiments were conducted in 0.1 M PBS (pH 7.4) using DPV. The current responses for kanamycin in the absence and presence of other antibiotics were obtained (Fig. 6A(a)). It was observed that in the presence of these interferents, the change in current was negligible, indicating that the selectivity of the developed aptasensor for kanamycin was good. To further confirm the selectivity of this assay, the cross-reactivity of other kanamycin analogues was tested. The experimental results are shown in Fig. 6A(b). As evident from Fig. 6A(b), kanamycin (10 mmol L−1) showed a much stronger current response, while almost negligible electrochemical changes were detected for any of the kanamycin analogues (100 mmol L−1) tested in its mixture. Because the current response was induced by the specific interaction of the aptamer and the target molecule, the results showed that these aptamers could only recognize kanamycin and could not combine with the kanamycin analogues. All of the above results confirmed that there was insignificant cross-reactivity for other kanamycin analogues and the developed aptasensor could be used to determinate kanamycin with high specificity. Therefore, the corresponding current responses of kanamycin analogues were almost negligible.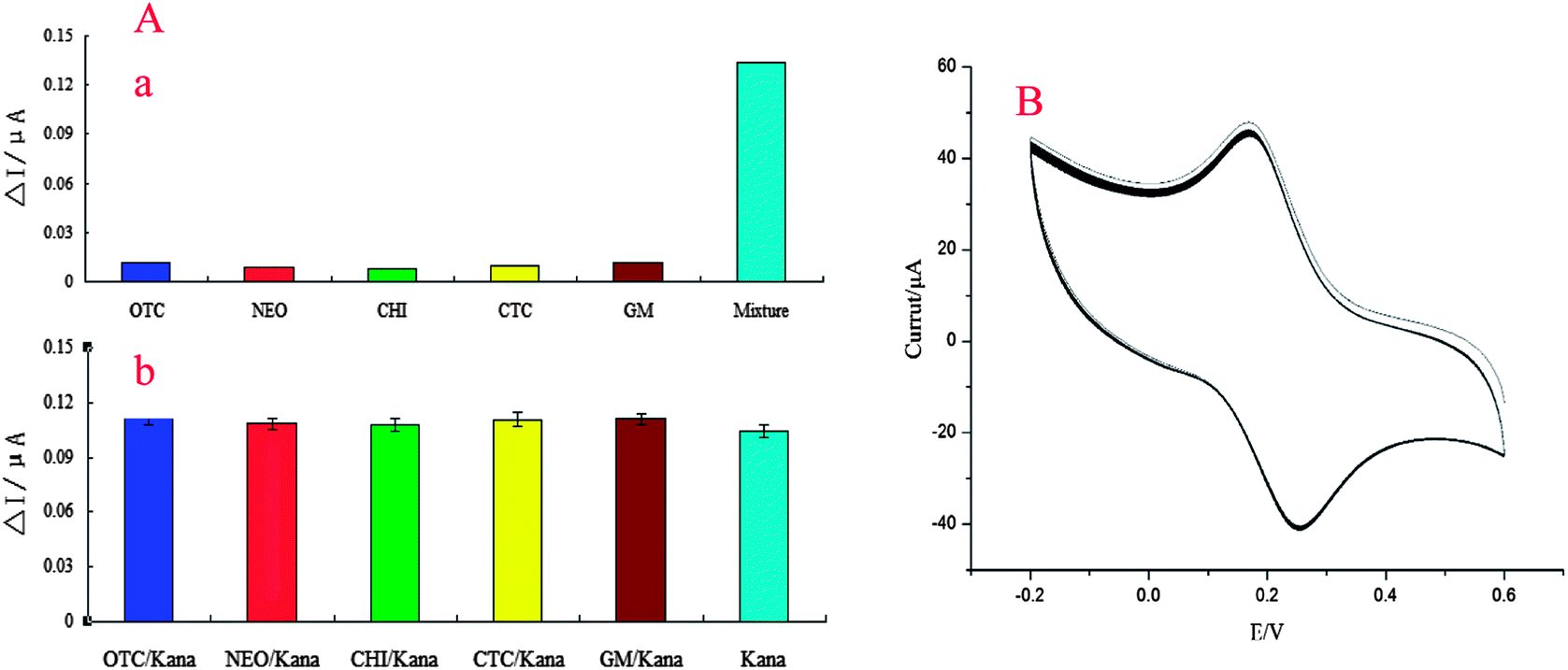 | ||
| Fig. 6 (A) Specificity of the aptasensor; and (B) stability analysis of the aptasensor. | ||
The reproducibility of the aptasensor was conducted by determining 10 μmol L−1 kanamycin solution with six aptasensors which were fabricated following the same procedure. The relative standard deviation (RSD) of 3.8% was obtained, which indicated that the aptasensor had good reproducibility.
To investigate the repeatability of the aptasensor, five aptasensors fabricated independently under the same conditions were examined. After using the five aptasensors for 3 times continuously, the RSD of 4.2% was observed. This indicated that the aptasensor had good repeatability.
Stability is a key parameter for the application and development of the sensor. Under optimal conditions, the aptasensor was measured by CV for a 60-cycle successive scan, and a 2.63% deviation of the initial response was observed (Fig. 6B). This result indicated that the proposed aptasensor had high stability. To investigate further the stability of the sensor, we prepared four aptasensors and stored them for 15 days at 4 °C. Then we used them to detect the 10 μmol L−1 kanamycin. The current response decreased by about 7%, which suggested that the developed aptasensor possessed good stability.
3.6. Determination of kanamycin in real samples
Although the proposed sensor showed good selectivity towards kanamycin, it is worth exploring the analytical utility of the sensor for practical application. The detection of kanamycin in milk is of considerable interest, since milk is one of the most heavily regulated products in the food industry because of the risk of it containing veterinary medicine residues. The milk samples used in this study were all purchased from a supermarket in China. The milk sample was firstly filtered through a sterile Millipore™ membrane (0.20 μm)49 and diluted five times with PBS. Further, kanamycin standard solution was spiked into the diluted milk, making the final concentrations of 0.05, 0.1, 0.5, 1.0, 2.0 μM kanamycin, and then experiments were carried out according to the aforementioned optimized conditions for kanamycin detection with the developed aptamer sensor. The kanamycin concentration recoveries were between 97.18 and 103.1% (Table 2), which clearly indicated that the aptasensor was suitable for the detection of kanamycin in real milk samples.| Milk found/μM | Added/μM | Total found/μM | Recovery (%) |
|---|---|---|---|
| Not detected | 0.05 | 0.0512 | 102.4 |
| Not detected | 0.1 | 0.0987 | 98.7 |
| Not detected | 0.5 | 0.4972 | 99.44 |
| Not detected | 1 | 1.1031 | 103.1 |
| Not detected | 2 | 1.9435 | 97.18 |
4. Conclusions
In this paper, we have developed an ultrasensitive and highly specific electrochemical aptasensor for kanamycin detection based on the aptamer/target/aptamer configuration. A detection limit down to 5.8 × 10−9 M for kanamycin has been achieved by the reaction of H2O2 and HQ catalyzed by HRP. This could be ascribed to improvement of the conductivity between the graphene nanosheets in the GR–AuNPs due to introduction of the AuNPs, fast charge transfer from the MWCNTs–CoPc composite to the graphene sheets and AuNPs due to unique electrochemical properties of the MWCNTs–CoPc, and the good biocompatibility of the AuNPs for HRP. The aptasensor possessed high sensitivity, good reproducibility and was cost-effective. In addition, the proposed sensor was successfully applied for kanamycin detection in milk samples. It could be a promising tool for use in food analysis and clinical diagnosis.Acknowledgements
This work was supported by the National Natural Science Foundation of the People's Republic of China (no. 31171700 and 31101296), the National High Technology Research and Development Program of China (National 863 Program of China) (no. 2012AA101604), the Natural Science Foundation of Shandong Province (no. ZR2010DQ025) and the Shandong Province Higher Educational Science and Technology Program (no. J10LB14).References
- J. Wirmer and E. Westhof, Methods Enzymol., 2006, 415, 180–202 CAS.
- R. Oertel, V. Neumeister and W. Kirch, J. Chromatogr., A, 2004, 1058, 197–201 CAS.
- E. Kaale, A. V. Schepdael, E. Roets and J. Hoogmartens, Electrophoresis, 2003, 24, 1119–1125 CrossRef CAS PubMed.
- M. Frasconi, R. Tel-Vered, M. Riskin and I. Willner, Anal. Chem., 2010, 82, 2512–2519 CrossRef CAS PubMed.
- J. L. Yan, Russ. J. Electrochem., 2008, 44, 1334–1338 CrossRef CAS.
- E. M. G. Loomans, J. Wiltenburg, M. Koets and A. Amerongen, J. Agric. Food Chem., 2003, 51, 587–593 CrossRef CAS PubMed.
- Y. X. Zhou, W. J. Yang, L. Y. Zhang and Z. Y. Wang, J. Liq. Chromatogr. Relat. Technol., 2007, 30, 1603–1615 CrossRef CAS.
- R. Althaus, M. I. Berruga, A. Montero, M. Roca and M. P. Molina, Anal. Chim. Acta, 2009, 632, 156–162 CrossRef CAS PubMed.
- D. J. Berry, J. Chromatogr., 1987, 385, 337–341 CrossRef CAS.
- S. D. Jayasena, Clin. Chem., 1999, 45, 1628–1650 CAS.
- J. A. Hansen, J. Wang, A. N. Kawde, Y. Xiang, K. V. Gothelf and G. Collins, J. Am. Chem. Soc., 2006, 128, 2228–2229 CrossRef CAS PubMed.
- J. Wang, A. Munir, Z. Li and H. S. Zhou, Biosens. Bioelectron., 2009, 25, 124–129 CrossRef CAS PubMed.
- M. Liss, B. Petersen, H. Wolf and E. Prohaska, Anal. Chem., 2002, 74, 4488–4495 CrossRef CAS.
- D. Xu, X. Yu, Z. Liu, W. He and Z. Ma, Anal. Chem., 2005, 77, 5107–5113 CrossRef CAS PubMed.
- H. Z. Huang and X. R. Yang, Colloids Surf., A, 2003, 226, 77–86 CrossRef CAS.
- V. Pavlov, B. Shlyahovsky and I. Willner, J. Am. Chem. Soc., 2005, 127, 6522–6523 CrossRef CAS PubMed.
- X. Yan, Z. Cao, M. Kai and J. Lu, Talanta, 2009, 79, 383–387 CrossRef CAS PubMed.
- H. Xu, X. Mao, Q. Zeng, S. Wang, A. N. Kawde and G. Liu, Anal. Chem., 2008, 81, 669–675 CrossRef PubMed.
- M. Mir, M. Vreeke and I. Katakis, Electrochem. Commun., 2006, 8, 505–511 CrossRef CAS PubMed.
- K. Ikebukuro, C. Kiyohara and K. Sode, Biosens. Bioelectron., 2005, 20, 2168–2172 CrossRef CAS PubMed.
- S. Centi, S. Tombelli, M. Minunni and M. Mascini, Anal. Chem., 2007, 79, 1466–1473 CrossRef CAS PubMed.
- A. Ambrosi, F. Airò and A. Merkoçi, Anal. Chem., 2009, 82, 1151–1156 CrossRef PubMed.
- A. A. Rowe, E. A. Miller and K. W. Plaxco, Anal. Chem., 2010, 82, 7090–7095 CrossRef CAS PubMed.
- Y. Zhu, J. I. Son and Y. B. Shim, Biosens. Bioelectron., 2010, 26, 1002–1008 CrossRef CAS PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS PubMed.
- C. Wang, L. Zhang and Z. H. Guo, Microchim. Acta, 2010, 169, 1–6 CrossRef CAS.
- T. T. Baby, S. S. J. Aravind, T. Arockiadoss, R. B. Rakhi and S. Ramaprabhu, Sens. Actuators, B, 2010, 145, 71–77 CrossRef CAS PubMed.
- J. M. Gong, T. Zhou, D. D. Song and L. Z. Zhang, Sens. Actuators, B, 2010, 150, 491–497 CrossRef CAS PubMed.
- W. J. Lian, S. Liu, J. H. Yu, X. R. Xing, J. Li, M. Cui and J. D. Huang, Biosens. Bioelectron., 2012, 38, 163–169 CrossRef CAS PubMed.
- K. F. Zhou, Y. H. Zhu, X. L. Yang, J. Luo, C. Z. Li and S. R. Luan, Electrochim. Acta, 2010, 55, 3055–3060 CrossRef CAS PubMed.
- Q. Xu, C. Mao, N. N. Liu, J. J. Zhu and J. Sheng, Biosens. Bioelectron., 2006, 22, 768–773 CrossRef CAS PubMed.
- C. C. Huang, Y. F. Huang, Z. Cao, W. Tan and H. T. Chang, Anal. Chem., 2005, 77, 5735–5741 CrossRef CAS PubMed.
- E. E. Ferapontova, V. G. Grigorenko, A. M. Egorov, T. Borchers, T. Ruzgas and L. Gorton, Biosens. Bioelectron., 2001, 16, 147–157 CrossRef CAS.
- D. Tomanek, Curr. Appl. Phys., 2002, 2, 47–49 CrossRef.
- S. Frank, P. Poncharal and Z. L. Wang, Science, 1998, 280, 1744–1746 CrossRef CAS.
- Y. Lin, S. Taylor, H. Li, K. A. S. Fernando, L. Qu, W. Wang, L. Gu, B. Zhou and Y. P. Sun, J. Mater. Chem., 2004, 14, 527–541 RSC.
- H. S. Yin, Y. L. Zhou, J. Xu, S. Y. Ai, L. Cui and L. S. Zhu, Anal. Chim. Acta, 2010, 659, 144–150 CrossRef CAS PubMed.
- D. A. Geraldo, C. A. Togo, J. Limson and T. Nyokong, Electrochim. Acta, 2008, 53, 8051–8057 CrossRef CAS PubMed.
- K. M. Song, M. Cho, H. Jo, K. Min, S. H. Jeon, T. Kim, M. S. Han, J. K. Ku and C. Ban, Anal. Biochem., 2011, 415, 175–181 CrossRef CAS PubMed.
- Y. Wang, R. Yuan, Y. Q. Chai, Y. L. Yuan, L. J. Bai and Y. H. Liao, Biosens. Bioelectron., 2011, 30, 61–66 CAS.
- C. F. Ding, Y. Ge and J. M. Lin, Biosens. Bioelectron., 2010, 25, 1290–1294 CrossRef CAS PubMed.
- D. B. Zhu, X. M. Zhou and D. Xing, Biosens. Bioelectron., 2010, 26, 285–288 CrossRef CAS PubMed.
- R. A. A. Muzzarelli, P. Ilari and M. Petrarulo, Int. J. Biol. Macromol., 1994, 16, 177–180 CrossRef CAS.
- B. L. Su, D. P. Tang, Q. F. Li, J. Tang and G. N. Chen, Anal. Chim. Acta, 2011, 692, 116–124 CrossRef CAS PubMed.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef CAS.
- J. Liu, A. G. Rinzler, H. Dai, J. H. Hafner, R. K. Bradley, P. J. Boul, A. Lu, T. Iverson, K. Shelimov, C. B. Huffman, F. Rodriguez-Macias, S. T. Y-Seon, R. Lee, D. T. Colbert and R. E. Smalley, Science, 1998, 280, 1253–1256 CrossRef CAS.
- Y. Zhu, P. Chandra, K. M. Song, C. Ban and Y. B. Shim, Biosens. Bioelectron., 2012, 36, 29–34 CrossRef CAS PubMed.
- H. Watanabe, A. Satake, Y. Kido and A. Tsuji, Analyst, 1999, 124, 1611–1615 RSC.
- C. Z. Yu, Y. Z. He, G. N. Fu, H. Y. Xie and W. E. Gan, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 333–338 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |