 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A luminescence-based assay of UDP-sugar producing pyrophosphorylases†
Daniel
Decker
a,
Stina
Lindberg
b,
Jonas
Eriksson
b and
Leszek A.
Kleczkowski
*a
aDepartment of Plant Physiology, Umeå Plant Science Centre, Umeå University, 90187 Umeå, Sweden. E-mail: Leszek.kleczkowski@plantphys.umu.se; Fax: +46 (0)7866676; Tel: +46 (0)786 5474
bLaboratories for Chemical Biology Umeå (LCBU), Department of Chemistry, Umeå University, 90187 Umeå, Sweden
First published on 31st October 2013
Abstract
A coupled luminescence assay was applied to monitor pyrophosphate (PPi) production by either purified barley UDP-glucose pyrophosphorylase (UGPase) or purified Leishmania UDP-sugar pyrophosphorylase (USPase). In the assay, the PPi produced by the pyrophosphorylases was converted to ATP by ATP-sulfurylase, and the ATP produced was linked to luminescent light formation through the action of firefly luciferase. The assay allowed for a quantitative measurement of UGPase and USPase activities, down to a pmol per min level. The activities were linear with time and proportional to the amount of the enzyme added, and were neither affected by Pi nor by DTT. For UGPase, Km values with UTP and Glc-1-P were 0.14 and 0.26 mM, respectively, whereas for USPase the respective Km values with UTP, Glc-1-P and Gal-1-P were 0.4, 2.9 and 3.9 mM. Possible applications of the luminescence-based assay for not only UDP-sugar producing pyrophosphorylases, but also other types of pyrophosphorylases are discussed.
Introduction
UDP-sugars are used in hundreds of glycosylation reactions, serving as substrates for synthesis of cell wall polysaccharides, soluble oligosaccharides (e.g. sucrose, trehalose), glycoproteins, glycolipids, etc., and they represent the most important precursors for biomass production in nature.1 In most cases, the production of UDP-sugars is derived from the respective sugar-1-P via the action of UTP-dependent pyrophosphorylases.2 They can be divided into several classes, depending on homologies between their amino acid sequences and tertiary structures, and on the nature of sugar-1-P serving as a substrate.3 They include UDP-Glc pyrophosphorylase (UGPase), which is fairly specific for Glc-1-P;2a,4 UDP-sugar pyrophosphorylase (USPase), which can use a variety of sugar-1-P as substrates;1b,5 and UDP-N-acetylgalactosamine pyrophosphorylase, which uses acetylated amino–sugar–phosphates and, sometimes, Glc-1-P.6 The reactions catalyzed by the pyrophosphorylases are freely reversible and they can be assayed both in the direction of UDP-sugar synthesis (synthesis reaction) and UDP-sugar utilization (pyrophosphorolysis reaction).Spectrophotometric enzyme assays continuously monitoring NAD(P)H formation “coupled” to either Glc-1-P or UTP formation from UDP-Glc or UDP-sugar, respectively, have been used to assay the pyrophosphorolytic reaction of UGPase and USPase.5c,7 For the synthesis reaction, a common assay involves quantification of inorganic phosphate (Pi) formed after hydrolysis of pyrophosphate (PPi).4c,8 The drawback of this system is that it cannot be monitored continuously during the reaction and it cannot be used for Pi-containing samples. Assays continuously monitoring either UDP-Glc or PPi formation have also been described. One of them uses purified UDP-Glc dehydrogenase (UGDH) to “couple” the formation of UDP-Glc by UGPase to NADH synthesis,9 and the other monitors NAD formation “coupled” to PPi synthesis by UGPase in the presence of purified PPi-dependent phosphofructokinase (PPFK) and three other enzymes.10 The drawback of these assays is that commercially available UGDH and PPFK have relatively low activities and thus their use in routine pyrophosphorylase assays can be very expensive. Assays of the synthesis reaction using chromatographic quantification of UDP-sugars were also reported.4b,5a
We report here a continuous assay for UGPase and USPase, based on a luminescence approach, where production of PPi by the pyrophosphorylases is “coupled” to light production by the ATP-sulfurylase–firefly luciferase system. Several characteristics of this assay are presented (including its use for Km determination of the pyrophosphorylases), along with discussion of its possible applications.
Materials and methods
Production of purified UGPase and USPase
Recombinant barley UGPase was heterologously expressed in Escherichia coli and purified to homogeneity, as detailed earlier.11 The enzyme was stored at −20 °C in the buffer containing 0.1 M sodium phosphate (pH 8.0), 300 mM NaCl, 250 mM imidazole and 25% glycerol. An expression construct containing cDNA of Leishmania major USPase (LmjF17.1160) was order-made by GenScript, CA, USA. The construct contained a nucleotide sequence that was optimized for bacterial expression. The full coding sequence was cloned into prokaryotic expression vector pET23d+ (Novagen) in fusion with a poly-His affinity tag. The construct was sequenced on both strands using a primer walking strategy with unlabelled primers (Cybergene, Huddinge, Sweden) and BigDye Terminator Cycle sequencing kit (Perkin Elmer), and transformed into BL21 (DE3) E. coli cells. Expression of USPase was induced for 20 h at 20 °C by 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Subsequently, the cells were lysed by sonication and, after centrifugation, the His-tagged USPase was purified from the soluble fraction under native conditions on a cobalt-affinity column (TALON Metal Affinity Resin) according to the manufacturer's instructions (Clontech). The eluate containing purified USPase was desalted on an NAP-10 column (GE Healthcare) that was equilibrated and eluted with 100 mM Tris (pH 7.8) and 10 mM MgCl2. The purified USPase was subsequently snap-frozen in liquid nitrogen for storage. Details of bacteria transformation and induction/purification of USPase were as earlier described for plant UGPase,12 and protein content was determined spectrophotometrically at 595 nm using the Bio-Rad Assay kit, with bovine serum albumin (BSA) as standard.The USPase protein, purified on the Co2+-affinity column, was at least 95% pure as judged by SDS-PAGE (ESI, Fig. 1†). The specific activity of USPase (measured with PPi and UDP-Glc as substrates)12 was 170 μmol min−1 per mg protein, which is similar to the value of 157 μmol min−1 mg−1 reported earlier for Leishmania USPase.5c Based on SDS-PAGE, the purified recombinant USPase has a molecular mass of ca. 67 kDa (ESI, Fig. 1†), which is identical to that earlier determined for Leishmania USPase,5c and in the same range as USPases from pea and Arabidopsis (66 and 70 kDa, respectively).5a,b,13
Assays of UGPase and USPase activities by luminescence
Production of PPi by either of the pyrophosphorylases was quantified using “coupled” reactions of ATP-sulfurylase and firefly luciferase, the latter producing luminescent light. Assays were carried out in white polystyrene 384-well microplates (Corning), where the total volume per well was 15 μl. For both UGPase and USPase assays, the standard reaction mixtures contained 100 mM Tris (pH 7.5), 5 mM Mg-acetate, 0.1% BSA, 5 μM adenosine 5′-phosphosulfate (APS), 1 mM dithiothreitol (DTT), 0.25 mM UTP, 0.1 mU ATP-sulfurylase (New England Biolabs, USA), 1.5 μl 10× SL reagent (prepared according to manufacturer's instruction, BioThema AB, Handen, Sweden), containing luciferin and luciferase, and aliquots of either UGPase or USPase. Reactions were initiated with 0.5 mM Glc-1-P, and run at room temperature for approx. 40–60 min. The rate was determined by quantification of luminescence, using a Wallac 1420 VICTOR2 plate reader (Perkin Elmer). In parallel, assays were run in the absence of sugar-1-P to establish the background rate, originating mostly from the presence of APS (sulfurylase substrate) and UTP (pyrophosphorylase substrate). The background accounted for about 10–20% of the overall rate in the presence of sugar-1-P, and it was routinely subtracted to reflect true activity of a given pyrophosphorylase.Kinetic studies
Assays to determine Km values of UGPase and USPase were carried out essentially as described above for the standard reaction mixture, with the exception of substrate concentrations. In assays of UGPase, either Glc-1-P was kept constant at 0.5 mM, whereas UTP was varied (0.12–0.4 mM), or UTP was kept constant at 0.5 mM and Glc-1-P was varied (0.15–0.5 mM). The Km values of USPase were determined for each of the alternative substrates – Glc-1-P and Gal-1-P. With Glc-1-P, either it was kept constant at 6 mM and UTP was varied (0.2–0.6 mM), or UTP was kept constant at 0.6 mM and Glc-1-P was varied (1.1–4.0 mM). With Gal-1-P, either it was kept constant at 2.4 mM and UTP was varied (0.21–0.6 mM), or UTP was kept constant at 0.6 mM and Gal-1-P was varied (0.58–1.1 mM). In all cases, assays were done at least three times for each experimental point, and the variation was always less than 5%.Results and discussion
Characterization of the luminescence assay
We present here a method for determination of UGPase and USPase activities, based on a coupled luminescence assay that is linked to PPi production by the pyrophosphorylases. The principles of the PPi-dependent luminescence were earlier described as the so-called ELIDA (enzymatic luminometric inorganic pyrophosphate detection assay) method,14 and were incorporated into the DNA sequencing technology (pyrosequencing).15 In the assay for pyrophosphorylases, the generated PPi is momentarily converted into ATP by ATP-sulfurylase, followed by the equally rapid conversion of ATP into light by firefly luciferase. This allows for continuous monitoring (at 562 nm) of the pyrophosphorylase reaction with the use of a luminescence detector, and can be easily quantified.We used purified barley UGPase to evaluate the applicability of the luminescence assay system to assay UDP-sugar producing pyrophosphorylases. The assays of UGPase were roughly linear with time (Fig. 1A) and the rates were proportional to the amount of UGPase up to 2.4 pg of the protein (Fig. 1B). In some cases, the reaction rate appeared non-linear during first 15–20 min of the assay (Fig. 1A), and for activity calculations we routinely took the slopes recorded between 20 and 60 min. The sensitivity of the luminescence-linked assay was very high, down to a pmol per min level. The assay was at least a thousand times more sensitive than spectrophotometric assays monitoring the pyrophosphorolytic direction and assays of Pi released from PPi in the synthesis direction.4c,7a
 | ||
| Fig. 1 Time- (A) and enzyme-amount- (B) dependence of the reaction of barley UGPase using the luminescence-based assay system. Conditions and content of all assays were as described for standard assay in Materials and methods. For (B), slopes from (A) were taken and plotted versus the amount of UGPase. RLU, relative luminescence unit. | ||
Neither Pi in a range up to 10 mM nor DTT (up to 10 mM) had any effects on assays of the pyrophosphorylases (data not shown). Inorganic phosphate determination is the basis of the most common assay for quantification of the synthesis reaction of pyrophosphorylases,8 and samples containing endogenous Pi would interfere with that assay. DTT at low concentration (1 mM) is usually required to stabilize light emission of the ELIDA system,14,15 and it was also present in our assays of the pyrophosphorylases. However, the fact that DTT at concentrations as high as 10 mM did not have any additional effects seems important since some of the UDP-sugar producing pyrophosphorylases are redox regulated,16 and high concentrations of DTT are frequently used in such studies to produce reduced proteins. Also, since the pH optima for the sulfurylase and luciferase reactions were reported as relatively broad and flat at the pH 7.0–8.5 range,17 the assay pH for the pyrophosphorylases (pH 7.5) is unlikely to interfere with the efficiency of those coupling enzymes.
Substrate kinetics for UGPase and USPase
We have used the luminescence assay to determine the Km values for substrates of UGPase and USPase. In this way we could test the robustness of the assay system to a range of substrate concentrations and compare the results to those obtained earlier using other assays. On double-reciprocal plots, the kinetics of UGPase were linear when either Glc-1-P or UTP were varied (Fig. 2), and the estimated Km values with those compounds were 0.26 and 0.14 mM, respectively. These values were similar to those earlier determined for the barley UGPase using Pi-detection assay system4c and, generally, for other UGPases.2a,4a,7a,10,18 In plants, physiological levels of Glc-1-P and UTP are believed to be an order of magnitude higher than their Km's with UGPase, suggesting that the enzyme is near saturated with its substrates in vivo.18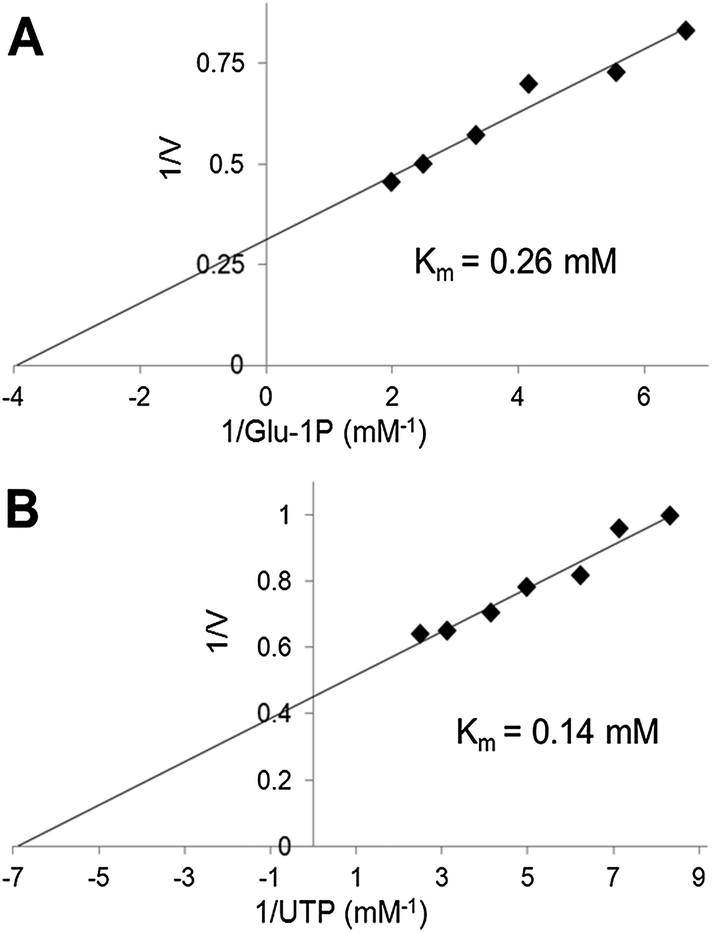 | ||
| Fig. 2 Substrate kinetics of barley UGPase determined using the luminescence system. In the assays, the varied range for Glc-1-P was from 0.15 to 0.5 mM, with UTP constant at 0.5 mM (A), whereas when UTP was varied (0.12 to 0.4 mM), Glc-1-P was kept at 0.5 mM (B). | ||
For USPase, the Km values with Glc-1-P and UTP were 2.9 and 0.42 mM, respectively, and the respective Km values with Gal-1-P and UTP were 3.9 and 0.42 mM (Fig. 3). Kinetics with all substrates, estimated from double-reciprocal plots, were linear throughout the whole range of substrate concentrations. The determined Km values with Glc-1-P, Gal-1-P and UTP were similar, or on the same order of magnitude, to those determined earlier for Leishmania USPase, based on detection of Pi from PPi formed during the reaction5c or on quantification of nucleotides and nucleotide-sugars.6bLeishmania belongs to single-celled eukaryotic pathogens where USPase may be involved in production of galactoconjugates important for the virulence of the pathogen.19 On the other hand, plant USPases generally have lower Km values with Glc-1-P or Gal-1-P when compared to USPases from single-celled pathogens, and their Km values may be as low as 0.2 mM.1b,5a,b,13 These differences may partly be the result of distinct experimental approaches to measure substrate kinetics of the enzyme or, more likely, they may reflect evolutionary changes in the enzyme structure to adapt to various metabolic environments.
 | ||
| Fig. 3 Substrate kinetics of Leishmania major USPase with Glc-1-P and UTP as a varied substrate (A and B, respectively), and with Gal-1-P and UTP as a varied substrate (C and D, respectively) determined using the luminescence system. In the assays, the varied range for sugar-1-P was 1.1–4.0 mM (Glc-1-P) or 0.58–1.1 mM (Gal-1-P), with UTP constant at 0.6 mM (A and C), whereas when UTP was varied (0.2–0.6 mM or 0.21–0.6 mM), Glc-1-P or Gal-1-P were kept at 6 and 2.4 mM, respectively (B and D). | ||
Perspectives
The results have suggested that the luminescence assay system can be used as a reliable method to accurately quantify the reaction rate and to study kinetics of the UDP-sugar producing pyrophosphorylases. Presumably the assay can also be employed to determine the activities of other nucleotide sugar pyrophosphorylases, e.g. GDP-mannose pyrophosphorylase20 and GDP-L-fucose pyrophosphorylase,21 which use GTP (but not UTP) and mannose-1-P and L-fucose-1-P, respectively, as their substrates. On the other hand, since firefly luciferase uses ATP as the substrate,22 the luminescence assay cannot be used to measure activities of ATP-dependent ADP-glucose pyrophosphorylase, a key enzyme of starch formation.23The luminescence-based assay for UDP-sugar producing pyrophosphorylases can potentially be used in high-throughput screening applications, e.g. when screening chemical libraries for inhibitors. One advantage is the detection wavelength (562 nm) that is away from the 300–400 nm range common for many compounds in the chemical libraries. Indeed, several luminescence-based kits are commercially available and have been applied to popular drug targets (e.g.ref. 24). Given the fact that UDP-sugars, and UDP-Glc in particular, serve as precursors in a plethora of glycosylation reactions in all cells and tissues, identifying specific inhibitors of enzymes synthesizing UDP-sugars would be of value, e.g. for studies on the in vivo roles of each of those enzymes.
Abbreviations
| APS | Adenosine 5′-phosphosulfate |
| BSA | Bovine serum albumin |
| DTT | Dithiothreitol |
| ELIDA | Enzymatic luminometric inorganic pyrophosphate detection assay |
| Gal | Galactose |
| Glc | Glucose |
| Pi | Inorganic phosphate |
| PPFK | PPi-dependent phosphofructokinase |
| PPi | Pyrophosphate |
| UGDH | UDP-Glc dehydrogenase |
| UGPase | UDP-Glc pyrophosphorylase |
| USPase | UDP-sugar pyrophosphorylase |
Acknowledgements
This research was funded by the Swedish Research Council (to L.A.K.).References
- (a) T. Kotake, C. Hirosawa, Y. Ando and Y. Tsumuraya, Plant Biotechnol., 2010, 27, 231 CrossRef CAS; (b) L. A. Kleczkowski, D. Decker and M. Wilczynska, Plant Physiol., 2011, 156, 3 CrossRef CAS PubMed.
- (a) D. S. Feingold and G. A. Barber, in Methods in plant biochemistry, ed. P. M. Dey and J. B. Harborne, Academic Press, London, 1990, vol. 2, p. 39 Search PubMed; (b) L. A. Kleczkowski, S. Kunz and M. Wilczynska, Crit. Rev. Plant Sci., 2010, 29, 191 CrossRef CAS.
- L. A. Kleczkowski, M. Geisler, E. Fitzek and M. Wilczynska, Biochem. J., 2011, 439, 375 CrossRef CAS PubMed.
- (a) L. A. Kleczkowski, Phytochemistry, 1994, 37, 1507 CrossRef CAS; (b) Y. Okazaki, M. Shimojima, Y. Sawada, K. Toyooka, T. Narisawa, K. Mochida, H. Tanaka, F. Matsuda, A. Hirai, M. Y. Hirai, H. Ohta and K. Saito, Plant Cell, 2009, 21, 892 CrossRef CAS PubMed; (c) D. Decker, M. Meng, A. Gornicka, A. Hofer, M. Wilczynska and L. A. Kleczkowski, Phytochemistry, 2012, 79, 39 CrossRef CAS PubMed.
- (a) T. Kotake, D. Yamaguchi, H. Ohzono, S. Hojo, S. Kaneko, H. K. Ishida and Y. Tsumuraya, J. Biol. Chem., 2004, 279, 45728 CrossRef CAS PubMed; (b) L. A. Litterer, J. A. Schnurr, K. L. Plaisance, K. K. Storey, J. W. Gronwald and D. A. Somers, Plant Physiol. Biochem., 2006, 44, 171 CrossRef CAS PubMed; (c) S. Damerow, A. C. Lamerz, T. Haselhorst, J. Fuhring, P. Zarnovican, M. von Itzstein and F. H. Routier, J. Biol. Chem., 2010, 285, 878 CrossRef CAS PubMed.
- (a) C. Peneff, P. Ferrari, V. Charrier, Y. Taburet, C. Monnier, V. Zamboni, J. Winter, M. Harnois, F. Fassy and Y. Bourne, EMBO J., 2001, 20, 6191 CrossRef CAS PubMed; (b) T. Yang and M. Bar-Peled, Biochem. J., 2010, 429, 533 CrossRef CAS PubMed.
- (a) R. G. Hansen, G. J. Albrecht, S. T. Bass and L. L. Seifert, Methods Enzymol., 1966, 8, 248 CrossRef CAS; (b) I. Ciereszko, H. Johansson and L. A. Kleczkowski, J. Plant Physiol., 2005, 162, 343 CrossRef CAS PubMed; (c) M. Meng, M. Geisler, H. Johansson, J. Harholt, H. V. Scheller, E. J. Mellerowicz and L. A. Kleczkowski, Plant Cell Physiol., 2009, 50, 998 CrossRef CAS PubMed.
- H. Aoyama, A. D. M. Cavagis, E. M. Taga and C. V. Ferreira, Phytochemistry, 2001, 58, 221 CrossRef CAS.
- L. Elling and M. R. Kula, Biotechnol. Appl. Biochem., 1991, 14, 306 CAS.
- L. Elling, Phytochemistry, 1996, 42, 955 CrossRef CAS.
- L. A. Kleczkowski, F. Martz and M. Wilczynska, Phytochemistry, 2005, 66, 2185 CrossRef PubMed.
- M. Meng, E. Fitzek, A. Gajowniczek, M. Wilczynska and L. A. Kleczkowski, Biochim. Biophys. Acta, 2009, 1794, 1734 CrossRef CAS PubMed.
- T. Kotake, S. Hojo, D. Yamaguchi, T. Aohara, T. Konishi and Y. Tsumuraya, Biosci., Biotechnol., Biochem., 2007, 71, 761 CrossRef CAS.
- P. Nyrén and A. Lundin, Anal. Biochem., 1985, 151, 504 CrossRef.
- M. Ronaghi, M. Uhlen and P. Nyrén, Science, 1998, 281, 363 CrossRef CAS.
- L. I. Martinez, C. V. Piattoni, S. A. Garay, D. E. Rodrigues, S. A. Guerrero and A. A. Iglesias, Biochimie, 2011, 93, 260 CrossRef CAS PubMed.
- (a) E. Hanna, K. F. Ng, I. J. MacRae, C. J. Bley, A. J. Fisher and I. H. Segel, J. Biol. Chem., 2004, 279, 4415 CrossRef CAS PubMed; (b) W. Van Leeuwen, M. J. M. Hagendoom, T. Ruttink, R. Van Poecke, L. H. W. Van Der Plas and A. R. Van Der Krol, Plant Mol. Biol. Rep., 2000, 18, 143 CrossRef.
- M. Meng, M. Wilczynska and L. A. Kleczkowski, Biochim. Biophys. Acta, 2008, 1784, 967 CrossRef CAS PubMed.
- A. C. Lamerz, S. Damerow, B. Kleczka, M. Wiese, G. van Zandbergen, J. Lamerz, A. Wenzel, F. F. Hsu, J. Turk, S. M. Beverley and F. H. Routier, Glycobiology, 2010, 20, 872 CrossRef CAS PubMed.
- H. Hashimoto, A. Sakakibara, M. Yamasaki and K. Yoda, J. Biol. Chem., 1997, 272, 16308 CrossRef CAS PubMed.
- T. Kotake, S. Hojo, N. Tajima, K. Matsuoka, T. Koyama and Y. Tsumuraya, J. Biol. Chem., 2008, 283, 8125 CrossRef CAS PubMed.
- W. D. McElroy, Proc. Natl. Acad. Sci. U. S. A., 1947, 33, 342 CrossRef CAS.
- L. A. Kleczkowski, FEBS Lett., 1999, 448, 153 CrossRef CAS.
- R. C. Durk, K. Singh, C. A. Cornelison, D. K. Rai, K. B. Matzek, M. D. Leslie, E. Schafer, B. Marchand, A. Adedeji, E. Michailidis, C. A. Dorst, J. Moran, C. Pautler, L. L. Rodriguez, M. A. McIntosh, E. Rieder and S. G. Sarafianos, PLoS One, 2010, 5, e15049 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. 1. See DOI: 10.1039/c3ay41811a |
| This journal is © The Royal Society of Chemistry 2014 |