Digital drug delivery: on–off ultrasound controlled antibiotic release from coated matrices with negligible background leaching
Misty L.
Noble
a,
Pierre D.
Mourad
b and
Buddy D.
Ratner
*a
aDepartment of Bioengineering, University of Washington, Seattle, WA 98195, USA. E-mail: ratner@uweb.engr.washington.edu
bApplied Physics Laboratory and Department of Neurosurgery, University of Washington, Seattle, WA 98195, USA. E-mail: pierre@apl.washington.edu
First published on 12th February 2014
Abstract
Hydrogels such as crosslinked poly(2-hydroxyethyl methacrylate) (pHEMA) have been used extensively in controlled release drug delivery systems. Our previous work demonstrated an ultrasound (US)-responsive system based on pHEMA coated with a self-assembled multilayer of C12–C18 methylene chains. The resulting coating was predominantly crystalline and relatively impermeable, forming an US-activated switch that controlled drug release on-demand, and kept the drug within the matrix in the absence of US. The device, as developed did, however, show a low background drug-leaching rate independent of US irradiation. For some applications, it is desirable to have very low or zero background release rates. This was achieved here by a combination of new processing steps, and by co-polymerizing HEMA with a relatively hydrophobic monomer, hydroxypropyl methacrylate (HPMA). These advances produced systems with undetectable ciprofloxacin background release rates that are capable of US-facilitated drug release – up to 14-fold increases relative to controls both before and after US exposure. In addition, these observations are consistent with the hypothesis that US-mediated disorganization of the coating allows a transient flux of water into the matrix where its interaction with bound and dissolved drug facilitates its movement both within and out of the matrix.
Introduction
An ultrasound (US)-responsive system was previously developed for on-demand delivery of insulin, ciprofloxacin or other agents.1–3 In this system, a drug-containing polymeric monolith, poly(2-hydroxyethyl methacrylate) [pHEMA], was coated with a self-assembled multilayer (SAM) coating of long methylene chains. Conceptually, the resulting coating restricted the drug molecules to within the matrix in the absence of US and permitted the release of drugs with exposure to US. The molecules released from this pHEMA-based matrix system (insulin,2 and ciprofloxacin3 (cipro)) retained their bioactivity and their potency. After US application, the methylene chains re-organized to a relatively impermeable self-assembled coating – this coating allowed only a very low level release compared to the release triggered by US.These US-responsive systems have shown low but measurable background release rates, which can be potentially used for applications requiring local delivery of drugs, such as for prophylactic4–6 and analgesic7–9 treatments after surgery and for drug-eluting stents10–12 for which a meaningful background release rate could provide an additional therapeutic benefit. For certain clinical applications, such as diabetes and insulin management, chemotherapy and others, it is important to have a system that will only dispense drugs on-demand with negligible or even zero background release. We refer to this as digital drug delivery – release is either “on” or “off.” Such a system would allow better control of the release kinetics (each pulse or “on” event releases a defined bolus of drug), maximize retention of drug within the matrix, and reduce unnecessary exposure of surrounding tissue to the drug.
This system can potentially be used on or within long-term implants or biomaterials that might develop device-centered infections or biofilms after varying periods of implantation. Norris et al. demonstrated that this system can significantly reduce the accumulation of Pseudomonas aeruginosa biofilms in flow cell studies using a low intensity US source.3 However, its high background cipro release rate can potentially promote point mutations in some bacterial strains, which can lead to increased resistance and associated complications.13–17 Hence, building a completely latent system is more desirable. If infection develops, an external stimulus such as US can then be used to trigger release from the latent system resulting in substantial local concentrations of antibiotic, often well above minimum therapeutic levels. Exposure of this drug delivery system to US at regular intervals could replace current antibiotic regimens of oral or systemic antibiotics for at least a two-week period.18,19 Thus, this on-demand system has the potential for delivering antibiotics locally at the site of infection, for example, the surfaces of many biomaterials and medical devices.
Results and discussion
Hydrogels, such as pHEMA, are water-swollen, cross-linked polymeric structures commonly used in biomaterials because of their high water content and rubbery consistency that is similar to that of living tissues. They also have good permeability properties, which allow small molecules to be extracted efficiently. Unreacted monomers, initiators and other organic solvents involved in polymerization can be removed and washed away with repeated rinses in water.20 Similarly, lyophilized cipro particles, which were incorporated during the polymerization of pHEMA, can also be washed away during this rinsing step (∼10%), especially those near the pHEMA surface.Analysis shows successful surface modification
With the dibutyl tin dilaurate catalytic system, the hydroxyl-isocyanate reaction is found to be fast and efficient resulting in the formation of stable urethane bonds as shown in Scheme 1. Within 5 minutes of the reaction, almost all of the available hydroxyl groups in the surface zone had already reacted with the isocyanate in solution as determined by X-ray photoelectron spectroscopy (XPS), which quantitatively determines the atomic composition of the samples. Table 1 lists the elemental surface composition of pHEMA modified with C18-isocyanate for 0, 5, 15, and 30 minutes obtained from XPS.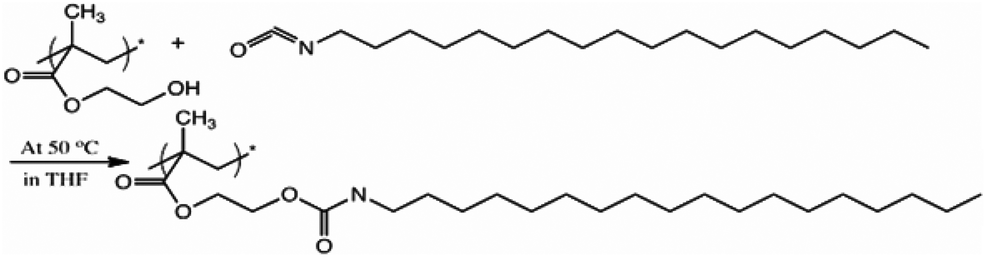 | ||
| Scheme 1 Surface modification with isocyanate. The surface –OH groups of pHEMA were reacted with C18-isocyanate to form a hydrophobic barrier. | ||
| Elemental composition (in %) | |||
|---|---|---|---|
| C | O | N | |
| Uncoated pHEMA (theory) | 66.7 | 33.3 | 0.0 |
| 0 min | 70.1 ± 0.9 | 27.5 ± 2.5 | 1.2 ± 1.7 |
| C18-coated pHEMA (theory) | 83.3 | 13.3 | 3.3 |
| 5 min | 86.1 ± 0.3 | 9.7 ± 0.9 | 4.2 ± 0.7 |
| 15 min | 84.8 ± 1.1 | 10.6 ± 0.4 | 4.0 ± 0.2 |
| 30 min | 85.1 ± 0.2 | 11.2 ± 0.7 | 3.8 ± 0.5 |
Theoretically, uncoated pHEMA should have 66.7% carbon (C), and 33.3% oxygen (O) and isocyanate-reacted samples should have 83.3% C, 13.3% O, and 3.3% nitrogen (N). Increasing %N in the surface suggests attachment of the isocyanate to the surface hydroxyl groups. Within 5 minutes, the %C is 86.1%, which is more than the theoretical value of complete surface modification with C18 chains. %O and %N are 9.7% and 4.2%, respectively, which are also close to the theoretical values of 13.3% and 3.3%. Furthermore, the values of %C, %O, and %N did not significantly change in coatings longer than 5 minutes. Surface analysis of different pHEMA-co-HPMA polymers was not carried out since they are too similar in their chemical structures – differing by only one methylene group (i.e., one C, since hydrogen is not directly measured in XPS).
High resolution carbon C1s scans showed that there are three different carbon species that can be resolved on the surface: hydrocarbon carbon (CH or CC), which has a binding energy (BE) of 285 eV, ether carbon (C–O), which has a 286.5 eV BE, and carbonyl carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), which has a 289.3 eV BE. As shown in Fig. 1, the relative amounts of carbon species changed substantially within 5 minutes. There is a significant increase in the CH carbon, while the C–O and CO decrease with longer reactions. Similar to the survey scans, there is minimal change in the high resolution carbon C1s spectra of samples coated longer than 5 minutes.
O), which has a 289.3 eV BE. As shown in Fig. 1, the relative amounts of carbon species changed substantially within 5 minutes. There is a significant increase in the CH carbon, while the C–O and CO decrease with longer reactions. Similar to the survey scans, there is minimal change in the high resolution carbon C1s spectra of samples coated longer than 5 minutes.
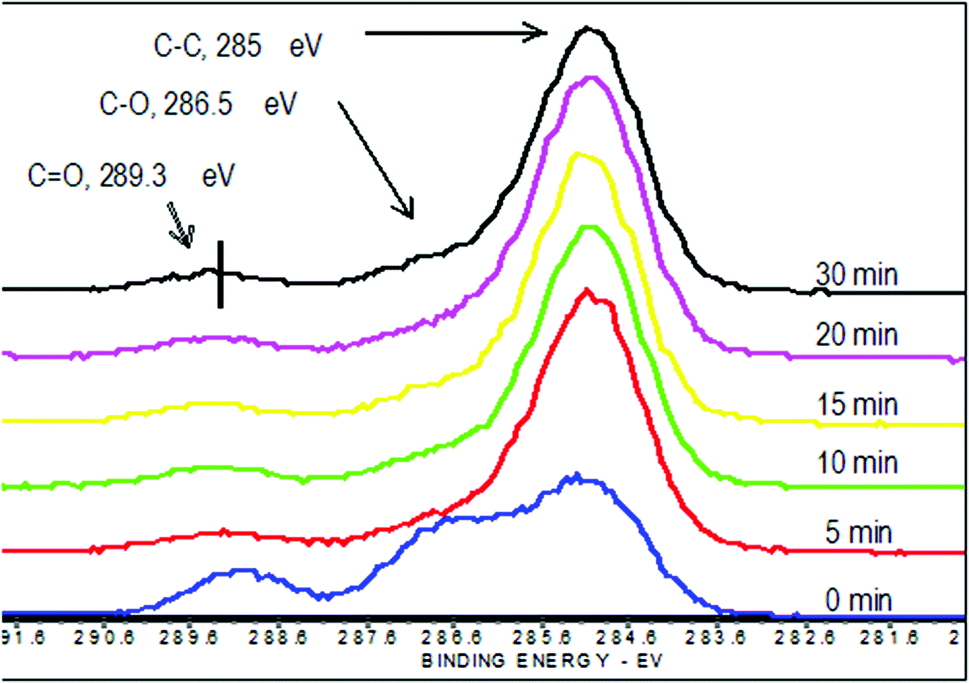 | ||
| Fig. 1 High resolution carbon C1s spectra of the coated and uncoated pHEMA matrices obtained using XPS. The three peaks represent the difference carbon species on the modified surface: hydrocarbon (CH or CC) at 285 eV, ether carbon (C–O) at 286.5 eV, and carbonyl carbon (CO) at 289.3 eV. | ||
XPS has a sampling depth of approximately 80 Å. If only the outermost surface monolayer of pHEMA was being reacted with the isocyanate, we would see a spectrum that would be a convolution of pHEMA and the pHEMA adduct with C18-isocyanate (HEMA-C18). Since the XPS spectra are characteristic of HEMA-C18 at all reaction times, this suggests that within 5 minutes, the reaction has penetrated to 80 Å. The absence of fluorine (F) signal from the survey spectra is consistent with few or no cipro particles in the top 80 Å of all coated and uncoated pHEMA samples. Since longer reaction times do not change the spectra, it is possible that the reaction penetrates deeper into the pHEMA matrix with time. Similar studies have shown that no additional alkyl groups were observed on the surface of samples reacted with isocyanate for longer than 30 minutes.21 The samples coated longer than 30 minutes were more swollen and eventually degraded in the isocyanate-THF solution. Degradation was more apparent in more hydrophobic samples, i.e., samples with more HPMA component.
Qualitative analysis using Fourier transform infrared-attenuated total reflectance (FTIR-ATR, or also ATR) spectroscopy showed the transformation of pHEMA surface to the modified alkyl-rich surface. Asymmetric stretching (st as) peaks, such as –O–CO at 1170 cm−1 and O–C–C at 1080 cm−1, which are characteristic of a methacrylate (pHEMA) surface, were observed in the uncoated sample as shown in Fig. 2a. The intensity of these peaks decreased upon reacting with the isocyanate. A doublet around 1275 cm−1 was observed in the uncoated samples, and was eventually replaced by a singlet at 1250 cm−1 that became more evident when hydroxyl groups were reacted with the isocyanate to form a urethane bond. This was monitored by increasing peak intensity of the N–CO–O stretching at 1250 cm−1 (st as) and 1060 cm−1 (symmetric, st sy). In addition, as alkyl chains were added into the surface by the isocyanate reaction, peaks characteristic of CH2 stretching at 2920 cm−1 (asym) and 2860 cm−1 (sym) emerged as illustrated in Fig. 2b. With longer reaction time, the peaks not only intensified, but they also shifted. The CH2 asymmetric stretching (st as) peak shifted from the 2922 cm−1 to 2918 cm−1 indicating improved packing, i.e., a more crystalline order.22 In this study, it was observed that as the reaction time is increased, it resulted in better packing and organization of the alkyl chains. However, similar studies have shown that no additional alkyl groups were observed on the surface of samples reacted with isocyanate for longer than 30 minutes.21 Samples coated longer than 30 minutes appeared to swell and eventually degraded in the isocyanate-THF solution. Degradation was more apparent in more hydrophobic samples, i.e., samples with more HPMA component. This is consistent with reaction of the –OH units in the bulk of the polymer, in contrast to at the surface, leading to excessive swelling that mechanically disrupts the network structure.
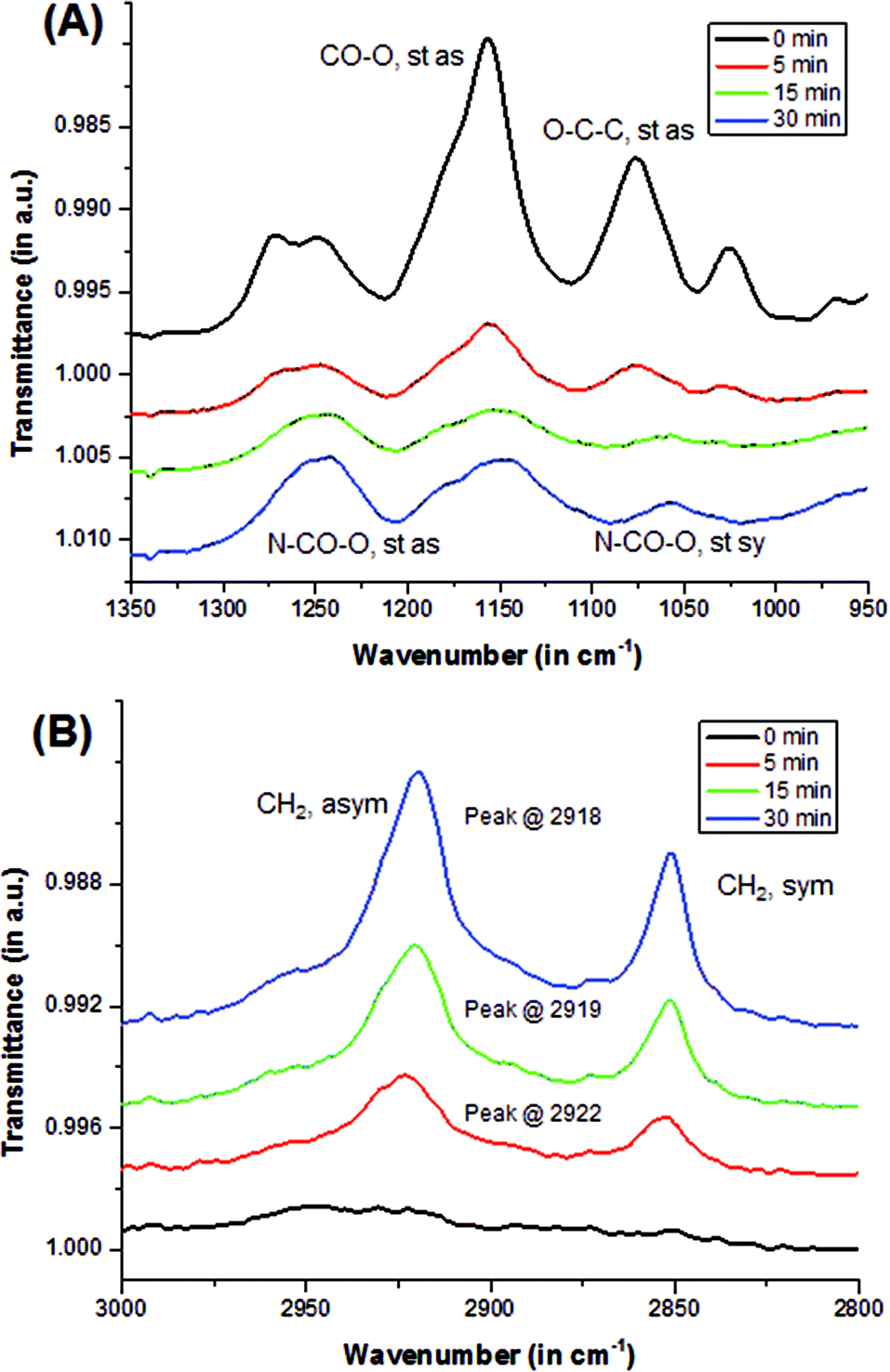 | ||
| Fig. 2 FTIR-ATR spectra showing the transformation of pHEMA surface to the modified n-alkyl surface. (A) With increasing reaction time, peaks attributed to pHEMA decrease, while peaks attributed to the hydrophobic, long alkyl chain coated surface increase. (B) Peaks attributed to methylene chains shift with longer coating times indicating a change in crystallinity. | ||
Addition of HPMA and a methylene coating results in reduced swelling and lowered release
To investigate how the addition of the more hydrophobic HPMA monomer affects the swelling properties of the coated and uncoated matrices, water contents were determined at pre-determined time points. The amount of water in the samples was calculated using Equation (1):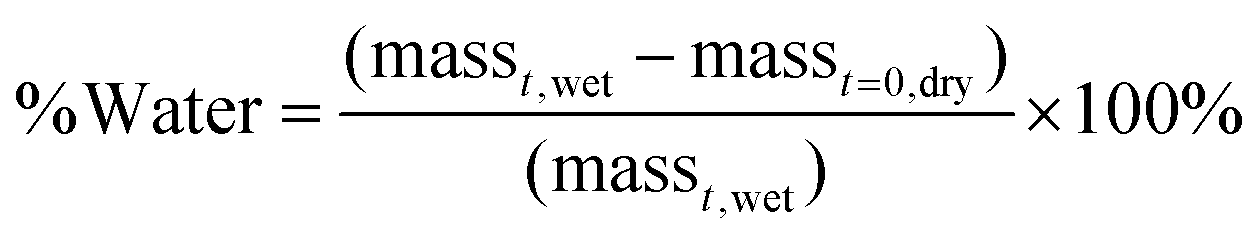 | (1) |
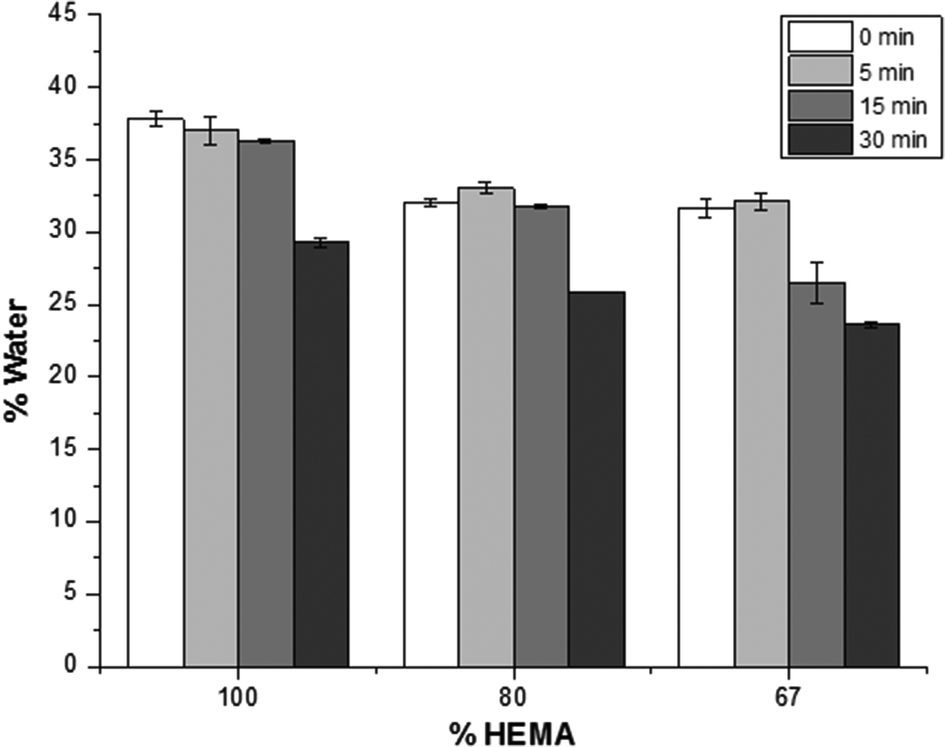 | ||
| Fig. 3 The water content in the coated and uncoated pHEMA-co-HPMA matrices after 24 hours. The swelling behavior of these pHEMA-co-HPMA matrices are directly related to the amount of HPMA in the matrices, as well as the length of the isocyanate reaction time. | ||
A two-factor analysis of variance (ANOVA) with replication determined that the amount of water in the polymer samples is statistically different (p < 0.001) for each polymeric composition. In this study, the swelling behaviour of the samples is related to the amount of HPMA in the matrices: with increasing HPMA in the composition, the resulting polymer becomes more hydrophobic, and the water content decreased. Likewise, the methylene chain coating also affected the amount of water in the samples as ascertained using the two-way ANOVA (p < 0.001). For each polymer composition, samples were reacted with C18-isocyanate. With longer reaction time, i.e. a more densely packed and thicker methylene chain coating, the influx of water into the matrix is reduced. Furthermore, it was also determined that there is an interaction between the polymer composition and the time of the methylene coating reaction (p < 0.001).
Release kinetics of cipro from the polymer matrices were measured to determine if the modification in the matrix composition and the isocyanate coating protocol was successful. Cipro release rates from the polymer matrices were calculated using Equation (2), and plotted as shown in Fig. 4.
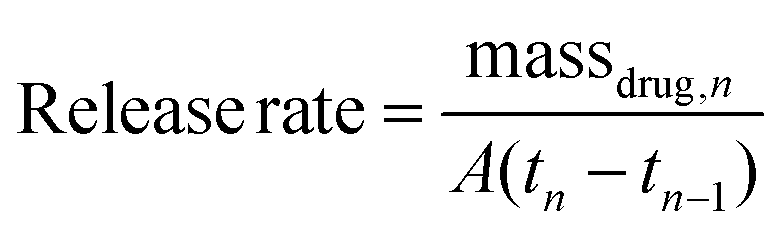 | (2) |
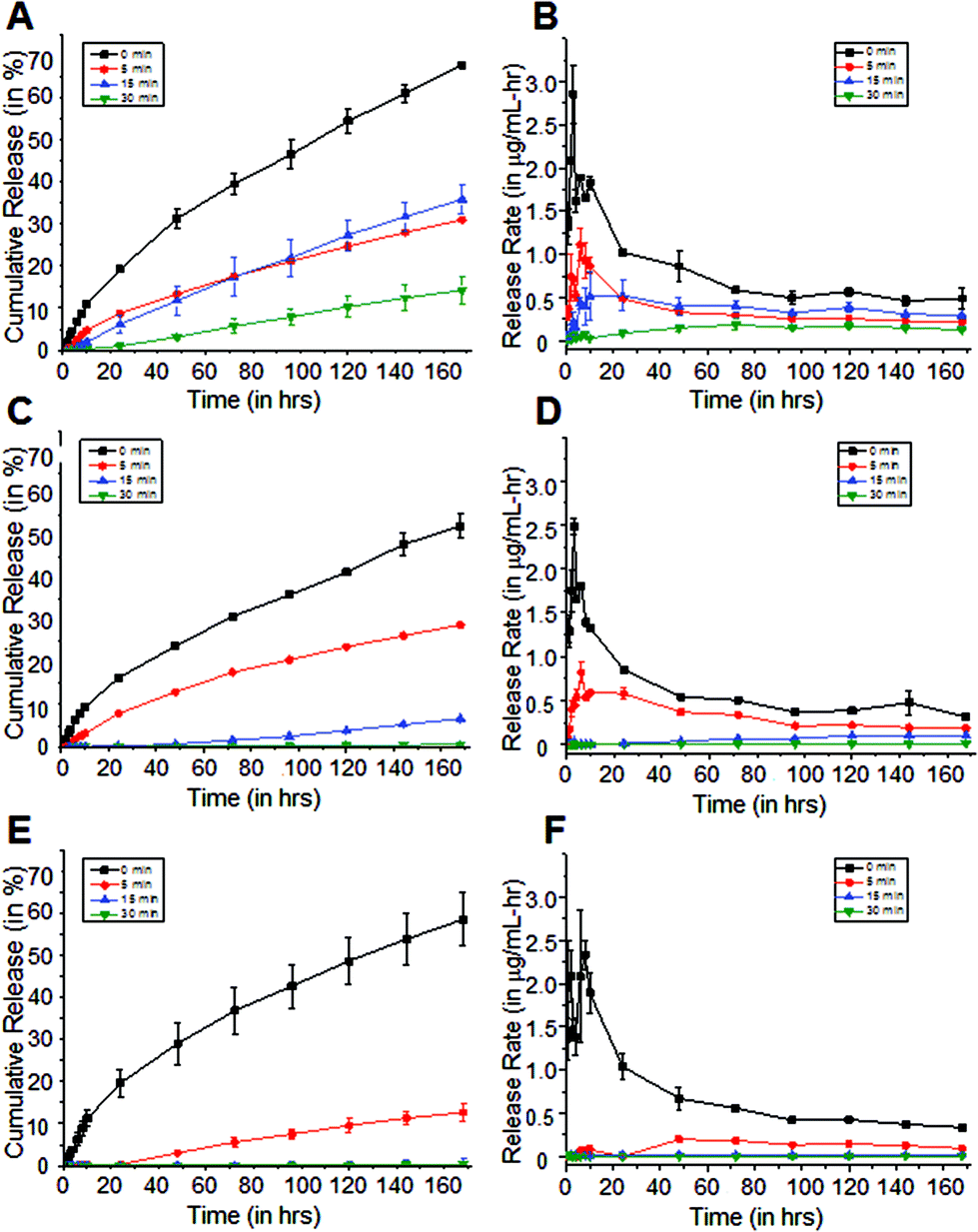 | ||
Fig. 4 Cipro cumulative release and release rates from coated pHEMA-co-HPMA matrices with the following compositions: (A, B) 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, (C, D) 80:20, and (E, F) 67:33 HEMA:HPMA vol/vol ratio. Release is dependent on hydrophobicity of the matrix, as well as its coating. The limit of detection is 0.057 μg mL−1 determined by UV-visible spectrophotometry at λ = 270 nm. Error bars indicate SD (n = 3–5). :0, (C, D) 80:20, and (E, F) 67:33 HEMA:HPMA vol/vol ratio. Release is dependent on hydrophobicity of the matrix, as well as its coating. The limit of detection is 0.057 μg mL−1 determined by UV-visible spectrophotometry at λ = 270 nm. Error bars indicate SD (n = 3–5). | ||
It was determined that the burst release, which is commonly observed in swelling-based drug delivery systems, was significantly reduced with longer reaction time coatings. Furthermore, it was also determined that release rates were dependent on the HPMA concentration in the sample matrices, which can then be related to the swelling. To better understand the drug release from these polymer matrices, mechanisms proposed for similar swellable polymer-based systems were considered. In particular, Siepmann and Peppas reviewed several mathematical models of release of low molecular weight molecules from hydroxypropyl methylcellulose.25 The authors proposed that steep water concentration gradients were formed initially when the dry polymer is immersed in water, allowing the polymer to imbibe water. In the dry state, the diffusion coefficients of most molecules in the polymer matrix are low, but once the polymer absorbs water, these diffusion coefficients increase to near their values in water. Further absorption of water leads to polymer swelling resulting in changes in the polymer and drug concentration, and increasing the physical dimensions of the system. Then, the drug dissolves and diffuses out of the matrix with a rate dependent on the concentration in the swollen polymer matrix.
The hydrophobic methylene coating slows the absorption of water in the matrices as shown in Fig. 3. It serves as a rate-limiting barrier that inhibits water from entering the matrix and restricts the solutes from diffusing out. However once water is imbibed, it was observed that it is more difficult to control the further absorption of water. This is evident in the cipro release profiles from 100% and 80% pHEMA matrices. The passive release rates from both coated and uncoated 100% and 80% pHEMA matrices are higher than those from more hydrophobic matrices that have thicker methylene coatings (Fig. 4). Although it was determined by surface analysis that the longer reaction time coatings have organized methylene chains, water was still able to penetrate the methylene coating after a certain amount of time, causing the polymer to undergo a glass-to-gel transition as it starts getting more hydrated and thus, can release trapped drug more easily. Addition of HPMA in the matrix composition was found to be necessary to obtain a slower hydrating, swelling and drug releasing polymer.
Exposure to 20 kHz ultrasonic pulses results in release of cipro from pHEMA-co-HPMA matrices
To demonstrate the response of the polymer matrices to external stimulation, samples were exposed to 20 kHz US. The ultrasonic probe was immersed into the solution, thereby delivering concentrated acoustic energy into the samples. The result of this acoustic energy within the fluid was observable cavitation as indicated by temperature increases in the fluid, and the resultant temporary perturbation of the methylene coating on these pHEMA-co-HPMA matrices, as shown by transient release of drug from the polymer matrices. US intensity was estimated using a calorimetry method,26,27 such that: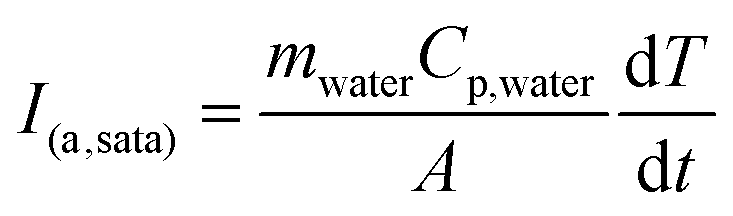 | (3) |
:0, 80:20, and 67:33 pHEMA-co-HPMA matrices respectively. 20 kHz US was applied for 5 minutes, followed by a 10 minute rest period.
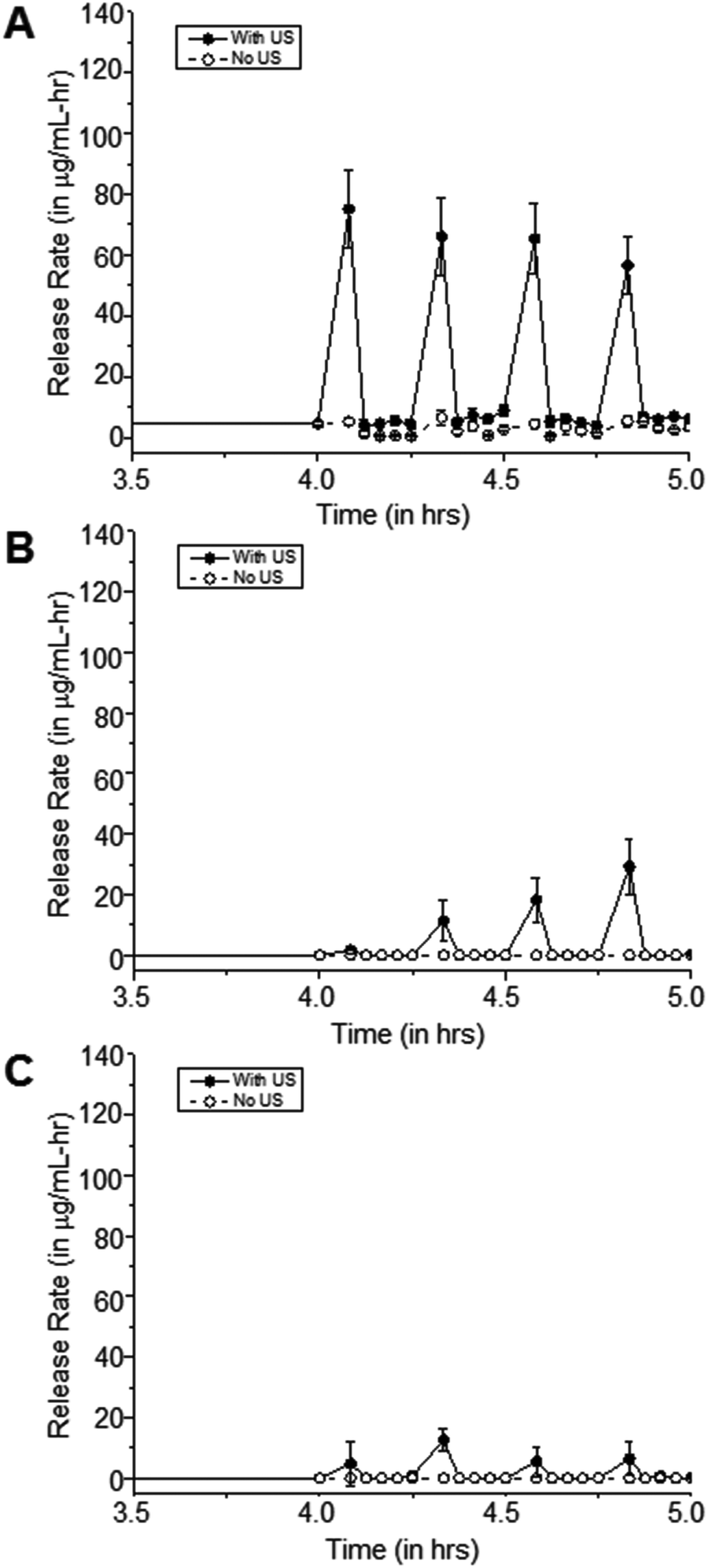 | ||
| Fig. 5 Release rates of ciprofloxacin from 15-minute coated (A) 100:0, (B) 80:20, and (C) 67:33 pHEMA-co-HPMA matrices when exposed to 20 kHz US (50% duty cycle) at 15 minute intervals. Limit of detection is 0.057 μg mL−1 as determined by UV-visible spectrophotometry at λ = 270 nm. Error bars indicate SD (n = 3–5). | ||
The effects of US on cipro release are more apparent in the hydrophilic samples, such as 100% pHEMA, compared to the more hydrophobic pHEMA-co-HPMA copolymers, which is consistent with the previous findings that the amount of water in the polymer matrices plays a major role in how US can release the trapped molecules from the matrix. The limited amount of water in the matrix and the inherent hydrophobicity of the pHEMA-co-HPMA can attenuate the US,28 such that US could have only acted on the outer part of the sample. It can be hypothesized that the absence of water prevents the oscillations of cavitation nuclei that allows enhance drug release.29
Duty cycles affect US-controlled cipro release from polymer matrices
Exposure of cipro-loaded 100% pHEMA matrices to 20 kHz US triggered high and reproducible release rates. In our experimental set-up, we obtained concentrations beyond the minimum inhibitory concentration (MIC), 0.125 μg mL−1, per US exposure with no clearance. This motivated their use in a study to determine how varying US duty cycle can affect drug release. The ultrasonic probe was programmed to emit US at different duty cycles, therefore producing different intensities. Using a calorimetry method,26,27 it was determined that the time average intensities at 50% duty cycle is about 4.5 W cm−2 and about 2.3 W cm−2 at 25% duty cycle. A statistical analysis, ANOVA, was performed on each of the release data sets. It was determined that the release rates resulting from US exposures at 25% and 50% duty cycles are statistically different from (a) the background release rates prior to US exposure (p < 0.05), (b) the background release rates in between each US pulse (p < 0.05), and (c) the release rates from the controls, i.e., US exposures at 0% duty cycle. Furthermore, the release rates due to exposure to US with 25% and 50% US duty cycles are also statistically different from each other (p < 0.001) – see Fig. 6. This indicates that the release rates are energy dose-dependent – the more energy delivered into the solution, the faster the release of the trapped molecules.30 Interestingly, the background release rates prior to US exposure were statistically different from the background release rates after each US exposure for each of the 0-minute, 5-minute and 15-minute coated matrices, indicating that the methylene coating in most instances did not fully re-heal after each exposure. However, it is possible that with more time in between pulses of US, this might allow the methylene coating to re-heal.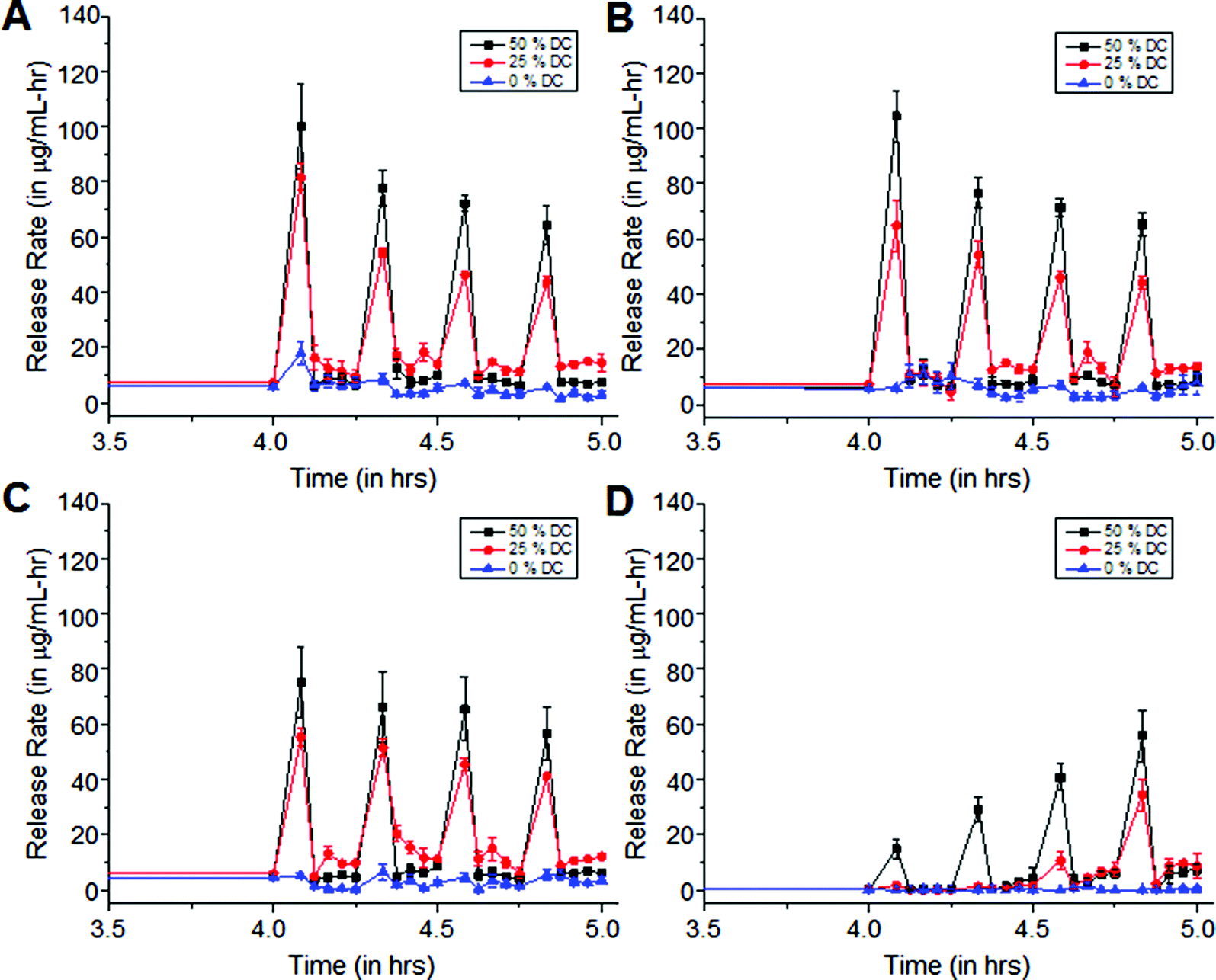 | ||
| Fig. 6 Release rates of ciprofloxacin from (A) 0-minute, (B) 5-minute, (C) 15-minute and (D) 30-minute coated pHEMA matrices when exposed to different US intensities as a function of duty cycle. Limit of detection is 0.057 μg mL−1 determined by UV-visible spectrophotometry at λ = 270 nm. Error bars indicate SD (n = 3–5). | ||
When water was hindered from penetrating the matrix, even after the hydration period (pre-US exposure) as in the 30-minute coated samples shown in Fig. 6d, the release rates were found to increase with subsequent US exposures. We hypothesize that the methylene chain coating in this case was relatively thick and highly ordered, causing it to behave as a very efficient rate-limiting barrier hindering water imbibition into the matrix. This, in turn, significantly slowed the dissolution and diffusion of the drug out of the matrix. But, with each subsequent US exposure, more water entered the polymer matrix, dissolving the trapped drug molecules and eventually transporting them out of the polymer. The background release rates prior to and after each of the first two US exposures do not differ statistically from each other or from the control sample (p < 0.05). This indicates that for the 30-minute isocyanate exposure case, the methylene chain coating could quickly re-heal. However, after the third and fourth US exposure, statistically significant background leaching rates relative to control were observed, consistent with the hypothesis that increased water uptake across the coating allowed the polymer to swell and enhanced cipro release, as in the cases shown in Fig. 6a–c.
Longer intervals between US pulses shows background returns to baseline
Fig. 7 shows such a study, where samples were exposed to 20 kHz US at 50% duty cycle for 5 minutes at 24-hour intervals rather than 10-minute intervals. As with the previous study, background release rates were also measured in between pulses, and all results were compared to the control case with no US application. Relative to the control samples, the differences in release rates of cipro from the 0-minute, 5-minute, and 15-minute coated samples after exposure to US were determined to be statistically significant (p < 0.05), on average.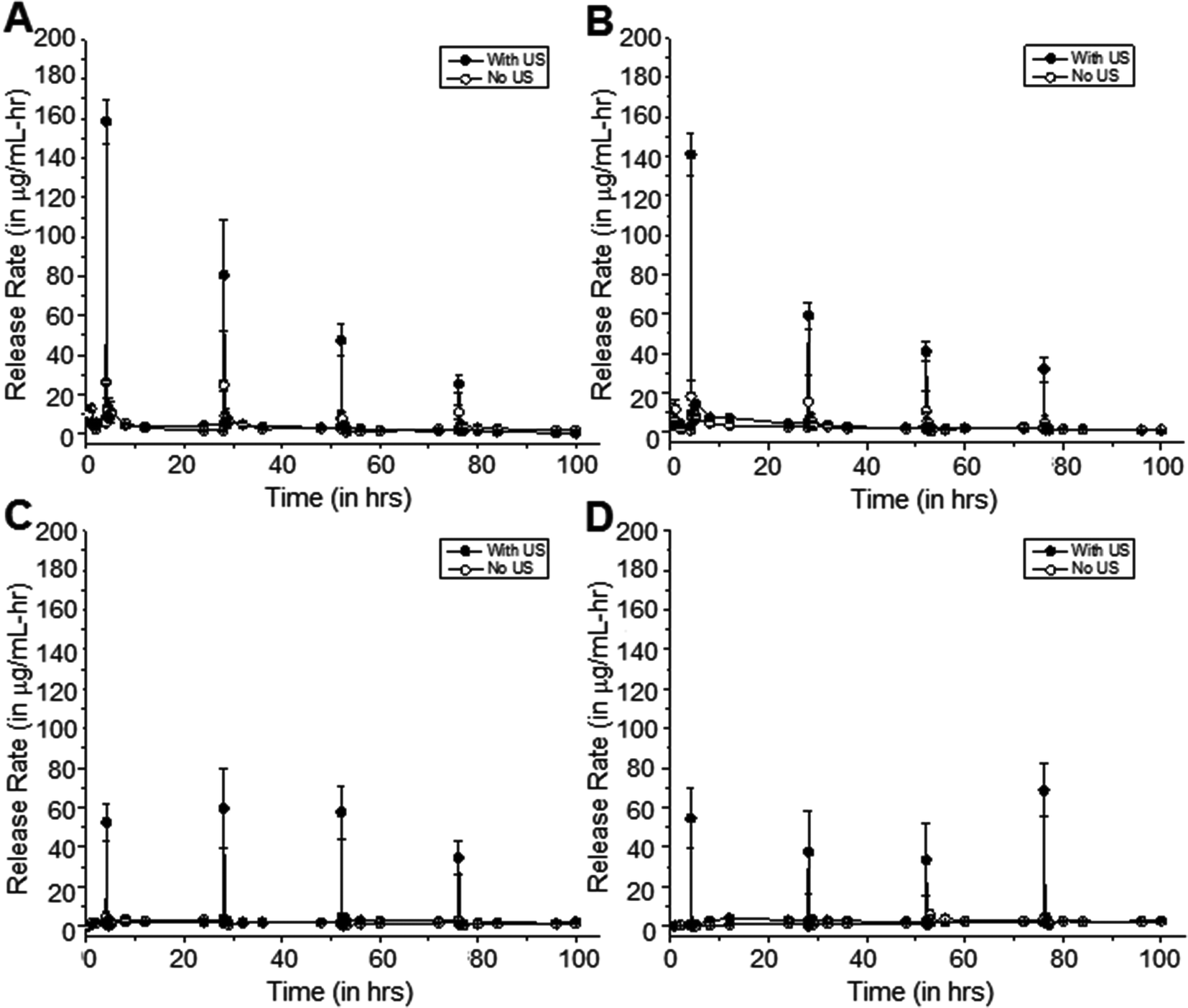 | ||
| Fig. 7 Release rates of cipro from (A) 0-minute, (B) 5-minute, (C) 15-minute and (D) 30-minute coated pHEMA matrices when exposed to 20 kHz US at 24-hour intervals. Limit of detection is 0.057 μg mL−1 determined by UV-visible spectrophotometry at λ = 270 nm. Error bars indicate SD (n = 3–5). | ||
Similarly, the differences in background release rates of cipro from samples after exposure to US are not statistically significant, on average, from that of the controls (p > 0.05). This shows that with enough time in between pulses, the methylene coating can re-heal and background release rates can return to low pre-US release rates. This observation is more apparent in samples that are more hydrated, i.e., 0-minute and 5-minute coating. In some instances, these 5-minute US exposures resulted in a constant release rate as illustrated in 15-minute coated samples showing reproducible release with every US exposure (Fig. 6 and 7c). In the other cases, the release rates due to US exposure start to decrease with each successive pulse as in Fig. 7a and 7b. This could be due to the decreasing amount of solubilized drug molecules in the water within the matrix with each successive pulse.
The release rates of cipro from 30-minute coated samples exposed to US at 24-hour intervals is shown in Fig. 7d. A substantial amount of cipro was released after each US exposure, but the background rates before and after US events remain unchanged (p > 0.05).
Fig. 8 shows the average release rates normalized to the uncoated pHEMA background rates at different duty cycles. It was determined that release rates from uncoated pHEMA samples to 25% and 50% duty cycle US enhanced drug release up to 10 and 14 times the background release rate, respectively. This confirmed previous observations, namely that enhanced mass transport is observed at higher doses of US, and that higher intensities are necessary to disrupt well-packed methylene coatings in order to produce the same enhanced effect.31–33
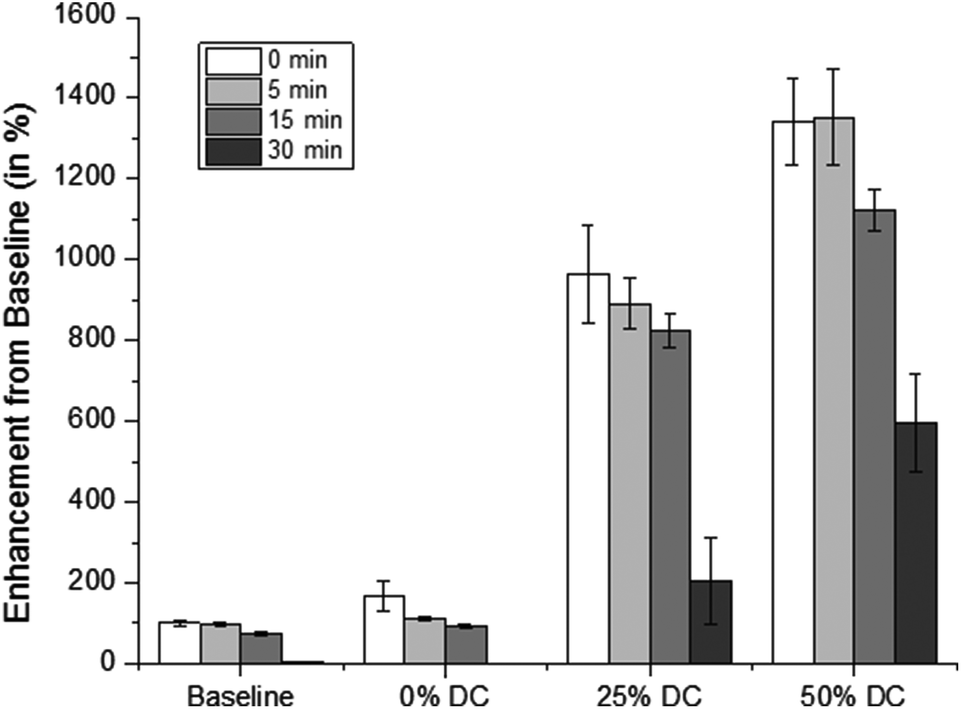 | ||
| Fig. 8 Exposure of the coated samples to 20 kHz US results in significant increase of drug release in comparison to the background rates. Each of the release rates were normalized relative to the background release rate prior to US exposure. The baseline release rates for the coated samples were normalized against the baseline release rate for the uncoated sample. | ||
Prior to US exposure, samples were rehydrated so that water could swell and plasticize the matrix, thereby dissolving the drug and forming “pockets” containing dissolved drugs within the swollen matrix. With exposure to US, cavitation in and near the matrix, and/or heating of the matrix and surrounding fluid could drive release. For example, cavitation and the ensuing acoustic streaming could disrupt the coating surrounding the matrix, and thereby enhance the influx of more water into the matrix and the diffusion or convective transport of the drug out of the matrix and into the external solution. The cipro concentration levels reached during US exposure of these pHEMA-co-HPMA matrices are several times higher than the MIC (0.125 μg mL−1) for planktonic cells. Even with the reduced swelling and release from the pHEMA-co-HPMA matrices, and 30-minute coated pHEMA (from Fig. 6d) samples at 1.73 μg mL−1, it is still at least 15 times the MIC, which is similar to the peak serum levels (1.97 to 5.39 μg mL−1) achieved with an oral dose of 750 mg prescribed to patients suffering from bone and joint infections.34
Furthermore, US-induced temperature rise could enhance the mobility of the polymer chains and the surrounding solvent that would also enhance transport as described by free volume theory.35,36 The theory is based on the presence of void space or unoccupied volume between flexible polymer chains. When the solution heats up, the increased mobility of the polymer chains results in the creation of free-volume permitting penetrant jumps into the new void region.36
These studies were performed at room temperature, and it is possible that release may be enhanced if these studies were performed at 37 °C, or implanted in vivo. However, modification with C18 was chosen because its of higher “melting” temperature, >70 °C, compared to shorter alkyl counterparts.37 It was shown that modifying pHEMA with C12 instead of C18, and exposing them to higher temperatures instead of US pulses showed considerable cipro release at 40 °C and even more at 60 °C. While, none to minimal cipro release from 30-minute coated pHEMA was detected at 40 °C and 60 °C, respectively.38 This indicates that cipro release in vivo should not be substantial, unless it is exposed to US.
Conclusions
A controlled release system must have predictable dose/time release characteristics – an understanding of the transport mechanism involved in the movement of drug molecules out of the polymer matrix assists with prediction of release characteristics. Based on surface analysis, we have optimized attachment of an assembled methylene-chain coating via the formation of urethane bonds from the reaction of the –OH groups of pHEMA and long n-aliphatic chain isocyanates. Longer reaction times led to better packing and organization of these surface-immobilized chains that led, in turn, to reduced water uptake affecting subsequent drug release. Copolymerization of the matrix polymer with a more hydrophobic monomer, HPMA, resulted in significant reduction of the cipro background release rates from the coated matrices, and ultimately, the formation of a cipro-loaded latent drug delivery system with negligible background release rates. Exposure to different US protocols showed that the US was sufficient to consistently disrupt the methylene coatings, even those with the densest packing. The US-mediated drug release rates could readily exceed 10 times that of the background and control release rates. When US was applied in rapid succession – every 10 minutes – the samples with less methylene coatings (the 0, 5, and 15-minute-coated samples) exhibited significant background release rates that were nonetheless low compared to that induced at the time of US application. For the dense coating (the 30-minute coated samples), the background release rates could return to the pre-US release rates even after US exposure, though not consistently. With more time between US applications – 24 hours rather than 10 minutes – successful drug release was followed by negligible background release rates in all cases. This indicates that the methylene coating can re-heal on time scales greater than ten minutes, and that US energy sufficient to release drugs from the methylene coated pHEMA matrices does not permanently disrupt the coating.This paper demonstrates the feasibility of creating a pHEMA-based antibiotic delivery system that can be latent until exposure to an external stimuli, such as US. This system could be an alternative vehicle for local antibiotic therapy for biofilm-associated or device-centered infections.
Experimental
Hydrogel preparation
All chemicals were used as received. Ophthalmic grade 2-HEMA (Cat. no. 04675) and tetraethylene glycol dimethacrylate (TEGDMA) (Cat. no. 02654) were obtained from Polysciences, Inc. (Warrington, PA). 2-Hydroxypropyl methacrylate (HPMA) (Cat. no. M-120) was obtained from Scientific Polymer Products, Inc (Ontario, NY). Ethylene glycol, ammonium persulfate, sodium metabisulfite, tetrahydrofuran, octadecyl isocyanate (C18-isocyanate), and dibutyl tin dilaurate were obtained from Sigma (St. Louis, MO). Ciprofloxacin hydrochloride was supplied by Bayer (New Haven, CT).The preparation of cipro-loaded pHEMA has been described previously.1,2 Briefly, cipro was first dissolved in deionized water, then immediately frozen and lyophilized for 48 hours. The resulting powder was ground to 90 μm particles, and 100 mg of the lyophilized cipro was incorporated into the monomer mixtures of HEMA and HPMA (5 mL:0 mL (100:0 HEMA-co-HPMA), 4 mL:1 mL (80:20), and 3.33 mL:1.67 mL (67:33) vol/vol ratio), ethylene glycol (1.5 mL), water (1 mL), and TEGDMA (0.23 mL), which was used as a cross-linking agent. Aqueous solutions of ammonium persulfate (40% w/v, 0.5 mL) and sodium metabisulfite (15% w/v, 0.5 mL) were added to initiate free radical polymerization. The mixture was then cast between two clean glass plates, separated by 0.015′′ Teflon spacers, and left to polymerize overnight. 10 mm round disks were punched from the polymer sheets, and washed in water for 4 h to remove unreacted monomers and initiators. Then, the samples were vacuum-dried for at least 48 h.
Surface modification with octadecyl isocyanate
Previously, Kwok described the surface modification of the pHEMA with C18-isocyanate.1,2 Changes to the protocols were made to increase the efficiency of the reaction. Under an anhydrous nitrogen atmosphere (glove box), a stock solution of 6 mL C18-isocyanate, 180 μL dibutyl tin dilaurate, and 90 mL anhydrous THF was prepared. 10 mL of this isocyanate solution was then aliquoted into 15 mL pressure vessels that contained a stir bar and dry cipro-loaded pHEMA disks. The vessels were removed from the glove box, and placed in a 50 °C oil bath. After allowing the samples to react for 0, 5, 15, and 30 minutes, they were retrieved, transferred to fresh THF, and sonicated for 2 minutes to remove any adsorbed isocyanate on the surface. Samples were blown dry with nitrogen, and then vacuum-dried overnight.Surface characterization
X-ray photoelectron spectroscopy (XPS) is technique used to quantitatively measure the atomic composition of a surface. An X-ray source is used to irradiate a sample, resulting in the emission of photoelectrons that correspond to specific elements, indicated by their binding energies. Survey and C1s high resolution scans were performed for samples at depths between 50 and 80 Å using a Surface Science Instrument S-Probe with an Al K-alpha X-ray source. Two 800 μm spots were analyzed on each sample, and the spectra obtained were analyzed using Service Physics ESCA VB data reduction software to identify and calculate the area under each peak using a linear background function. The binding energies were normalized to the C1s binding energy (BE) of 285 eV. From the high resolution carbon C1s scans, various carbon species were identified by resolving the carbon C1s peak into separate Gaussian peaks using a least-squares fitting routine in the SSI software. Each peak was referenced to the hydrocarbon peak at 285 eV to measure BE shifts. Peaks were fitted with a 100% Gaussian line function and a Shirley function was utilized for the background model.To support the quantitative data obtained from XPS, Fourier transform infrared-attenuated total reflectance (FTIR-ATR, or also ATR) spectroscopy was used to determine alkyl chain orientation and packing. FTIR-ATR measurements were carried out using Harrick's GATR single angle reflection accessory in conjunction with a Bruker Tensor spectrometer. A spectrum was collected with a minimum of 80 scans, 4 cm−1 resolution, and 65° incident angle. The modification of the surface by the isocyanate reaction was determined by monitoring peaks in the 1250 cm−1 and 1060 cm−1 regions corresponding to N–CO–O stretching from the resulting urethane bond, and the 1170 cm−1 and 1080 cm−1 regions corresponding to CO–O and O–C–C stretching from the methacrylate bonds. In addition, a shift of the CH2 stretching peak, around 2920 cm−1 indicated a change in packing order.
Swelling studies
After 48 hours of vacuum-drying, samples were weighed and placed in 2 mL of phosphate buffered saline (PBS) at room temperature. After 24 hours, the samples were removed from PBS, blotted with Kimwipes, and weighed. The amount of water in the sample was calculated using eqn (1).Release studies
The amount of cipro released in 2 mL of PBS at room temperature at specified time points was determined by measuring the absorbance of the solution using a plate reader equipped with a UV-vis detector (SAFIRE II, Tecan Systems Inc, San Jose, CA). The absorbance at λmax = 270 nm was plotted against known concentrations (between 0.1 μg mL−1 and 1 μg mL−1), and the best-fit equation was obtained. The limit of detection was found to be 0.057 μg mL−1. Based on this calibration, the concentration of the solution and the mass of released cipro was determined, and the release rate was calculated using eqn (2).Ultrasound release studies
A 20 kHz ultrasonic liquid processor (VCX 500, Sonics and Materials) with a 3 mm transducer tip was used as the US source. The amplitude was set at 20% of the maximum tip displacement. The transducer tip was immersed in the PBS solution, and positioned about 5 mm from the polymer sample. For the first study, the sonicator was pulsed at three different duty cycles: 50% (1 s on, 1 s off), 25% (1 s on, 3 s off), and 0%. The increase in the water temperature at the end of each US exposure was recorded, and the estimated time average intensity was calculated using eqn (3). After allowing the samples to re-hydrate in PBS for 4 hours, the samples were exposed to US for 5 minutes, and the change in temperature of the solution was recorded. After which, samples were moved to fresh buffer every 2.5 minutes for 10 minutes, and then the samples were exposed to US again. The samples were exposed to US for four times total (four cycles: 5 minutes on, 10 minutes off).In the second study, 20 kHz US at 50% duty cycle was applied for 5 minutes, and samples were moved to fresh buffer every 15 minutes for the first hour, and at 4 hours, 8 hours, 16 hours, and 24 hours after the previous pulse. Similarly, the samples were exposed to US for four times total, and the increase in temperature of the solution was recorded. The concentration of the solution, the amount of cipro released and the release rates from the samples were determined as described above using Equation (2). A two-sample ANOVA and t-tests were used to determine if the differences in the release rates from different samples were statistically significant.
Acknowledgements
This work is supported by grants from the National Science Foundation (EEC-9529161 and EEC-0121881) and the National Institutes of Health Biomaterials Training Grant (GC201-03-Z2406).References
- C. S. Kwok, P. D. Mourad, L. A. Crum and B. D. Ratner, Biomacromolecules, 2000, 1, 139–148 CrossRef CAS.
- C. S. Kwok, P. D. Mourad, L. A. Crum and B. D. Ratner, J. Biomed. Mater. Res., 2001, 57, 151–164 CrossRef CAS.
- P. Norris, M. Noble, I. Francolini, A. M. Vinogradov, P. S. Stewart, B. D. Ratner, J. W. Costerton and P. Stoodley, Antimicrob. Agents Chemother., 2005, 49, 4272–4279 CrossRef CAS PubMed.
- J. M. Saul, M. D. Ellenburg, R. C. de Guzman and M. Van Dyke, J. Biomed. Mater. Res., Part A, 2011, 98, 544–553 CrossRef PubMed.
- E. De Giglio, S. Cometa, M. A. Ricci, D. Cafagna, A. M. Savino, L. Sabbatini, M. Orciani, E. Ceci, L. Novello, G. M. Tantillo and M. Mattioli-Belmonte, Acta. Biomater., 2011, 7, 882–891 CrossRef CAS PubMed.
- S. Garty, R. Shirakawa, A. Warsen, E. M. Anderson, M. L. Noble, J. D. Bryers, B. D. Ratner and T. T. Shen, Invest. Ophthalmol. Vis. Sci., 2011, 52, 6109–6116 CAS.
- S. Heilmann, S. Kuchler, C. Wischke, A. Lendlein, C. Stein and M. Schafer-Korting, Int. J. Pharm., 2013, 444, 96–102 CrossRef CAS PubMed.
- G. Deepa, A. K. Thulasidasan, R. J. Anto, J. J. Pillai and G. S. Kumar, Int. J. Nanomedicine, 2012, 7, 4077–4088 CAS.
- I. M. El-Sherbiny and H. D. Smyth, Mol. Pharm., 2012, 9, 269–280 CrossRef CAS PubMed.
- S. Petersen, J. Hussner, T. Reske, N. Grabow, V. Senz, R. Begunk, D. Arbeiter, H. K. Kroemer, K. P. Schmitz, H. E. Meyer Zu Schwabedissen and K. Sternberg, J. Mater. Sci. Mater. Med., 2013, 24, 2589–2600 CrossRef CAS PubMed.
- G. Zelichenko, D. Steinberg, G. Lorber, M. Friedman, B. Zaks, E. Lavy, G. Hidas, E. H. Landau, O. N. Gofrit, D. Pode and M. Duvdevani, J. Endourol., 2013, 27, 333–337 CrossRef PubMed.
- N. Daneshpour, R. Collighan, Y. Perrie, P. Lambert, D. Rathbone, D. Lowry and M. Griffin, Eur. J. Pharm. Biopharm., 2013, 83, 106–113 CrossRef CAS PubMed.
- T. Bjarnsholt, APMIS Suppl., 2013, 1–51 CrossRef PubMed.
- A. K. Seth, M. R. Geringer, S. J. Hong, K. P. Leung, T. A. Mustoe and R. D. Galiano, J. Surg. Res., 2012, 178, 330–338 CrossRef PubMed.
- G. Cammarota, M. Sanguinetti, A. Gallo and B. Posteraro, Aliment. Pharmacol. Ther., 2012, 36, 222–230 CrossRef CAS PubMed.
- K. M. Jorgensen, T. Wassermann, P. O. Jensen, W. Hengzuang, S. Molin, N. Hoiby and O. Ciofu, Antimicrob. Agents Chemother., 2013, 57, 4215–4221 CrossRef PubMed.
- B. Jerman, M. Butala and D. Zgur-Bertok, Antimicrob. Agents Chemother., 2005, 49, 3087–3090 CrossRef CAS PubMed.
- R. Davis, A. Markham and J. A. Balfour, Drugs, 1996, 51, 1019–1074 CrossRef CAS PubMed.
- E. L. Schuck, A. Dalhoff, H. Stass and H. Derendorf, Infection, 2005, 33(Suppl 2), 22–28 CrossRef CAS PubMed.
- E. Brynda, M. Stol, V. Chytry and I. Cifkova, J. Biomed. Mater. Res., 1985, 19, 1169–1179 CrossRef CAS PubMed.
- E. M. Anderson, M. L. Noble, S. Garty, H. Ma, J. D. Bryers, T. T. Shen and B. D. Ratner, Biomaterials, 2009, 30, 5675–5681 CrossRef CAS PubMed.
- E. A. Disalvo, A. M. Bouchet and M. A. Frias, Biochim. Biophys. Acta, 2013, 1828, 1683–1689 CrossRef CAS PubMed.
- D. E. Gregonis, C. M. Chen and J. D. Andrade, in Hydrogels for Medical and Related Applications, ed. J. D. Andrade, ACS Symposium Series, Washington, DC, 1976, pp. 88–104 Search PubMed.
- B. D. Ratner and I. F. Miller, J. Biomed. Mater. Res., 1973, 7, 353–367 CrossRef CAS PubMed.
- J. Siepmann and N. A. Peppas, Adv. Drug Delivery Rev., 2001, 48, 139–157 CrossRef CAS.
- T. J. Mason, Practical Sonochemistry: User's Guide to Applications in Chemistry and Chemical Engineering, Ellis Horwood, West Sussex, 1991 Search PubMed.
- V. Zderic, S. Vaezy, R. W. Martin and J. I. Clark, Ultrasound Med. Biol., 2002, 28, 823–829 CrossRef.
- C. Aschkenasy and J. Kost, J. Control. Release, 2005, 110, 58–66 CrossRef CAS PubMed.
- B. E. Polat, D. Hart, R. Langer and D. Blankschtein, J. Control. Release, 2011, 152, 330–348 CrossRef CAS PubMed.
- M. S. Aw and D. Losic, Int. J. Pharm., 2013, 443, 154–162 CrossRef CAS PubMed.
- L. S. Liu, J. Kost, A. D'Emanuele and R. Langer, Macromolecules, 1992, 25, 123–128 CrossRef CAS.
- I. Lavon and J. Kost, J. Control. Release, 1998, 54, 1–7 CrossRef CAS.
- C. M. Agrawal, M. E. Kennedy and D. M. Micallef, J. Biomed. Mater. Res., 1994, 28, 851–859 CrossRef CAS PubMed.
- M. B. Kays, B. R. Overholser, B. A. Mueller, S. M. Moe and K. M. Sowinski, Am. J. Kidney Dis., 2003, 42, 1253–1259 CrossRef CAS PubMed.
- J. S. Vrentas, J. L. Duda and H. C. Ling, J. Polym. Sci., 1985, 23, 275–288 CAS.
- N. A. Peppas and S. R. Lustig, in Hydrogels in Medicine and Pharmacy, ed. N. A. Peppas, CRC Press, Inc., Boca Raton, FL, 1986, vol. I. Fundamentals, pp. 57–83 Search PubMed.
- C. S. Kwok, PhD Thesis, University of Washington, 2001 Search PubMed.
- M. L. Noble, PhD Thesis, University of Washington, 2009 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |