Characterization of a multilayer heparin coating for biomolecule presentation to human mesenchymal stem cell spheroids†
J.
Lei
a,
L. T.
McLane
b,
J. E.
Curtis
bc and
J. S.
Temenoff
*cd
aWoodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, Georgia, USA
bSchool of Physics, Georgia Institute of Technology, Atlanta, Georgia, USA
cWallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, Georgia, USA. E-mail: johnna.temenoff@bme.gatech.edu
dParker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA, USA
First published on 19th March 2014
Abstract
Mesenchymal stem cell (MSC) therapies have the potential to treat many pathologies, however, controlling cell fate after implantation remains challenging. We have used a multilayer technology to graft a range of 5 μg mL−1–5 mg mL−1 heparin onto the surface of MSC aggregates. Heparin coating does not affect cell viability (seen through LIVE/DEAD staining) and cell anti-inflammatory properties (seen through co-culture with activated monocytes) and facilitates sequestration by coated cells of a growth factor (TGF-β1) that remains bioactive. This system can maximize the therapeutic potential of MSC-based treatments because the cell surface-loaded protein could both signal the cells to influence transplanted cell fate and be released into the surrounding environment to help repair injured tissue.
Introduction
Mesenchymal stem cells (MSCs) have been used in cell therapy to regenerate injured tissue and treat inflammation resulting from a wide range of pathologies such as cardiovascular disease, myocardial infarction, osteoarthritis, brain and spinal cord injury, diabetes, Crohn's disease and graft versus host disease.1,2 These multipotent adult progenitor cells are capable of differentiating into bone, cartilage, fat, muscles, tendons/ligaments, and other connective tissue cell types, which can be mediated through local soluble factors and/or mechanical stimulation.3 In addition to their differentiation capacity, MSCs have the ability to secrete cytokines and growth factors that can promote angiogenesis, modulate extracellular matrix (ECM) remodelling, affect immune cell activation or suppression (immunomodulation) and promote cell recruitment.1–4 Recently, there is growing evidence that, when aggregated into spheroids, MSCs have enhanced anti-inflammatory properties over single MSCs grown in monolayers.5 These anti-inflammatory effects are useful in treating inflammatory pathologies and regulating the damage that can be caused by inflammation in an acute injury.Upon injection or placement into an injured environment, MSCs are able to respond to the milieu surrounding them and secrete the appropriate mediators to repair the tissue or begin to differentiate to a tissue-specific cell type.4,6 However, MSC-based therapies may be rendered ineffective due to the lack of control over cell fate upon administration into a complex, pathological environment. Further compounding the problem is the fact that the injured environment may contain many proteases or highly inflammatory molecules that can harm the injected cells.7
Controlled differentiation of stem cells has been achieved by cell seeding on scaffolds that mimic the architecture and mechanical properties of the native extracellular matrix in tissues in the presence of soluble factors.8 However, fibrous scaffolds do not retain ECM secreted by cells, which can limit their ability to fully repair tissue.9 Additionally, processing that requires specialized equipment to synthesize scaffold materials (i.e. electrospinning) makes this method difficult to scale up for cell therapies. Finally, scaffold-based technologies cannot be utilized as a minimally invasive injectable treatment, and thus, require a full surgical implantation for cell delivery.9,10
Another method of controlled differentiation is by encapsulating cells in injectable hydrogels.11–13 Hydrogels can be functionalized to be biodegradable, contain motifs that drive differentiation, and provide a 3D environment that can retain secreted extracellular matrix molecules.12 However, limitations of using a hydrogel system include the polymerization requirement that typically involves an extra radical initiator that could potentially be cytotoxic to encapsulated cells, the excess void space of the material which increases the required volume injected and the lack of temporal and spatial control of degradation, which may be required for full tissue integration.14
In response to these shortcomings, we aim to develop a thin, conformal coating method for MSC spheroids to allow simultaneous injection of both cells and a therapeutic agent to a site of injury. This system uses multilayer deposition of biotin and avidin to graft heparin onto cell surfaces.15,16 Heparin is a negatively charged glycosaminoglycan that can sequester and release positively charged proteins.17 It has been used as an anti-coagulant, as a drug delivery system and as a component of hydrogels to help drive differentiation in multiple cell types, including MSCs and embryonic stem cells.18–20 It is envisioned that the negative charge on this coating will allow for preloading of a growth factor of interest to both guide stem cell differentiation, as well as release the loaded biomolecule to facilitate local tissue repair. These coating layers are advantageous over using scaffolds or bulk hydrogels for guiding cell fate because of their simple assembly through nontoxic interactions (i.e. noncovalent, electrostatic) and precise tuning of thickness, composition and permeability, ultimately allowing for control over coating degradation (and concomitant biomolecule release).21–23 Moreover, such cell coating technologies would greatly enable injectable therapies, as there is no excess material that needs to be delivered with the cells. Toward developing this coating methodology, the objectives of this study were to characterize the coating and loaded protein via fluorescence microscopy and a particle exclusion assay, determine cell viability after coating, investigate MSC anti-inflammatory properties after coating, and determine the bioactivity of a growth factor (TGF-β1) loaded on cell surfaces.
Methods
Cell culture
MSCs derived from bone marrow aspirates were obtained from The Texas A&M Health Sciences Center. Cryopreserved MSCs were thawed and expanded in alpha minimum essential medium (α-MEM, Mediatech, Herndon, VA) containing 16.5% fetal bovine serum (FBS, Atlanta Biological, Atlanta, GA), 2 mM L-glutamine (Mediatech), 100 units mL−1 penicillin and 100 μg mL−1 streptomycin (Mediatech). Media were changed every 2–3 days until 80% confluent and were used at passage 3 or 4. Human THP-1 monocytes (ATCC, Manassas, VA) were expanded in RPMI 1640 (Mediatech) containing 10% FBS, 2 mM L-glutamine, 100 units mL−1 penicillin and 100 μg mL−1 streptomycin and 0.05 mM 2-β-mercaptoethanol (Sigma, St. Louis, MO). Media were replaced every 4 days until seeding in co-culture systems. Mink lung epithelial cells (MLECs, ATCC) were thawed and expanded in Dulbecco's modification of Eagle's medium (DMEM) with 4.5 g mL−1 glucose, 10% FBS, 2 mM L-glutamine and 200 μg mL−1 Geneticin® (G418, Gibco, Grand Island, NY). Media were exchanged every 2–3 days until cells were 80% confluent and were used at passage 29.MSC spheroid formation
To make spheroids, cells were forced into aggregation via centrifugation.24 Briefly, a single cell suspension (3–6 × 106 cells mL−1) was added to AggreWell™ agarose microwell inserts (400 μm wide wells; ∼6000 wells per insert) and was centrifuged (200 rcf; 5 minutes) to aggregate cells within the microwells. After 24 hours, MSC aggregates were rinsed from the microwells and cultured thereafter either encapsulated in a 1.5% alginate layer (approximately 3 mm thick) crosslinked in 100 mM calcium chloride contained in a 10 cm petri dish (Corning, Tewksburg, MA) for up to 14 days or in a rotary orbital suspension culture for up to 3 days to inhibit agglomeration of individual aggregates (approximately 1500 spheroids per 10 mL media per petri dish; 65 rpm for rotary suspension only).Multilayer coating and subsequent cell viability
MSCs and MLECs were cultured until 80% confluent and lifted using 0.05% trypsin (Mediatech). Cells were washed in phosphate buffered solution (PBS, Life Technologies, Grand Island, NY) 2 times and then modified with heparin by multilayer assembly of biotin and avidin layers.15,16 Cells were first cultured in 4 mM EZ-Link Sulfo-NHS-LC-Biotin (Pierce, Rockford, IL) in PBS, followed by 0.5 mg mL−1 avidin in PBS (Life Technologies, Carlsbad, CA), and lastly in 5 μg mL−1, 1 mg mL−1 or 5 mg mL−1 biotin-conjugated heparin in PBS. Each incubation was performed in an Ultra-Low Attachment Surface plate (Corning) for 30 minutes at 37 °C. For imaging studies, cells were incubated in a biotin–heparin solution that has been tagged with AlexaFluor 633 (Invitrogen). To load coated surfaces with a protein after the heparin layer, cells were incubated for 30 minutes in a 90 μg mL−1 fluorescein isothiocyanate (FITC)-tagged histone (Sigma) solution for imaging studies or 3 ng mL−1 or 3 μg mL−1 TGF-β1 (Peprotech, Rocky Hill, NJ) solution for MLEC luminescence studies. Once multilayer deposition of all layers was complete, all cells were washed in PBS and formed into spheroids (500 or 1000 cells per spheroid) as previously described. Spheroids were stained with LIVE/DEAD solution (1 μM calcein AM and 1 μM ethidium homodimer-1) to assess cell viability. Images were taken using a Zeiss LSM 700 LSM confocal microscope and channels were merged and projections were flattened in ImageJ (NIH, Bethesda, MD).Particle exclusion assay
To characterize the pericellular matrix (PCM), 3 μm passivated (covalently modified with methoxypolyethylene glycol amine) polystyrene microspheres (Life Technologies, supplied by Curtis laboratory) were used in a particle exclusion assay on a monolayer of MSCs.25 Beads with diameter of 3 μm are of the same order of magnitude in size as other particles excluded by the pericellular matrix, as seen in previous experiments.25 MSCs were seeded at 500 cells cm−2 on 12 mm glass coverslips and cultured for 24 hours. Cells were then modified via multilayer deposition with 5 mg mL−1 biotin–heparin as described above and then suspended in PBS. Control samples were handled identically, but not treated with the multilayer coating. After coating, 20 μL of the bead solution and 180 μL of PBS were added to a custom imaging mold (Teflon) containing seeded MSCs. Beads were allowed to settle for 10 minutes. Bright field imaging was performed using a Zeiss LSM 700 confocal microscope. The effective thickness of the PCM was measured as a separation between the beads and the cell surface using ImageJ (3 measurements per cell).Monocyte anti-inflammatory co-culture assay
To examine anti-inflammatory effects of spheroid culture with and without coating, MSCs were co-cultured with LPS-activated human monocytes.5,26,27 MSCs were coated and formed into spheroids as previously described and cultured in a rotary orbital suspension for 3 days. MSC monolayer and 1000-cell spheroids (coated and noncoated) were then indirectly co-cultured with THP-1 monocytes in a 12-well Transwell system (Corning) at a ratio of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MSCs to 400000 THP-1 monocytes. All cells were suspended in RPMI 1640 media. Monocytes were activated by addition of lipopolysaccharide (LPS, 400 ng mL−1) to the media. After 16 hours, the MSC monolayer and the spheroid supernatant were collected and tumour necrosis factor (TNF)-α levels were quantified via ELISA (R&D Systems, Minneapolis, MN).
000 MSCs to 400000 THP-1 monocytes. All cells were suspended in RPMI 1640 media. Monocytes were activated by addition of lipopolysaccharide (LPS, 400 ng mL−1) to the media. After 16 hours, the MSC monolayer and the spheroid supernatant were collected and tumour necrosis factor (TNF)-α levels were quantified via ELISA (R&D Systems, Minneapolis, MN).
Mink lung epithelial cell luminescence assay
MLECs were used as a reporter cell line to study the effects of the coating on TGF-β1 signalling to cells. MLECs were coated with heparin at the concentration of 5 mg mL−1 and then loaded with TGF-β1 at concentrations of 0 ng mL−1, 3 ng mL−1 and 3 μg mL−1 by incubating heparin-coated cells in TGF-β1 solutions for 30 minutes, similar to the previously discussed coating experiment. Noncoated controls were also exposed to TGF-β1 at the previously mentioned concentrations using identical methods. After coating and growth factor loading, all cells were formed into aggregates (500 cells per aggregate) using the technique previously described. At 24 hours, cells were transferred to a 96-well plate (150000 cells per well) and ONE-GloTM Luciferase Assay buffer was added to each well using manufacturer's protocols. Luminescence was measured using a plate reader (BioTek Synergy H4 Hybrid Multi-Mode Microplate Reader, Winooski, VT). Simultaneously, to study the bioactivity of released protein, supernatant from aggregates was also collected 24 hours after aggregate formation. 100 μL aliquots of each supernatant sample were suspended over noncoated MLECs plated in monolayer at 50000 cells cm−2 in a 96-well plate and cultured for 24 hours. The following day, 100 μL of ONE-GloTM Luciferase Assay buffer was added according to manufacturer's protocols and luminescence levels were measured using a plate reader (BioTek).
Statistical analysis
All values were reported as mean ± standard deviation. For statistical analysis, one or two factor analysis of variance (ANOVA) was used to determine statistical significance of groups, and Tukey's post hoc multiple comparison test with significance set at p < 0.05 indicated a significance between individual samples. Analysis was performed using Minitab (v.15.1, State College, PA).Results
MSC spheroid coating
MSCs were successfully coated using a multilayer technique with deposition of biotin and avidin to graft molecules of interest (heparin and a protein) onto their surfaces and subsequently formed into spheroids (Fig. 1A). Using a dimethyl methylene blue assay, it was seen that biotinylated heparin was 90.2% ± 3.9% sulphated when compared to native heparin. Additionally, nuclear magnetic resonance analysis revealed that the sugar structure was 53% conjugated with biotin at the carboxyl groups. The viability of the resulting coated spheroids does not appear to be affected when compared to the viability of noncoated spheroids, as most of the cells in the aggregates in all cases were with calcein dye, indicating that the cells are alive (Fig. 1B). A higher initial coating concentration results in more heparin being grafted to cell surfaces throughout the entire spheroid when compared to spheroids coated with heparin at a lower concentration, seen by fluorescent quantification via image analysis (Fig. 1C & D). Heparin fluorescence decreases over 14 days, resulting in approximately a 40% loss of heparin from all three groups when compared to the amount of heparin found on surfaces at day 1 (Fig. 1D). After coating, a model positively-charged protein, histone (∼19 kDa, pI = 8.5), tagged with FITC, was loaded onto spheroid cell surfaces. It was seen that histone was present on cell surfaces at day 1 and remained up to day 7. Without the intermediate biotin and avidin layers, heparin was not grafted to cell surfaces, resulting in minimal protein loading on cell surfaces (Fig. 1E).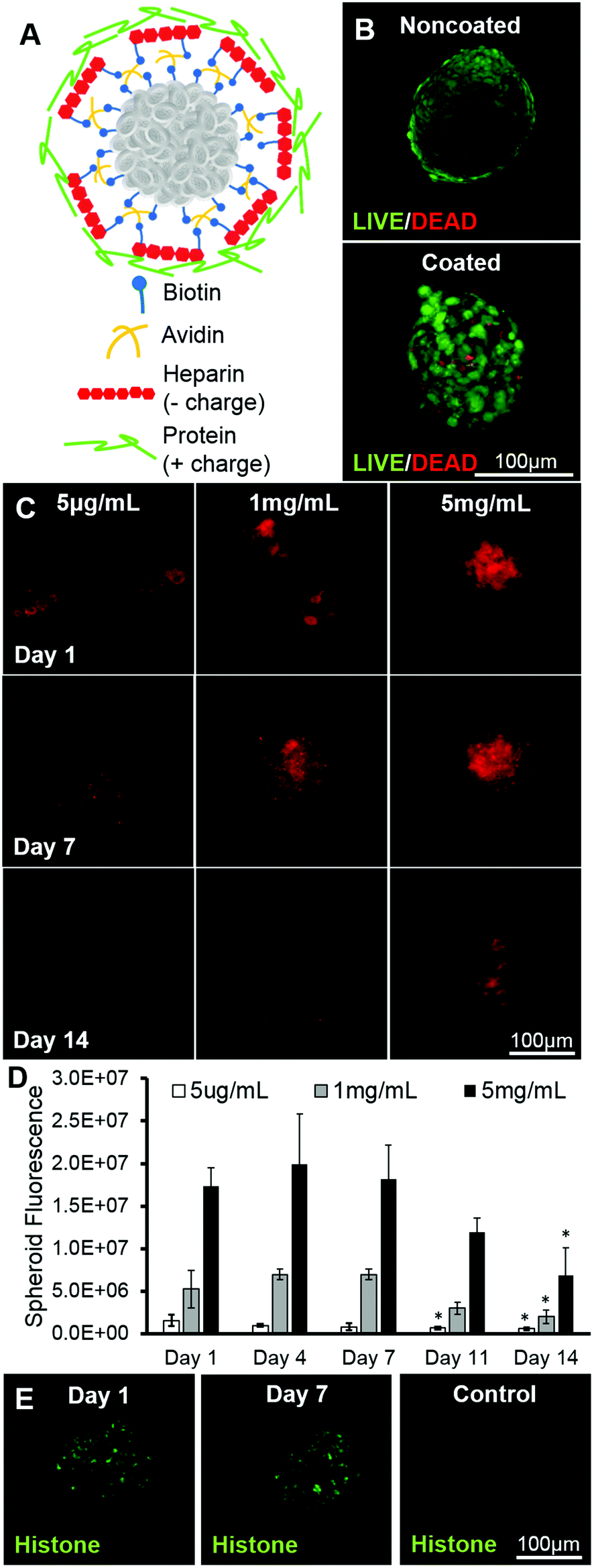 | ||
| Fig. 1 Characterization of heparin coating and histone loading via layer-by-layer deposition on cell surfaces. (a) Schematic of coating. (b) High cell viability in both coated and noncoated spheroids as visualized with flattened image of LIVE/DEAD staining. (c) Flattened images of Alexafluor 633-tagged heparin coating at 5 μg mL−1, 1 mg mL−1 and 5 mg mL−1 coating concentrations over 14 days. (d) Quantification of fluorescence/spheroid from tagged heparin over 14 days. (e) FITC-histone loaded on heparin-coated cells at day 1 (left) and day 7 (middle) and non-heparin coated control cells at day 1 (right). *Statistically different from day 1 levels at the same concentration; p < 0.05; n = 10–12. | ||
Particle exclusion assay
Many cell surfaces are decorated with surface bound hyaluronan (a long, linear glycosaminoglycan) that acts to densely aggregate native proteoglycans (aggrecan, versican) possessing a very high negative charge. This hierarchical assembly of bottlebrushes anchored to the cell surface makes up the PCM.28,29 We performed a particle exclusion assay to determine whether the native PCM is altered after multilayer coating of cell surfaces. Immediately after coating MSCs in monolayer (day 0), there was no PCM observed around coated cell membranes, as evidenced by the lack of separation between the beads and cell membrane, as opposed to the noncoated MSCs in which a PCM thickness of approximately 6 μm was observed (Fig. 2A). 24 hours after coating (day 1), coated samples still did not indicate the presence of any PCM, while noncoated samples remained similar to the previous day. Both immediately after coating (day 0) and 24 hours after coating (day 1), noncoated samples had a significantly thicker PCM than coated samples (Fig. 2B).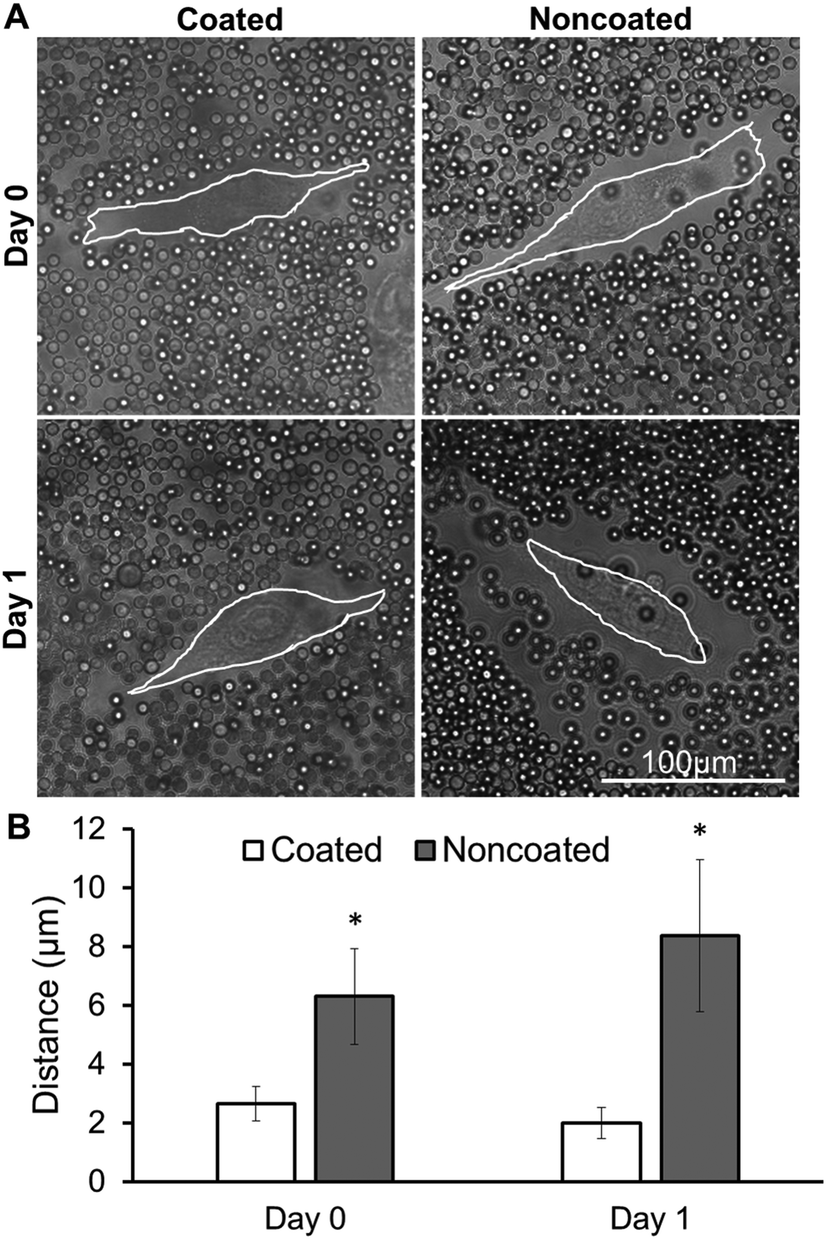 | ||
| Fig. 2 Particle exclusion analysis of coated and noncoated MSCs. (a) Phase images of coated and noncoated MSCs in monolayer with 3 μm passivated polystyrene microspheres. White lines outline cell surfaces. (b) Quantification of PCM thickness (distance between cell surface and microsphere layer). *Statistically different from coated samples on the same day; p < 0.05; n = 18. | ||
Monocyte anti-inflammatory co-culture assay
In this experiment, the effects of the heparin coating on MSC spheroid anti-inflammatory properties were studied through a monocyte co-culture assay. Both noncoated and coated spheroids were able to attenuate the level of secreted TNF-α from LPS-activated monocytes significantly more than MSCs seeded in monolayer, as seen by greater changes in TNF-α levels normalized to MSC DNA content (Fig. 3). No difference was seen between the two spheroid groups.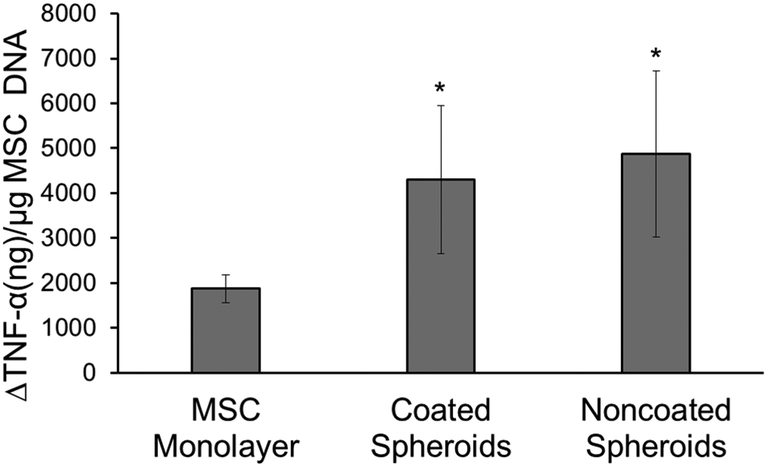 | ||
| Fig. 3 Degree of attenuation of TNF-α secretion by LPS-activated monocytes in co-culture with MSCs in monolayer, coated MSC spheroids, or noncoated spheroids (reported as a change from levels of TNF secreted from activated monocytes not in co-culture with MSCs divided by MSC amount in each culture as determined by picogreenTM assay). *Statistically different from MSC monolayer; p < 0.05; n = 5. | ||
TGF-β1 bioactivity (MLEC assay)
To study the biological activity of a protein localized to cell surfaces via heparin coating, MLECs were used as a biological reporter of loaded TGF-β1 activity. MLECs are a cell type that has been transfected with a luciferase reporter gene fused to plasminogen activator inhibitor-1 (PAI-1), which is expressed in response to TGF-β1. It has been seen that TGF-β1 activity results in a dose-dependent increase in the quantified luciferase activity.30 First, it was seen that MLECs can be coated with heparin and formed into aggregates using the previously utilized methods, without having detrimental effects on cell viability (Fig. 4A & B). When heparin was not present, there is minimal protein localized to cell surfaces (Fig. 4C); however, when the heparin layer was present, a positively charged model protein (histone) was observed on the surfaces of the cells (Fig. 4D). Noncoated MLECs exhibited a dose-dependent response to TGF-β1 exposure. Coated MLECs had significantly higher luminescence reported when compared to noncoated samples at each loading concentration after 24 hours (Fig. 4E).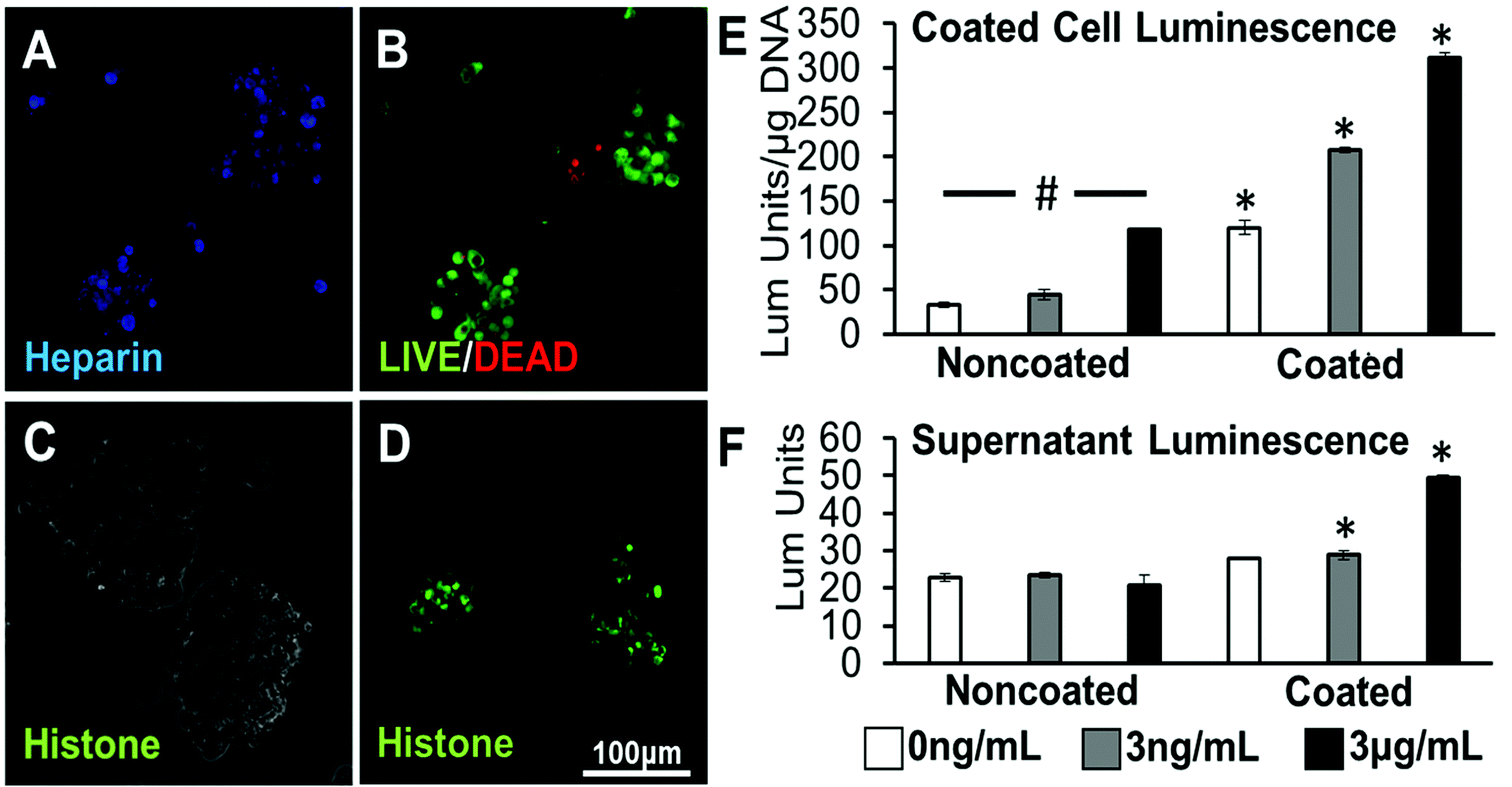 | ||
| Fig. 4 Coated MLEC aggregates loaded with TGF-β1. (a) Flattened image of Alexafluor 633-tagged heparin coating on MLEC aggregates after layer-by-layer deposition of biotin and avidin. (b) High cell viability in coated aggregates as visualized with flattened image of LIVE/DEAD staining. (c) No FITC-tagged histone is observed on control MLEC aggregates without heparin coating (phase image). (d) Flattened image of FITC-tagged histone loaded on heparin-coated aggregates. (e) Luminescence response of noncoated and coated MLEC aggregates after loading. (f) Luminescence response resulting from released supernatant from noncoated and coated MLEC aggregates collected 24 hours after TGF-β1 loading and aggregate formation. *Statistically different from noncoated samples at the same concentration; #statistically different from all other concentration samples within the same coating treatment; p < 0.05; n = 4. | ||
In a second bioactivity assay, the supernatant collected from noncoated and coated MLEC aggregates 24 hours after protein loading was suspended over a different set of MLECs plated in 96-well plates. The supernatant collected from all noncoated spheroid groups 24 hours after formation elicited a low level of luminescence response that was not significantly different from each other. However, supernatants taken from TGF-β1 loading concentrations of 3 ng mL−1 or 3 μg mL−1 elicited a luminescence response significantly higher than their noncoated counterparts (Fig. 4F).
Discussion
In this study, a multilayer assembly technique was applied to form a conformal coating of heparin on MSCs, with the ultimate goal of grafting a biomolecule of interest on cell surfaces that may control cell behaviours, such as differentiation. A similar technique was used previously to graft unmodified heparin or thrombomodulin onto the surface of pancreatic islets to prevent blood-mediated inflammation or coagulation upon injection into the bloodstream.15,16 We have used a modification of this system to sequester a protein of interest onto the surface of cells that may act as a stimulus to guide cell fate after delivery. In this method, biotin is functionalized with an NHS group that targets cell surface amines. The grafted biotin can then bind avidin with high affinity and specificity. In the following layer, while heparin can electrostatically interact with the positively charged avidin, heparin has also been further functionalized with a biotin group that can interact with the presented avidin with higher affinity. As the outer layer, heparin's highly negative charge, resulting from the presence of multiple sulphate groups, facilitates the sequestration of positively charged biomolecules via electrostatic interactions.19The amount of heparin grafted on cells can be tuned using different initial concentrations in solution. Given a fixed number of cells per spheroid, the coating concentrations chosen for this study represented levels that were below the theoretical saturation of heparin loaded onto the cell surfaces as a packed single layer (5 μg mL−1), close to the saturation level (1 mg mL−1), and well above the saturation level (5 mg mL−1) (calculations based on a cell diameter of 7.5 μm, cell surface area and heparin diameter of 10 nm).31 While it may be expected that the amount of heparin grafted would plateau near saturation at the 1 mg mL−1 concentration, the fact that increase in fluorescence beyond saturation concentration was observed (Fig. 1C) suggests that multiple layers of heparin can be deposited onto the surface, perhaps mediated through both the avidin–biotin–heparin interactions and electrostatic interactions between avidin and heparin. These results indicate that it is possible to tune the amount of heparin that can be grafted on to cell surfaces, and thus, ultimately control the amount of biomolecules being sequestered.
Over time, the heparin coating appeared to be removed from cell surfaces, as seen by lower levels of quantified fluorescence at later timepoints. While this loss could be attributed to several factors, we have observed that fluorescence of the same heparin coating in an acellular system does not decrease over time when placed in media (ESI Fig. 1†). This leads us to believe that the loss of heparin is cell-mediated via secretion of enzymes that cleave heparin or via cell membrane turnover. Non-biotin-tethered heparin molecules could also be released due to diffusion into the surrounding media or exchange of ions with other charged molecules.32 For all three concentrations, there was a 40% loss of heparin by day 14 when compared to the original amount on day 1. However, the rates of decrease were different, as a higher percentage of heparin was lost on earlier days for cells coated with 5 μg mL−1, indicating that the kinetics of heparin loss are different depending on starting concentrations (Fig. 1D). This discrepancy in release kinetics lends support to the idea of multiple heparin layers being present when coated with a concentration above the theoretical saturation concentration, some of which may be less tightly bound through electrostatic interactions and therefore diffuses away from cell surfaces more quickly. To further characterize the heparin coating, the model protein, fluorescent histone, was used to image protein sequestration. Heparin coating facilitates the localization of a model positively charged protein, seen through imaging of histone, indicating that a biomolecule can be presented to MSCs via the heparin coating that may then act on to direct cell fate.
The PCM is composed predominantly of chondroitin sulphate-rich proteoglycans and hyaluronan. In its native form, the PCM can extend several microns from the cell surface and can play a role in cell interaction with the environment and cell adhesion.25,29,33 It is important to study the presence of the PCM after the coating procedure to better understand what is being presented on the surface of the cells and how that may affect cell behaviour.34 The decrease in PCM thickness seen in coated cells compared to noncoated cells, observed in the particle exclusion assay, suggests that the coating procedure removed or collapsed (via crosslinking) the natural PCM found on cell surfaces.
It was also observed that, 24 hours after coating, an appropriate amount of time allowed the PCM to grow to its natural state in media;25 the native PCM did not grow back on coated cells (Fig. 2B). This finding confirms that using our method, the resulting heparin coating can exist on cell surfaces for at least 1 day without being overwhelmed by the natural matrix, potentially allowing for control of loaded biomolecules that are presented to the cells during that time, while simultaneously maintaining a high concentration of the loaded biomolecule near cell surface receptors for efficient signalling.
It has been shown that MSCs have anti-inflammatory properties and are able to act as modulators of lymphocytes.35,36 MSCs in spheroid form have enhanced anti-inflammatory characteristics when compared to single MSC from monolayer culture.5 The results from the co-culture experiments demonstrate that the intrinsic anti-inflammatory properties of the MSCs are not affected during the coating process or by the coating itself as both spheroid samples exhibited enhanced properties over MSCs in monolayer (Fig. 3). It has been shown that aggregation of MSCs results in the production of prostaglandin E2 (PGE2), tumour necrosis factor-inducible gene 6 protein (TSG-6) and stanniocalcin-1 (STC-1), molecules that can act on activated monocytes to decrease TNF-α secretion.27,37 PGE2 is a small molecule derivative of arachidonic acid and TSG-6 and STC-1 are large anti-inflammatory proteins (29 kDa and 25 kDa, respectively) that are not expected to penetrate through multilayers of negatively charged heparin.38–40 However, in this assay, the highly negatively charged coating did not alter the MSCs ability to attenuate TNF-α production by monocytes, suggesting that sufficient anti-inflammatory signalling was still able to occur. On the other hand, clinical evidence has also shown that heparin administration has benefitted patients with pathologies that are associated with strong inflammatory response, such as asthma, ulcerative colitis and burns.41,42 A proposed mechanism of this anti-inflammatory property is inhibition of the transcription factor nuclear factor-κB (NF-κB) that promotes the expression of pro-inflammatory cytokines in monocytes.43 Surprisingly, it was observed that a heparin coating on MSC spheroids does not increase the anti-inflammatory properties in this study when compared to noncoated spheroids. A potential explanation is that heparin may not be released from cell surfaces in high enough concentrations after 3 days in culture to act on activated monocytes,43 or that the decrease in MSC anti-inflammatory secretion was compensated for by a direct anti-inflammatory effect of heparin, resulting in no overall change from noncoated spheroids. Further studies need to be performed in order to fully study the effects of the heparin coating on changes to the anti-inflammatory secretome of MSC spheroids.
For this coating system to be effective in promoting cell differentiation, the protein must remain bioactive to act on cells or the surrounding environment. Therefore, after ascertaining that the coating was not deleterious to viability or anti-inflammatory properties of MSCs, we used a reporter cell line to study bioactivity of a growth factor (TGF-β1) sequestered to the heparin. MLECs are cell lines that are known to express PAI-1 in response to the specific presentation of TGF-β1 in the media or on cell surfaces. In these cells, the PAI-1 gene has been fused with a luciferase firefly reporter. Addition of TGF-β1 results in a dose-dependent increase of luciferase activity in the lysed cells, rendering this assay to be highly specific and sensitive.30,44 Utilizing the heparin coating, TGF-β1 was localized to the surfaces of MLECs and aggregates were formed with the coated cells (Fig. 4A–D). When noncoated, aggregated MLECs were exposed to TGF-β1, the short term exposure during the coating process resulted in a dose-dependent increase in luminescence, representing a cellular response to TGF-β1. This phenomenon reveals that aggregation of these cells does not impair their inherent response to TGF-β1 and that short term (30 minutes) exposure can activate luciferase expression.
While it is noted that the heparin coating itself, without TGF-β1 exposure, can upregulate luciferase activity (Fig. 4E), it has not been previously studied if heparin directly affects the MLEC cell type. It is known that biotin (used in the coating procedure) can act as a coenzyme that is involved in fatty acid synthesis, glucogenesis and catabolism of branched amino acids, but is unlikely to affect PAI-1 expression due to its specificity to TGF-β1.45,46 When assaying the aggregates themselves, it is evident that the combination of TGF-β1 loading and heparin coating results in a luminescence response from the cells within the aggregate that is significantly higher than the luminescence response from noncoated samples and coated, non-loaded samples (Fig. 4E) and that there are no effects of soluble heparin on activation of this cell line (ESI Fig. 2†). While promising, further studies with a cell type that is not activated by the coating procedure are necessary to fully confirm the ability of the coating to facilitate signalling of a loaded growth factor to the cells.
With the coating present, the supernatant collected from coated aggregates that were loaded with either concentration of TGF-β1 can elicit a luminescence response from reporter MLECs that is significantly higher than noncoated samples treated at the same loading concentration (Fig. 4F). These results provide evidence that the presence of heparin may help preserve the bioactivity of the TGF-β1, as previous studies have shown that heparin complexes can protect proteins from enzymatic degradation and may potentiate presentation to its target receptor.19,47,48 Such results are striking because in a control experiment, it was seen that when soluble heparin was present in the media during MLEC exposure to TGF-β1, luminescence response by the cells was significantly lower (ESI Fig. 3†), suggesting that soluble heparin may not preserve bioactivity of TGF-β1 in this assay and may even interfere with or inhibit signalling to the cells. Taken together, these data indicate that TGF-β1 can be released from the heparin coating in a bioactive form. This coating technique is advantageous over delivering soluble heparin with growth factors because the coating allows release over a longer period (in this case, at least 1 day), thus providing a sustained effect of preserving bioactivity of growth factors over time to interact with surrounding tissues. In contrast, the addition of soluble heparin will likely diffuse away and only have an effect immediately upon administration.
Based on these results, it is envisioned that the bioactive protein may be able to “signal out” to the neighbouring cells and the surrounding environment to encourage regeneration, as well as potentially “signal in” and act on the cells to which it is sequestered to promote differentiation or other functions. Thus, this multifaceted system may be used to both prime cells for cell-mediated tissue regeneration and modulate the damaged tissue environment after implantation.
Conclusions
Through these studies, we have developed a multilayer coating system to graft heparin and facilitate protein sequestration onto cell surfaces. Results demonstrate that MSC spheroids can be coated without negatively affecting cell viability and anti-inflammatory properties. Positively-charged proteins bind preferentially to the coated cells and an assay with a reporter cell line suggests that TGF-β1 remains bioactive after sequestration and release. In the future, this simple and efficient method of presenting growth factors to stem cell aggregates may have significant implications in enabling local signalling between transplanted cells and surrounding tissue post-injection. Moreover, the ease of the chemistries employed makes this technique amenable to attaching other bioactive molecules (antibodies, enzymes) to stem cell aggregate surfaces. As such, the methods presented here represent an exciting platform with sufficient flexibility to maximize the therapeutic effect of injected stem cells in ways that could be tailored to the needs of a particular pathology.Acknowledgements
We would like to acknowledge Melissa Goude for assistance with culture of spheroids, Dr Todd McDevitt and the Stem Cell Engineering Center for use of the AggreWellsTM to form cell aggregates and for providing the spheroid image, and Dr Thomas Barker and Dr Ashley Carson Brown for help with the MLEC assay. The human MSCs employed in these studies were provided by the Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine at Scott & White through a grant from NCRR of the NIH, Grant #P40RR017447. This work was also supported by an NSF Integrated Graduate Education and Research Traineeship fellowship and the NIH Cell and Tissue Engineering Training Program to J. L. and funding from NSF (DMR 1207045) to J. S. T.Notes and references
- J. Ankrum and J. M. Karp, Trends Mol. Med., 2010, 16, 203 CrossRef PubMed.
- M. E. Bernardo and W. E. Fibbe, Cell Stem Cell, 2013, 13, 392 CrossRef CAS PubMed.
- A. I. Caplan and J. E. Dennis, J. Cell Biochem., 2006, 98, 1076 CrossRef CAS PubMed.
- A. M. Dimarino, A. I. Caplan and T. L. Bonfield, Front Immunol, 2013, 4, 201 Search PubMed.
- T. J. Bartosh, J. H. Ylostalo, A. Mohammadipoor, N. Bazhanov, K. Coble, K. Claypool, R. H. Lee, H. Choi and D. J. Prockop, Proc. Nat. Acad. Sci. U. S. A., 2010, 107, 13724 CrossRef CAS PubMed.
- B. E. Strauer, Circulation, 2003, 107, 929 CrossRef.
- A. Papadopoulou, M. Yiangou, E. Athanasiou, N. Zogas, P. Kaloyannidis, I. Batsis, A. Fassas, A. Anagnostopoulos and E. Yannaki, Ann. Rheum. Dis., 2012, 71, 1733 CrossRef PubMed.
- W. J. Li, R. Tuly, X. Huang, P. Laquerriere and R. S. Tuan, Biomaterials, 2005, 26, 25 Search PubMed.
- S. Grad, L. Kupcsik, K. Gorna, S. Gogolewski and M. Alini, Biomaterials, 2003, 24, 5163 CrossRef CAS.
- E. Farell, F. J. O'Brien, P. Doyle, J. Fischer, I. Yannas, B. A. Harley, B. O'Connell, P. J. Prendergast and V. A. Campbell, Tissue Eng., 2006, 12, 459 CrossRef PubMed.
- D. S. Benoit, A. R. Durney and K. S. Anseth, Biomaterials, 2007, 28, 66 CrossRef CAS PubMed.
- C. C. Lin and K. S. Anseth, Pharm. Res., 2009, 26, 631 CrossRef CAS PubMed.
- C. G. Williams, T. K. Kim, A. Taboas, A. L. Malik, P. Manson and J. Elisseeff, Tissue Eng., 2004, 9 Search PubMed.
- C. N. Salinas and K. S. Anseth, J. Dent. Res., 2009, 88, 681 CrossRef CAS PubMed.
- J. T. Wilson, C. A. Haller, Z. Qu, W. Cui, M. K. Urlam and E. L. Chaikof, Acta Biomater., 2010, 6, 1895 CrossRef CAS PubMed.
- S. Cabric, J. Sanchez, T. Lundgren, A. Foss, M. Felldin, R. Kallen, K. Salmela, A. Tibell, G. Tufveson, R. Larsson, O. Korsgren and B. Nilsson, Diabetes, 2007, 56, 2008 CrossRef CAS PubMed.
- S. M. Cool and V. Nurcombe, J. Cell Biochem., 2006, 99, 1040 CrossRef CAS PubMed.
- D. S. Benoit and K. S. Anseth, Acta Biomater., 2005, 1, 461 CrossRef PubMed.
- I. Capila and R. J. Linhardt, Angew. Chem., Int. Ed., 2002, 41, 390 CrossRef CAS.
- Y. Liang and K. L. Kiick, Acta Biomater., 2014, 10, 1588 CrossRef CAS PubMed.
- P. T. Hammond, Adv. Mater., 2004, 16, 1271 CrossRef CAS PubMed.
- C. R. Kinnane, G. K. Such, G. Anteguera-Garcia, Y. Yan, S. J. Dodds, L. M. Liz-Marzan and F. Caruso, Biomacromolecules, 2009, 10, 2839 CrossRef CAS PubMed.
- J. F. Quinn, A. P. Johnston, G. K. Such, A. N. Zelikin and F. Caruso, Chem. Soc. Rev., 2007, 36, 707 RSC.
- A. M. Bratt-Leal, A. H. Nguyen, K. A. Hammersmith, A. Singh and T. C. McDevitt, Biomaterials, 2013, 34, 7227 CrossRef CAS PubMed.
- L. T. McLane, P. Chang, A. Granqvist, H. Boehm, A. Kramer, J. Scrimgeour and J. E. Curtis, Biophys. J., 2013, 104, 986 CrossRef CAS PubMed.
- F. Tanaka, K. Tominaga, M. Ochi, T. Tanigawa, T. Watanabe, Y. Fujiwara, K. Ohta, N. Oshitani, K. Higuchi and T. Arakawa, Life Sci., 2008, 83, 771 CrossRef CAS PubMed.
- J. Ylostalo, T. J. Bartosh, K. Coble and D. J. Prockop, Stem Cells, 2012, 30, 2283 CrossRef CAS PubMed.
- M. Cohen, E. Klein, B. Geiger and L. Addadiz, Biophys. J., 2003, 85, 1996 CrossRef CAS.
- W. Knudson and C. B. Knudson, J. Cell Sci., 1991, 99, 227 Search PubMed.
- M. Abe, J. G. Harpel, C. N. Metz, I. Nunes, D. J. Loskutoff and D. B. Rifkin, Anal. Biochem., 1994, 216, 276 CrossRef CAS PubMed.
- G. Pavlov, S. Finet, K. Tatarenko, E. Korneeva and C. Ebel, Eur. Biophys. J., 2003, 32, 437 CrossRef CAS PubMed.
- T. Miller, M. C. Goude, T. C. McDevitt and J. S. Temenoff, Acta Biomater., 2014, 10, 1705 CrossRef CAS PubMed.
- F. L. Goldberg and B. P. Toole, J. Cell Biol., 1984, 99, 2114 CrossRef.
- K. Rilla, R. Tiihonen, A. Kultti, M. Tammi and R. Tammi, J. Histochem. Cytochem., 2008, 56, 901 CrossRef CAS PubMed.
- B. Amelia, C. Sturgeon, M. Siatskas, K. Ferrer, K. McIntosh, S. Patil, W. Hardy, S. Devine, D. Ucker, R. Deans, A. Maoseley and R. Hoffman, Exp. Hematol., 2002, 30, 42 CrossRef.
- H. Yagi, A. Soto-Gutierrez, B. Parekkadan, Y. Kitagawa, R. G. Tompkins, N. Kobayashi and M. L. Yarmush, Cell Transplant, 2010, 19, 667 Search PubMed.
- T. J. Bartosh, J. H. Ylostalo, N. Bazhanov, J. Kuhlman and D. J. Prockop, Stem Cells, 2013, 31, 2443 CrossRef CAS PubMed.
- T. H. Lee, H. G. Wisniewski and J. Vilcek, J. Cell Biol., 1992, 116, 545 CrossRef CAS.
- P. Kalinski, J. Immunol., 2012, 188, 21 CrossRef CAS PubMed.
- J. Kanellis, R. Bick, G. Garcia, L. Truong, C. C. Tsao, D. Etemadmoghadam, B. Poindexter, L. Feng, R. J. Johnson and D. Sheikh-Hamad, Am. J. Physiol. Renal Physiol., 2004, 286 Search PubMed.
- A. Gorski, M. Waskik, M. Nowaczyk and G. Korczak-Kowalska, FASEB J., 1991, 5, 2287 CAS.
- E. Young, Thromb. Res., 2008, 122, 743 CrossRef CAS PubMed.
- H. Hochart, P. V. Jenkins, O. P. Smith and B. White, Br. J. Haematol., 2006, 133, 62 CrossRef CAS PubMed.
- S. Amatayakul-Chantler, S. W. Qian, K. KGakenheimer, E. P. Bottinger, A. B. Roberts and M. B. Sporn, J. Biol. Chem., 1994, 269, 27687 CAS.
- K. Balamurugan, A. Ortiz and H. M. Said, Am. J. Physiol. Gastrointest. Liver Physiol., 2003, 285, G73 CAS.
- H. M. Said, A. Ortiz, E. McCloud, D. Dyer, M. P. Moyer and S. Rubin, Am. J. Physiol. Cell Physiol., 1998, 275, 1365 Search PubMed.
- D. S. Bramono, S. Murali, B. Rai, L. Ling, W. T. Poh, Z. X. Lim, G. S. Stein, V. Nurcombe, A. J. van Wijnen and S. M. Cool, Bone, 2012, 50, 954 CrossRef CAS PubMed.
- M. Mitsi, K. Forsten-Williams, M. Gopalakrishnan and M. A. Nugent, J. Biol. Chem., 2008, 283, 34796 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60271k |
| This journal is © The Royal Society of Chemistry 2014 |