Smart hydrogels as functional biomimetic systems
Han L.
Lim
,
Yongsung
Hwang
,
Mrityunjoy
Kar
and
Shyni
Varghese
*
Department of Bioengineering, University of California, San Diego, La Jolla, CA 92093, USA. E-mail: svarghese@ucsd.edu; Fax: +1-858-534-5722; Tel: +1-858-822-7920
First published on 12th February 2014
Abstract
Stimuli-responsive (smart) hydrogels have attracted widespread attention as biomimetic systems due to their ability to respond to subtle changes in external and internal stimuli ranging from physical triggers such as temperature and electric field to chemical triggers like glucose and pH. Besides their intriguing behavior, the main interest in such smart hydrogels lies in their potential industrial and biomedical applications. Some of these applications include injectable biomaterials, tunable surfaces for cell sheet engineering, sensors, and actuators. In this review, we discuss the fundamental principles underlying the stimuli-responsive behavior of hydrogels and how these properties have led to major technological innovations. We also review recent advancements in the field of hydrogels, including self-healing and stimuli-responsive degradation in hydrogels. We conclude by providing a perspective on the potential use of smart hydrogels as multifunctional, bioactuating systems for cell and tissue engineering.
 Han Liang Lim | Han Liang Lim graduated from the Department of Bioengineering at University of California, San Diego in 2009 and embarked on his graduate education in 2011 after serving two years in the Singapore Armed Forces. He is currently pursuing his doctoral research under the guidance of Professor Shyni Varghese in the Department of Bioengineering at University of California, San Diego. |
 Yongsung Hwang | Yongsung Hwang is a postdoctoral researcher at the Department of Bioengineering at University of California San Diego. He received his B.S. degree in Materials Science from Sungkyunkwan University in 2006 and his M.S./Ph.D. degrees in Materials Science from University of California San Diego in 2007 and 2011, respectively. His current research focuses on developing bioengineered microenvironments using synthetic polymer-based biomaterials to understand the role of cell–matrix interactions on human pluripotent stem cells for musculoskeletal tissue regeneration. |
 Mrityunjoy Kar | Mrityunjoy Kar received his BSc (2005) and MSc (2007) from Vidyasagar University, India. He received his Ph.D. in 2013 from CSIR-National Chemical Laboratory, India in Polymer and Nanomaterial Science under the guidance of Dr Sayam Sen Gupta. Currently he is working as a postdoctoral researcher at University of California, San Diego with Dr Shyni Varghese. His research interests are focused on the synthesis of degradable hydrogels, polymer and nanomaterial based biomaterials for tissue engineering and biomedical applications. |
 Shyni Varghese | Dr Shyni Varghese is an Associate Professor in the Department of Bioengineering and an Affiliate Faculty of the NanoEngineering Department, Institute of Engineering in Medicine, and the Materials Science program at University of California, San Diego. Dr Varghese's laboratory works at the interface of bioinspired materials and stem cell engineering. Specifically, her laboratory focuses on understanding the role of physicochemical cues of the extracellular matrix in tissue regeneration (with a focus on the musculoskeletal system) and disease progression (with a focus on cancer metastasis and fibrosis). |
1. Introduction
Advancements in the field of hydrogel science and technology have gained considerable momentum in the past decade due to their biomimetic nature and widespread biomedical applications. These hydrogels have been envisioned to have applications as smart diagnostics,1,2in vivo delivery vehicles,3,4 and scaffolds for cell and tissue engineering.5 Hydrogels, composed of a network of polymer chains imbibed with large quantities of water, have been regarded as biomimetic systems owing to their structural resemblance to soft tissues.6 This biomimetic nature is further reinforced by findings that some hydrogels can reversibly undergo deformation and change their size and shape responding to external stimuli such as temperature,7–9 pH,8–10 electric and magnetic fields,11,12 and chemical triggers,13 much like living systems.The stimuli-responsive properties in hydrogels are mainly controlled by polymer–polymer and polymer–solvent interactions.14 Specifically, the fundamental interactions that control stimuli-induced swelling–deswelling transitions in hydrogels are ionic interactions,15 hydrophobic interactions,16 hydrogen bonding,17 and van der Waals forces.18 These interactions function independently or in concert to manifest stimuli-responsive behavior in hydrogels. Interestingly, the same secondary interactions have been identified to be responsible for some of the unique functions of biological systems such as molecular recognition.19 In addition to sensitivity and responsiveness, recent studies have introduced new design principles and strategies to endow hydrogels with other biomimetic features such as self-healing.20 Hydrogels have also been engineered to have functional groups and structural features to create dynamic and tunable extracellular matrix-like environments for cell culture.21 In this review, we focus on the fundamental principles that govern stimuli-responsiveness in several smart hydrogels and highlight some of the major inventions in the field.
2. An overview of stimuli-responsive hydrogels
2.1 Temperature-responsive hydrogels
Amongst the smart hydrogels, temperature-responsive hydrogels are the most studied.22 These hydrogels are characterized by the ability to undergo reversible volume-phase transitions (VPT) in response to subtle changes in their surrounding temperature.23 The temperature-dependent deformation of these hydrogels has been attributed to a critical balance of hydrophilic and hydrophobic moieties within the polymer network.24,25 Poly(N-isopropylacrylamide) (pNIPAm) is by far the most widely studied temperature-responsive polymer; homopolymer pNIPAm hydrogels undergo reversible transition from a swollen state to a collapsed state around the lower critical solution temperature (LCST) of ∼32 °C.26,27 Unlike upper critical solution temperature (UCST), the LCST type of phase behavior is anomalous and counter intuitive, where the polymer precipitates upon heating. The LCST behavior in polymers has been attributed to the presence of specific interactions between the polymer and solvent molecules. This allows the entropy and the enthalpy change during mixing to be negative due to formation of specific configurations and interactions between the components of the mixture. Hence, the free energy of mixing is negative at lower temperatures and becomes positive upon increasing the temperature. Thus, phase separation occurs upon heating.It has been shown that this delicate balance of hydrophilic and hydrophobic interactions determines the LCST, the temperature at which the hydrogel undergoes VPT.25 In the case of pNIPAm hydrogels, the amide groups contribute to the hydrophilic interactions while isopropyl groups and the hydrocarbon backbone contribute to hydrophobic interactions.28 This presents a unique opportunity to tailor temperature-responsive hydrogels with different LCSTs.29 As shown previously, an increase in hydrophilic moieties (or a decrease in hydrophobic moieties) results in an increase in the LCST, while a decrease in hydrophilic moieties (or an increase in hydrophobic components) leads to a decrease in the LCST.30 The ability of these temperature-responsive hydrogels to transition from a hydrophilic to hydrophobic state corresponding to changes in temperature has been harnessed to create injectable materials31–34 and smart surfaces.35
2.2 pH-responsive hydrogels
Another well-studied stimuli-responsive hydrogel is the pH-sensitive hydrogel, where hydrophilic networks undergo volume deformations in response to changes in the surrounding pH.36 The building blocks of such hydrogels are polymers that are weakly acidic or basic, such as poly(acrylic acid) or chitosan.37,38 The governing principle behind the sensitivity of pH-responsive hydrogels is their ability to dissociate and associate with hydrogen ions depending on the pH of the aqueous environment. These pH-dependent functional groups carry different charges depending on the protonation state and hence contribute differently to the osmotic pressure of the hydrogel.39 For example, when the pH of the solvent is higher than the pKa of a poly(acrylic acid) hydrogel, the carboxylic acid groups are deprotonated and carry a negative charge, contributing significantly to the osmotic pressure within the network. In this state, the hydrogel is capable of imbibing large quantities of water and swells immensely. Another important factor that increases swelling is the long-range repulsion of like charges along the polymer chain.40 Conversely, when the pH of the solution is lower than the pKa of the hydrogel, the carboxylic groups are protonated, which contributes much less to the osmotic pressure of the network. As a result, the hydrogel shrinks. Since this protonation–deprotonation is reversible, the swelling/collapse of hydrogels can be easily reversed by changing the pH of the surrounding solution.2.3 E-field-responsive hydrogels
Electric-field (E-field)-responsive hydrogels exhibit fast response and deform under physiologically relevant conditions, such as exposure to ion currents generated by streaming potentials in the body. These hydrogels have long been suggested to be an ideal system to mimic various biological functions like motility.41 E-field-responsive hydrogels have been extensively studied as artificial muscles due to the many parallels the two exhibit, such as flexing/bending.42 E-field-responsive hydrogels are synthesized by crosslinking polyionic chains and as a result usually exhibit much larger swelling ratios than their non-E-field-responsive counterparts.43 Upon the application of an E-field, mobile counter ions in the solution can bind to the polymer network and accumulate differentially within the hydrogel such that an opposing potential difference is generated across the gel. This electrical polarization causes the osmotic pressure to vary spatially within the gel and leads to anisotropic swelling.44 For a hydrogel strip with a high aspect ratio, this E-field-induced swelling causes the hydrogel to bend, and this propensity for reversible bending was used in a study by Osada et al. to demonstrate the remarkable potential of synthetic materials to exhibit a walking movement akin to a living organism.45E-field-responsive hydrogels have also demonstrated bi-directional bending in a constant E-field.46 Findings from our research show that under constant electrical stimuli, certain E-field-responsive hydrogels would first bend towards the cathode before relaxing back to their original state and then continue to bend towards the anode.47 To explain this phenomenon, we developed a theoretical model based on previous studies by Doi et al.48 Results from our simulations suggest that the crosslinking density plays a critical role in regulating the directionality of bending. As E-field-responsive hydrogels undergo swelling, the concentration of immobile ions decreases while the mobile counter-ions in solution continue to accumulate within the hydrogel. This leads to a reversal in bending as the free ion concentration plays more significant roles in governing swelling via osmotic pressure than the ionic groups in the polymer. This was validated in experiments using hydrogels with varying crosslink densities (Fig. 1).47 In accordance with the theoretical predictions, hydrogels with high crosslinking density only exhibited uni-directional bending in contrast to hydrogels with low crosslinking density, which exhibited bi-directional bending. The advancements in the field enable tailoring of E-field-responsive hydrogels to exhibit desired swelling behaviors and kinetics for applications such as micro-actuators to control flow in microfluidic devices or to achieve site/directional specific delivery of targeted molecules.
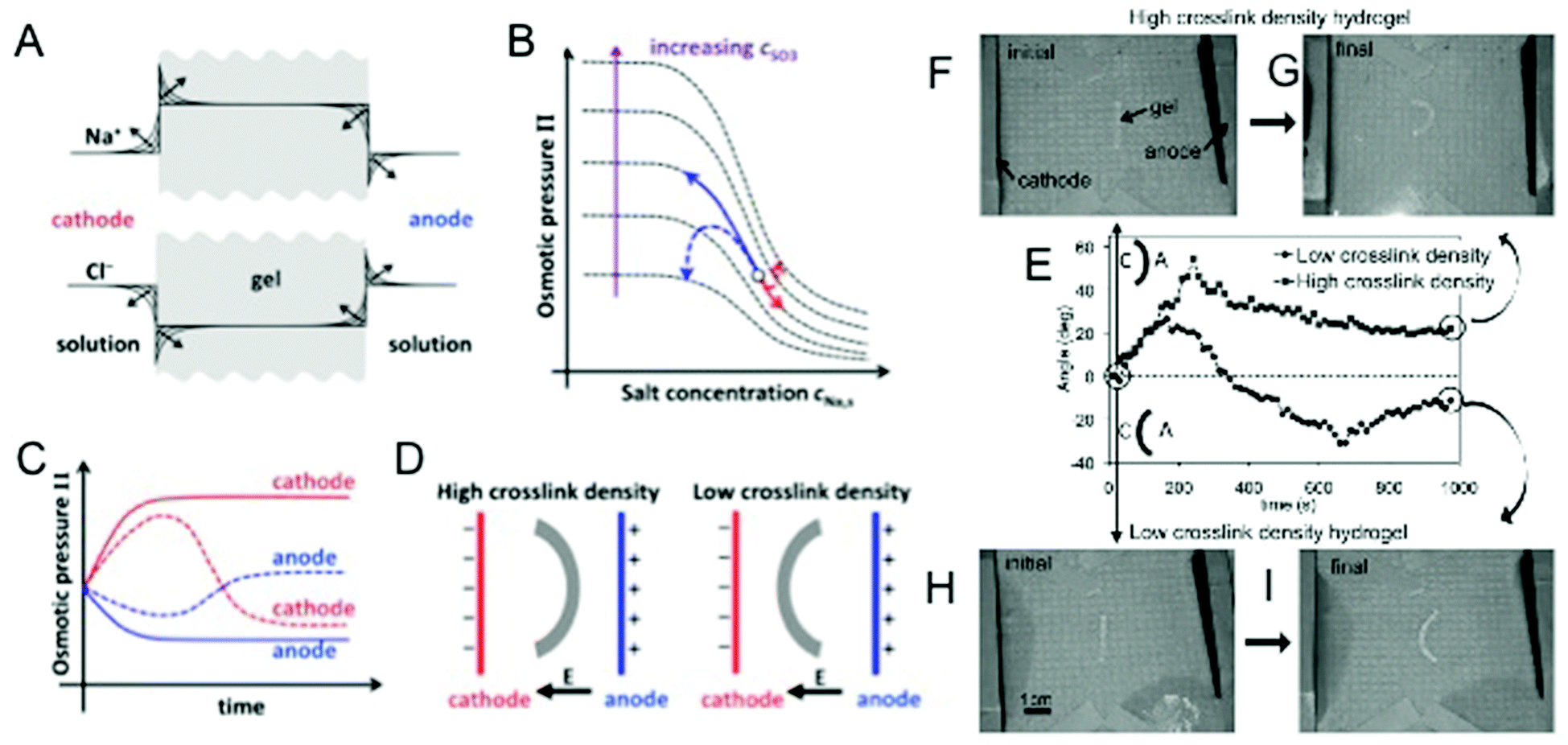 | ||
| Fig. 1 E-field responsive hydrogels. (A) Expected accumulation of Na+ and depletion of Cl− ions at the cathode-side hydrogel boundary and depletion of Na+ and accumulation of Cl− ions at the anode-side hydrogel boundary with time. (B) Variation of osmotic pressure with solution Na+ concentration for different concentrations of anionic groups (SO3−) within the gel. Solid arrows indicate the anticipated (from Doi theory) monotonic swelling and shrinkage of a high crosslink density hydrogel at the anode (black) and cathode (gray) sides, respectively. Dashed arrows indicate the non-monotonic swelling behavior (from modification of Doi theory) expected from a low crosslink density hydrogel as a result of swelling and shrinkage of the gel causing large changes in the concentration of anionic groups within the gel. (C) Time evolution of the osmotic pressure of a high and low crosslinked hydrogel at the anode and cathode sides. Solid and dashed lines correspond to trajectories for high and low crosslink density hydrogels, respectively. (D) Steady-state bent conformation of high and low crosslinked hydrogels predicted by our modified theory. (E) Time evolution of the bending angle for low crosslink density (circles) and high crosslink density (squares) hydrogels. Schematic of the bending configuration for positive and negative angles is given, where A and C represent the anode and the cathode, respectively. The initial and final steady-state configurations of the high crosslink density hydrogel (F, G) and low crosslink density hydrogels (H, I) are also shown. Adapted from ref. 47. | ||
2.4 Light-responsive hydrogels
Light is another external stimulus that has been extensively studied in the field of stimuli-responsive hydrogels. Light-responsive hydrogels are often prepared by incorporating light sensitive functional groups into the hydrogel networks. Most of the initial studies have utilized the ability of light-sensitive chromophores such as azobenzenes to undergo reversible cis–trans isomerization in the presence of UV light to create light-responsive hydrogels.49,50 The inter-conversion of the immobilized azobenzene groups between the two photoisomers induces macroscopic, reversible volume changes of the hydrogel network. In addition to isomerization, studies have employed light-activated dimerization to also create responsive hydrogels.51,52 Similarly, immobilization of photocleavable moieties into the network endowed the hydrogels with photosensitivity. Photocleavable groups, such as triphenylmethane leuco derivatives, undergo photo-dissociation into ion pairs in the presence of UV light.53 This photo-dissociation of the functional moieties that are incorporated within the hydrogel network results in an increase in osmotic pressure (as well as electrostatic repulsion) and thereby gives rise to light-induced swelling of the hydrogels. Removal of the stimulus causes the triphenylmethane leuco derivatives to revert back to their non-ionic state, allowing the hydrogel to deswell back to its original size and shape. Likewise, by introducing a visible light-active chlorophyllin choromophore into pNIPAm hydrogels, Suzuki et al. created a visible-light sensitive hydrogel. Here, the choromophore absorbs the light, which is then dissipated as heat, increasing the local temperature. This change in temperature then induces collapse of the pNIPAm network.54 More recently, Takashima et al. engineered a light-responsive hydrogel by using host–guest interactions between a-cyclodextrin and photo-active azobenzene derivatives.55 The supramolecular structure formed through the host–guest interactions was shown to exhibit reversible macroscopic deformations when exposed to ultraviolet light at 365 nm or visible light at 430 nm.3. Applications of stimuli-responsive hydrogels
3.1 Smart hydrogels as actuators
One of the unifying features amongst stimuli-responsive hydrogels is their ability to undergo reversible, discontinuous, and large volume changes when subjected to external or internal stimuli.56 This ability of smart hydrogels to exhibit reversible “on–off” swelling behavior in the presence of physiologically relevant cues has been utilized to develop in vitro models mimicking various attributes of living systems such as motility and actuating functions.57 In recent years, a number of studies have employed the actuating function of smart hydrogels for various applications.Javey and colleagues have harnessed the temperature-responsive properties of PNIPAm hydrogels to induce controlled 3-dimensional folding in a synthetic system.58 In this study, pNIPAm hydrogels embedded with single walled carbon nanotubes were used as hinges of a low density polyethylene (LDPE) sheet that was designed to fold into a box. Increasing the temperature above the LCST of pNIPAm induced folding of the LDPE sheet from a 2-D structure into a 3-D box, while decreasing the temperature to below the LCST caused the box to unfold in the opposite manner. Such systems may be utilized to induce folding/unfolding in closed systems where any form of contact with the scaffold is prevented, such as for the delivery of larger particles in the macroscale.
The ability of stimuli-responsive hydrogels to undergo reversible swelling–deswelling in response to subtle changes in external stimuli makes them an ideal candidate for switchable valves such as in microfluidic devices. One of the most commonly used strategies involves incorporation of E-field-responsive hydrogels within the space between the electrodes of microfluidic channels.59 When exposed to a potential difference across the electrodes, these hydrogels undergo swelling many times higher than their original size, which then blocks the channels and subsequently prevents the fluid from flowing through them. Smart hydrogel-based actuators provide an alternative to complex pneumatic and peripheral devices.60,61 The E-field-induced swelling–deswelling of hydrogels has been used in different bioMEMs systems with varying applications, such as sequencing, drug testing, and lab-on-a-chip.62 Furthermore, studies by Kwon et al. have shown that microfluidic systems containing E-field-responsive hydrogel could be functional in cell culture media conditions without having any detrimental effects on the cells.63 We have shown that hydrogels that exhibit bending dynamics under physiological E-fields can be used as electro-chemo-mechano bioactuating scaffolds for stem cell culture.47 When exposed to a pulsatile ‘on–off’ E-field of 0.5 V mm−1 corresponding with magnitudes similar to those of membrane potentials,64 the hydrogel responded with oscillatory volumetric strains similar to the deformation generated in the cartilage. Human mesenchymal stem cells cultured in these hydrogels showed improved chondrogenic differentiation compared to control cultures grown in the absence of E-field stimulation.
In a recent study, Whitesides and colleagues utilized the ability of hydrogels to exhibit oscillatory swelling–deswelling dynamics to create transparent, high-speed, large strain actuators.65 These hydrogel-assisted systems were shown to function as variable capacitors, as their swelling–deswelling dynamics changed the resistance of the hydrogel and the distance between the electrodes, thus changing the voltage separation across the electrodes. This electromechanical transduction was achieved without any electrochemical reactions within the device. The large strains have also been used to create a completely transparent loudspeaker that produces sound over the entire audible range.
In an elegant study, Dong et al. took a step further and created a prototype of an artificial cornea by harnessing the ability of stimuli-responsive hydrogels to oscillate between swollen and collapsed states with subtle changes in environmental pH.66,67 Artificial optical systems developed have thus far been only able to focus on objects at different distances through physical displacement of the lenses. Unlike the conventional artificial systems, the microlens developed with the integration of pH-responsive hydrogels recapitulated the ability of the human eye to focus on objects at different lengths. The pH-responsive hydrogel synthesized from poly(acrylic acid) was used to regulate the height of a circular aperture where oil and water interface. Depending on the pH, the hydrogel will either be in a swollen or collapsed state, which then dictates the height and the geometry of the liquid meniscus between the oil and water interface, thus allowing light from different distances to be focused (Fig. 2).67 For example, in an acidic buffer, the hydrogel is in its collapsed state, which causes the aqueous layer to bulge out from the circular aperture. This allows light from further distances to be focused as it passes through the lens. In contrast, in a swollen state, the height of the aperture is increased, which then causes the lens to concave inward, allowing light from a closer distance to be focused. The ability of such systems to be powered by other forms of stimuli-responsive hydrogels was also highlighted, such as temperature-responsive pNIPAm hydrogels that could be utilized to achieve the same effect.68 Beyond recapitulating biological functions in a synthetic system, such systems mimicking the ability of the eye to focus light from different distances can yield applications in optical sensing in modern medical diagnostics and microfluidic devices.
 | ||
| Fig. 2 pH responsive poly(acrylic acid) hydrogels used to create and control the focusing of a liquid microlens. (A) An optical image of the device. Scale bar, 1.0 mm. (B) The contracted state of the hydrogel ring at pH 2.0. Scale bar, 500 μm. (C) The expanded state of the hydrogel ring at pH 12.0, where the red and black dashed lines represent the boundaries of the inside and outside periphery of the hydrogel ring, respectively. Scale bar, 500 μm. (D–G) Optical images of the operation of the two-pixel pH-sensitive liquid microlens array, showing snapshots at indicated times. Left, microlens A, 2-(dimethylamino)ethyl methacrylate (DMAEMA); right, microlens B, poly (acrylic acid). Scale bar, 500 μm. (H) Conceptual diagram showing the monitoring of different areas in space by lenses A and B. The blue lines represent the image planes. The red and black curves represent the oil–water interfaces of the two microlenses at two pH values, n and m, respectively. For microlens A, the focal point changes from fn (red point) to fm (black point) and the object plane moves from On (red line) to Om (black line), as the pH value of its surrounding fluid changes from n to m. Microlens B exhibits opposite effects (f′n to f′m and O′n to O′m) when simultaneously exposed to this same local environmental fluid. Reprinted with permission from ref. 67. | ||
While most of the above studies have focused on external stimuli to achieve oscillatory behavior in hydrogel systems, studies have also shown that autonomous mechanical oscillations in hydrogels can be achieved by integrating the innate oscillatory behavior of the Belousov–Zhabotinsky (BZ) reaction within the hydrogel network.69–71 In the BZ reaction, a rubidium complex is incorporated into a hydrogel, which catalyzes a redox reaction between substrates that causes the oxidation state of the rubidium to oscillate between Ru2+ and Ru3+. The substrates used to drive the BZ reaction are malonic acid and bromate ions, which act as the reducing agent and the oxidizing agent, respectively. When incorporated into a temperature-responsive hydrogel, such as a pNIPAm hydrogel, the oscillation of the oxidation state of the Ru ions causes the hydrogel to exhibit volumetric self-oscillation. Though such an oscillating behavior has been utilized in many applications, such as inducing peristaltic movement across the surface of the gel for the transportation of cargo, extending such systems to biomedical applications is challenging due to substrate incompatibility in physiologically relevant settings.72–74
Aizenberg and colleagues have developed an alternate approach involving continuous chemo-thermo-mechano feedback events to induce self-oscillations in hydrogels.75 The design of the system involved creating high aspect ratio micropillars of pNIPAm, which were designed to collapse from their upright to bent configuration at temperatures above their LCST. This was achieved by immersing pNIPAm micropillars in a solution containing two immiscible liquids with the aqueous layer on the bottom. The lower region of the micropillars and their catalyst functionalized tip were in contact with the aqueous layer and the reactant-containing organic layer, respectively. Initially, the hydrogel pillars swell to manifest an upright configuration, which facilitates catalytic reaction of reagents in the organic layer. The resulting exothermic reaction elevates the temperature of the surrounding liquid above the LCST of the hydrogel, causing the micropillars to collapse thereby preventing further reaction at the functionalized tip. As the heat dissipates, the temperature of the surrounding environment decreases below the LCST and the micropillars regain the upright configuration to resume the catalytic reaction. This catalyst-mediated exothermic reaction enables the hydrogel to exhibit a self-regulated oscillatory function for as long as the reagents are present in the organic layer. Aizenberg and colleagues have also demonstrated the viability of pH-responsive hydrogels to function in a similar manner, showing that the self-oscillatory behavior can be tuned to the different stimuli present in the environment by using hydrogels sensitive to that stimulus.76
While the aforementioned describes the actuating function of stimuli-responsive hydrogels exhibiting oscillating swelling–deswelling kinetics, Feinberg et al. utilized temperature dissolution–precipitation of pNIPAm molecules to create cell films that can function as living actuators.77 Similar to cell sheet engineering, the pNIPAM molecules were used to create a temperature-dependent smart interface with varying levels of cell–substrate adhesion. The thin film constructs were built from rat ventricular cardiomyocytes using micro-patterned surfaces. Upon releasing from a thermally responsive surface, the muscular films with intact cell–cell contacts formed a functional 3D structure. The proof-of-concept study showed that these muscular thin sheets could be designed to function as actuators and soft-robotics due to the inherent contractile properties of cardiomyocytes. Specifically, the muscular thin films performed different biomimetic tasks such as gripping, pumping, walking, and swimming with a spatiotemporal control. The muscular, thin film-based actuators were demonstrated to generate stresses up to 4 mN mm−2. Although these proof-of-concept studies show remarkable potential for creating bioactuators, the challenge remains in translating these conceptual advancements into mainstream applications.
3.2 Biomolecular-responsive hydrogels as smart sensors
Biomolecular recognition plays an important role in regulating homeostasis in the body. To keep the body in equilibrium, different tissues quickly detect fluctuations in concentrations of important metabolites and consume or replenish these metabolites as needed.78 Loss of such function inevitably leads to severe chronic conditions, such as diabetes, where the body is not capable of responding appropriately to an increase in blood glucose concentrations.79 Hydrogels with molecular recognition abilities have been created from polymers that can exhibit dissolution–precipitation dynamics in the presence of specific biomolecules. Such stimuli-responsive hydrogels are useful for detecting different kinds of analytes and/or small molecules. For instance, Miyata et al. have developed a hydrogel, which can swell reversibly in response to an increase in the concentration of a specific antigen (rabbit IgG).80 The hydrogel was prepared by incorporating the antigen (rabbit IgG) and its corresponding antibody, goat anti-rabbit IgG (GAR IgG), into a polyacrylamide hydrogel such that the binding between the antibody and the antigen increased the crosslinking density within the network. As free antigen was introduced into the solution, the hydrogel swelled as the antigen–antibody binding was dissociated by the exchange of network-bound antigen with the free antigen, reducing the crosslinking of polymer chains in the network.Smart hydrogels have also been tailored to change their color in response to chemical triggers, allowing for the harnessing of an optical signal.81,82 These studies involved integration of crystalline colloidal arrays (CCA) with reversible swelling–deswelling of hydrogels, where the periodicity (or lattice spacing) of the CCA was tuned by temperature-induced volume changes of pNIPAm hydrogels.83 These mesoscopically periodic smart hydrogels diffract light at different wavelengths as dictated by the lattice spacing, giving rise to color change. Holtz et al. took this a step further to create smart glucose sensors by grafting glucose oxidase (GOx) and catalase onto the polystyrene colloids that make up the CCA.84 GOx catalyzes the oxidation of glucose and, in turn, becomes reduced. This reduced state carries an anionic charge, contributing to the osmotic pressure within the hydrogel, and induces the hydrogel to swell. Swelling of the gel then changes the periodicity of the CCA array, which manifests as a change in the visual signal from the gel. This enzymatic conversation of glucose to gluconic acid produces hydrogen peroxide as a byproduct, which is then converted into water and oxygen via catalase. In the absence of free glucose, the GOx returns to its uncharged state allowing the gel to deswell to its original state. The authors have shown that the CCA–pNIPAm system can produce detectable signals at a glucose concentration as low as 10−12 M and as high as 0.3 mM. This study was amongst the first few to demonstrate the harnessing of an optical signal from the volume deformations of a stimuli-responsive hydrogel, and soon more studies followed suit to create smart sensors with broader ranges and improved sensitivity and selectivity.
In recent years, smart hydrogel-based glucose sensors have opted to use glucose-binding phenylboronate functional groups due to their stability and the ease with which phenylboronate can be chemically manipulated.85 These hydrogels have been shown to exhibit volume deformations in response to changes within the range of physiological blood glucose concentration of 3.9 mM to 7.2 mM.86 To enhance the interactions between the glucose molecules and the polymer, multiple phenylboronate moieties were tethered to the hydrogel network.87 The polyvalent phenylboronate–glucose interactions were then used to modulate the hydrogel swelling kinetics. One of the limitations of this approach is that it takes hours for the concentration of glucose to fully diffuse into and equilibrate within a hydrogel of macroscopic thickness. To overcome the diffusion limitation of such hydrogels, Zhang et al. have used a layer-by-layer fabrication technique to create ultra-thin glucose-responsive hydrogels.88 Rapid diffusion of glucose across such ultra-thin hydrogels induces deformation of the hydrogels within minutes, thus improving sensitivity. The hydrogel is shown to respond to a glucose concentration range between 0 mM and 10 mM in a linear manner.
Apart from monitoring glucose levels, studies have shown that smart hydrogels are also capable of responding to changes in the concentrations of DNA.89 This is achieved by incorporating complementary nucleotide sequences of targeted DNA into the polymer network, such that the targeted DNA strands can anneal to it.90 As the targeted DNA is introduced into the gel via electrophoresis, it anneals to the complimentary strands in the hydrogel to form temporary crosslinks, resulting in hydrogel deformation. These hydrogels exhibit high specificity and sensitivity with a capability of detecting DNA concentrations in the picomolar range. These hydrogels have been touted to find applications in the development of novel diagnostics and genomic applications.
3.3 Cell and drug delivery
An area where stimuli-responsive hydrogels are extensively applied is drug delivery. Studies have shown that stimuli-responsive hydrogels, such as temperature and pH-sensitive hydrogels, can be used to retain and deliver bioactive molecules to the target in response to changes in temperature or pH.31,91–94 One such example is ophthalmic drug delivery, where drugs are conventionally delivered via eye drops.95 However, when delivered via this route, they exhibit short residence time in the eyeball, leading to low drug bioavailability and wastage of drug.96 This is further compounded by patient non-compliance when it comes to delivering drugs via eye drops.97 To overcome these limitations, strategies enabling a slow release of the drug into the eye have been devised. One straightforward approach is entrapping or immobilizing the drug within the contact lens which itself is a hydrogel.98 However, initial studies have shown that the drug retention could only be moderately prolonged by this approach. Hence, studies have resorted to incorporating drug-loaded nanocarriers within the contact lens to improve drug retention at room temperature and facilitate delivery of the drug at physiological temperatures. In such an approach, Jung et al. have shown that hydrogel-based contact lenses embedded with temperature-responsive nanogels exhibit higher retention rates and prolonged release of drug at 37 °C.94 In this study, the ophthalmic drug, timolol maleate, was first loaded into highly crosslinked propyl glyceryl methacrylate and ethylene glycol dimethacrylate nanogels. These drug-loaded nanocarriers were then incorporated into poly(hydroxyl methyl methacrylate) hydrogel contact lenses to achieve controlled release. The release profile of the drug from one such contact lens showed continued release over a period of 4 weeks, which is a significant improvement over other conventional methods.In addition to delivering biomolecules, the ability of temperature-responsive materials to undergo rapid phase transition (or increase in hydrophobic interactions) has been employed to create scaffolds for cell and tissue engineering.99 In a study by Tan et al., pNIPAm units were grafted onto hyaluronic acid (HA) molecules to create a temperature-responsive injectable system.100 The HA-co-pNIPAm polymer exhibited an LCST of 31 °C, where it manifested as a clear solution at temperatures below its LCST and formed a scaffold suitable for cell cultures at physiological temperature of 37 °C. Adipose-derived stem cells encapsulated within the HA-co-pNIPAm exhibited long-term viability (∼21 days) both in vitro and in vivo. Healy and colleagues have demonstrated that pNIPAm-based hydrogels support growth and differentiation of a number of cells including embryonic stem cells.101 These studies demonstrate the potential of temperature-responsive polymers such as pNIPAm to contribute to minimally invasive treatment strategies. In addition to pNIPAm, a number of synthetic materials with varying amounts of hydrophobic and hydrophilic groups have been developed to achieve in situ thermogelling properties.102,103
The pH sensitive hydrogels that exhibit varying levels of swelling responding to environmental pH are highly appealing to deliver therapeutic agents to specific regions.91,92 A number of studies have used pH-responsive nanogels as anti-cancer drug carriers to target tumors.104–106 Leveraging on the fact that tumors often tend to have a relatively lower pH (5.5–6.5) compared to the physiological pH (∼7.4), strategies involving pH-responsive nanogels have been utilized to improve drug retention at the higher physiological pH while releasing the drug from the hydrogel at a comparatively more acidic microenvironment of the tumor.8 In one such study, Zhang et al. created chitosan nanogels based on ionic interactions between the chitosan derivative N-[(2-hydroxy-3-trimethylammonium)propyl]chitosan chloride (HTCC) and sodium tripolyphosphate(TPP).91 These nanogels loaded with the anticancer drug, methotrexate disodium (MTX), were found to facilitate the drug release at pH 5 owing to their pH-induced swelling. Furthermore, the fact that the drug MTX is pH-sensitive and adapted a protonated state at pH 5 eliminated the potential interaction between the carrier and the drug, thereby enabling complete diffusion of the drug from the carrier. Such systems have attracted much attention and many strategies are currently being devised to improve the penetration of such nanogels into cells.
In addition to targeting sites in the human body with different pH to deliver encapsulated drugs, the ability of the pH-responsive hydrogels to change their swelling behavior in response to pH changes has also been harnessed to develop self-regulated insulin delivery systems.107 Such systems were initially conceived to be able to function as implantable artificial pancreas to aid in regulating blood glucose concentrations in diabetic patients. Klumb et al. developed an insulin delivery pump prototype based on pH-responsive hydrogels.108 Glucose oxidase and catalase were incorporated into a hydroxyethyl methacrylate-based hydrogel containing insulin to create a smart insulin delivery vehicle. Additionally, pendant amine groups were incorporated to bestow the hydrogel with pH sensitivity. As the concentration of glucose in the surrounding solution increases, the glucose oxidase-mediated conversion of glucose to gluconic acid results in a lowering of the pH in the local environment and induces the pH-responsive hydrogel to swell. This in turn increases the rate of insulin delivery from the hydrogel. On the other hand, a decrease in diffusion of glucose into the hydrogel will cause contraction and decrease the rate of insulin delivery. While such devices were able to elegantly employ a pH-responsive hydrogel to respond to an increase in glucose concentrations and release insulin, they suffer from the lack of reusability as they lose their functionality upon secreting the insulin embedded within.
Another stimulus that has been extensively studied in recent years is light. A number of studies have demonstrated the spatial and temporal versatility of light-sensitive hydrogels in drug and biomolecular delivery.109–113 A recent study by Griffin et al. has shown that by tuning the electronegativity in the photolabile nitrobenzyl group, one can alter the wavelength of the light that is responsible for the photolysis of the nitrobenzyl group, thus creating a photoselective system to deliver therapeutic agents on demand.113 Hydrogels containing these nitrobenzyl groups that are sensitive to different wavelengths can be used to deliver multiple biomolecules from the same carrier in a controlled manner.
Recently, stimuli responsive approaches have been extended to create cell delivery devices. For instance, Mooney and colleagues have shown that an alginate-based macroporous hydrogel (i.e., cryogel) embedded with iron oxide nanoparticles exhibits large deformations and volume changes responding to external magnetic fields and can be used as a remotely controlled cell delivery device.114 The pore sizes and interconnectivity within these macroporous cryogels can be tailored by freezing the cryogels at different temperatures,115 allowing encapsulation of small molecules to cells. The magnetic field-induced, large deformations are caused by the collapse of the pores in the cryogel, which squeezes water and cells out from the gel. Magnetic field-induced actuating resulted in a burst release of cells from the network, whereby one third of the seeded cells were expelled from the cryogel. Hydrogels containing photodegradable ortho-nitrobenzyl (o-NB) groups in the backbone have also been shown to deliver encapsulated cells when exposed to light. Griffin et al. conducted a study where a series of o-NB was designed to have varying degradation rates when exposed to light of fixed wavelengths.116 Very interestingly, the authors have shown that cells encapsulated in a PEG-based hydrogel with a spatially varying degradation profile can be preferentially released as some parts of the hydrogel network degrade more rapidly than the other.
3.4 Smart surfaces for cell cultures
The interfacial properties of surfaces such as hydrophobicity play an important role in modulating cell adhesion through adsorption and conformation of proteins.117–119 One unique aspect of temperature-responsive hydrogels is that it allows one to create tunable surfaces with varying levels of hydrophilicity (or hydrophobicity).35 An interesting application of such temperature-responsive smart surfaces is in cell sheet engineering for creating layered sheets of cells with intact cell–cell contacts.35,120–123 Cells are first grown to confluency on a pNIPAm-based surface, which provides a hydrophobic and adhesive surface to cells at 37 °C. At temperatures below its LCST, the surface undergoes transition from a hydrophobic to a hydrophilic state, weakening the cell–substrate interaction, which induces the detachment of the confluent cells in the form of an intact cell sheet (Fig. 3).124 Unlike traditional protease-mediated detachment of cells, this form of detachment preserves both adherens and gap junctions between the cells.125 This is important especially in myocardial sheet engineering where ionic currents involved in muscle contraction signaling are conducted through these gap junctions.126 Cell sheets maintaining such physiologically relevant features can be used as cell patches to promote healing and aid regeneration of a number of compromised tissues. Some of these tissues include skin,127 cornea,128,129 liver,130 periodontal ligament tissue,131 bladder,132 and cardiac muscle.2,133 This smart surface-mediated cell-sheet engineering could also be used to create cell-dense 3-D tissues.134,135 Such engineered 3-D tissues could circumvent many of the undesirable translational consequences that biomaterial-based scaffolds currently present; however, the application of smart surface-mediated cell sheet engineering to create tissues with significant thickness for treating critical size defects is yet to be realized. Nonetheless, cell sheet engineering via smart hydrogel surfaces offers an innovative strategy to move cell and tissue engineering from the bench to the clinic.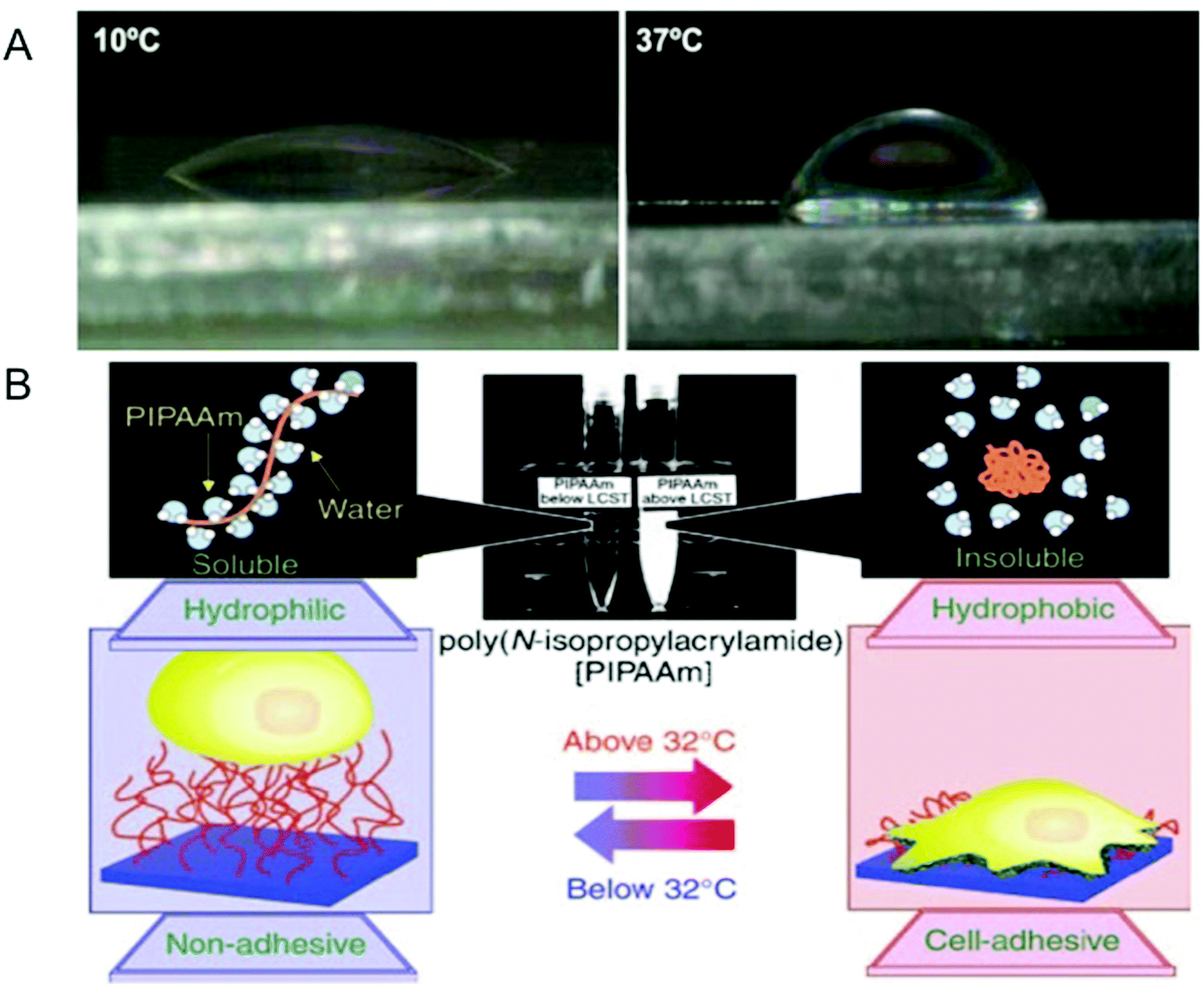 | ||
| Fig. 3 Modulation of surface properties using temperature-responsive hydrogels. (A) Temperature-dependent wettability of poly(N isopropylacrylamide) (pNIPAM)-based surfaces at 10 °C (left panel) and 37 °C (right panel). (B) Temperature-responsive culture dishes. The temperature-responsive pNIPAm exhibits a transition from hydrophobic to hydrophilic across its lower critical solution temperature (LCST) of 32 °C. The noninvasive harvest of cells as intact sheets, along with a deposited extracellular matrix, can be achieved by reducing the culture temperature. Reprinted with permission from ref. 2. | ||
While temperature-responsive smart surfaces remain the most extensively investigated, other stimuli responsive systems can also give rise to smart surfaces. An example is E-field-responsive hydrogels with pendant side chains capable of transitioning quickly between hydrophobic and hydrophilic properties.136 In the pioneering paper by Lahann et al., amphiphilic long chain carboxylic acids were conjugated via self-assembly onto a gold surface.137 In the absence of any stimulation, the charged residues repel each other creating a highly charged hydrophilic surface. When the system is exposed to an E-field, a positive charge is generated on the gold surface, resulting in the acidic residues bending backwards to create a hydrophobic surface. Thus, the on–off E-field alters the surface from a charged brush-like structure to a hydrophobic surface. Such smart surfaces could possibly be used in cell-sheet experiments, much like the temperature-responsive surfaces or in micro-bioseparation applications. Studies have also used hydrogels containing photocleavable groups to alter surface hydrophilic properties. Hydroxyethylacrylate hydrogels containing photolabile 2-nitrobenzyl acrylate moieties have been shown to exhibit altered mechanical and surface properties when exposed to UV light, which cleaves the photolabile groups.138 The authors have shown that these photosensitive materials can be used to manage cell adhesion and cell shape in a controlled manner.
3.5 Self-healing hydrogels
Another unique characteristic of living systems is their ability to undergo “healing” when damaged.139–141 Despite numerous advancements in the field, achieving self-healing in permanently crosslinked hydrogels remains a challenge due to the presence of water and irreversible crosslinks.142 A number of bio-inspired approaches have been used to encode hydrogels with a healing ability similar to biological tissues. For instance, Messersmith and colleagues have devised hydrogel systems with healing potential by following the principles governing the self-adhesive behavior of mussels (Fig. 4).143 Mussels are known to adhere very strongly onto rocky surfaces via byssus threads, a supramolecular adhesive protein containing a high number of 3,4-dihydroxy-L-phenylalanine (dopa) residues.143–145 Under basic conditions, the catechol groups within the dopa residues are capable of strong ligand interactions with ferric ions. As each ferric ion is capable of binding to 3 catechol groups, they can be used to form coordination-mediated crosslinks between dopa-containing surfaces. Hydrogels immobilized with catechol units self-heal in the presence of ferric ions similar to how mussels adhere to surfaces. Interestingly, catechol has also been shown to bind covalently through ferric ions under acidic conditions, allowing the gels to exhibit the same self-healing capabilities under these conditions.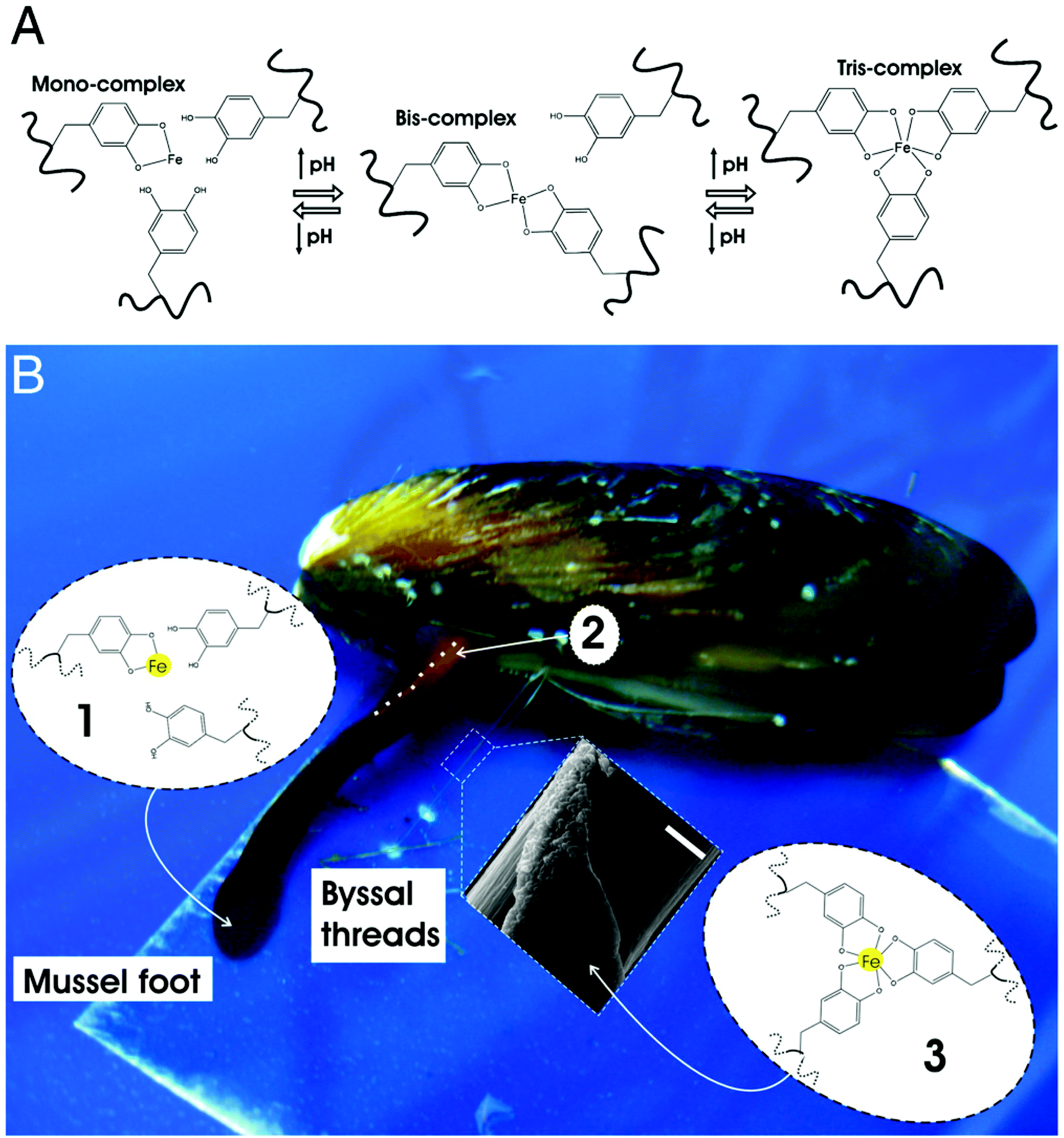 | ||
| Fig. 4 Mussel-inspired Dopa-Fe3+ cross-linking. (A) The pH-dependent stoichiometry of Fe3+-catechol complexes. (B) Schematic of the proposed crosslinking mechanism of byssal thread cuticle: (1) production and storage of Mytilus foot protein-1 (mfp-1) and Fe3+ in specialized cells of the epithelium lining the ventral groove of the mussel foot. Low pH (≤5) ensures mono-catechol-Fe3+ complexes (no cross-linking), (2) secretion and self-assembly of cuticle with the whole mussel thread in the ventral groove (outline of part of the ventral groove indicated with a dashed white line), (3) seawater exposure (pH ∼ 8) of nascent byssal thread drives immediate cuticle cross-linking via bis- and/or tris-catechol-Fe3+ complexes (inset shows a scanning electron micrograph of mussel thread with partial cuticle on top of fibrous core) (scale bar, 20 μm). Reprinted with permission from ref. 143. | ||
Sodium polyacrylate molecules forming physical hydrogels (through non-covalent interactions) when mixed with dendritic macromolecules and clay nanosheets were shown to exhibit self-healing upon contact.146 Freshly cut surfaces of these hydrogels were found to stick to each other when brought into contact with an interface strong enough to hold its own weight. Yet, the healing potential of the material was impeded when the surfaces were not freshly cut; hydrogel pieces left alone for more than a minute were not able to stick to each other when brought together.
Recently, we have designed chemically crosslinked hydrogels with the capacity for self-healing.20 To impart this self-healing potential, the chemically crosslinked networks were decorated with dangling side chains terminating with carboxyl groups that would mediate hydrogen bonding across two separate hydrogel pieces or across a rupture in the hydrogel. Our studies show that poly(acryloyl-6-aminocaproic acid)-based hydrogels can undergo rapid self-healing in an acidic solution (Fig. 5).20 A molecular simulation along with spectroscopy analyses has revealed that these dangling side chains can reach across the gel to form hydrogen bonds with amide groups, creating interleaved configurations. Experiments have shown that gels with longer or shorter dangling side chains failed to exhibit healing of equivalent strength. Such self-healing has also been demonstrated to be reversible, simply by reversing the pH of the solution. Chemically crosslinked poly(acryloyl-6-aminocaproic acid)-based hydrogels have also been shown to weld together in the presence of Cu2+ ions.147 Two separate pieces of a lightly crosslinked hydrogel can adhere to each other in the presence of Cu(II) ions to form a single hydrogel piece while keeping a weld line at the interface. The fusion of hydrogel pieces occurs by the formation of polymer–metal ions coordination complexes across the weld line.
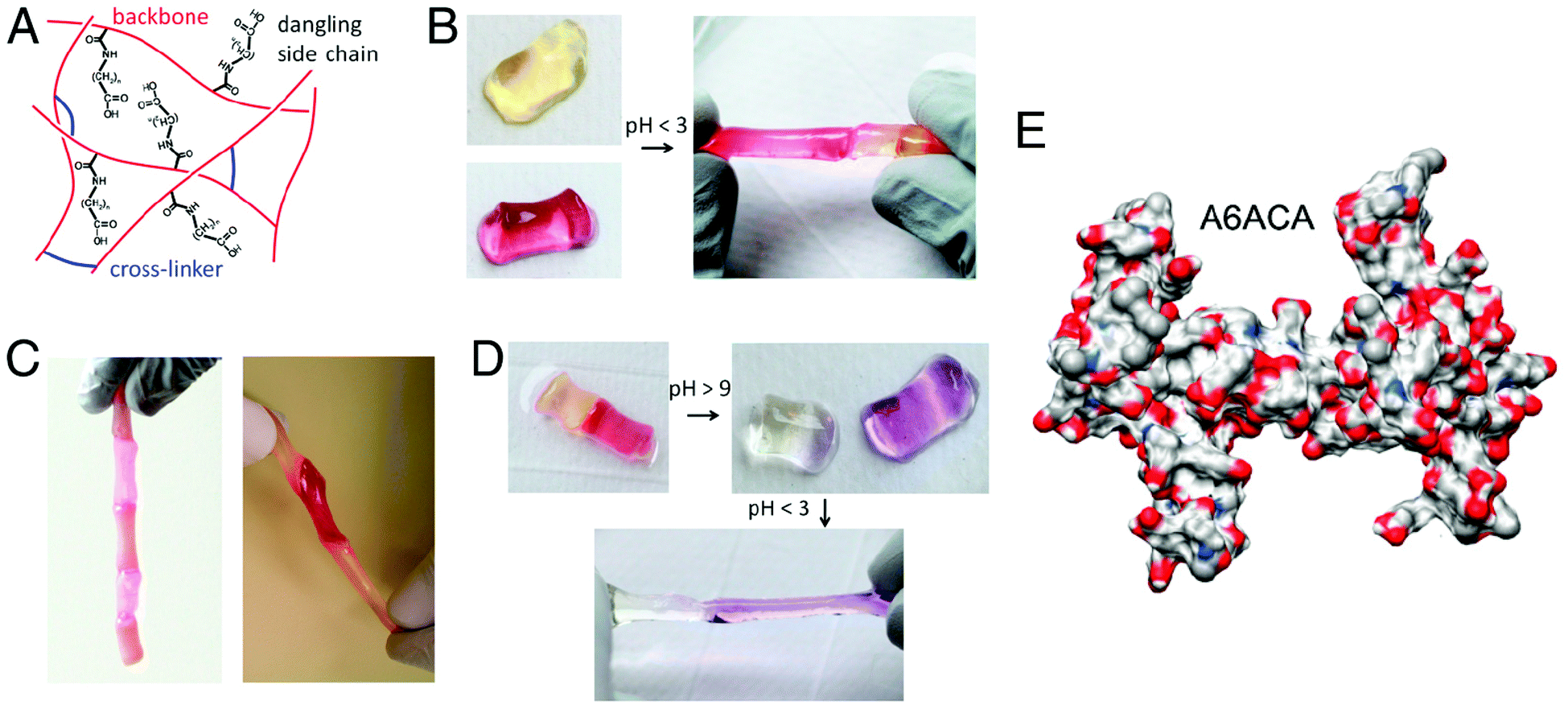 | ||
| Fig. 5 Self-healing of chemically cross-linked hydrogels. (A) Schematic illustration of the structure of self-healing A6ACA hydrogels containing dangling side chains terminating with a carboxyl group. (B) Deprotonated cylindrical hydrogels at pH 7.4 (left) healed in low-pH solution (pH ≤ 3) (right). The hydrogels are dyed yellow and maroon to allow for easily distinguished interface. (C) Healed hydrogels carrying their own weight(s) (left) and being stretched manually (right) illustrate the weld-line strength. (D) The healed hydrogels at low pH (left) separate after exposure to a high pH solution (with pH > 9) (right). The change in color is due to the reaction of the dyes with the NaOH solution. The separated hydrogels re-heal upon exposure to acidic solution (pH < 3). (E) Representative configuration of the A6ACA network obtained from molecular dynamics simulations, shown in terms of solvent excluded surface, illustrating the higher accessibility of the amide groups in the former network. Blue, red, light gray, and white colors correspond to the surfaces of nitrogen, oxygen, carbon, and hydrogen, respectively. Reprinted with permission from ref. 20. | ||
3.6 Stimuli-responsive biodegradation of hydrogel scaffolds
Perhaps the most significant contribution of hydrogels so far is in the areas of cell and tissue engineering.148–150 Hydrogels have been extensively used as a 3-D scaffold for cell culture and tissue engineering owing to their structural similarities to soft tissues.5 The extracellular matrix (ECM), a natural scaffold, is more than a structural support to the cells.151 The natural ECM provides a range of physicochemical cues to elicit signal transductions that influence cell functions.152 Hydrogel scaffolds have been endowed with various signaling cues, recapitulating the native tissue environment, to promote cell–matrix interactions and also to direct targeted cellular functions like proliferation and differentiation.153 There exist a plethora of reviews describing the advancements in the field describing the role of matrix properties in cellular behavior.154–156 Similar to physicochemical cues, the degradation kinetics of the scaffold play an important role in the properties and function of engineered tissue.157,158 Developments in the field have harnessed the stimuli-responsive properties in order to design hydrogels with precise and controlled degradation kinetics.Scaffold degradation kinetics plays a key role in various processes of encapsulated cells and functional properties of the engineered tissue.159,160 One of the reasons for this is that native tissues undergo a natural turnover as ECM is being degraded and deposited by the cells over and over again, and in order to mimic this dynamic environment, hydrogel scaffolds need to degrade at a rate mirroring the neo-tissue formation.161 Although hydrolytic degradation of hydrogel scaffolds remains the focus, a number of alternative and innovative strategies have been developed to degrade hydrogel scaffolds at a rate that mimics ECM remodeling.21,162 One of these approaches, pioneered by Hubbell and colleagues, involves integration of peptide units that are sensitive to secreted and cell-activated enzymes such as matrix metalloproteinases (MMP), plasmin, and elastase.163–167 The first example of such cell-responsive degradability in hydrogels was achieved through incorporation of MMP substrates found within the alpha chain of type I collagen. Studies have shown that hydrogels containing biodegradable moieties often promoted cell infiltration and tissue formation both in vitro and in vivo.168 Results from these studies demonstrate a strong correlation between the rate of scaffold degradation and tissue formation. Using a combinatorial approach, studies have identified MMP substrate sequences with varying sensitivity and specificity. Incorporation of MMP substrates with varying sensitivities into hydrogel networks can result in hydrogels with different rates of degradation.
Another important domain within hydrogel scaffolds with controlled-degradability is the use of light-sensitive groups to make hydrogel degradation sensitive to light.169,170 Anseth and colleagues have shown that hydrogels encoded with photosensitive nitrobenzyl ether-derived moieties underwent degradation in the presence of two-photon irradiation (Fig. 6).110 Manipulation of hydrogel properties remotely in a spatiotemporal manner provides a unique opportunity to understand the role of various physicochemical properties of the surrounding scaffold on cell functions.111 Kloxin et al. have shown that the temporal variation of poly(ethylene glycol) hydrogels functionalized with RGD moieties influence chondrogenic differentiation of encapsulated human mesenchymal stem cells.170 Such photo-sensitive systems that can be degraded in a selective and controlled manner by light can vary the mechanical properties of the biomaterial in real time to understand the role of matrix rigidity in various cellular behaviors.109 In one study, the authors showed that valvular interstitial cells (VIC) cultured on stiffer matrices exhibit higher activation compared to those cultured on softer matrices. More interestingly, softening the hydrogel through photodegradation reversed the phenotype, where the activated myofibroblasts converted back into a fibroblast-like phenotype. This suggests that the rigidity of the matrix directly and reversibly influences the extent of myofibroblast activation of the VIC. In another study, Singh et al. demonstrated the potential of photomodulation to activate gene expression in 3-D cell culture systems.171 Here, the authors have incorporated genetic inducers such as isopropyl β-D-1-thiogalactopyranoside (IPTG) into hydrogel networks through a photolabile linker. Cleavage of the linker with a UV light releases the immobilized IPTG from the hydrogel network which promoted enhanced green fluorescent protein (EGFP) expression in cells transfected with LTRi-EGFP, an IPTG responsive genetic switch. Beyond controlling the mechanical and structural properties of the hydrogel scaffolds, selective and spatially controlled photodegradation of pre-formed scaffolds could lead to the creation of voids and channels within the hydrogels to direct various cellular processes including migration, differential cell–matrix, and cell–cell interactions. Such remote manipulation of cell-laden hydrogels to create dynamic and tunable cell culture systems brings synthetic systems one step closer to physiologically relevant in vivo systems.
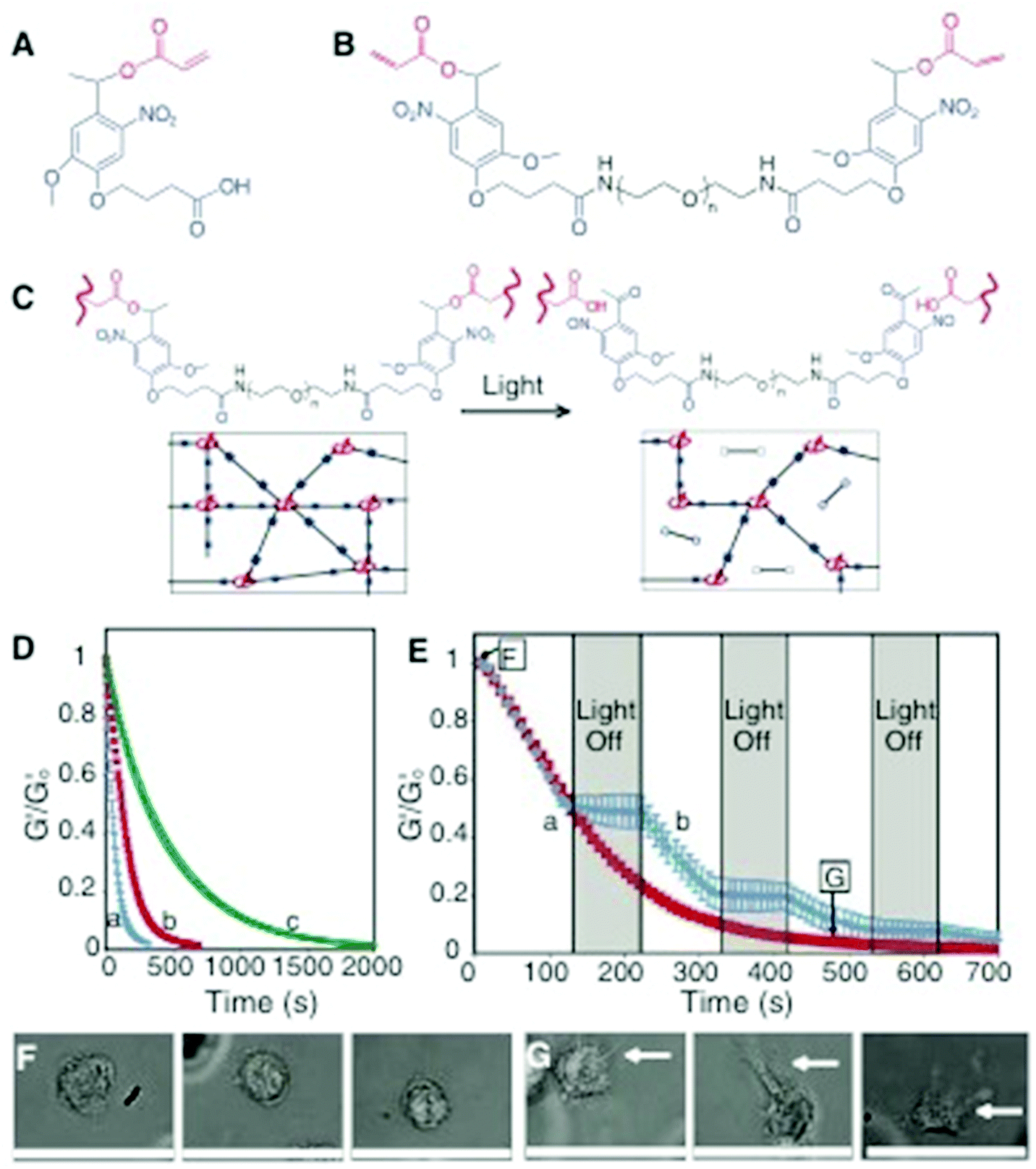 | ||
| Fig. 6 Photodegradable hydrogel synthesis and degradation for tuning gel properties. (A) The base photodegradable acrylic monomer was used to synthesize (B) the photodegradable cross-linking macromer (compound 1, Mn ∼ 4070 g mol−1), composed of PEG (black), photolabile moieties (blue), and acrylic end groups (red). (C) Compound 1 was copolymerized with PEGA (Mn ∼ 375 g mol−1), creating gels composed of poly(acrylate) chains (red coils) connected by PEG (black lines) with photolabile groups (solid blue boxes) (left). Upon irradiation, the photolabile moiety cleaves (open blue boxes), decreasing ρx and releasing modified PEG (right). (D) The physical structure of the hydrogel is degraded by photolysis, decreasing ρx and G’. The influence of irradiation on G′, normalized to G′o, was monitored with rheometry. The degradation rate was precisely controlled by different wavelengths and irradiation intensities: (a) 365 nm at 20 mW cm−2, (b) 365 nm at 10 mW cm−2, and (c) 405 nm at 25 mW cm−2. (E) The gel degradation was modulated by either (a) continuous or (b) periodic irradiation with 365 nm at 10 mW cm−2. The extent of degradation corresponding to the materials used in F and G is indicated. (F) Human mesenchymal stem cells (hMSCs) encapsulated within dense hydrogels exhibit a rounded morphology. (G) Irradiation (480 s, 365 nm at 10 mW cm−2) significantly degrades the gel (ρx/ρxo ∼ 0.04), promoting hMSC spreading after 3 days in culture. Scale bar, 50 μm. Reprinted with permission from ref. 110. | ||
4. Conclusion and future perspective
The studies discussed in this review primarily focus on stimuli-responsive hydrogels and their biomimetic functions. Nature has been an inspiration for the development of hydrogels with biomimetic functions. Knowledge gained from fundamental studies in conjunction with chemistry and fabrication have led to the development of synthetic hydrogels with unique functions such as motility, sensitivity, generation of sound, and healing. While most of these advancements remain proof-of-concept, some of these functional aspects have been harnessed towards biomedical applications such as drug delivery devices, cell sheet engineering, and for bioseparation. Stimuli-responsive hydrogels exhibiting actuating functions could be a model system to study the dynamic interaction of cells with the extracellular matrix; however, extending the application of these bioactuating systems to support 3-D culture of cells requires more sophisticated molecular designs. The ability to encode synthetic materials with biological activity will not only improve their functions but will also increase the number of their potential applications. Hydrogels with controlled degradation using photosensitive molecules are a critical step towards this direction. One focus of next generation bio-inspired materials could probably involve multifunctionality and/or cell responsive bioactuation. Such systems will not only advance the field of biomaterials, but also enable the development of a new generation of in vitro systems mimicking multiple aspects of living tissues. Beyond bridging the gap between synthetic and living systems, development in smart, multifunctional materials would advance the biomedical and industrial applications of stimuli-responsive hydrogels.References
- C. Geraths, E. H. Christen and W. Weber, Macromol. Rapid Commun., 2012, 33, 2103–2108 CrossRef CAS PubMed.
- Stimuli-Responsive Hydrogels and Their Application to Functional Materials, ed. R. Yoshida and T. Okano, Springer, New York, 2010 Search PubMed.
- P. W. Henderson, S. P. Singh, D. D. Krijgh, M. Yamamoto, D. C. Rafii, J. J. Sung, S. Rafii, S. Y. Rabbany and J. A. Spector, Wound Repair Regen., 2011, 19, 420–425 CrossRef PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS PubMed.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
- J. Elisseeff, Nat. Mater., 2008, 7, 271–273 CrossRef CAS PubMed.
- G. Grassi, R. Farra, P. Caliceti, G. Guarnieri, S. Salmaso, M. Carenza and M. Grassi, Am. J. Drug Deliv., 2005, 3, 239–251 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Deliv. Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- T. G. Park, Biomaterials, 1999, 20, 517–521 CrossRef CAS.
- Y. Qiu and K. Park, Adv. Drug Deliv. Rev., 2001, 53, 321–339 CrossRef CAS.
- K. Sawahata, M. Hara, H. Yasunaga and Y. Osada, J. Controlled Release, 1990, 14, 253–262 CrossRef CAS.
- R. Yoshida, K. Sakai, T. Okano and Y. Sakurai, Adv. Drug Deliv. Rev., 1993, 11, 85–108 CrossRef CAS.
- J. Dolbow, E. Fried and H. D. Jia, J. Mech. Phys. Solids, 2004, 52, 51–84 CrossRef CAS.
- S. H. Gehrke, Adv. Polym. Sci., 1993, 110, 81–144 CrossRef CAS.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS PubMed.
- Y. Maeda, T. Higuchi and I. Ikeda, Langmuir, 2000, 16, 7503–7509 CrossRef CAS.
- M. M. Prange, H. H. Hooper and J. M. Prausnitz, AIChE J., 1989, 35, 803–813 CrossRef CAS.
- F. Ilmain, T. Tanaka and E. Kokufuta, Nature, 1991, 349, 400–401 CrossRef CAS.
- B. Chen, S. Piletsky and A. P. Turner, Comb. Chem. High Throughput Screening, 2002, 5, 409–427 CrossRef CAS.
- A. Phadke, C. Zhang, B. Arman, C.-C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383–4388 CrossRef CAS PubMed.
- J. A. Burdick and W. L. Murphy, Nat. Commun., 2012, 3, 1269 CrossRef PubMed.
- L. E. Bromberg and E. S. Ron, Adv. Drug Deliv. Rev., 1998, 31, 197–221 CrossRef CAS.
- T. Tanaka, Phys. Rev. Lett., 1978, 40, 820–823 CrossRef CAS.
- A. K. Lele, S. K. Karode, M. V. Badiger and R. A. Mashelkar, J. Chem. Phys., 1997, 107, 2142–2148 CrossRef CAS PubMed.
- S. Varghese, A. K. Lele and R. A. Mashelkar, J. Chem. Phys., 2000, 112, 3063–3070 CrossRef CAS PubMed.
- K. Otake, H. Inomata, M. Konno and S. Saito, Macromolecules, 1990, 23, 283–289 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- K. Kubota, S. Fujishige and I. Ando, Polym. J., 1990, 22, 15–20 CrossRef CAS.
- M. V. Badiger, A. K. Lele, V. S. Bhalerao, S. Varghese and R. A. Mashelkar, J. Chem. Phys., 1998, 109, 1175–1184 CrossRef CAS PubMed.
- Y. Kaneko, R. Yoshida, K. Sakai, Y. Sakurai and T. Okano, J. Membr. Sci., 1995, 101, 13–22 CrossRef CAS.
- J. D. Kretlow, L. Klouda and A. G. Mikos, Adv. Drug Deliv. Rev., 2007, 59, 263–273 CrossRef CAS PubMed.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- H. Tan, C. M. Ramirez, N. Miljkovic, H. Li, J. P. Rubin and K. G. Marra, Biomaterials, 2009, 30, 6844–6853 CrossRef CAS PubMed.
- J.-P. Chen and T.-H. Cheng, Macromol. Biosci., 2006, 6, 1026–1039 CrossRef CAS PubMed.
- R. M. P. da Silva, J. F. Mano and R. L. Reis, Trends Biotechnol., 2007, 25, 577–583 CrossRef CAS PubMed.
- P. Gupta, K. Vermani and S. Garg, Drug Discov. Today, 2002, 7, 569–579 CrossRef CAS.
- J. C. Garbern, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2010, 11, 1833–1839 CrossRef CAS PubMed.
- J. P. Chen and T. H. Cheng, Macromol. Biosci., 2006, 6, 1026–1039 CrossRef CAS PubMed.
- S. K. De, N. R. Aluru, B. Johnson, W. C. Crone, D. J. Beebe and J. Moore, J. Microelectromech. Syst., 2002, 11, 544–555 CrossRef CAS.
- Y. Zhao, H. J. Su, L. Fang and T. W. Tan, Polymer, 2005, 46, 5368–5376 CrossRef CAS PubMed.
- S. Maeda, Y. Hara, T. Sakai, R. Yoshida and S. Hashimoto, Adv. Mater., 2007, 19, 3480–3484 CrossRef CAS.
- S. Ashley, Sci. Am., 2003, 289, 52–59 CrossRef CAS PubMed.
- T. Shiga, Y. Hirose, A. Okada and T. Kurauchi, J. Appl. Polym. Sci., 1993, 47, 113–119 CrossRef CAS.
- T. Tanaka, I. Nishio, S. T. Sun and S. Uenonishio, Science, 1982, 218, 467–469 CAS.
- Y. Osada, H. Okuzaki and H. Hori, Nature, 1992, 355, 242–244 CrossRef CAS.
- W. M. Lai, D. D. Sun, G. A. Ateshian, X. E. Guo and V. C. Mow, Biorheology, 2002, 39, 39–45 CAS.
- H. L. Lim, J. C. Chuang, T. Tuan, A. Aung, G. Arya and S. Varghese, Adv. Funct. Mater., 2011, 21, 55–63 CrossRef CAS PubMed.
- M. Doi, M. Matsumoto and Y. Hirose, Macromolecules, 1992, 25, 5504–5511 CrossRef CAS.
- R. Tomer and A. T. Florence, Int. J. Pharm., 1993, 99, R5–R8 CrossRef.
- G. S. Kumar and D. C. Neckers, Chem. Rev., 1989, 89, 1915–1925 CrossRef CAS.
- Y. L. Yu and T. Ikeda, Macromol. Chem. Phys., 2005, 206, 1705–1708 CrossRef CAS.
- H. Meng and J. L. Hu, J. Intell. Mater. Syst. Struct., 2010, 21, 859–885 CrossRef CAS PubMed.
- M. Irie and D. Kunwatchakun, Macromolecules, 1986, 19, 2476–2480 CrossRef CAS.
- A. Suzuki and T. Tanaka, Nature, 1990, 346, 345–347 CrossRef CAS.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- S. Maeda, Y. Hara, R. Yoshida and S. Hashimoto, Int. J. Mol. Sci., 2010, 11, 52–66 CrossRef CAS PubMed.
- X. B. Zhang, C. L. Pint, M. H. Lee, B. E. Schubert, A. Jamshidi, K. Takei, H. Ko, A. Gillies, R. Bardhan, J. J. Urban, M. Wu, R. Fearing and A. Javey, Nano Lett., 2011, 11, 3239–3244 CrossRef CAS PubMed.
- M. J. Bassetti, A. N. Chatterjee, N. R. Aluru and D. J. Beebe, J. Microelectromech. Syst., 2005, 14, 1198–1207 CrossRef.
- D. T. Eddington and D. J. Beebe, Adv. Drug Deliv. Rev., 2004, 56, 199–210 CrossRef CAS PubMed.
- R. H. Liu, Q. Yu and D. J. Beebe, J. Microelectromech. Syst., 2002, 11, 45–53 CrossRef CAS.
- S. Chaterji, I. K. Kwon and K. Park, Prog. Polym. Sci., 2007, 32, 1083–1122 CrossRef CAS PubMed.
- G. H. Kwon, Y. Y. Choi, J. Y. Park, D. H. Woo, K. B. Lee, J. H. Kim and S. H. Lee, Lab chip, 2010, 10, 1604–1610 RSC.
- S. Sundelacruz, M. Levin and D. L. Kaplan, Stem Cell Rev., 2009, 5, 231–246 CrossRef PubMed.
- C. Keplinger, J. Y. Sun, C. C. Foo, P. Rothemund, G. M. Whitesides and Z. Suo, Science, 2013, 341, 984–987 CrossRef CAS PubMed.
- L. Dong and H. Jiang, Appl. Phys. Lett., 2006, 89, 211120 CrossRef PubMed.
- L. Dong, A. K. Agarwal, D. J. Beebe and H. Jiang, Nature, 2006, 442, 551–554 CrossRef CAS PubMed.
- L. Dong, A. K. Agarwal, D. J. Beebe and H. R. Jiang, Adv. Mater., 2007, 19, 401–405 CrossRef CAS.
- R. Yoshida, Adv. Mater., 2010, 22, 3463–3483 CrossRef CAS PubMed.
- D. Suzuki and R. Yoshida, J. Phys. Chem. B, 2008, 112, 12618–12624 CrossRef CAS PubMed.
- R. Yoshida, T. Takahashi, T. Yamaguchi and H. Ichijo, J. Am. Chem. Soc., 1996, 118, 5134–5135 CAS.
- R. Yoshida and Y. Murase, Colloids Surf., B: Biointerfaces, 2012, 99, 60–66 CAS.
- Y. Murase, S. Maeda, S. Hashimoto and R. Yoshida, Langmuir, 2009, 25, 483–489 CrossRef CAS PubMed.
- S. Maeda, Y. Hara, R. Yoshida and S. Hashimoto, Angew. Chem., Int. Ed., 2008, 47, 6690–6693 CrossRef CAS PubMed.
- X. He, M. Aizenberg, O. Kuksenok, L. D. Zarzar, A. Shastri, A. C. Balazs and J. Aizenberg, Nature, 2012, 487, 214–218 CrossRef CAS PubMed.
- L. D. Zarzar, P. Kim and J. Aizenberg, Adv. Mater., 2011, 23, 1442–1446 CrossRef CAS PubMed.
- A. W. Feinberg, A. Feigel, S. S. Shevkoplyas, S. Sheehy, G. M. Whitesides and K. K. Parker, Science, 2007, 317, 1366–1370 CrossRef CAS PubMed.
- T. Miyata, T. Uragami and K. Nakamae, Adv. Drug Deliv. Rev., 2002, 54, 79–98 CrossRef CAS.
- R. Taylor and L. Agius, Biochem. J., 1988, 250, 625–640 CAS.
- T. Miyata, N. Asami and T. Uragami, Nature, 1999, 399, 766–769 CrossRef CAS PubMed.
- J. M. Weissman, H. B. Sunkara, A. S. Tse and S. A. Asher, Science, 1996, 274, 959–960 CrossRef CAS.
- Y. Takeoka and M. Watanabe, Langmuir, 2003, 19, 9104–9106 CrossRef CAS.
- Z. B. Hu, X. H. Lu and J. Gao, Adv. Mater., 2001, 13, 1708–1712 CrossRef CAS.
- J. H. Holtz and S. A. Asher, Nature, 1997, 389, 829–832 CrossRef CAS PubMed.
- C. J. Zhang, M. D. Losego and P. V. Braun, Chem. Mater., 2013, 25, 3239–3250 CrossRef CAS.
- X. Pan, X. Yang and C. R. Lowe, J. Mol. Recognit.: JMR, 2008, 21, 205–209 CrossRef CAS PubMed.
- K. Kataoka, H. Miyazaki, M. Bunya, T. Okano and Y. Sakurai, J. Am. Chem. Soc., 1998, 120, 12694–12695 CrossRef CAS.
- X. Zhang, Y. Guan and Y. Zhang, Biomacromolecules, 2012, 13, 92–97 CrossRef CAS PubMed.
- Y. Murakami and M. Maeda, Biomacromolecules, 2005, 6, 2927–2929 CrossRef CAS PubMed.
- H. Qi, M. Ghodousi, Y. Du, C. Grun, H. Bae, P. Yin and A. Khademhosseini, Nat. Commun., 2013, 4, 2275 Search PubMed.
- H. Zhang, S. Mardyani, W. C. Chan and E. Kumacheva, Biomacromolecules, 2006, 7, 1568–1572 CrossRef CAS PubMed.
- B. Kim and N. A. Peppas, Int. J. Pharm., 2003, 266, 29–37 CrossRef CAS.
- D. Kuckling, Colloid Polym. Sci., 2009, 287, 881–891 CAS.
- H. J. Jung and A. Chauhan, Biomaterials, 2012, 33, 2289–2300 CrossRef CAS PubMed.
- J. J. Kang Derwent and W. F. Mieler, Trans. Am. Ophthalmol. Soc., 2008, 106, 206–213 Search PubMed ; discussion 213–204.
- S. K. Kushwaha, P. Saxena and A. Rai, Int. J. Pharm. Invest., 2012, 2, 54–60 CrossRef CAS PubMed.
- A. L. Hennessy, J. Katz, D. Covert, C. Protzko and A. L. Robin, Ophthalmology, 2010, 117, 2345–2352 CrossRef PubMed.
- A. Guzman-Aranguez, B. Colligris and J. Pintor, J. Ocul. Pharmacol. Ther., 2013, 29, 189–199 CrossRef CAS PubMed.
- S. R. Sershen, S. L. Westcott, N. J. Halas and J. L. West, J. Biomed. Mater. Res., 2000, 51, 293–298 CrossRef CAS.
- H. Tan, C. M. Ramirez, N. Miljkovic, H. Li, J. P. Rubin and K. G. Marra, Biomaterials, 2009, 30, 6844–6853 CrossRef CAS PubMed.
- E. F. Irwin, R. Gupta, D. C. Dashti and K. E. Healy, Biomaterials, 2011, 32, 6912–6919 CrossRef CAS PubMed.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- L. J. Suggs and A. G. Mikos, Cell Transplant, 1999, 8, 345–350 CAS.
- D. Schmaljohann, Adv. Drug Deliv. Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- K. Raemdonck, J. Demeester and S. De Smedt, Soft Matter, 2009, 5, 707–715 RSC.
- J.-Z. Du, T.-M. Sun, W.-J. Song, J. Wu and J. Wang, Angew. Chem., Int. Ed., 2010, 122, 3703–3708 CrossRef.
- R. C. Mundargi, V. Rangaswamy and T. M. Aminabhavi, J. Microencapsul., 2011, 28, 384–394 CrossRef CAS PubMed.
- L. A. Klumb and T. A. Horbett, J. Controlled Release, 1992, 18, 59–80 CrossRef CAS.
- A. M. Kloxin, J. A. Benton and K. S. Anseth, Biomaterials, 2010, 31, 1–8 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- A. M. Kloxin, M. W. Tibbitt and K. S. Anseth, Nat. Protoc., 2010, 5, 1867–1887 CrossRef CAS PubMed.
- D. R. Griffin and A. M. Kasko, J. Am. Chem. Soc., 2012, 134, 13103–13107 CAS.
- D. R. Griffin, J. L. Schlosser, S. F. Lam, T. H. Nguyen, H. D. Maynard and A. M. Kasko, Biomacromolecules, 2013, 14, 1199–1207 CrossRef CAS PubMed.
- X. Zhao, J. Kim, C. A. Cezar, N. Huebsch, K. Lee, K. Bouhadir and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 67–72 CrossRef CAS PubMed.
- Y. S. Hwang, C. Zhang and S. Varghese, J. Mater. Chem., 2010, 20, 345–351 RSC.
- D. R. Griffin and A. M. Kasko, ACS Macro Lett., 2012, 1, 1330–1334 CrossRef CAS.
- Y. Mei, K. Saha, S. R. Bogatyrev, J. Yang, A. L. Hook, Z. I. Kalcioglu, S. W. Cho, M. Mitalipova, N. Pyzocha, F. Rojas, K. J. Van Vliet, M. C. Davies, M. R. Alexander, R. Langer, R. Jaenisch and D. G. Anderson, Nat. Mater., 2010, 9, 768–778 CrossRef CAS PubMed.
- R. Ayala, C. Zhang, D. Yang, Y. Hwang, A. Aung, S. S. Shroff, F. T. Arce, R. Lal, G. Arya and S. Varghese, Biomaterials, 2011, 32, 3700–3711 CrossRef CAS PubMed.
- C. W. Chang, Y. Hwang, D. Brafman, T. Hagan, C. Phung and S. Varghese, Biomaterials, 2013, 34, 912–921 CrossRef CAS PubMed.
- H. E. Canavan, X. Cheng, D. J. Graham, B. D. Ratner and D. G. Castner, Plasma Process. Polym., 2006, 3, 516–523 CrossRef CAS.
- A. K. A. S. Brun-Graeppi, C. Richard, M. Bessodes, D. Scherman and O.-W. Merten, Prog. Polym. Sci., 2010, 35, 1311–1324 CrossRef CAS PubMed.
- Y. Kumashiro, M. Yamato and T. Okano, Ann. Biomed. Eng., 2010, 38, 1977–1988 CrossRef PubMed.
- K. Tadakuma, N. Tanaka, Y. Haraguchi, M. Higashimori, M. Kaneko, T. Shimizu, M. Yamato and T. Okano, Biomaterials, 2013, 34, 9018–9025 CrossRef CAS PubMed.
- R. Yoshida and T. Okano, in Biomedical Applications of Hydrogels Handbook, ed. R. M. Ottenbrite, K. Park and T. Okano, Springer, New York, 2010, ch. 2, pp. 19–43 Search PubMed.
- H. E. Canavan, X. Cheng, D. J. Graham, B. D. Ratner and D. G. Castner, J. Biomed. Mater. Res., Part A, 2005, 75, 1–13 CrossRef PubMed.
- T. Shimizu, M. Yamato, A. Kikuchi and T. Okano, Tissue Eng., 2001, 7, 141–151 CrossRef CAS PubMed.
- M. Yamato, M. Utsumi, A. Kushida, C. Konno, A. Kikuchi and T. Okano, Tissue Eng., 2001, 7, 473–480 CrossRef CAS PubMed.
- K. Nishida, M. Yamato, Y. Hayashida, K. Watanabe, N. Maeda, H. Watanabe, K. Yamamoto, S. Nagai, A. Kikuchi, Y. Tano and T. Okano, Transplantation, 2004, 77, 379–385 CrossRef PubMed.
- K. Nishida, M. Yamato, Y. Hayashida, K. Watanabe, K. Yamamoto, E. Adachi, S. Nagai, A. Kikuchi, N. Maeda, H. Watanabe, T. Okano and Y. Tano, N Engl. J. Med., 2004, 351, 1187–1196 CrossRef CAS PubMed.
- M. Harimoto, M. Yamato, M. Hirose, C. Takahashi, Y. Isoi, A. Kikuchi and T. Okano, J. Biomed. Mater. Res., 2002, 62, 464–470 CrossRef CAS PubMed.
- T. Akizuki, S. Oda, M. Komaki, H. Tsuchioka, N. Kawakatsu, A. Kikuchi, M. Yamato, T. Okano and I. Ishikawa, J. Periodontal Res., 2005, 40, 245–251 CrossRef PubMed.
- Y. Shiroyanagi, M. Yamato, Y. Yamazaki, H. Toma and T. Okano, BJU Int., 2004, 93, 1069–1075 CrossRef CAS PubMed.
- T. Shimizu, M. Yamato, Y. Isoi, T. Akutsu, T. Setomaru, K. Abe, A. Kikuchi, M. Umezu and T. Okano, Circ. Res., 2002, 90, e40–e48 CrossRef CAS.
- Y. Haraguchi, T. Shimizu, T. Sasagawa, H. Sekine, K. Sakaguchi, T. Kikuchi, W. Sekine, S. Sekiya, M. Yamato, M. Umezu and T. Okano, Nat. Protoc., 2012, 7, 850–858 CrossRef CAS PubMed.
- T. Sasagawa, T. Shimizu, S. Sekiya, Y. Haraguchi, M. Yamato, Y. Sawa and T. Okano, Biomaterials, 2010, 31, 1646–1654 CrossRef CAS PubMed.
- M. Sha, D. Niu, Q. Dou, G. Wu, H. Fang and J. Hu, Soft Matter, 2011, 7, 4228–4233 RSC.
- J. Lahann, S. Mitragotri, T. N. Tran, H. Kaido, J. Sundaram, I. S. Choi, S. Hoffer, G. A. Somorjai and R. Langer, Science, 2003, 299, 371–374 CrossRef CAS PubMed.
- V. V. Ramanan, J. S. Katz, M. Guvendiren, E. R. Cohen, R. A. Marklein and J. A. Burdick, J. Mater. Chem., 2010, 20, 8920–8926 RSC.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- A. B. South and L. A. Lyon, Angew. Chem., Int. Ed., 2010, 122, 779–783 CrossRef.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS PubMed.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed.
- D. G. Barrett, D. E. Fullenkamp, L. He, N. Holten-Andersen, K. Y. C. Lee and P. B. Messersmith, Adv. Funct. Mater., 2013, 23, 1111–1119 CrossRef CAS PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- S. Varghese, A. Lele and R. Mashelkar, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 666–670 CrossRef CAS.
- A. Khademhosseini and R. Langer, Biomaterials, 2007, 28, 5087–5092 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- K. T. Nguyen and J. L. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS.
- T. Rozario and D. W. DeSimone, Dev. Biol., 2010, 341, 126–140 CrossRef CAS PubMed.
- N. F. Huang and S. Li, Ann. Biomed. Eng., 2011, 39, 1201–1214 CrossRef PubMed.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 CrossRef CAS PubMed.
- K. Saha, J. F. Pollock, D. V. Schaffer and K. E. Healy, Curr. Opin. Chem. Biol., 2007, 11, 381–387 CrossRef CAS PubMed.
- R. A. Marklein and J. A. Burdick, Adv. Mater., 2010, 22, 175–189 CrossRef CAS PubMed.
- P. A. Gunatillake and R. Adhikari, Eur. Cell. Mater., 2003, 5, 1–16 CAS ; discussion 16.
- J. F. Mano, G. A. Silva, H. S. Azevedo, P. B. Malafaya, R. A. Sousa, S. S. Silva, L. F. Boesel, J. M. Oliveira, T. C. Santos, A. P. Marques, N. M. Neves and R. L. Reis, J. R. Soc. Interface, 2007, 4, 999–1030 CrossRef CAS PubMed.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS PubMed.
- J. A. Hubbell and R. Langer, Nat. Mater., 2013, 12, 963–966 CrossRef CAS PubMed.
- M. E. Furth, A. Atala and M. E. Van Dyke, Biomaterials, 2007, 28, 5068–5073 CrossRef CAS PubMed.
- J. Patterson, M. M. Martino and J. A. Hubbell, Mater. Today, 2010, 13, 14–22 CrossRef CAS.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS PubMed.
- M. P. Lutolf, G. P. Raeber, A. H. Zisch, N. Tirelli and J. A. Hubbell, Adv. Mater., 2003, 15, 888–892 CrossRef CAS.
- D. Seliktar, A. H. Zisch, M. P. Lutolf, J. L. Wrana and J. A. Hubbell, J. Biomed. Mater. Res., Part A, 2004, 68, 704–716 CrossRef CAS PubMed.
- J. Patterson and J. A. Hubbell, Biomaterials, 2011, 32, 1301–1310 CrossRef CAS PubMed.
- T. P. Kraehenbuehl, L. S. Ferreira, A. M. Hayward, M. Nahrendorf, A. J. van der Vlies, E. Vasile, R. Weissleder, R. Langer and J. A. Hubbell, Biomaterials, 2011, 32, 1102–1109 CrossRef CAS PubMed.
- J. Patterson and J. A. Hubbell, Biomaterials, 2010, 31, 7836–7845 CrossRef CAS PubMed.
- M. A. Azagarsamy and K. S. Anseth, Angew. Chem., Int. Ed., 2013, 13803–13807 CrossRef CAS PubMed.
- M. W. Tibbitt, A. M. Kloxin, L. A. Sawicki and K. S. Anseth, Macromolecules, 2013, 46, 2785–2792 CrossRef CAS PubMed.
- A. Singh, T. L. Deans and J. H. Elisseeff, ACS Macro Lett., 2013, 2, 269–272 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |