Designing nanoparticle carriers for enhanced drug efficacy in photodynamic therapy†
Zhiqin
Chu‡
a,
Silu
Zhang‡
a,
Chun
Yin
b,
Ge
Lin
b and
Quan
Li
*a
aDepartment of Physics, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong. E-mail: liquan@phy.cuhk.edu.hk
bSchool of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong
First published on 19th March 2014
Abstract
Although nanoparticles (NPs) have been proposed as carriers for photosensitizers (PS) in photodynamic therapy (PDT), how the design parameters of nanocarriers affect the final drug efficacy remains unclear. By designing SiO2-PS NPs with specific features (such as enabling easy release of PS from the nanocarriers, or introducing plasmonic Au NPs in the vicinity of the PS), and comparing the respective efficacy of PS to that of the conventional dense SiO2-PS NPs (PS were tightly confined into the silica matrix), we identified that both PS trapping-in/releasing from the silica nanocarriers and the Au plasmonic effect were responsible for the PS efficacy variation. The mechanistic study also disclosed that the different NP configurations would affect the cellular death pathway. These findings provide a general guideline for the design of NP-based PDT applications.
Introduction
Photodynamic therapy (PDT) is an effective method for various superficial cancer therapies by the combination of administration and irradiation of photosensitizers (PS), which damage the cancer cells by producing reactive oxygen species (ROS).1–3 Despite the large number of different PS developed, many of them suffered from inefficient cellular uptake, chemical instability in a physiological environment, and consequently, low efficacy.4–6 In recent years, nanoparticles (NPs) have been found to enter cells easily via endocytosis,7,8 and thus have been proposed as effective drug carriers for various PS, aimed at enhanced drug bioavailability and efficacy.Silica NPs were one of the most popular candidates as the nanocarriers for PDT applications, due to their easily modifiable surface, excellent chemical stability and low cytotoxicity.9–11 Nevertheless, it has been found that simply trapping PS in silica NPs seldom resulted in PS efficacy enhancement in PDT,12–14 although enhanced cellular uptake of the PS was always reported.15,16
In a typical PDT process, the efficacy of PS is determined by both the type and total amount of produced ROS. One problem associated with PS loaded into NPs is the consequent trapping of the produced ROS—the presence of solid nanocarriers slows down the out-diffusion of the generated ROS. The inhibited ROS escaping from the nanocarriers to the cytoplasm would largely reduce their cell killing capability. In this regard, effective release of ROS (or PS themselves) from the silica matrix would improve the drug efficacy. On the other hand, one can expect that increasing the generation efficiency of ROS would always contribute to the efficacy enhancement.
In the present work, we have designed two specific SiO2-based nanocarriers loading PS to perform PDT in vitro, with one aiming at effective release of the ROS (or PS) upon NPs’ cellular uptake and the other addressing the issue of improving the generation efficiency of ROS. Methylene blue (MB), a typical PS with a wide range of therapeutic applications,17–19 was chosen as a model PDT drug molecule to be incorporated into the corresponding nanocarrier. The design of various nanocarriers for in vitro PDT study is shown in Fig. 1. Firstly, MB was uniformly incorporated into the silica matrix using a modified Stober's method.20 This type of conventional SiO2-MB NPs was characterized as tight trapping of MB into the nanocarrier with little drug leakage. We named it as the dense SiO2-MB NPs, and used it as one of the control samples in the following experiments. In the first approach, we fabricated a special type of “loose” SiO2-MB NPs.21 Compared to conventional dense silica NPs, the loose SiO2-MB NPs had higher ROS or MB diffusion capability as the NPs themselves would undergo self-decomposition accompanied by facile MB release in a physiological environment.21 In the second approach, we embedded MB in the surface layer of dense silica outside the Au nanorod (NR) core (denoted Au@(SiO2-MB) NPs), taking advantage of the surface plasmon enhanced ROS generation. We showed that both approaches improved the drug efficacy of MB. In addition, the detailed mechanisms of the drug efficacy enhancement were discussed.
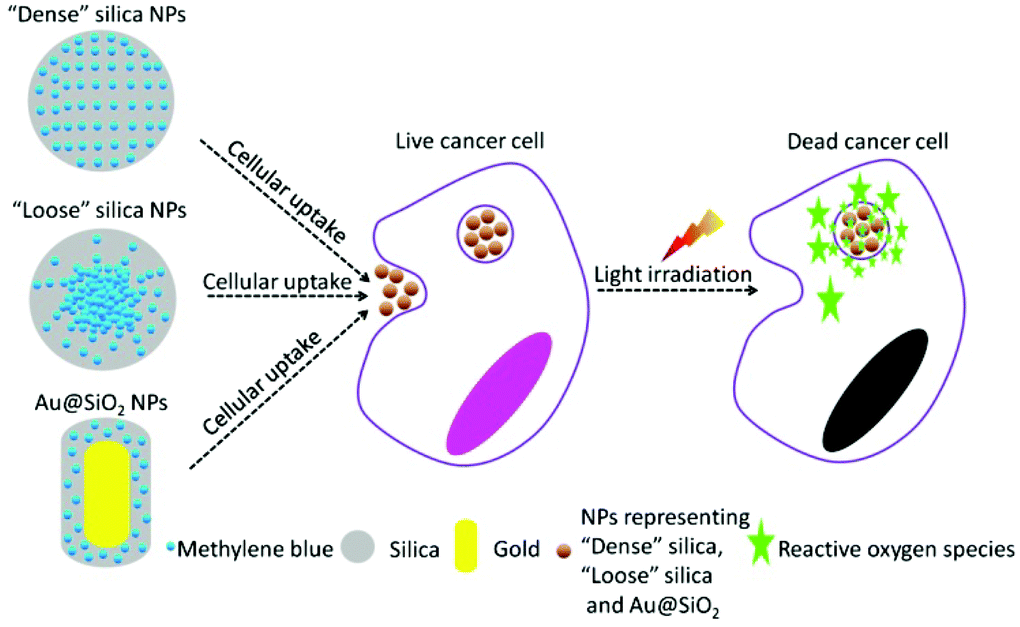 | ||
| Fig. 1 Schematic illustration of design of dense SiO2-MB, loose SiO2-MB and Au@(SiO2-MB) NPs for performing PDT in vitro. | ||
Results
The as-synthesized dense SiO2-MB NPs and loose SiO2-MB NPs were all spherical in shape with an average diameter of ∼80 nm (Fig. 2a and b). The as-synthesized Au@(SiO2-MB) NPs were uniform with a core/shell morphology (Fig. 2c). The Au NRs core had an average diameter of ∼40 nm and an aspect ratio close to 2, and the MB containing silica shell appeared uniform with a thickness of ∼40 nm. Evidence of successful MB incorporation into the SiO2 matrix can be found in the absorption spectra taken from these samples (Fig. 2d). SiO2 alone has little absorption in the wavelength range of 400–800 nm. MB itself had two absorption peaks at 665 nm and 600 nm, corresponding to its monomer and dimer absorption, respectively (Fig. 2d). When incorporated into the SiO2, its dimer form was significantly increased due to MB aggregation.21 The Au NRs themselves had two plasmonic absorption peaks with the transverse mode occurring at ∼522 nm and the longitudinal mode at longer wavelength, which was tunable (∼600 nm in the present case) by varying the aspect ratio of NRs. Compared to that of the Au NRs alone, incorporating MB into the SiO2 shell (in the Au@(SiO2-MB) NPs) only broadened the Au longitudinal SPR, without affecting much the spectrum line shape. This was mainly due to the position overlap between the Au longitudinal SPR and the MB dimer absorption.20 We then investigated the release behavior of MB molecules from these nanocarriers in phosphate buffered saline (PBS, pH ∼ 7.4) at 37 °C (mimicking the physiological conditions). One could observe that nearly 60% MB was released from the loose SiO2-MB NPs after 24 hours of incubation, while only less than 5% MB escaped from the dense SiO2-MB or Au@(SiO2-MB) NPs.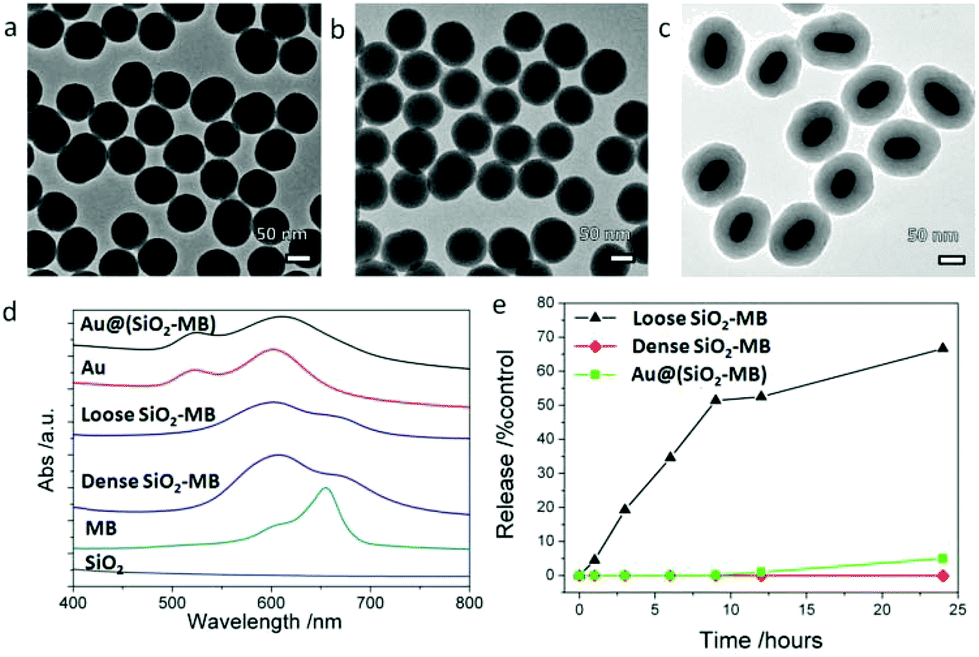 | ||
| Fig. 2 Characterization of dense SiO2-MB, loose SiO2-MB and Au@(SiO2-MB) NPs. Low magnification TEM images of (a) dense SiO2-MB NPs, (b) loose SiO2-MB NPs and (c) Au@(SiO2-MB) NPs; (d) absorption spectra taken from SiO2 NPs, MB alone, dense SiO2-MB, loose SiO2-MB, Au NRs and Au@(SiO2-MB) NPs in aqueous solution ([MB] = 5 μM for all samples); (e) evolution of the MB released from the NPs as a function of the immersing duration in PBS at 37 °C. | ||
Generally speaking, there were two major types of ROS generated by light irradiation of PS (MB in the present case), i.e., Type I and Type II ROS. Type I ROS was known as the free radicals, including primary species such as superoxide and secondary species such as hydroxyl radicals, while Type II ROS mainly referred to singlet oxygen.22 Since the cell responded differently to different types of ROS, we compared the major types and the corresponding amount of generated ROS by free MB or MB loaded into various nanocarriers in a semi-quantitative manner in aqueous solution.
Singlet oxygen sensor green (SOSG) was chosen as the detection agent for the generated singlet oxygen,23 whose amount was represented by the height of the fluorescent peak of SOSG at 525 nm. For the control sample (only SOSG in aqueous solution), a weak signal was observed and can be attributed to the background signal of singlet oxygen in the solution. An obvious fluorescent peak at 525 nm with high intensity (Fig. S1†) could be seen after 20 minutes of irradiation of the free MB containing sample. As a comparison, the amount of singlet oxygen generated from loose SiO2-MB NPs was barely half of that from free MB, while the amount of singlet oxygen detected in dense SiO2-MB NPs was similar to that from Au@(SiO2-MB) NPs, but it was merely half of that produced from loose SiO2-MB NPs (green bar in Fig. 3).
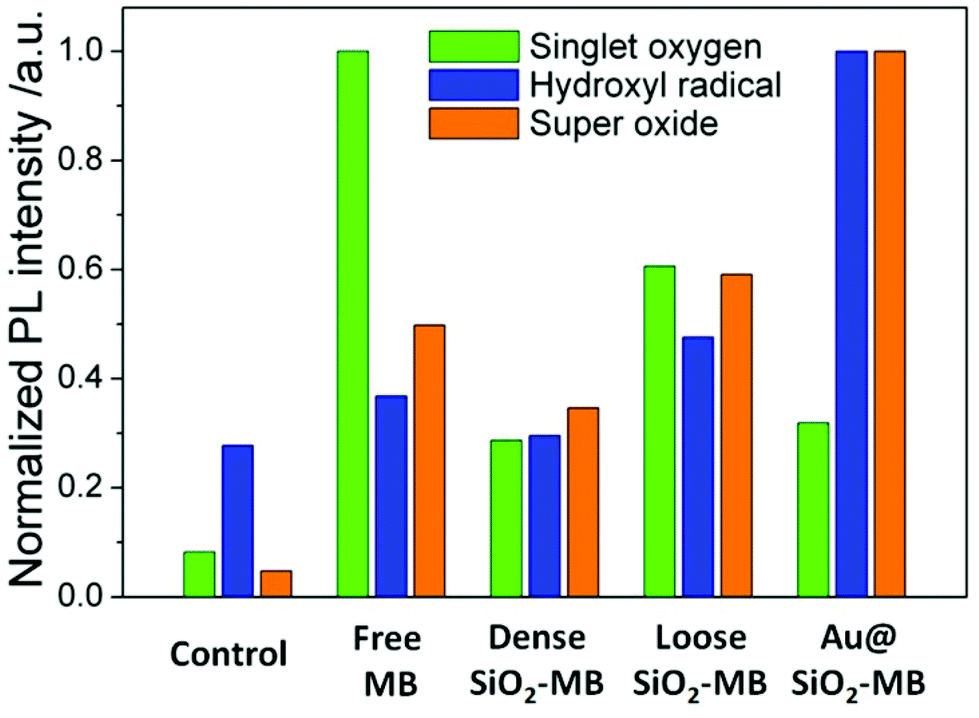 | ||
| Fig. 3 Quantitative comparison of various ROS generated including singlet oxygen, hydroxyl radical and superoxide in various MB loaded NPs in aqueous solution. The amount of specific ROS was represented by the height of the fluorescent peak of various ROS detection dyes in Fig. S1.† | ||
The generated hydroxyl radicals and superoxide were measured using detection dyes terephthalic acid (TA)24 and dihydroethidium (DHE),25 respectively. An obvious fluorescent peak at 425 nm (Fig. S1†), which indicated the presence of hydroxyl radicals, could be observed in the solution containing Au@(SiO2-MB) NPs after light irradiation for 20 minutes. Such peaks can also be observed in all other samples, but with all of their intensities only slightly higher than that of the control sample (blue bar in Fig. 3), indicating little generation of hydroxyl radicals from those samples without Au NR core. For superoxide radicals detected using DHE, both free MB, loose SiO2-MB, dense SiO2-MB, and Au@(SiO2-MB) NPs showed distinct fluorescent peak values at 586 nm (Fig. S1†), with the order of the peak intensity as Au@(SiO2-MB) NPs > loose SiO2-MB > free MB > dense SiO2 MB (orange bar in Fig. 3). We have also investigated the ROS produced by Au@SiO2 NPs, and found that the results were similar to those of the control sample, i.e., aqueous solution containing ROS detection dyes only (results not shown here).
In vitro results showed that all kinds of MB loaded NPs, including the dense SiO2-MB, loose SiO2-MB and Au@(SiO2-MB) NPs, entered the cells easily (Fig. 4a–c). The fact that these nanocarriers were always found in membrane bound vesicles (as shown in the insert of Fig. 4a–c) after cellular uptake suggested endocytosis as the major route for NPs’ cellular entry.26 For the PDT experiments carried out in vitro, the efficacy of free MB or MB loaded in various nanocarriers was firstly evaluated using the MTT assay at a concentration of [MB] = 5 μM (a value approximately equal to IC50 of free MB, see Fig. S2†) in HepG2 cells (Fig. 4d). Low cytotoxicity (∼85% viability) was observed for all samples in the dark. After light irradiation for 20 minutes, the cell viability dropped to 47% for free MB, 51% for dense SiO2-MB, 35% for loose SiO2-MB and 26% for Au@(SiO2-MB) NP treated samples (Fig. 4d). The concentration dependent effects of both loose SiO2-MB and Au@(SiO2-MB) NP treated cells were also demonstrated (Fig. 4e and 4f). Compared to free MB alone (IC50 was 4.70 μM (Fig. S2†)), both the loose SiO2-MB and the Au@(SiO2-MB) NPs largely enhanced the MB efficacy with IC50 of 2.63 μM (Fig. 4e) and 2.59 μM (Fig. 4f), respectively.
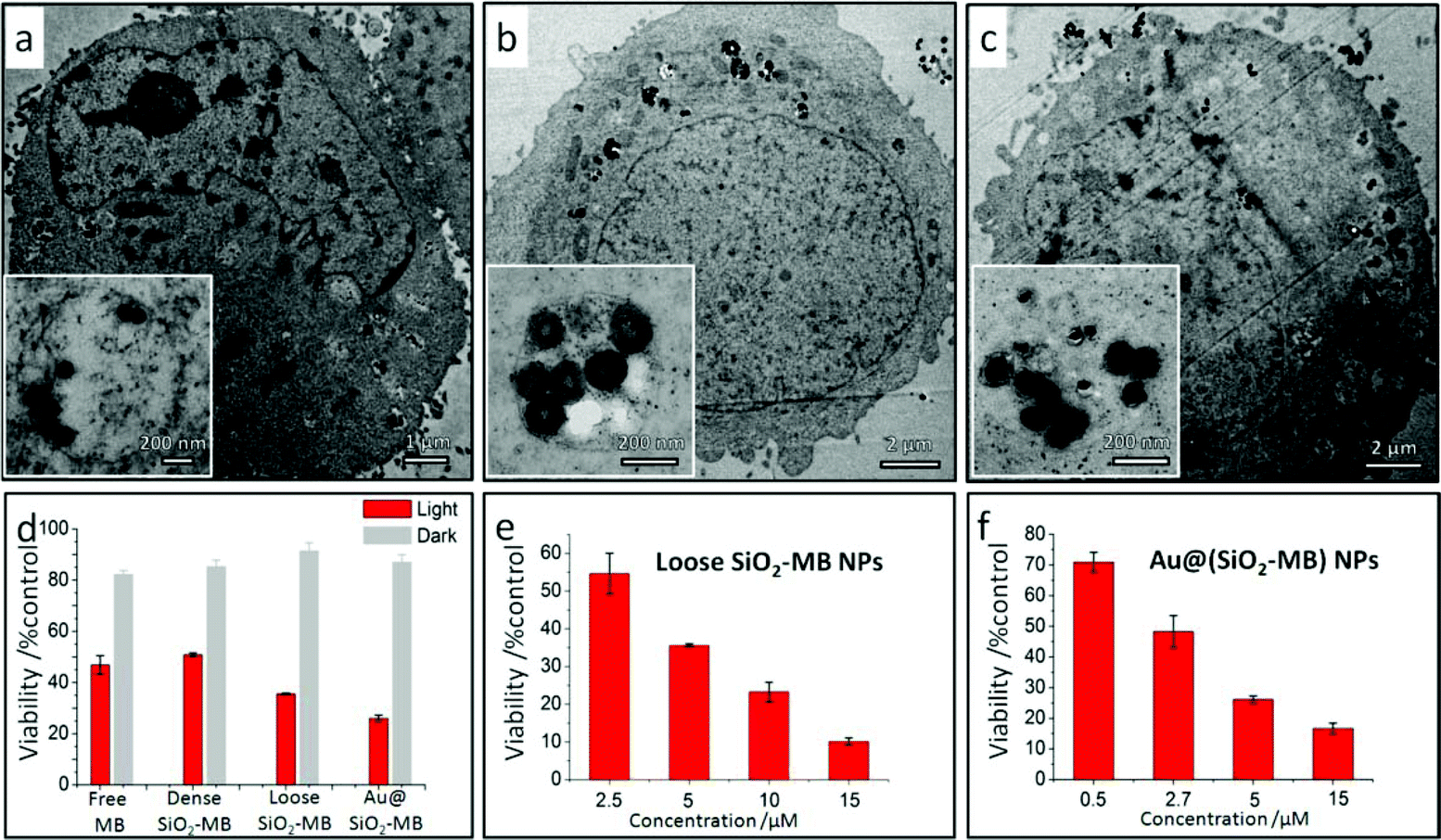 | ||
| Fig. 4 Intracellular locations of various MB loaded NPs upon cellular uptake, and the efficacy of free MB or various MB loaded NPs when performing PDT in vitro. TEM images showing a typical HepG2 cell after being incubated with (a) dense SiO2-MB, (b) loose SiO2-MB and (c) Au@(SiO2-MB) NPs for 24 hours; the insert is the magnified TEM image of one selected vesicle. (d) MTT results of the HepG2 cells treated with pure MB, dense SiO2-MB, loose SiO2-MB and Au@(SiO2-MB) NPs for 24 hours (the concentration of MB was fixed as 5 μM in all samples). MB concentration dependence of the HepG2 cell viability when incubated with (e) loose SiO2-MB NPs and (f) Au@(SiO2-MB) NPs for 24 hours; the feeding concentration of the NPs was kept the same for all experiments. All data are shown as mean ± SD (from three independent experiments) and significantly different (p < 0.05) from the control (analyzed by the Student's t-test). | ||
The presence of different types of dominant ROS suggested different death pathways for the cells after their incubation with free MB or MB loaded NPs (upon light irradiation). It was known that singlet oxygen was more likely to direct cells to apoptosis (including early and late apoptosis),27,28 while free radicals (including both hydroxyl radicals and superoxide) would lead cells to necrosis.29 In addition, higher ROS level was also more likely to induce necrosis than apoptosis.30 As the MTT assay only disclosed the overall cell viability, we took another approach to evaluate the cell death pathway. As the morphological features of the cells would vary when they undergo either apoptosis or necrosis,31 we compared the cell morphology after their being treated with free MB and MB loaded NPs using light microscopy. In free MB, dense SiO2-MB NP and loose SiO2-MB NP treated samples, cells with a shrinking membrane as well as their detaching from the substrate were commonly observed, which features were consistent with the apoptosis pattern31 (Fig. 5b–d). As a comparison, most of the cells remained attached to the substrate but with massive production of bubbles when treated with Au@(SiO2-MB) NPs (Fig. 5e). The bubble formation, which was a feature of necrosis,31 can be observed in cells treated with Au@(SiO2-MB) NPs after only a few minutes of light irradiation (Fig. S3†). These light microscopy results indicated that apoptosis was dominant in cells treated with free MB, dense SiO2-MB NPs and loose SiO2-MB NPs, while necrosis served as the predominant death route for Au@(SiO2-MB) NP treated cell samples.
 | ||
| Fig. 5 Morphological change of cells treated with various samples after in vitro PDT treatment. Light microscopy images of HepG2 cells treated with (a) medium (control), (b) free MB, (c) dense SiO2-MB, (d) loose SiO2-MB and (e) Au@(SiO2-MB) NPs for 24 hours (the concentration of MB was fixed as 5 μM for all samples), followed by light irradiation for 20 minutes and further incubation with NP-free medium for another 24 hours. The scale bar is 100 μm. | ||
Discussions
The experimental results suggested that trapping MB into the dense SiO2 matrix suppressed all types of generated ROS (Fig. 3). This was mainly due to the difficulty in ROS's out-diffusion from the NPs—MB was trapped in the dense SiO2 matrix, and ROS was always firstly generated in the vicinity of MB and then diffused to elsewhere.20 This was particularly important when the lifetime of a specific ROS was short, such as the singlet oxygen and hydroxyl radical.32 Compared to dense SiO2-MB NPs, loose SiO2-MB NPs showed enhanced generation of all types of ROS examined. This was caused by the loose and fragmentable features of the loose SiO2-MB NPs,21 resulting in easier diffusion of the generated ROS.However, one should note that the amount of singlet oxygen generated by the loose SiO2-MB NPs was also lower than that of free MB, contrary to the case of hydroxyl radical and superoxide. This was not too surprising, as the incorporation of MB into the SiO2 matrix would promote MB dimer formation at the expense of the MB monomer.33 While the MB monomer mainly produced singlet oxygen under light irradiation, the excitation of the dimer led to a large amount of free radical (e.g., hydroxyl radical and superoxide) generation.34 In fact, this phenomenon held true for all MB-loaded SiO2 NPs as shown in Fig. 3. Compared to singlet oxygen, which mainly caused apoptosis, the free radicals (e.g., hydroxyl radicals and superoxide) were more likely to cause necrosis, and thus acted as more effective agents for cell killing in PDT.29 It was also important to note that apoptosis (the major pattern of cell death in free MB treated samples) was not an effective cell death pathway, as it did not necessarily lead to cell death.35 In addition, cancer cells may develop resistance to apoptosis.36 These then provided a reasonable explanation for the observed cell viability difference (Fig. 4d), i.e., the loose SiO2-MB NPs showed the highest cell death rate when compared to free MB and dense SiO2-MB NPs, although the amount of singlet oxygen generated by loose SiO2-MB NPs was merely half of that of the free MB.
Although the MB (or generated ROS) was also trapped in the case of Au@(SiO2-MB) NPs, such a SiO2 shell was quite thin (∼40 nm). On the other hand, the introduction of Au NRs brought in the plasmonic effect. With the MB dimer absorption matching the surface plasmon resonance energy of the Au core (Fig. 2d) and the spatial confinement of MB in the vicinity of Au, MB absorption and thus the corresponding generation efficiency of ROS were significantly increased due to the plasmonic enhancement effect,20 especially for the type I ROS (i.e. hydroxyl radicals and superoxide) (Fig. 3). Here one can exclude the contribution of Au@SiO2 only, as we found that the ROS produced by Au@SiO2 NPs was similar to those of the control sample, i.e., aqueous solution containing ROS detection dyes only. Nor did the presence of Au@SiO2 decrease the cell viability when irradiated with light under the described experimental conditions (results not shown here). The enhanced generation of type I ROS due to the Au plasmonic effect was responsible for the most effective PDT in vitro (Fig. 4d). Therefore, the observed cell viability decrease in Au@(SiO2-MB) NP treated samples resulted from both plasmonic enhanced generation of ROS and the predominant type I ROS produced by MB dimers.
Conclusions
In conclusion, we found that incorporation of MB into the silica matrix would bring in a number of advantages, i.e., the dominance of dimer instead of monomer would produce predominant type I ROS, which were more efficient agents responsible for cell death. Nevertheless, the traditional dense silica nanocarriers would reduce the ROS production due to their difficult diffusion out of the silica matrix. We tackled this problem by designing loose SiO2-MB NPs, in which facile diffusion of MB and generated ROS was realized via self-decomposition of such nanocarriers, and an enhanced drug efficacy was demonstrated. We also showed that increasing the generation efficiency of ROS by introducing the Au plasmonic effect served as an alternative route for enhanced drug efficacy. Such mechanisms provided a general guideline for the optimum design of NP-based drug delivery for PDT.Experimental
Preparation of various MB loaded NPs
The loose SiO2-MB NPs were synthesized using a conventional method21 with modified parameters. In a typical procedure, 2.5 mg MB was firstly added to a mixture of 75 ml ethanol and 3.4 ml 25% ammonia–water solution; after that, 0.08 ml TEOS was added. The loose SiO2-MB NPs were obtained after 24 hours of stirring, and washed several times before their being dried. The dense SiO2-MB NPs were synthesized using a published method;21 briefly, 2.5 mg of MB was added to a mixture of 50 ml ethanol with 5 ml 25% ammonia–water solution; after that 0.2 ml TEOS was added. The ammonia amount is 2.27 vol%, which is more than double of that in synthesizing self-decomposable NPs (<1.08 vol%). Such a recipe was known to generate dense SiO2-MB NP, in which the MB can be stably trapped in SiO2.The growth of Au NRs was firstly conducted using a seed-mediated method.20 The as-grown NRs can be shortened by oxidation to the required aspect ratio. Pegylation of Au NRs (40 ml) was realized by mixing them with freshly prepared aqueous mPEG-SH solution (1 mM, 2 ml; NANOCS, America) in a 30 °C water bath overnight. Growth of the Au@(SiO2-MB) NPs was similar to that of Au@SiO2 NPs reported in the literature.37 In a typical procedure, 7.5 ml as-prepared PEG-stabilized Au NRs (in ethanol) were mixed with 2.3 ml deionized H2O and 0.15 ml 30% ammonia–water solution; after that, 50 μl MB stock solution (10 mM in ethanol) was added before 20 μl TEOS was finally introduced. The resulting NPs were washed and dried at 65 °C for further use.
The general morphology, size, and the size distribution of various NPs were characterized using transmission electron microscopy (TEM, Philips CM120). All of the UV/Vis absorption spectra were acquired using a HitachiU-3501 UV-visible-NIR spectrophotometer.
Quantifying the ROS generation of various MB loaded NPs in a cuvette
To study ROS generation in a cuvette, three different reagents were selected to detect the singlet oxygen (singlet oxygen sensor green (SOSG), Ex: 488 nm), hydroxyl radical (terephthalic acid (TA), Ex: 315 nm) and superoxide (dihydroethidium (DHE), Ex: 470 nm), respectively. The free MB, dense SiO2-MB, loose SiO2-MB and Au@(SiO2-MB) NPs were respectively dispersed in a cuvette with deionized water, followed by addition of a suitable amount of the specific ROS detection dye, and then irradiated with 590 nm LED light for 20 minutes. Finally, the fluorescence signals of different ROS detection dyes in the cuvette were measured using a fluorescence spectrophotometer (Hitachi, FL7000). In all experiments, deionized water containing only the corresponding ROS detection dyes was used as the controls. The fluorescence intensity of various ROS detection dyes was finally normalized by the concentration of MB (the concentration of MB was fixed at 5 μM in all samples).Introduction of various MB loaded NPs into the cells for performing PDT in vitro
HepG2, the human liver carcinoma cells, were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2.0 g l−1 sodium bicarbonate, 0.1 g l−1 streptomycin sulfate, 0.06 g l−1 penicillin G and 5.958 g l−1 HEPES. The cells were maintained in a standard, cell culture incubator at 37 °C under a humidified atmosphere with 5% CO2.All of the NPs were sterilized by steaming at 115 °C (NPs in powder form) for 2 hours before they were dispersed in the medium and introduced into the cells, which had already been seeded and incubated for 24 hours. The concentration of NPs used in this study can be represented by that of Au NRs, which was calculated to be 0.34 nM from its UV/Vis absorption.5
A home-built 590 nm LED (the emission profile is shown in Fig. S4†) was employed for the in vitro PDT study. The LED was aligned directly under the sample wells (96-well plates) to obtain uniform irradiation of the cells. The power density of the LED is 5 mW cm−2, which is too weak to cause any irradiation damage to the cells (Fig. S5†).
Characterization of the cells
For all transmission electron microscopy studies, the NP-fed cells were fixed with typical procedures published elsewhere.26 A microtome (Leica, EM UC6) was then used to cut the cured cell cube (in Spurr resin (Electron Microscopy Sciences, USA)) into thin slices (70–90 nm in thickness). The samples were collected on 300-mesh copper TEM grids for observation.The cell viability was measured 24 hours after irradiation using the MTT assay. The 24 hours delay was designed to account for both apoptosis and necrosis mechanisms of cell death.7 The significance of all data was determined by Student's t-test for all in vitro studies; p < 0.05 was deemed as significant for all data compared to the control.
The morphology change of HepG2 cells was observed with a light microscope (Olympus CKX41, Japan). The cells were initially seeded into a 6 well plate at the initial density of 1 × 105 ml−1 (2 ml) in DMEM medium with 10% FBS. After allowing 24 hours for cell attachment, the original medium was discarded and the free MB/MB loaded NP containing medium was added to the cell samples ([MB] = 5 μM) dosing for 24 hours. Finally, the cells were observed 24 hours after irradiation using a light microscope with a 10× objective lens.
Acknowledgements
The authors are grateful for financial support from a CUHK direct grant (project no. 4053074).References
- P. Huang, J. Lin, X. S. Wang, Z. Wang, C. L. Zhang, M. He, K. Wang, F. Chen, Z. M. Li, G. X. Shen, D. X. Cui and X. Y. Chen, Adv. Mater., 2012, 24, 5104–5110 CrossRef CAS PubMed.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- C. A. Robertson, D. H. Evans and H. Abrahamse, J. Photochem. Photobiol., B, 2009, 96, 1–8 CrossRef CAS PubMed.
- P. Huang, J. Lin, D. Yang, C. Zhang, Z. Li and D. Cui, J. Controlled Release, 2011, 152(Suppl 1), e33–e34 CrossRef CAS PubMed.
- J. Lin, S. Wang, P. Huang, Z. Wang, S. Chen, G. Niu, W. Li, J. He, D. Cui, G. Lu, X. Chen and Z. Nie, ACS Nano, 2013, 7, 5320–5329 CrossRef CAS PubMed.
- H. Y. Yang, F. Y. Wang and Z. Y. Zhang, Dyes Pigm., 1999, 43, 109–117 CrossRef CAS.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- H. L. Tu, Y. S. Lin, H. Y. Lin, Y. Hung, L. W. Lo, Y. F. Chen and C. Y. Mou, Adv. Mater., 2009, 21, 172 CrossRef CAS PubMed –177.
- P. Couleaud, V. Morosini, C. Frochot, S. Richeter, L. Raehm and J. O. Durand, Nanoscale, 2010, 2, 1083–1095 RSC.
- P. Huang, J. Lin, S. J. Wang, Z. J. Zhou, Z. M. Li, Z. Wang, C. L. Zhang, X. Y. Yue, G. Niu, M. Yang, D. X. Cui and X. Y. Chen, Biomaterials, 2013, 34, 4643–4654 CrossRef CAS PubMed.
- V. Simon, C. Devaux, A. Darmon, T. Donnet, E. Thienot, M. Germain, J. Honnorat, A. Duval, A. Pottier, E. Borghi, L. Levy and J. Marill, Photochem. Photobiol., 2010, 86, 213–222 CrossRef CAS PubMed.
- B. Zhao, J. J. Yin, P. J. Bilski, C. F. Chignell, J. E. Roberts and Y. Y. He, Toxicol. Appl. Pharmacol., 2009, 241, 163–172 CrossRef CAS PubMed.
- C. Compagnin, L. Bau, M. Mognato, L. Celotti, G. Miotto, M. Arduini, F. Moret, C. Fede, F. Selvestrel, I. M. R. Echevarria, F. Mancin and E. Reddi, Nanotechnology, 2009, 20 Search PubMed.
- I. I. Slowing, J. L. Vivero-Escoto, C. W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. S. Y. Lin, Adv. Funct. Mater., 2007, 17, 1225–1236 CrossRef CAS PubMed.
- B. Coulibaly, A. Zoungrana, F. P. Mockenhaupt, R. H. Schirmer, C. Klose, U. Mansmann, P. E. Meissner and O. Muller, PLoS One, 2009, 4, e5318 Search PubMed.
- M. Oz, D. E. Lorke and G. A. Petroianu, Biochem. Pharmacol., 2009, 78, 927–932 CrossRef CAS PubMed.
- R. H. Schirmer, H. Adler, M. Pickhardt and E. Mandelkow, Neurobiol. Aging, 2011, 32 Search PubMed.
- Z. Chu, C. Yin, S. Zhang, G. Lin and Q. Li, Nanoscale, 2013, 5, 3406–3411 RSC.
- S. L. Zhang, Z. Q. Chu, C. Yin, C. Y. Zhang, G. Lin and Q. Li, J. Am. Chem. Soc., 2013, 135, 5709–5716 CrossRef CAS PubMed.
- M. Valko, D. Leibfritz, J. Moncol, M. T. D. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS PubMed.
- C. Flors, M. J. Fryer, J. Waring, B. Reeder, U. Bechtold, P. M. Mullineaux, S. Nonell, M. T. Wilson and N. R. Baker, J. Exp. Bot., 2006, 57, 1725–1734 CrossRef CAS PubMed.
- K. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, Electrochem. Commun., 2000, 2, 207–210 CrossRef CAS.
- Y. Zhang, K. Aslan, M. J. R. Previte and C. D. Geddes, Appl. Phys. Lett., 2007, 91 Search PubMed.
- Z. Q. Chu, Y. J. Huang, Q. Tao and Q. Li, Nanoscale, 2011, 3, 3291–3299 RSC.
- I. E. Kochevar, M. C. Lynch, S. G. Zhuang and C. R. Lambert, Photochem. Photobiol., 2000, 72, 548–553 CrossRef CAS.
- C. P. Lin, M. C. Lynch and I. E. Kochevar, Exp. Cell Res., 2000, 259, 351–359 CrossRef CAS PubMed.
- Y. H. Kim, E. Y. Kim, B. J. Gwag, S. Sohn and J. Y. Koh, Neuroscience, 1999, 89, 175–182 CrossRef CAS.
- S. V. Lennon, S. J. Martin and T. G. Cotter, Cell Prolif., 1991, 24, 203–214 CrossRef CAS.
- S. Rello, J. C. Stockert, V. Moreno, A. Gamez, M. Pacheco, A. Juarranz, M. Canete and A. Villanueva, Apoptosis, 2005, 10, 201–208 CrossRef CAS PubMed.
- L. M. Rossi, P. R. Silva, L. L. R. Vono, A. U. Fernandes, D. B. Tada and M. S. Baptista, Langmuir, 2008, 24, 12534–12538 CrossRef CAS PubMed.
- D. B. Tada, L. L. R. Vono, E. L. Duarte, R. Itri, P. K. Kiyohara, M. S. Baptista and L. M. Rossi, Langmuir, 2007, 23, 8194–8199 CrossRef CAS PubMed.
- H. C. Junqueira, D. Severino, L. G. Dias, M. S. Gugliotti and M. S. Baptista, Phys. Chem. Chem. Phys., 2002, 4, 2320–2328 RSC.
- B. Fadeel, B. Gleiss, K. Hogstrand, J. Chandra, T. Wiedmer, P. J. Sims, J. I. Henter, S. Orrenius and A. Samali, Biochem. Biophys. Res. Commun., 1999, 266, 504–511 CrossRef CAS PubMed.
- A. Ashkenazi, Nat. Rev. Cancer, 2002, 2, 420–430 CrossRef CAS PubMed.
- C. Fernandez-Lopez, C. Mateo-Mateo, R. A. Alvarez-Puebla, J. Perez-Juste, I. Pastoriza-Santos and L. M. Liz-Marzan, Langmuir, 2009, 25, 13894–13899 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00024b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |