DOI:
10.1039/C4BM00028E
(Paper)
Biomater. Sci., 2014,
2, 1120-1127
A fluorescently labelled sialic acid for high performance intraoperative tumor detection
Received
29th January 2014
, Accepted 12th March 2014
First published on 1st May 2014
Abstract
Surgical resection is widely used for tumor treatment, necessitating approaches for the precise locating of elusive tumor foci. We report the high performance detection of tumors in mice with fluorescein-isothiocyanate (FITC) labelled sialic acid (FITC-SA), a fluorescent monosaccharide with low cytoxicity. Analysis of mice intravenously injected with FITC-SA revealed high target-to-background fluorescence ratios in subcutaneous tumors and liver tumor implants with 0.2–5 mm diameters, which are significantly below the clinical threshold of minimal residual cancer (∼1 cm clearance). Extracellular FITC-SA is quickly cleared from circulation whereas the intracellular FITC-SA could be metabolically incorporated into glycoproteins via a cellular sialylation pathway. Compared with FITC-SA-laden nanoparticles, free FITC-SA is preferentially and quickly taken up by tumors in mice and displays high tumor-to-background signal contrast, suggesting the potential for fluorescence directed surgical ablation of tumors.
Introduction
Surgical resection remains a major modality for tumor treatment. Incomplete removal of tumor foci, largely dependent on visual inspection by the surgeon, leads to tumor relapses. As such, methods that could direct surgeons to disseminated or evasive tumor foci are valuable for cancer surgery.1 Optical probes that could selectively accumulate within tumors are being actively explored for fluorescence guided cancer surgery. To achieve high tumor-to-background signal ratios, optical dyes are often conjugated with tumor-targetable carriers such as folate or monoclonal antibodies, etc.2 The utility of tumor-targeting optical probes has been validated in clinical trials, e.g. imaging of epithelial ovarian cancer with FITC-tagged folate.2c
Sialic acids, a family of natural derivatives of N-acetyl-neuraminic acid,3 are the common termini of cell surface glycans involved in host–pathogen interactions and cell–cell adhesions, etc.4 Overexpression of cell surface sialic acids has been correlated with the metastatic potential of a number of tumors, and cell surface sialic acid has been a target for drug delivery.5 Substrate promiscuity of the sialic acid biosynthetic pathway has been employed for the biochemical engineering of cellular sialoproteins with various exogenous N-acyl mannosamines,6 which are enzymatically condensed with pyruvate to give unnatural sialic acids for the subsequent sialylation of glycoproteins.7 The disadvantage associated with the metabolic labelling approach is the lack of cell-type or tissue specificity, where unnatural sialic acids were found to be present in different tissues in animals treated with N-acyl mannosamine.8 Analogous to N-acyl mannosamine, sialic acids with C-9 substitutions supplemented in culture medium could also be taken up by cells and then incorporated into cellular sialoproteins.9 To date, the use of sialic acid derivatives for tumor imaging in animals has been largely unexplored.10
We herein report the high performance tumor illumination in mice with 9-fluoresccinylthioureido-9-deoxy-N-acetylneuraminic acid (FITC-SA), which is preferentially and quickly taken up by tumors in vivo (Fig. 1). Nanomedicine holds great promise for targeted drug delivery. For comparison, FITC-SA-laden boronic acid-functionalized silica nanoparticles were constructed and assessed for their efficacy to target tumors in mice.
 |
| | Fig. 1 Schematic diagram of FITC-SA based tumor imaging. FITC-SA was preferentially taken up into tumors and then incorporated into cellular proteins through an endogenous sialylation pathway. | |
Results and discussion
Incorporation of FITC-SA into mammalian cells
It was previously shown that sialic acids with bulky substitutions at the C-9 position could be effectively incorporated into cell surface glycoproteins on B cells.9a In addition, CMP activated FITC-SA has been shown to be the functional donor of sialyltransferases and used to transfer FITC-SA to endogenous glycoproteins in permeabilized Chinese hamster ovary cells.11 On the basis of these observations, we examined the feasibility of illuminating tumors in mice by systemic administration of FITC-SA.
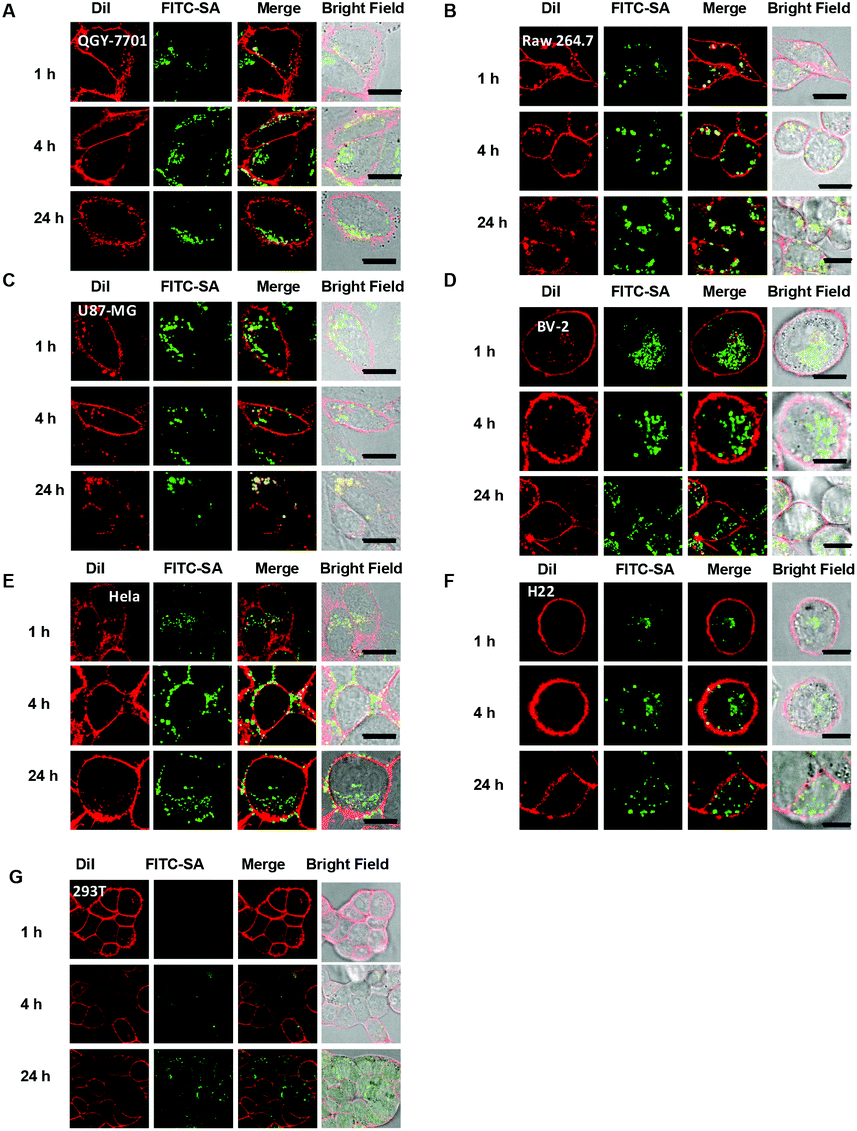
To probe cell type-dependent uptake of FITC-SA, Raw 264.7 macrophages, human epithelial carcinoma (HeLa), human hepatocellular carcinoma (QGY-7701), human primary glioblastoma (U87-MG), H22 hepatocellular carcinoma, murine microglial cells (BV-2) and human embryonic kidney 293 cells, a normal tissue cell line, were respectively cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with FITC-SA and DiI, which is a plasma membrane specific dye. The intracellular fluorescence was visualized as a function of incubation time by confocal fluorescence microscopy. It was shown that intense FITC signals were clearly present in all cell lines examined (Fig. 2), suggesting that FITC-SA supplemented in culture medium could be effectively taken up by a broad spectrum of tumor cell lines and normal tissue cells. Images of individual cells at different stages of incubation revealed the punctate and discrete fluorescence of FITC-SA is mostly confined within the cytosol of BV-2 cells, whereas the cytosolic FITC-SA in Raw 264.7, QGY-7701, and U87-MG cells largely relocates to cell membranes upon prolonged incubation, as indicated by the increased colocalization of the FITC-SA signal with the DiI fluorescence (Fig. 2).
 |
| | Fig. 2 Tempo-spatial distribution of FITC-SA within mammalian cells. Raw 264.7, HeLa, QGY-7701, U87-MG and BV-2 cells were respectively incubated in DMEM containing FITC-SA (100 μM) for 1–24 h, followed by staining with DiI (10 μM) for 10 min and then analysis by fluorescence confocal microscopy to pinpoint the subcellular locations of FITC-SA. The cell membrane was stained by DiI. Overlay of FITC-SA (green signal) with DiI (red signal) reveals colocalization, as indicated by the yellow areas. Bars: 10 μm. | |
The tempo-spatial alterations of the intracellular fluorescence patterns shown in Fig. 2 suggest that FITC-SA could be incorporated into endogenous glycoproteins which are further sorted to distinct subcellular locations (e.g. cell membrane) in a cell-type dependent manner. Confocal fluorescence microscopic images of HeLa, U87-MG, and BV-2 cells co-stained with FITC-SA, Lyso-Tracker Blue DND-22, Mito-Tracker Deep Red, and Golgi-Tracker Red show the presence of FITC-SA within the Golgi, whereas no obvious FITC-SA signals are present in lysosomes and mitochondria (Fig. 3B), which is consistent with the fact that cytosolic CMP-SA is transported into the Golgi apparatus for protein sialylation. To further prove metabolic incorporation of FITC-SA into cellular proteins, HeLa cells pretreated with FITC-SA and DiI were permeabilized and fixed with formaldehyde and then incubated in fresh DMEM for 12 h to remove unbound FITC-SA within cells. As shown in Fig. 4, no staining of the cell membrane with DiI was observed in cells treated with formaldehyde, confirming disruption of the functional cell plasma membrane. The bright FITC-SA signal remaining in treated cells indicates covalent incorporation of FITC-SA into cellular proteins (Fig. 4). Collectively, these results show that FITC-SA is compatible with the cellular sialylation pathway, which enables metabolic labelling of host cells with FITC-SA.
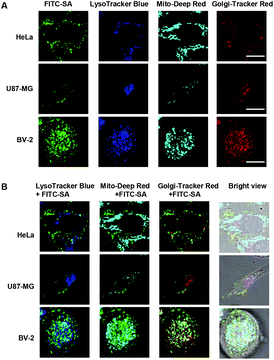 |
| | Fig. 3 Subcellular locations of FITC-SA. HeLa, U87-MG and BV-2 cells were respectively incubated in DMEM containing FITC-SA (100 μM) for 4 h, stained with Golgi-Tracker Red (333 μg ml−1) in DMEM at 4 °C for 30 min, and then incubated in DMEM with LysoTracker Blue DND-22 (1 μM) and Mito-Tracker Deep Red (500 nM) for 30 min at 37 °C with 5% CO2. The cells were analyzed by fluorescence confocal microscopy to pinpoint the subcellular locations of FITC-SA (A). Overlay of FITC-SA signals with those of Mito-Deep Red and Golgi-Tracker Red reveals partial colocalization of FITC-SA with Golgi-Tracker Red, which is indicated by the yellow areas. Bars: 10 μm. | |
 |
| | Fig. 4 Metabolic incorporation of FITC-SA into cellular proteins. HeLa cells prestained with FITC-SA (100 μM) were digested by trypsin and then cultured in DMEM for 12 h. The cells were treated with or without formaldehyde (4%) for 15 min in PBS, incubated in fresh DMEM for 12 h to allow the clearance of free FITC-SA, and stained with DiI (10 μM) for 10 min and then imaged by fluorescence confocal microscopy. The cell membrane stained with DiI was shown in red and the intracellular FITC-SA was shown in green. | |
Detection of subcutaneous tumors with FITC-SA
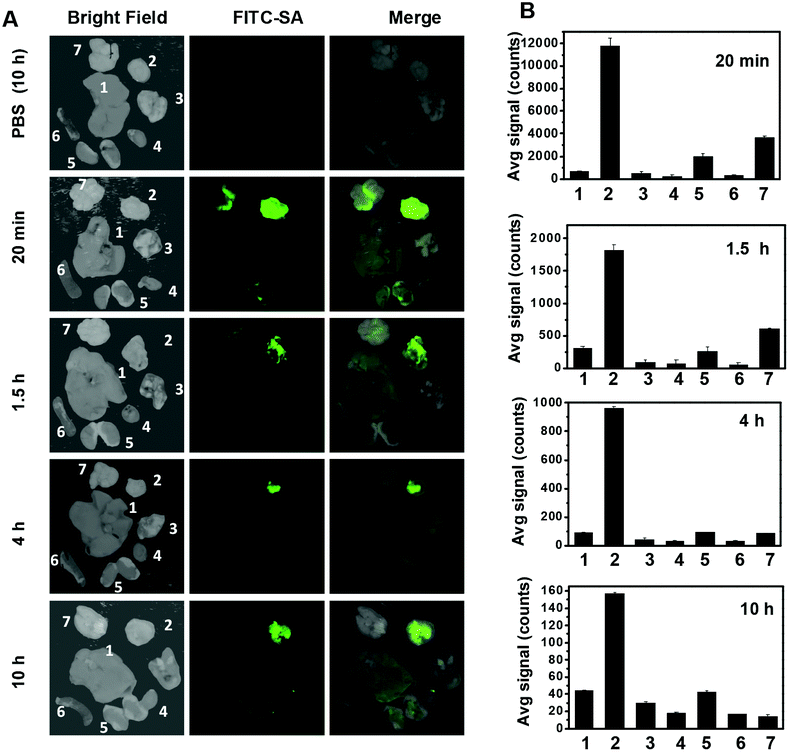
Effectively taken up by different tumor cell lines in vitro, FITC-SA was further assessed for its capability to accumulate in subcutaneous tumors in mice. ICR mice were inoculated subcutaneously with H22 hepatocellular carcinoma cells and then maintained for 5–10 days to allow the development of tumor xenografts. FITC-SA was injected via the tail vein into a cohort of tumor-bearing mice. The mice were sacrificed at 20 min, 1.5 h, 4 h and 10 h following injection. The tumors and representative organs were dissected and then probed by ex vivo fluorescence analysis. An intense FITC-SA signal is observed in tumors within 20 min after administration of FITC-SA (Fig. 5A), suggesting quick tumoral uptake of FITC-SA from the bloodstream. The fluorescence in the brain, also observed 20 min after injection, quickly diminished to background levels 4 h postinjection, meanwhile no obvious accumulation of FITC-SA was identified in other healthy organs examined (Fig. 5A). The tumor-to-organ fluorescence ratios remained high up to 10 h postinjection (Fig. 5B).
 |
| | Fig. 5 High-efficiency illumination of subcutaneous tumors with FITC-SA. ICR mice bearing subcutaneous tumors were intravenously injected with FITC-SA (50 mg kg−1) or PBS (100 μl) via the tail vein and then sacrificed 20 min, 1.5 h, 4 h and 10 h post-injection. (A) Ex vivo fluorescence images of the dissected tumors and representative organs. (B) The bar graph shows the tissue distributions of the FITC-SA fluorescence. The organs were shown in the following sequence: liver (1), tumor (2), lung (3), heart (4), kidney (5), spleen (6), and brain (7). | |
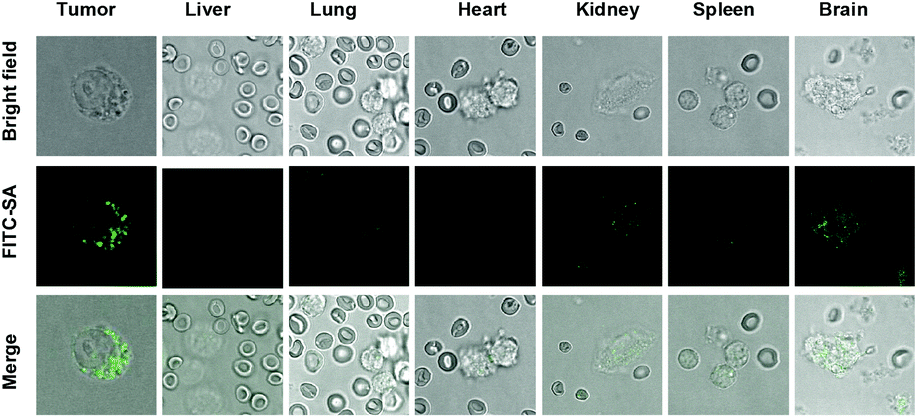
To probe the location of FITC-SA within tumors, cells were released from the excised tumors and organs by digestion with trypsin, and then visualized by confocal fluorescence microscopy. Fig. 6 revealed the intracellular fluorescence of FITC-SA within cells from tumors, and brain etc., confirming transportation of FITC-SA from the bloodstream into tumor cells. Taken together, the fast and preferential uptake of FITC-SA by tumors in vivo and the resultant high tumor-to-background signal ratios suggests the utility of FITC-SA for low background intraoperative tumor detection.
 |
| | Fig. 6 Cellular uptake of intravenously injected FITC-SA in mice. Mice bearing subcutaneous tumors were intravenously injected with FITC-SA (50 mg kg−1) and then sacrificed 2.5 h postinjection. The tumors and representative organs were dissected, minced and digested with trypsin-EDTA solution (0.125%) for 30 min. The liberated cells were visualized by confocal fluorescence microscopy. | |
Illumination of liver tumor implants in mice with FITC-SA
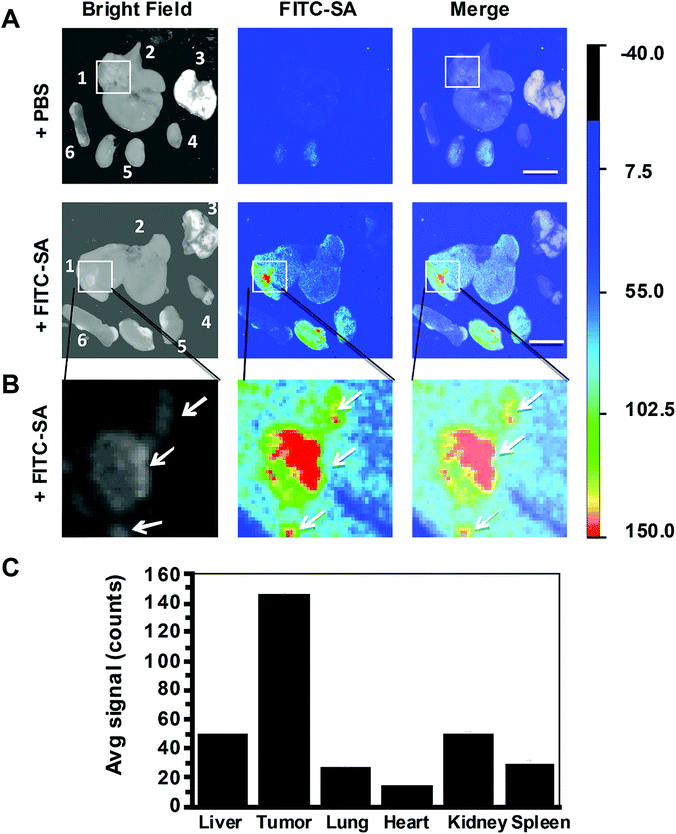
Hepatocellular carcinoma, the predominant form of liver cancer, is a common and lethal malignancy claiming more than 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lives each year.12 Cytoreduction remains one of the major approaches for liver cancer therapy. Residual tumor foci due to incomplete surgical removal often lead to tumor relapses. Encouraged by the selective imaging of subcutaneous tumors with FITC-SA, we further evaluate the efficacy of FITC-SA to illuminate liver tumor foci in mice. ICR mice with tumor implants of H22 hepatocellular carcinoma in the liver were intravenously administered with FITC-SA. The liver and selected organs were excised 17 h after injection. Ex vivo fluorescence analysis revealed intensive fluorescence from tumor foci in the liver and low levels of signals in neighbouring liver tissue and other healthy organs (Fig. 7). The tumor implants with diameters of 0.2–5 mm could be clearly discerned with FITC-SA. The resolution is significantly below the minimal residual cancer (<1 cm) limit pursued by surgeons.13 The high tumor-to-background fluorescence contrasts and the detection of sub-millimeter tumors further corroborate the potential of FITC-SA for fluorescence guided tumor surgery.
000 lives each year.12 Cytoreduction remains one of the major approaches for liver cancer therapy. Residual tumor foci due to incomplete surgical removal often lead to tumor relapses. Encouraged by the selective imaging of subcutaneous tumors with FITC-SA, we further evaluate the efficacy of FITC-SA to illuminate liver tumor foci in mice. ICR mice with tumor implants of H22 hepatocellular carcinoma in the liver were intravenously administered with FITC-SA. The liver and selected organs were excised 17 h after injection. Ex vivo fluorescence analysis revealed intensive fluorescence from tumor foci in the liver and low levels of signals in neighbouring liver tissue and other healthy organs (Fig. 7). The tumor implants with diameters of 0.2–5 mm could be clearly discerned with FITC-SA. The resolution is significantly below the minimal residual cancer (<1 cm) limit pursued by surgeons.13 The high tumor-to-background fluorescence contrasts and the detection of sub-millimeter tumors further corroborate the potential of FITC-SA for fluorescence guided tumor surgery.
 |
| | Fig. 7 High performance illumination of liver tumors with FITC-SA. ICR mice with liver tumor implants were intravenously injected with FITC-SA (50 mg kg−1) or PBS (100 μl) via the tail vein and then sacrificed 17 h postinjection. (A) Ex vivo fluorescence images of the excised livers and selected organs; (B) tumor foci highlighted in the square were enlarged for clarity; (C) the bar graph shows the fluorescence intensity of FITC-SA in tumor foci vs. surrounding liver tissue and other organs. Bar: 1 cm. | |
Tissue or cell-specific delivery of functional sugars is of significant interest in biomedical studies of cellular glycosylation. In contrast with N-acyl mannosamines widely used for the metabolic remodelling of sialoconjugates, sialic acid derivatives, albeit taken up by cells in vitro, are less well explored for animal studies. N-Acetyl-3-[18F]fluoroneuraminic acid and N-acetyl-2-deoxy-2,3-di-[18F]fluoroneuraminic acid were found to show limited tumor accumulation in mice as determined by positron emission tomography.10 Recently, folate-decorated liposomes were employed for selective and enhanced delivery of azido-sialic acid into folate receptor-overexpressing cells.14 The unexpected high performance detection of tumors with FITC-SA in mouse models is likely due to a combination of a distinct tumor-targeting property of FITC-SA, a sialic acid derivative with C-9 substitution, relative to [18F]-substituted sialic acids,10 and high sensitivity of fluorometry over positron emission tomography. It is reported that hours are required for efficient cell surface glycan labeling with acetylated N-acylmannosamine, the metabolic precursor of sialic acids.7 In the case of intraoperative tumor detection, FITC-SA inside cells, or later metabolically displayed on the cell surface, effectively contributes to detectable intra-tumoral signals, which might account for the observed fluorescecne within tumors at 20 min postinjection.
FITC-SA is compatible for intraoperative tumor imaging owing to exposure of cancerous tissues, which circumvents the limited tissue penetration of fluorescein fluorescence. In addition, fluorescein, the optical reporter of FITC-SA, provides bright and green emission bioorthogonally distinct from the autofluorescence located in the visible-to-red region. Although the molecular or physiological factors underlying the selective tumor uptake of FITC-SA in vivo remain elusive at this stage, the high tendency of FITC-SA to accumulate in tumors suggests the use of sialic acids with appropriate modifications for low background tumor detection.
Construction and characterization of FITC-SA loaded mesoporous silica nanoparticles
Nanomedicine holds enormous potential for targeted drug delivery. Nanoscaled systems are actively being explored for tumor targeting due to the enhanced permeability and retention (EPR) effect.15 Akin to hydrazone and orthoesters widely explored for lysosomal pH promoted drug release,16 boronic acid–cis diol esters are prone to acidic pH mediated hydrolysis. To compare the performance of nanovector mediated tumor imaging, FITC-SA was loaded into boronic acid-displaying mesoporous silica nanoparticles (MSN) and then applied for tumor imaging.
MSN was capped with [3-(O-boronobenzyl)aminopropyl]-triethoxysilane (BA-APTS) in anhydrous DMF under reflux for 24 h to give BA@M which was further modified with poly(ethylene glycol) to give BA@M-P (Scheme 1). BA@M and BA@M-P were respectively incubated with excess FITC-SA in DMF and then harvested by centrifugation. Analysis shows that the hydrodynamic diameters of sugar-laden MSN remained largely unaffected while the surface potentials slightly decreased upon complexation with FITC-SA (Fig. 8), indicating anionic FITC-SA was largely encapsulated into the pores of silica particles. To assess the pH mediated release of MSN-captured FITC-SA, FITC-SA@BA@M and FITC-SA@BA@M-P were separately incubated in sodium phosphate buffers of pH 4.5 or pH 7.5. At fixed time intervals, portions of the mixtures were centrifuged, and the amounts of FITC-SA remaining in the silica particles were quantified by UV-vis spectrometry. It was shown that levels of FITC-SA within MSN incubated at acidic pH decreased dramatically relative to those at pH 7.5 (Fig. 9), indicating the applicability of acidic pH mediated hydrolysis of boronic acid complexed FITC-SA.
 |
| | Scheme 1 Boronic acid-displaying MSN for intracellular delivery of FITC-SA. Sugar-laden MSN undergoes acidic pH promoted hydrolysis of the boronate-diol ester. | |
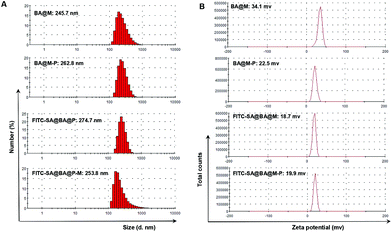 |
| | Fig. 8 Dynamic light scattering (A) and zeta potential analysis (B) of sugar-laden MSN as compared to sugar-free MSN. | |
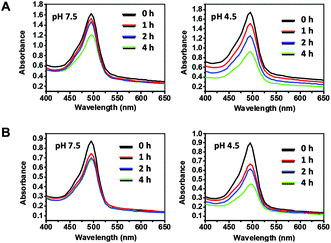 |
| | Fig. 9 pH dependent release of FITC-SA from sugar-loaded MSN. FITC-SA@BA@M (A) (10 mg ml−1) or FITC-SA@BA@M-P (B) (10 mg ml−1) incubated at Na2HPO4–H3PO4 buffer (100 mM, pH 4.5 or 7.5) for 0–4 h. A portion of the mixture (100 μl) was centrifuged. The supernatant free of nanoparticles was removed and the pellets were resuspended in Na2HPO4–H3PO4 buffer (100 mM, pH 7.5) and then analyzed by UV-vis absorbance spectroscopy. | |
As the major acidic cellular vesicles, lysosomes are frequently explored to promote therapeutic release from endocytosed nanocarriers. As MSNs are often internalized into lysosomes,17 HeLa cells were cultured with FITC-SA-laden MSN in DMEM and then stained with DiI and LysoTracker Blue. Strong fluorescence of FITC-SA was identified inside treated cells. The partial colocalization of FITC-SA with LysoTracker Blue, which selectively stains lysosomes, proves sugar-loaded MSN could be internalized into lysosomes. The rise of the extralysosomal FITC-SA signal within cells upon further incubation indicates the release of FITC-SA from the nanocarriers (Fig. 10).
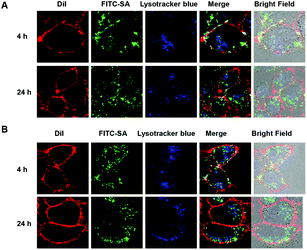 |
| | Fig. 10 Uptake of FITC-SA-laden MSN into HeLa cells. HeLa cells were cultured for 24 h in DMEM supplemented with FITC-SA@BA@M (50 μg ml−1) (A) or FITC-SA@BA@M-P (50 μg ml−1) (B) for 4 and 24 h. The cells were stained with DiI (10 μM) in DMEM for 10 min, incubated with LysoTracker Blue DND-22 (1 μM) for 30 min, and then analyzed by confocal fluorescence microscopy. Overlay of green fluorescein signals and red DiI signals reveals colocalization where yellow areas were presented. | |
Tumor detection with FITC-SA-laden MSN
To evaluate the in vivo performance of the sugar-laden MSN, FITC-SA@BA@M and FITC-SA@BA@M-P were respectively injected via the tail vein into ICR mice harbouring subcutaneous tumors. The mice were sacrificed 10 and 22 h postinjection and the biodistribution of the nanoprobes was assessed by measuring fluorescence emission in the dissected organs and tumors. As shown in Fig. 11, tumor-associated fluorescence was negligible at 10 h and became evident by 22 h postinjection in mice treated with FITC-SA@BA@M, whereas no obvious fluorescence could be discerned in tumors in mice treated with FITC-SA@BA@M-P. Hepatocytes efficiently capture nanoscaled materials,18 which likely contributes to the bright fluorescence observed in the liver (Fig. 11). Compared with FITC-SA, sugar-laden MSN displays slow kinetics of accumulation into tumors and low tumor-to-normal organ fluorescence ratios (Fig. 11).
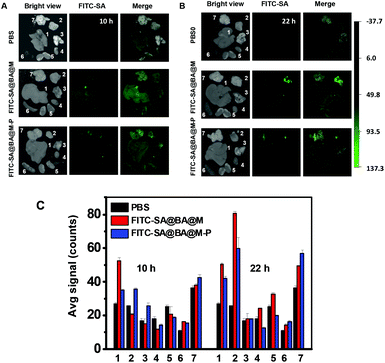 |
| | Fig. 11 Detection of subcutaneous tumors in mice with FITC-SA@BA@M or FITC-SA@BA@M-P. ICR mice with H22 subcutaneous tumors were respectively injected with FITC-SA@BA@M (10 mg kg−1), FITC-SA@BA@M-P (10 mg kg−1) or PBS (100 μl) via the tail vein. The mice were sacrificed 10 h and 22 h postinjection. Ex vivo fluorescence images of the subcutaneous tumors and representative organs from mice treated with FITC-SA@BA@M (A) or FITC-SA@BA@M-P (B). The bar graph shows FITC-SA fluorescence intensity in tumor vs. healthy organs, shown in the following sequence: liver (1), tumor (2), lung (3) heart (4), kidney (5) and spleen (6) and brain (7). | |
Cytotoxicity of FITC-MSN and FITC-SA-loaded MSN
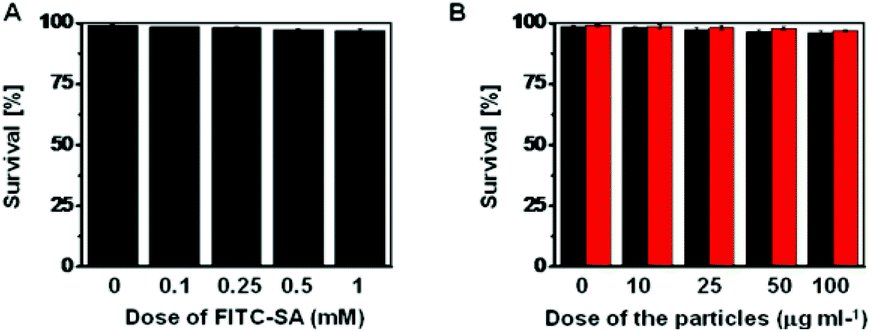
The cytotoxicity of FITC-SA, FITC-SA@BA@M and FITC-SA@BA@M-P was evaluated in HeLa cells by the Trypan Blue exclusion test. No detrimental effects on cell viability were observed at doses up to 1 mM for FITC-SA or 100 μg ml−1 for FITC-SA-loaded MSN after incubation for 24 h (Fig. 12), suggesting that FITC-SA and boronic acid-displaying MSN are of low cell toxicity.
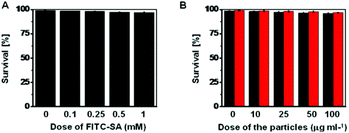 |
| | Fig. 12
In vitro cytotoxicity of FITC-SA, FITC-SA@BA@M or FITC-SA@BA@M-P. HeLa cells were respectively cultured in DMEM medium containing various levels of FITC-SA (A, 0–1 mM), FITC-SA@BA@M (B, in black, 0–100 mg ml−1) or FITC-SA@BA@M-P (B, in red, 0–100 mg ml−1) for 24 h. The cell number and cell viability were determined by the trypan blue exclusion assay. | |
Conclusions
We demonstrate the use of FITC-SA for high performance illumination of subcutaneous tumors and liver tumors in mice via quick and selective tumor uptake of systemically administered FITC-SA. Tissue-specific incorporation of functional sugars is valuable for a number of applications. For instance, the avidity of tumors for glucose over normal tissues underlies the use of [18F]-2-fluoro-2-deoxyglucose (18FDG) for tumor prognosis.19 In contrast with 18FDG with a half life of 109 min, FITC-SA is also a simple and structurally defined monosaccharide with superior chemical and physical stability under physiological conditions. The demonstrated advantageous in vivo tumor-targeting properties of FITC-SA over sugar-laden silica nanoparticles, including effective resolution of mm-level tumor foci, high tumor-to-background signal ratios and fast imaging kinetics, not only validate its clinical potential for fluorescence guided intraoperative tumor detection, but also suggest the promising use of sialic acids with appropriate modifications for a number of biomedical studies, such as manipulating tumor surface sialylation in living animals.
Experimental
Material and methods
LysoTracker Blue DND-22 and MitoTracker Deep Red were purchased from Invitrogen. Golgi-Tracker Red and DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) were purchased from Beyotime. Fluorescein isothiocynate isomer I (FITC), Dulbecco's Modified Eagle Medium (DMEM) and fetal bovine serum (FBS) were purchased from Sigma-Aldrich. FBS was inactivated by heating at 60 °C for 30 min. FITC-SA was synthesized according to a published procedure.11a All other chemicals were used as received from Alfa Aesar. QGY-7701 cells, U-87 MG cells, HeLa cells, Raw 264.7 cells and BV-2 cells were obtained from American Type Culture Collection and grown at 37 °C under 5% CO2 in DMEM.
The fluorescence spectra and UV-vis absorbance spectra were recorded on a spectrofluorimeter (Spectramax M5, Molecular Device). Dynamic light scattering and Zeta potential analysis of the nanoparticles were performed on a Zetasizer Nano ZS (ZEN3500, Malvern). Confocal microscopic images were obtained on a Leica SP5 using the following filters: λex@543 nm and λem@560–625 nm for DiI; λex@543 nm and λem@580–650 nm for Golgi-Tracker Red; λex@633 nm and λem@700–790 nm for MitoTracker Deep Red; λex@488 nm and λem@500–530 nm for FITC-SA. Fluorescence images were merged by Photoshop CS 6.0. Graphs were processed with Origin 8.5 software. Fluorescence quantification of the organs of mice were performed on a Carestream FX PRO in vivo imaging system using an excitation filter of 470 nm and an emission filter of 535 nm. Data were analyzed with Carestream MI SE. ICR mice were purchased from Xiamen University Laboratory Animal Center. H22 hepatocellular carcinoma cells were collected from the peritoneal cavity of tumor-bearing mice and then used for innoculation of tumors in ICR mice. All animal experiments were performed in accordance with the guidelines of Xiamen University's Animal Care and Use Committee.
Preparation of BA@M and BA@M-P
3-Aminopropyltrimethoxysilane (APTS, 6 ml) was incubated in dichloromethane (20 ml) containing 2-bromomethylbenzylboronic acid (3 g) for 1 h. The resulting precipitate (BA-ABPS) was filtered and the organic layer was concentrated by evaporation to afford BA-APTS. To distilled water (480 ml) was added N-cetyltrimethylammonium bromide (1.0 g), aqueous sodium hydroxide solution (2.0 M, 3.6 ml) and tetraethoxysilane (4.8 ml). The mixture was vigorously stirred at 80 °C for 1 h and then centrifuged at 10 000 rpm for 5 min. The pellet was washed with distilled water and methanol, and then refluxed in methanol (180 ml) containing hydrochloric acid (12.1 M, 10 ml) for 24 h. The mixture was centrifuged and the pellet was subjected to repeated suspension in methanol by sonication and centrifugation to afford MSN. BA-APTS (600 mg) was dissolved in anhydrous DMF (4 ml) and then added dropwise into anhydrous DMF (50 ml) containing MSM (600 mg). The mixture was stirred for 20 h at 80 °C and then centrifuged at 10000 rpm for 10 min. The pellet was washed with methanol to afford BA@M. mPEG-SPA (300 mg) was added into anhydrous DMF (25 ml) containing BA@M (300 mg) and triethylamine (300 μl). The mixture was sonicated for l h and then centrifuged to give BA@M-P which was further washed with DMF and then stored in DMSO before use.
Complexation of FITC-SA with B@M and B@M-P
B@M (150 mg) and B@M-P (150 mg) were respectively added to methanol (25 ml) containing FITC-SA (50 mg). The mixtures were refluxed for 30 min, maintained at rt overnight with gentle stirring and then centrifuged to afford FITC-SA@B@M and FITC-SA@B@M-P. FITC-SA@B@M and FITC-SA@B@M-P were resuspended in DMSO (20 mg ml−1) for subsequent analysis.
Characterization of FITC-SA laden MSN
B@M, B@M-P, FITC-SA@BA@M and FITC-SA@B@M-P were respectively spiked in distilled water to a final concentration of 200 μg ml−1. The mixtures were sonicated and then analyzed by a Zetasizer Nano ZS (ZEN3500, Malvern) to obtain their hydrodynamic sizes and Zeta potentials.
pH dependent release of FITC-SA from sugar-laden MSN
FITC-SA@BA@M (10 mg) or FITC-SA@BA@M-P (10 mg) was added to Na2HPO4–H3PO4 buffer (100 mM, 1 ml, pH 4.5 or 7.5). The mixtures were sonicated and then maintained at rt with stirring. At fixed time points (0, 1, 2, 4 h), 100 μl of the mixture was aliquoted and centrifuged at 10000 rpm for 10 min. The supernatant free of nanoparticles was removed and the pellets were resuspended in Na2HPO4–H3PO4 buffer (1 ml, 100 mM, pH 7.5) and then analyzed for UV-vis absorbance.
Cellular uptake of FITC-SA and FITC-SA laden MSN
Raw 264.7, QGY-7701, U87-MG, HeLa and BV-2 cells were respectively seeded on 35 mm glass-bottom dishes (NEST) and incubated for 24 h in DMEM supplemented with 10% FBS. The cells were spiked with one of the following species: FITC-SA (100 μM), FITC-SA@BA@M (50 μg ml−1) or FITC-SA@BA@M-P (50 μg ml−1) for 1–24 h. The cells were washed with PBS (1 ml), and then stained with DiI (10 μM) for 10 min. The cells incubated with FITC-SA laden MSN were stained with LysoTracker Blue DND-22 (1 μM) for 30 min and then analyzed by confocal fluorescence microscopy to determine the intracellular location of FITC-SA and DiI.
Determination of subcellualr location of FITC-SA
HeLa, U87-MG and BV-2 cells were respectively cultured in DMEM for 24 h and then treated with FITC-SA (100 μM) for 4 h. The cells were then stained with Golgi-Tracker Red (333 μg ml−1) at 4 °C for 30 min according to the manufacturer's protocol, and cultured with LysoTracker Blue DND-22 (1 μM) and MitoTracker Deep Red (500 nM) for 30 min in DMEM at 37 °C with 5% CO2. The cells were analyzed by fluorescence confocal microscopy to determine the intracellular locations of FITC-SA and the cell markers.
Fixation of cells
HeLa cells were stained with FITC-SA (100 μM) for 4 h and then digested by trypsin. The cells were harvested and then cultured for 12 h in DMEM. The cells were stained with DiI (10 μM) in DMEM for 10 min and treated with or without formaldehyde (4%) for 15 min in PBS. The treated cells were incubated in fresh DMEM for 12 h and then imaged by fluorescence confocal microscopy for DiI and FITC-SA signals.
Cytotoxicity of FITC-SA and FITC-SA laden MSN
HeLa cells were respectively cultured for 24 h in DMEM containing various levels of FITC-SA (0–1 mM), FITC-SA@BA@M (0–100 μg ml−1) or FITC-SA@BA@M-P (0–100 μg ml−1). The cell number and cell viability were determined by trypan blue exclusion.
Imaging of subcutaneous tumors in mice with FITC-SA
ICR mice were xenografted in the flank by subcutaneous injections of H22 cells (1 × 106). At 5–10 days after the transplantation, FITC-SA (50 mg kg−1) or PBS (100 μl) were respectively injected intravenously via the tail vein into tumor-bearing mice. At 20 min, 1.5 h, 4 h or 10 h following injection, the mice were anesthetized. The tumors and selected organs were excised, washed with PBS and then subjected to ex vivo analysis for the fluorescence intensity of FITC-SA.
Intracellular distribution of FITC-SA in subcutaneous tumors
ICR mice with subcutaneous tumors were injected intravenously via the tail vein with FITC-SA (50 mg kg−1). At 2.5 h following injection, the mice were anesthetized. The tumors and selected organs were excised, washed with PBS and then minced to 3–5 mm with sterile scissors, and then incubated in Trypsin-EDTA solution (0.125%) for 30 min. The liberated cells were visualized by confocal fluorescence microscopy.
Imaging of subcutaneous tumors in mice with FITC-SA laden MSN
FITC-SA@B@M (10 mg kg−1), FITC-SA@B@M-P (10 mg kg−1) and PBS were respectively intravenously injected into the aforementioned ICR mice bearing subcutaneous tumor implants. At 10 h and 22 h postinjection, the mice were anesthetized. The tumors and selected organs were excised, washed with PBS and then subjected to ex vivo analysis to determine the fluorescence intensity of FITC-SA.
Detection of liver tumors in mice with FITC-SA
ICR mice xenografted in the liver with H22 cells were obtained from the animal center of Xiamen University. At 5–10 days after transplantation, the mice were injected intravenously via the tail vein with FITC-SA (50 mg kg−1 in mice) or PBS (100 μl). After 17 h, the mice were anesthetized. The liver and healthy organs were excised, washed with PBS and then subjected to ex vivo analysis to determine the fluorescence intensity of FITC-SA.
Acknowledgements
Dr. S. Han was supported by grants from 973 program 2013CB93390, NSF China (21272196), PCSIRT, and the Fundamental Research Funds for the Central Universities (2011121020); Dr. J. Han was supported by grants from NSF China (30830092, 30921005, 91029304, 81061160512).
Notes and references
- Q. T. Nguyen and R. Y. Tsien, Nat. Rev. Cancer, 2013, 13, 653 CrossRef CAS PubMed.
-
(a) Q. T. Nguyen, E. S. Olson, T. A. Aguilera, T. Jiang, M. Scadeng, L. G. Ellies and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4317 CrossRef CAS PubMed;
(b) Y. Urano, D. Asanuma, Y. Hama, Y. Koyama, T. Barrett, M. Kamiya, T. Nagano, T. Watanabe, A. Hasegawa, P. L. Choyke and H. Kobayashi, Nat. Med., 2009, 15, 104 CrossRef CAS PubMed;
(c) G. M. van Dam, G. Themelis, L. M. Crane, N. J. Harlaar, R. G. Pleijhuis, W. Kelder, A. Sarantopoulos, J. S. de Jong, H. J. Arts, A. G. van der Zee, J. Bart, P. S. Low and V. Ntziachristos, Nat. Med., 2011, 17, 1315 CrossRef CAS PubMed;
(d) X. Wu, Y. Tian, M. Yu, B. Li, J. Han and S. Han, Biomater. Sci., 2014 10.1039/C4BM00007B.
- T. Angata and A. Varki, Chem. Rev., 2002, 102, 439 CrossRef CAS PubMed.
-
(a) B. E. Collins and J. C. Paulson, Curr. Opin. Chem. Biol., 2004, 8, 617 CrossRef CAS PubMed;
(b) Y. Pilatte, J. Bignon and C. R. Lambre, Glycobiology, 1993, 3, 201 CrossRef CAS PubMed;
(c) T. A. Springer, Cell, 1994, 76, 301 CrossRef CAS.
-
(a) R. J. Bernacki and U. Kim, Science, 1977, 195, 577 CAS;
(b) J. Dennis, C. Waller, R. Timpl and V. Schirrmacher, Nature, 1982, 300, 274 CrossRef CAS;
(c) A. Matsumoto, N. Sato, K. Kataoka and Y. Miyahara, J. Am. Chem. Soc., 2009, 131, 12022 CrossRef CAS PubMed;
(d) J. Wang, Z. Zhang, X. Wang, W. Wu and X. Jiang, J. Control. Release, 2013, 168, 1 CrossRef CAS PubMed.
-
(a) S. J. Luchansky, S. Goon and C. R. Bertozzi, ChemBioChem, 2004, 5, 371 CrossRef CAS PubMed;
(b) C. Oetke, R. Brossmer, L. R. Mantey, S. Hinderlich, R. Isecke, W. Reutter, O. T. Keppler and M. Pawlita, J. Biol. Chem., 2002, 277, 6688 CrossRef CAS PubMed;
(c) C. Oetke, S. Hinderlich, R. Brossmer, W. Reutter, M. Pawlita and O. T. Keppler, Eur. J. Biochem., 2001, 268, 4553 CrossRef CAS.
-
(a) L. K. Mahal, K. J. Yarema and C. R. Bertozzi, Science, 1997, 276, 1125 CrossRef CAS;
(b) E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef CAS;
(c) A. Varki, FASEB J., 1991, 5, 226 CAS.
-
(a) H. Kayser, R. Zeitler, C. Kannicht, D. Grunow, R. Nuck and W. Reutter, J. Biol. Chem., 1992, 267, 16934 CAS;
(b) J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873 CrossRef CAS PubMed;
(c) A. A. Neves, H. Stockmann, R. R. Harmston, H. J. Pryor, I. S. Alam, H. Ireland-Zecchini, D. Y. Lewis, S. K. Lyons, F. J. Leeper and K. M. Brindle, FASEB J., 2011, 25, 2528 CrossRef CAS PubMed.
-
(a) S. Han, B. E. Collins, P. Bengtson and J. C. Paulson, Nat. Chem. Biol., 2005, 1, 93 CrossRef CAS PubMed;
(b) R. E. Kosa, R. Brossmer and H. J. Gross, Biochem. Biophys. Res. Commun., 1993, 190, 914 CrossRef CAS.
- K. Ishiwata, T. Ido, T. Nakajima, H. Ohrui, I. Kijima-Suda and M. Itoh, Int. J. Rad. Appl. Instrum. B, 1990, 17, 363 CrossRef CAS.
-
(a) H. J. Gross and R. Brossmer, Eur. J. Biochem., 1988, 177, 583 CrossRef CAS;
(b) H. J. Gross, Eur. J. Biochem., 1992, 203, 269 CrossRef CAS.
-
(a) D. Y. Kim and K. H. Han, Liver Cancer., 2012, 1, 2 CrossRef PubMed;
(b) P. Ferenci, M. Fried, D. Labrecque, J. Bruix, M. Sherman, M. Omata, J. Heathcote, T. Piratsivuth, M. Kew, J. A. Otegbayo, S. S. Zheng, S. Sarin, S. S. Hamid, S. B. Modawi, W. Fleig, S. Fedail, A. Thomson, A. Khan, P. Malfertheiner, G. Lau, F. J. Carillo, J. Krabshuis and A. Le Mair, J. Clin. Gastroenterol., 2010, 44, 239 CrossRef PubMed;
(c) M. Sherman, Semin. Liver Dis., 2010, 30, 3 CrossRef CAS PubMed.
-
(a) M. Partridge, S. R. Li, S. Pateromichelakis, R. Francis, E. Phillips, X. H. Huang, F. Tesfa-Selase and J. D. Langdon, Clin. Cancer Res., 2000, 6, 2718 CAS;
(b) K. Pantel and T. J. Moss, Cytotherapy, 1999, 1, 53 CrossRef CAS.
- R. Xie, S. Hong, L. Feng, J. Rong and X. Chen, J. Am. Chem. Soc., 2012, 134, 9914 CrossRef CAS PubMed.
-
(a) H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Control Release, 2000, 65, 271 CrossRef CAS;
(b) V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131 CrossRef CAS PubMed.
-
(a) R. V. Chari, Acc. Chem. Res., 2008, 41, 98 CrossRef CAS PubMed;
(b) M. C. Garnett, Adv. Drug Delivery Rev., 2001, 53, 171 CrossRef CAS;
(c) A. Kakinoki, Y. Kaneo, Y. Ikeda, T. Tanaka and K. Fujita, Biol. Pharm. Bull., 2008, 31, 103 CrossRef CAS;
(d) T. Etrych, M. Jelinkova, B. Rihova and K. Ulbrich, J. Control Release, 2001, 73, 89 CrossRef CAS;
(e) Z. Huang, X. Guo, W. Li, J. A. MacKay and F. C. Szoka Jr., J. Am. Chem. Soc., 2006, 128, 60 CrossRef CAS PubMed;
(f) E. R. Gillies, A. P. Goodwin and J. M. Frechet, Bioconjugate Chem., 2004, 15, 1254 CrossRef CAS PubMed;
(g) W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharm., 2010, 7, 1913 CrossRef CAS PubMed.
-
(a) J. Lu, M. Liong, Z. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1794 CrossRef CAS PubMed;
(b) J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S. Lin, Small, 2010, 6, 1952 CrossRef CAS PubMed;
(c) X. Wu, S. Wu, L. Yang, J. Han and S. Han, J. Mater. Chem., 2012, 22, 17121 RSC.
-
(a) T. Kasuya and S. Kuroda, Expert Opin. Drug Deliv., 2009, 6, 39 CrossRef CAS PubMed;
(b) J. D. Perkins, Liver Transpl., 2007, 13, 167 Search PubMed.
- P. Som, H. L. Atkins, D. Bandoypadhyay, J. S. Fowler, R. R. MacGregor, K. Matsui, Z. H. Oster, D. F. Sacker, C. Y. Shiue, H. Turner, C. N. Wan, A. P. Wolf and S. V. Zabinski, J. Nucl. Med., 1980, 21, 670 CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.