 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bottom-up assembly of a surface-anchored supramolecular rotor enabled using a mixed self-assembled monolayer and pre-complexed components†
Josep
Puigmartí-Luis
a,
Wojciech J.
Saletra
a,
Asensio
González
b,
David B.
Amabilino
*a and
Lluïsa
Pérez-García
*bc
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus Universitari, 08193 Bellaterra, Catalonia, Spain. E-mail: amabilino@icmab.es; Fax: +34 935 805 729; Tel: +34 935 801 853
bFacultat de Farmàcia, Universitat de Barcelona, 08028 Barcelona, Catalonia, Spain
cInstitut de Nanociència i Nanotecnologia, Universitat de Barcelona, 08028 Barcelona, Catalonia, Spain. E-mail: mlperez@ub.edu
First published on 14th October 2013
Abstract
Three different routes to rotor-type systems on a gold surface provide sparse and dense layers of rotors with best control exerted using mixed ordered monolayers that guide the creation of the potential molecular machine components from solution.
The controlled construction of functional systems by bottom-up surface modification1–4 is very relevant for the preparation of synthetic molecular machinery.2 While the thrust to study molecular motion at the (sub)molecular level on surfaces has led to the design of individual artificial molecular machines,3 little is known about the geometry of anchoring on a surface. Specific anchoring is desirable, especially for rotor systems in which a rigid axle is oriented perpendicularly to a surface with the mobile component parallel to it.4 Our aim is to use bottom-up approaches for the solution-based preparation and study of supramolecular rotors (Fig. 1) as potential components of machine-type systems, using the axial coordination of large π-functional molecules – which can be readily imaged using scanning probe microscopy and act as rotators – bound to axles linking them to a surface which is the stator component.
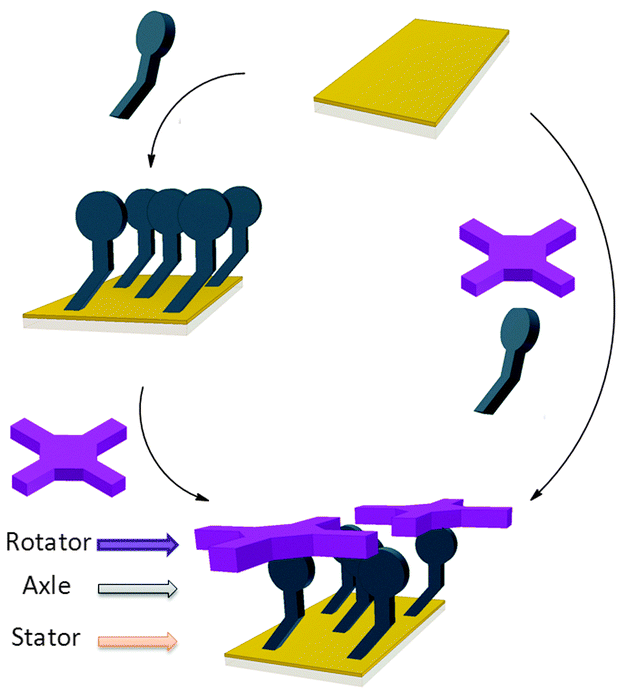 | ||
| Fig. 1 Representation of the bottom-up approaches employed here for the formation of supramolecular rotors on surfaces. | ||
As the large π-functional molecule, here we use a zinc(II) porphyrin known to complex pyridyl type ligands in an axial way in solution5 and surfaces.6 Metalloporphyrins are interesting components in molecular machinery,7 and their large dimensions allow imaging of their motions in certain cases.8 Here, we anchor porphyrin rotator motifs on a gold surface (which is the stator) with an axle that binds the rotator: the orientation of the axle is influenced by the organisation of the self-assembled monolayer (SAM). We foresee the monolayer being necessary to prevent the tipping of the axle and “crashing” of the rotator into the surface. It also turned out in our studies that the structure of the monolayer is essential to efficient binding, and the orientation of the groups involved in the rotor must be controlled.
The axle component we used in our study is pyridin-4-yl-methanethiol (1, Fig. 2). As the “filling layer” between the axle components we foresaw dodecanethiol (2) and/or molecules of 1 that are not complexed to the eventual rotor. SAMs of 1 and 2 chemisorbed on Au(111) surfaces investigated separately by Scanning Tunnelling Microscopy (STM) in air (see ESI†) showed disordered and ordered layers, respectively. When the monolayer of 1 was treated with a toluene solution of 3, features which could correspond to the complex were observed, but extremely rarely, presumably because the pyridyl rings are not oriented appropriately for coordination to take place. As expected no adsorption of the porphyrin onto the monolayer of 2 was observed either. UV-visible absorption spectroscopy indicated efficient binding of 3 by 1 in solution (ESI†). A very interesting situation occurs in toluene – the solvent used for deposition –wherein the attempted titration experiment showed an increase in the absorptivity of the solution as the pyridine derivative was added (ESI†). This effect is a result of increased solubility caused by complexation of the porphyrin which is sparingly soluble in this solvent in its pure form, and indicates the benefits of axial coordination for the processing of this kind of material.
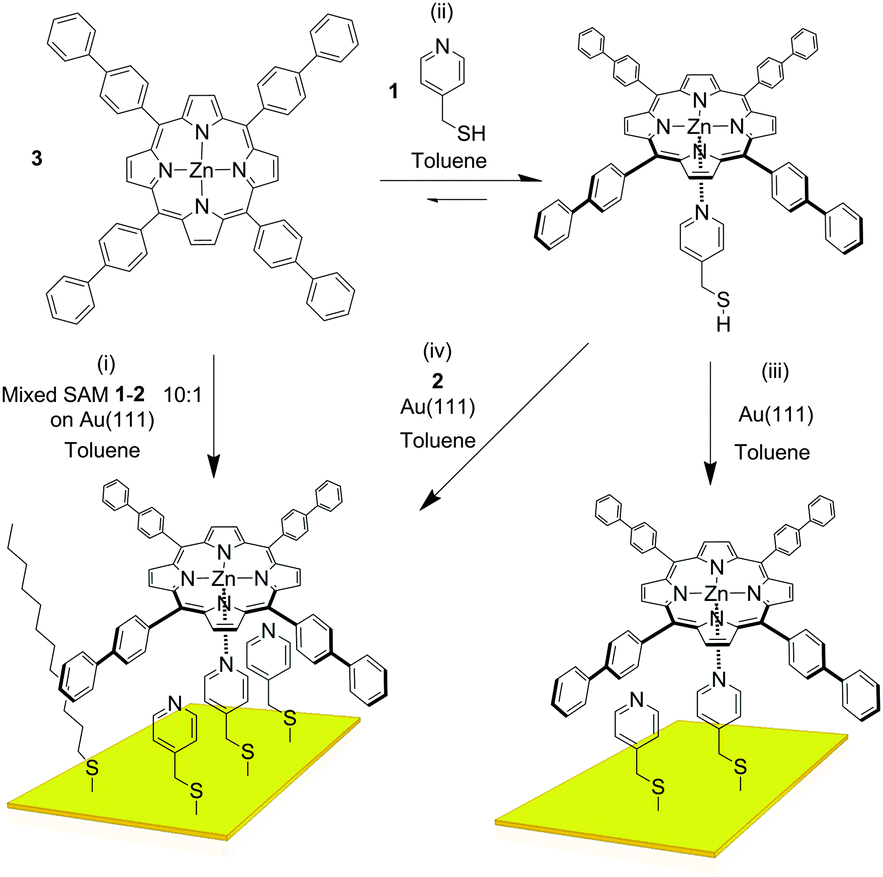 | ||
Fig. 2 Structure of 3 and schematic views showing the different coordination processes and reactions used here. (i) Coordination of 3 with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixed SAM of 1 and 2; (ii) coordination of 3 with 1 in solution; (iii) and (iv) functionalisation of Au(111) substrates with a toluene solution containing the 3–1 complex, and with a toluene solution containing a mixture of 1, 2, and 3, respectively. :1 mixed SAM of 1 and 2; (ii) coordination of 3 with 1 in solution; (iii) and (iv) functionalisation of Au(111) substrates with a toluene solution containing the 3–1 complex, and with a toluene solution containing a mixture of 1, 2, and 3, respectively. | ||
In order to improve the orientation of 1 with respect to the gold surface, SAMs of 1 co-deposited with 2 were prepared and studied by STM at room temperature. Unlike the SAM of pyridine derivative 1 (ESI†), the 1:1 mixed SAM has a quite well organized structure, displaying striped domains and brighter spots presumably associated with the pyridyl rings of 1 (see ESI†). These striped features were never seen in monolayers of the pyridyl compound on its own. It seems to be logical to assume that this higher degree of organization of 1 is induced by 2. A similar effect has been observed in functionally distinct mixtures of other thiolated moieties on gold.9 The mixed SAM formed by 1 and 2 from a solution ratio of 10:1, respectively, gives images from the STM experiment with remarkably good resolution (Fig. 3). Molecules of 1 line up showing an ordered spatial confinement of the pyridine moieties along the gold lattice. The distance between consecutive pyridyl moieties is approximately 0.9 nm (Fig. 3), contrasting with the separation between molecules of 2 in its SAMs (0.5 nm). The spacing between rows of molecules of 1 is approximately 1.1 nm. These lattice parameters are not consistent with a lying-down conformation of the thiol, where the distance between rows would be expected to be 50% greater,10 but imply a tilted orientation. The pyridine striped-line structure coexists with locally segregated 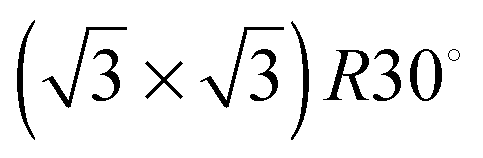 structures assigned to domains of 2 (yellow arrows in Fig. 3A). We are not aware of any other experimental evidence showing the great influence that a minority densely packed alkylthiol SAM, such as 2, can have on the organization and orientation of a ligand based thiolated molecule (in this case 1), even when small amounts of alkylthiol are employed. The effect is potentially important, because the capacity of the surface-anchored ligand to bind solution borne compounds to give complexes will be dramatically affected by its orientation.
structures assigned to domains of 2 (yellow arrows in Fig. 3A). We are not aware of any other experimental evidence showing the great influence that a minority densely packed alkylthiol SAM, such as 2, can have on the organization and orientation of a ligand based thiolated molecule (in this case 1), even when small amounts of alkylthiol are employed. The effect is potentially important, because the capacity of the surface-anchored ligand to bind solution borne compounds to give complexes will be dramatically affected by its orientation.
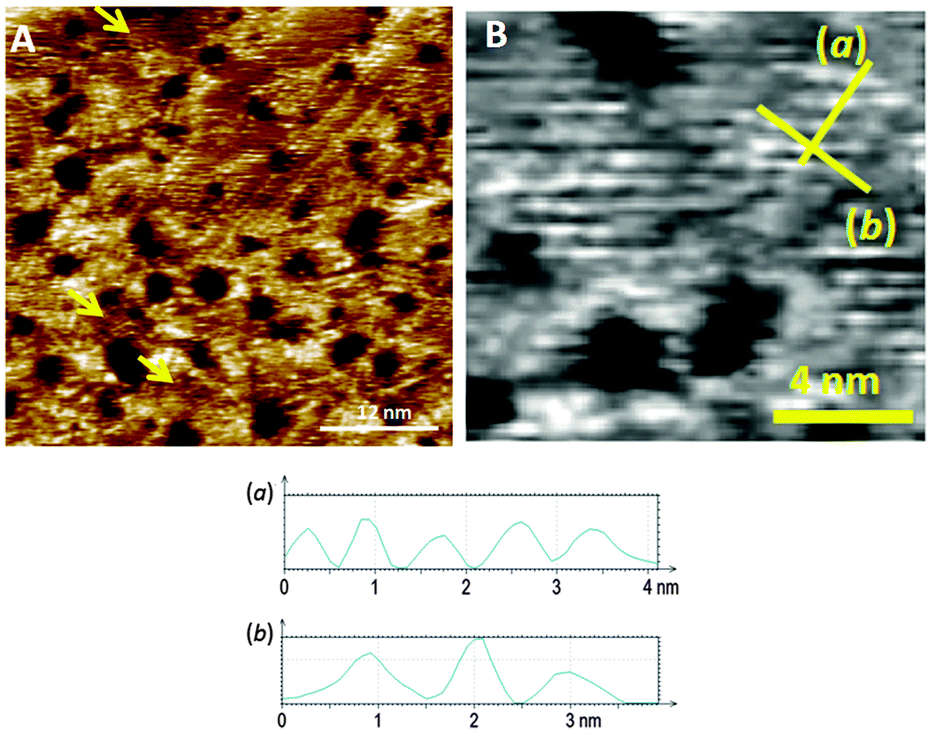 | ||
| Fig. 3 (A) STM image of a mixed SAM comprising 1 and 2 at a solution deposition ratio of 10:1 (Iset = 50 pA, Vbias = −0.8 V). Yellow arrows indicate areas of monolayers formed by 2. (B) Expanded region of the SAM incorporating 1 in image B, with lines (a) and (b) corresponding to the profiles shown below the image. | ||
With this optimized mixed SAM in hand the coordination studies were conducted using the pyridyl units for complexation of a zinc(II) porphyrin derivative with C4 symmetry (3, Fig. 2).11 In a typical experiment, a 10:1 mixed SAM was immersed for twelve hours in a toluene solution of 3 (1× 10−4 M). The samples were subsequently rinsed exhaustively with toluene to remove non-specifically physisorbed molecules of 3 before STM analysis was performed on the dry monolayer.
Favourable coordination of one building block to the other on the surface was demonstrated by both mass spectrometry (ESI†) and STM (Fig. 4). Bright protrusions of high tunnelling current and with uniform diameter (approximately 4 nm) and contrast were observed by STM. These higher conductive areas correspond to the porphyrin cores (that appear as disk-like protrusions with no sub-molecular resolution) coordinated to the pyridine groups in the monolayer. The vertically oriented molecules of 1 are assumed to coordinate porphyrin molecules in an azimuthal-rotor configuration where rotation about the nitrogen–zinc bond could enhance significantly the 3–1 image-size (the calculated diameter of the porphyrin is 2.4 nm, and the observed one is approximately 4 nm), in accord with literature data showing pivoting motion of a molecule around a central pivot point on Au(111).12 Importantly, no visualization of bright protrusions was possible with SAMs of either 1 or 2 after 12 hours of immersion in a toluene solution of 3 (1 × 10−4 M), corroborating its poor coordination ability assigned to the lack of order in the monolayer of pure 1 on Au(111), and confirms that the bright protrusions arise from a specific binding to the SAM rather than to non-specific adsorption.
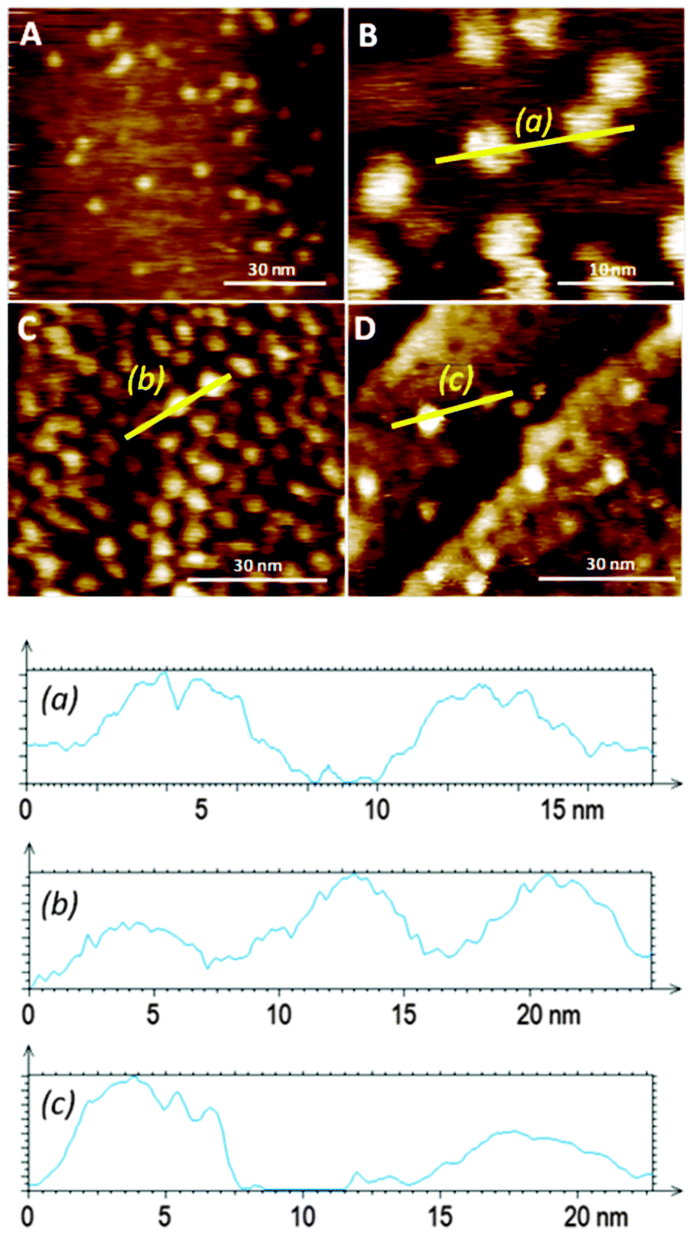 | ||
| Fig. 4 Typical wide area STM images of 3–1 complexes formed on the mixed SAM of 1 and 2 at a 10:1 ratio (A and B), and of Au(111) functionalized with toluene solutions containing the 3–1 complex (C) and a toluene solution containing a mixture of 1 (1 × 10−5 M), 2 (1 × 10−6 M), and 3 (1 × 10−4 M) (D), respectively. All STM images were recorded at Iset = 50 pA and Vbias = −0.80 V. Profiles (a), (b) and (c) correspond to the yellow lines in images B, C and D, respectively. | ||
The distribution of the adsorbed molecules of 3 on the surface is sparse and with no apparent order even at short length scales. Therefore, even though defined nanoscale anchoring sites are apparently present in 10:1 mixed SAMs, high steric energy barriers may exist during the complexation of 3 from solution, thereby leading to randomly distributed 3–1 complexes in the SAM. Evidence of the negative allosteric effect during the coordination of molecules of 3 on the 10:1 mixed SAM was provided by directly functionalizing Au(111) with a solution of the 3–1 complex in toluene.
When an Au(111) surface was exposed to a toluene solution of the 3–1 complex for 12 hours the adsorption of the rotor system is more efficient than prior formation of a monolayer followed by rotator attachment. Direct 3–1 adsorption on gold is manifested by a high density of bright disc-like protrusions similar to the 3–1 complexes observed from the adsorption of 3 to the 10:1 mixed SAMs (Fig. 4). In this layer some of the apparent protrusions corresponding to the porphyrin appear to be brighter and wider than others (the duller ones are similar in size and shape to those seen in the adsorption of 3 to the mixed SAM of 1 and 2), pointing to non-homogeneous binding sites for the rotator and possibly to different dynamics in these positions. The larger size is probably a result of greater mobility in this SAM which is likely to be more poorly packed than the prefabricated one. This feature emphasizes the great influence that the bottom-up approach can have on the monolayer structure and homogeneity.
When a direct functionalisation of Au(111) was performed with a mixture of 1 (1 × 10−5 M), 2 (1 × 10−6 M), and 3 (1 × 10−4 M), an inferior number of bright protrusions, assigned to 3–1, were observed (Fig. 4D) when compared with the attachment of the 3–1 complex alone. It would be expected that diffusion is greater in the case of 2 – which would prompt its preferential transport through the fluid and chemisorption to the metal surface – when compared with 3–1 – whose adsorption on gold from the solution would be disfavored relatively. Similarly to the direct adsorption of the axle–rotator complex on the gold surface, different porphyrin environments are detected by the STM measurements, with some showing high tunneling current and large dimensions indicative of proximity to the gold surface. The surface density is of the order of that formed by the adsorption of 3 onto the mixed SAM.
These results show that mixed SAMs – that are ordered, thanks to the very low content of the ordering component – allow the formation of specific anchoring/recognition areas, and that the three routes performed led to different outcomes, the most efficient being the adsorption of a pre-formed supramolecular rotor, implying that an ordered SAM is not necessarily an ideal site for adsorption. The specific case of the formation of rotor features on the surface makes possible the preparation of a number of systems which could reveal new physical phenomena at the nanoscale.13
This research was supported by the MINECO, Spain (projects CTQ2010-16339 and TEC2011-29140-C03-02) and the DGR, Catalonia (Project 2009 SGR 158). The authors warmly acknowledge Dr Daniel Ruiz-Molina for the loan of the STM head. JPL thanks the MINECO for a Ramón y Cajal contract (RYC-2011-08071). We thank the CSIC for OA funding.
Notes and references
- (a) M. Koepf, F. Cherioux, J. A. Wytko and J. Weiss, Coord. Chem. Rev., 2012, 256, 2872 CrossRef CAS PubMed; (b) S. Fabiano and B. Pignataro, Chem. Soc. Rev., 2012, 41, 6859 RSC.
- M. M. Boyle, R. A. Smaldone, A. C. Whalley, M. W. Ambrogio, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2011, 2, 204 RSC.
- (a) J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611 CrossRef CAS; (b) W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25 CrossRef CAS PubMed; (c) V. Balzani, A. Credi and M. Venturi, ChemPhysChem, 2008, 9, 202 CrossRef CAS PubMed.
- (a) G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281 CrossRef CAS PubMed; (b) H. L. Tierney, C. J. Murphy, A. D. Jewell, A. E. Baber, E. V. Iski, H. Y. Khodaverdian, A. F. McGuire, N. Klebanov and E. C. H. Sykes, Nat. Nanotechnol., 2011, 6, 625 CrossRef CAS PubMed; (c) U. G. E. Perera, F. Ample, H. Kersell, Y. Zhang, G. Vives, J. Echeverria, M. Grisolia, G. Rapenne, C. Joachim and S. W. Hla, Nat. Nanotechnol., 2013, 8, 46 CrossRef CAS PubMed.
- (a) G. C. Vogel and B. A. Beckmann, Inorg. Chem., 1976, 15, 483 CrossRef CAS; (b) G. M. Mamardashvili and O. M. Kulikova, Russ. J. Coord. Chem., 2006, 32, 756 CrossRef CAS.
- (a) F. Da Cruz, K. Driaf, C. Berthier, J.-M. Lameille and F. Armand, Thin Solid Films, 1999, 349, 155 CrossRef CAS; (b) Q. Ferreira, L. Alcácer and J. Morgado, Nanotechnology, 2011, 22, 435604 CrossRef PubMed; (c) A. Dreas-Wlodarczak, M. Müllneritsch, T. Juffmann, C. Cioffi, M. Arndt and M. Mayor, Langmuir, 2010, 26, 10822 CrossRef CAS PubMed; (d) G. Salassa, M. J. J. Coenen, S. J. Wezenberg, B. L. M. Hendriksen, S. Speller, J. A. A. W. Elemans and A. W. Kleij, J. Am. Chem. Soc., 2012, 134, 7186 CrossRef CAS PubMed.
- (a) K. P. Ghiggino, J. A. Hutchison, S. J. Langford, M. J. Latter, M. A. P. Lee, P. R. Lowenstern, C. Scholes, M. Takezaki and B. E. Wilman, Adv. Funct. Mater., 2007, 17, 805 CrossRef CAS; (b) A. Guenet, E. Graf, N. Kyritsakas and M. W. Hosseini, Chem.–Eur. J., 2011, 17, 6443 CrossRef CAS PubMed.
- (a) J. Otsuki, Y. Komatsu, D. Kobayashi, M. Asakawa and K. Miyake, J. Am. Chem. Soc., 2010, 132, 6870 CrossRef CAS PubMed; (b) S. C. Yan, Z. J. Ding, N. Xie, H. Q. Gong, Q. Sun, Y. Guo, X. Y. Shan, S. Meng and X. H. Lu, ACS Nano, 2012, 6, 4132 CrossRef CAS PubMed; (c) D. Lensen and J. A. A. W. Elemans, Soft Matter, 2012, 8, 9053 RSC.
- G. Pace, A. Petitjean, M.-N. Lalloz-Vogel, J. Harrowfield, J.-M. Lehn and P. Samorì, Angew. Chem., Int. Ed., 2008, 47, 2484 CrossRef CAS PubMed.
- L.-J. Wan, Y. Hara, H. Noda and M. Osawa, J. Phys. Chem. B, 1998, 102, 5943 CrossRef CAS.
- O. Penon, F. Marsico, D. Santucci, L. Rodríguez, D. B. Amabilino and L. Pérez-García, J. Porphyrins Phthalocyanines, 2012, 16, 1293 CrossRef CAS.
- A. E. Baber, H. L. Tierney and E. C. H. Sykes, ACS Nano, 2008, 2, 2385 CrossRef CAS PubMed.
- (a) A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19 RSC; (b) M. von Delius and D. A. Leigh, Chem. Soc. Rev., 2011, 40, 3656 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental methods. See DOI: 10.1039/c3cc44794d |
| This journal is © The Royal Society of Chemistry 2014 |