Enantioselective desymmetrization of prochiral 1,3-dinitropropanes via organocatalytic allylic alkylation†
Soumya Jyoti
Singha Roy
and
Santanu
Mukherjee
*
Department of Organic Chemistry, Indian Institute of Science, Bangalore 560 012, India. E-mail: sm@orgchem.iisc.ernet.in; Fax: +91-80-2360-0529; Tel: +91-80-2293-2850
First published on 23rd October 2013
Abstract
An enantioselective desymmetrization of prochiral 1,3-dinitropropanes has been developed which proceeds via enantiogroup differentiating organocatalytic allylic alkylation. Densely functionalized products with two vicinal stereocenters were obtained generally with good to excellent diastereoselectivity (up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and superb enantioselectivity (up to >99:1 er).
:1 dr) and superb enantioselectivity (up to >99:1 er).
Rapid construction of molecular complexity with step-economy remained the ultimate motivation behind the discovery and development of new synthetic strategies.1 C–C bond forming desymmetrization, proceeding through enantiogroup differentiating reactions, can create functional diversity within a molecule with the generation of multiple stereocenters in a single step.2
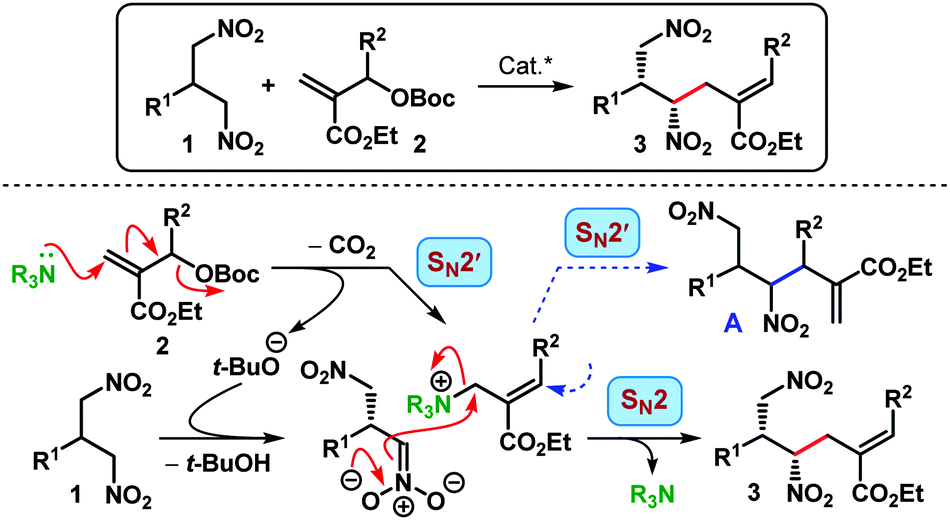
Nitroalkanes are a very important class of synthetic building blocks owing to the potential utility of the nitro group as a precursor to various other functionalities, especially amines.3 However, synthetic difficulties associated with higher nitroalkane homologs impose a restriction on their utility as pronucleophiles in the synthesis. In contrast, 2-substituted 1,3-dinitropropanes (1, Scheme 1) represent an easily accessible class of prochiral compounds.4 Enantiogroup differentiating reactions involving the nitromethyl group of such 1,3-dinitropropanes could potentially generate highly functionalized compounds with more than one stereocenter. Yet, compared to the extensive applicability of simple nitroalkanes in asymmetric catalysis,3a 1,3-dinitropropanes have largely been overlooked.5 The single example utilizing 1,3-dinitropropanes as the nucleophilic component has been reported by Jørgensen et al. for an enantioselective Michael–aldol cascade route to pentasubstituted cyclohexanes.6
 | ||
| Scheme 1 Enantioselective desymmetrization of prochiral 1,3-dinitropropanes and the mechanistic hypothesis. | ||
Inspired by the pioneering studies in the area of Pd-catalyzed allylic alkylative desymmetrization,7 we charted a similar strategy for the desymmetrization of 1,3-dinitropropanes. We envisioned the reaction of a suitable allylating agent with one of the enantiotopic nitromethyl groups under the control of a chiral catalyst (Scheme 1). The use of simple nitroalkanes as nucleophiles for allylic alkylation is well precedented, both for Pd-catalysis7a,8 and organocatalysis.9 We became particularly interested in the report by Lu and co-workers concerning an organocatalytic enantioselective allylic alkylation of nitroalkanes using allylic carbonates (2, Scheme 1) derived from Morita–Baylis–Hillman10 adducts.9b This type of allylic carbonates has recently come out as a very popular reagent for a variety of asymmetric allylic alkylation11 and annulation reactions.12,13 Reminiscent of Lu's case,9b we expected the reaction to proceed via an initial SN2′ attack of a Lewis base catalyst (NR3) to the allylic carbonate 2 followed by another SN2-type displacement with the active nucleophile. Generation of an active nucleophile through enantiogroup differentiating deprotonation of 1,3-dinitropropane as well as the regioselectivity of the nucleophilic attack (SN2 vs. SN2′) were anticipated to be controlled by a suitably placed Brønsted acidic functionality within the catalyst.9b
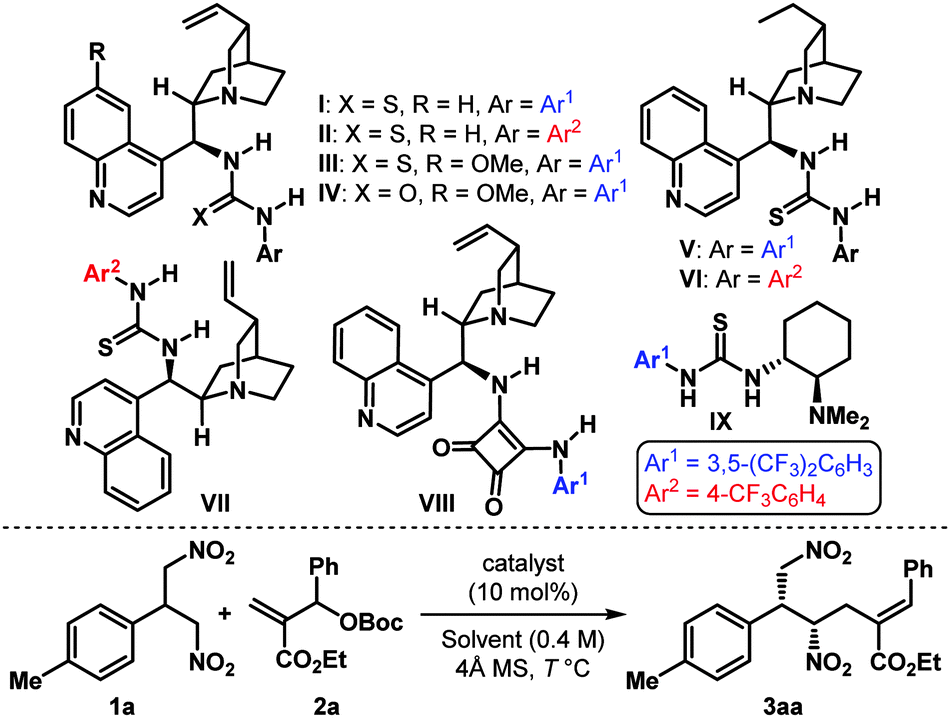
Accordingly, we began our investigation with the screening of some well-established bifunctional catalysts containing a Lewis basic tertiary amine and a Brønsted acidic (thio)urea/squaramide moiety.14,15 The allylic alkylation of p-tolyl substituted 1,3-dinitropropane 1a with phenyl substituted allylic carbonate 2a in toluene at 25 °C was chosen as the model reaction (Table 1). Gratifyingly the cinchonidine-derived catalyst I having the 3,5-bis(trifluoromethyl)phenyl substituent provided the monoallylated product 3aa with good enantioselectivity and acceptable diastereoselectivity (entry 1). More importantly, the reaction was found to be completely regioselective, as no regioisomeric product (A in Scheme 1) could be detected. The use of the 4 Å MS as an additive improved the reaction rate considerably (entry 2), presumably due to its tert-butanol sequestering ability. The corresponding thiourea II containing a 4-trifluoromethylphenyl moiety improved the er but did so at the expense of the diastereomeric outcome of the reaction (entry 3). Quinine-derived catalysts III–IV were less efficient, and so were cinchonidine-derived squaramide VIII as well as Takemoto catalyst IX16 (entries 4–5, 8–9). However, dihydrocinchonidine-derived catalysts V–VI displayed efficacy comparable to their dehydro analogs I–II (entries 6–7). At this point, a detailed solvent, concentration and temperature optimization15 established II as the catalyst of choice: a welcoming development considering the easier accessibility of II over V. Using only 10 mol% of II in 1,2-dichloroethane at −10 °C, 3aa was obtained with high dr (10:1) and excellent er of 98.5:1.5 (entry 17). It must be emphasized that the reaction requires only slight excess (1.1 equiv.) of the allylating agent 2a and a very small amount of solvent (conc. 2.0 M).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Solvent | T/°C (t/h) | Conv.b (%) | drb | erc |
| a Reactions were carried out using 1.0 equiv. of 1a and 1.1 equiv. of 2a. b Conversion and diastereomeric ratio (dr) determined by 1H-NMR analysis of the crude reaction mixture. c Enantiomeric ratio (er) determined by HPLC analysis using a stationary phase chiral column (see ESI). d Reaction without molecular sieves. e Solvent conc. 1.0 M. f Solvent conc. 2.0 M. | ||||||
| 1d | I | Toluene | 25 (48) | >95 | 4.5:1 |
95.5:4.5 |
| 2 | I | Toluene | 25 (14) | >95 | 5.8:1 |
95:5 |
| 3 | II | Toluene | 25 (40) | >95 | 3.7:1 |
96.5:3.5 |
| 4 | III | Toluene | 25 (48) | 97 | 5:1 |
94:6 |
| 5 | IV | Toluene | 25 (36) | 97 | 5.4:1 |
87:13 |
| 6 | V | Toluene | 25 (48) | >95 | 4.9:1 |
96.5:3.5 |
| 7 | VI | Toluene | 25 (12) | >95 | 6:1 |
96:4 |
| 8 | VIII | Toluene | 25 (48) | 55 | 4.3:1 |
89.5:10.5 |
| 9 | IX | Toluene | 25 (48) | 95 | 3:1 |
13:87 |
| 10 | II | PhCF3 | 25 (40) | >95 | 7:1 |
97.5:2.5 |
| 11 | II | CHCl3 | 25 (30) | >95 | 7.8:1 |
96.5:3.5 |
| 12 | II | CH2Cl2 | 25 (14) | >95 | 6.8:1 |
97:3 |
| 13 | II | (CH2Cl)2 | 25 (40) | >95 | 7.8:1 |
>97:3 |
| 14e | II | (CH2Cl)2 | 25 (8) | >95 | 7.7:1 |
97:3 |
| 15e | II | (CH2Cl)2 | 0 (48) | >95 | 9.5:1 |
>97.5:2.5 |
| 16f | II | (CH2Cl)2 | −20 (60) | 86 | 10:1 |
>98:2 |
| 17f | II | (CH2Cl)2 | −10 (48) | >95 | 10:1 |
98.5:1.5 |
The optimized reaction conditions (Table 1, entry 17) were then applied to the desymmetrization of various 2-substituted 1,3-dinitropropanes (1a–o) with 2a as the allyl source (Table 2A). A wide range of substituents with diverse steric and electronic demand were well accommodated under the reaction conditions to deliver the desymmetrized products generally with good diastereoselectivities and excellent enantioselectivities. These include aromatic (3aa–ia), heteroaromatic (3ja–la), branched and linear aliphatic (3ma–na) as well as olefinic (3oa) substituents. However, in some cases where accessibility of the nucleophilic nitromethyl group was counteracted by the steric factor (3ga, 3ma–na), higher reaction temperatures were required. Most strikingly, N-unprotected indole substituted dinitropropane (1l) worked exceptionally well and highlights the chemoselectivity of the nitromethyl group over potentially nucleophilic indolyl NH.17 The same is true for 3ga, where the sterically well accessible alcohol functionality on the dinitro substrate (1g) was left untouched. Furthermore, the amenability of the reaction conditions to various allylic carbonates having substituents with different steric and electronic requirements (Table 2B) underscores the generality of this protocol. It must be mentioned that in all these cases only (E)-olefinic products could be isolated. The relative and absolute stereochemistry of the product was established by X-ray diffraction analysis of the single crystal obtained for 3ka (Table 2A).
|
|
|||||
|---|---|---|---|---|---|
| R1 | t/h | Yield (%) | dr | er | |
| a Reactions were performed on a 0.2 mmol scale under an argon atmosphere. b Yields correspond to the isolated yield of the products after column chromatography. c Diastereomeric ratio (dr) determined by 1H-NMR analysis of the crude reaction mixture. d Enantiomeric ratio (er) determined by HPLC analysis using a stationary phase chiral column (see ESI). e Reaction conducted at 10 °C. f Reaction conducted at 5 °C. | |||||
(A)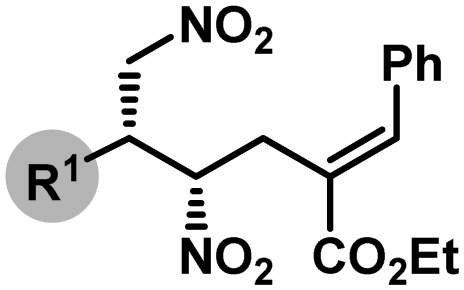 |
4-MeC6H4 (3aa) | 48 | 74 | 10:1 |
98.5:1.5 |
| Ph (3ba) | 48 | 75 | 8:1 |
97:3 |
|
| 4-ClC6H4 (3ca) | 55 | 73 | 9:1 |
97.5:2.5 |
|
| 2-BrC6H4 (3da) | 34 | 84 | 10:1 |
>98.5:1.5 |
|
| 2-FC6H4 (3ea) | 48 | 83 | 16:1 |
98:2 |
|
| 2,4-Cl2C6H3 (3fa) | 48 | 76 | >20:1 |
98:2 |
|
| 2-(CH2OH)C6H4 (3ga)e | 45 | 73 | 3:1 |
99:1 |
|
| 1-Naphth (3ha) | 43 | 83 | 13:1 |
98.5:1.5 |
|
|
72 | 78 | 15:1 |
98:2 |
|
| 2-Furyl (3ja) | 72 | 74 | 9:1 |
97:3 |
|
| 2-Thienyl (3ka) | 60 | 73 | 5:1 |
98:2 |
|
| 3-Indolyl (3la)f | 65 | 79 | 8:1 |
>99:1 |
|
| c-Hex (3ma)f | 72 | 67 | 5:1 |
>99:1 |
|
| n-Pent (3na)f | 72 | 73 | 8:1 |
97.5:2.5 |
|
(E)-PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH (3oa) CH (3oa) |
60 | 77 | 8:1 |
97:3 |
|
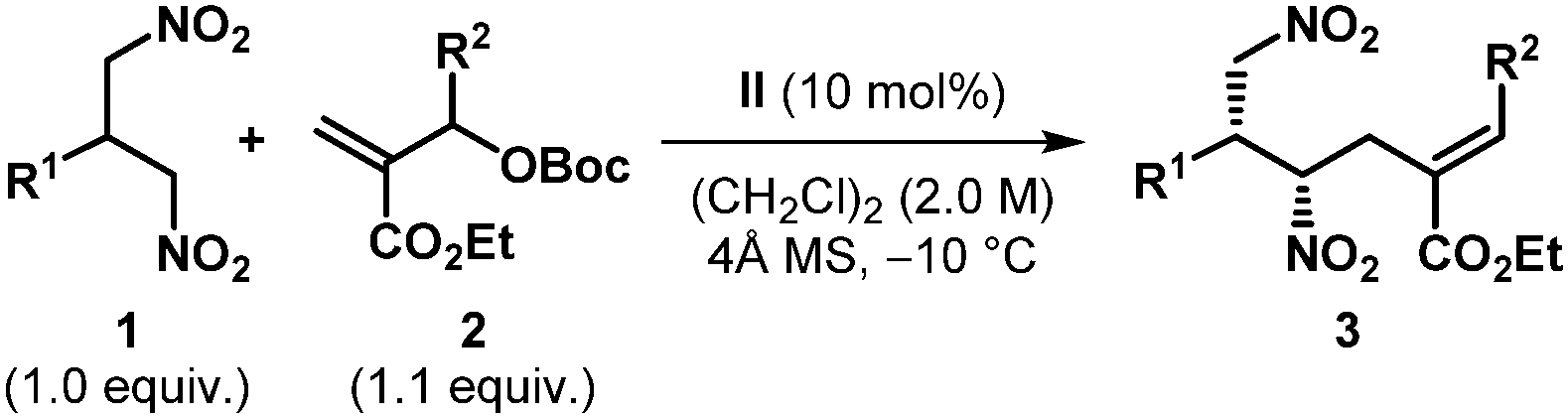
|
|
|---|
(B)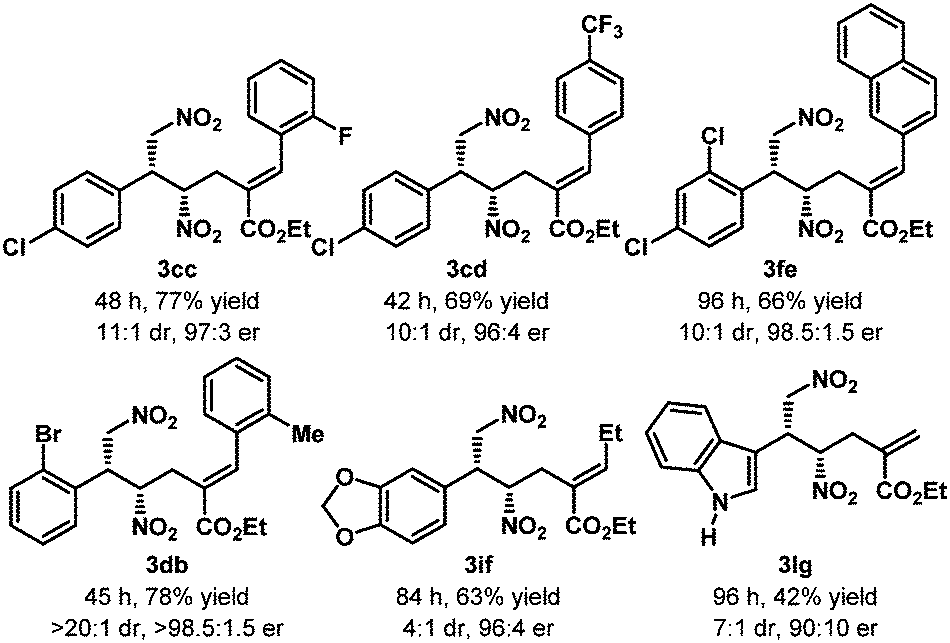 |
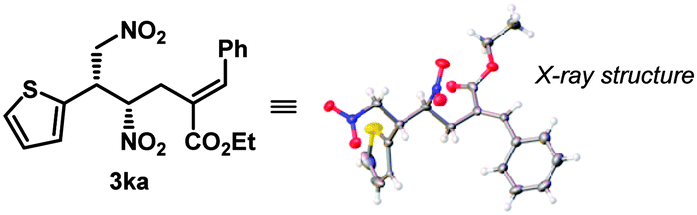
The other product antipode could be accessed simply by using the pseudoenantiomeric cinchonine-derived catalyst VII (see Table 1) and under our standard reaction conditions, ent-3aa was obtained with outstanding enantioselectivity (>99:1 er), albeit with marginally reduced dr in 61% yield (Scheme 2).
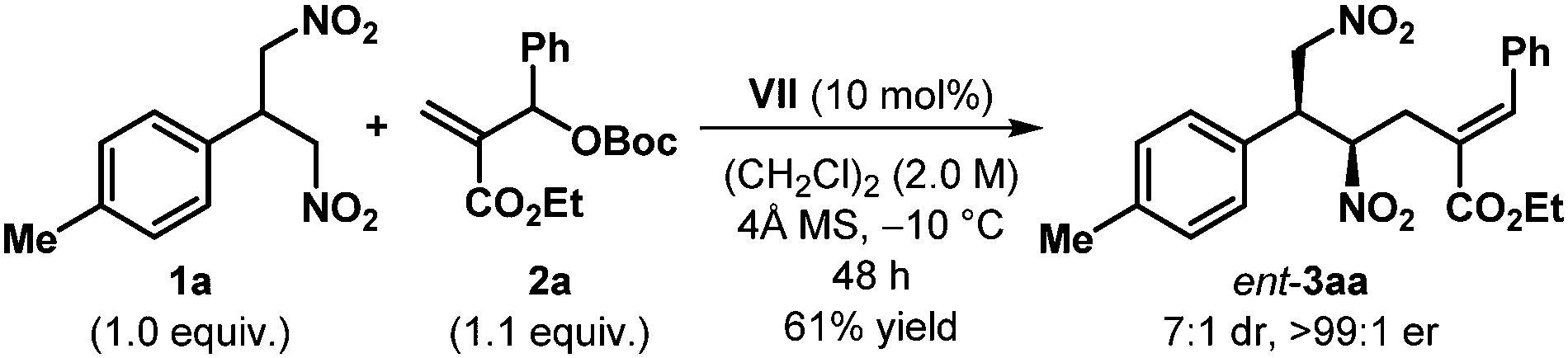 | ||
| Scheme 2 Desymmetrization using the pseudoenantiomeric catalyst VII. | ||
As the products are densely functionalized and contain functional groups in their highest oxidation states, subjecting them to reductive conditions can generate structurally and stereochemically diverse motifs, as demonstrated in Scheme 3. Treatment of 3ca with Zn–AcOH led to the complete reduction of both the nitro groups; the reaction with the proximal ester moiety then followed to generate, after Boc protection, the pyrrolidinone derivative 4 with an exocyclic double bond in good yield. Reduction of the ester with DIBAL-H afforded the corresponding alcohol 5, which upon exposure to Al(Hg), produced the dihydropyrazole derivative 6, as evident from its X-ray structure; this further confirmed the stereochemistry of the product.
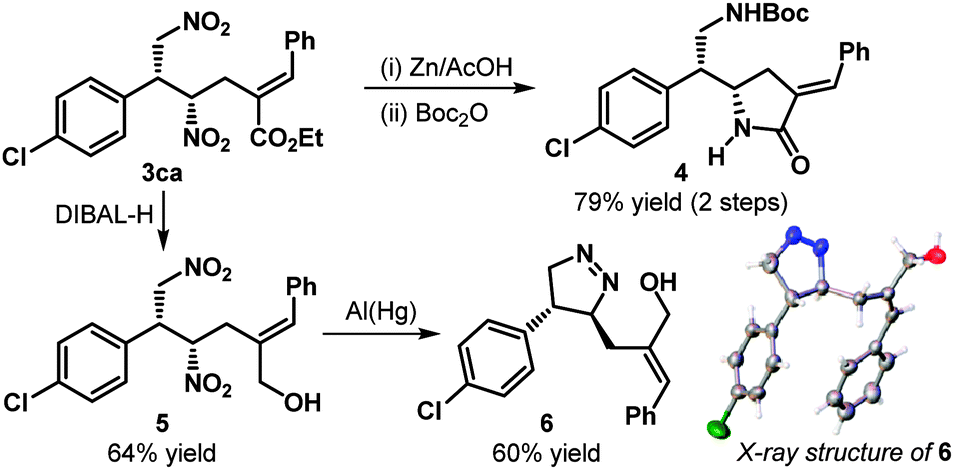 | ||
| Scheme 3 Synthetic transformations involving 3ca. | ||
In summary, we have developed a catalytic enantioselective desymmetrization of prochiral 1,3-dinitropropanes, which proceeds via an enantiogroup differentiating allylic alkylation reaction. Functionalized allylic carbonates were used as the allyl source and the densely functionalized products consisting of two vicinal stereocenters were obtained through acyclic stereocontrol generally with good to excellent diastereoselectivity (up to >20:1 dr) and superb enantioselectivity (up to >99:1 er). The utility of the products was demonstrated through a few synthetic manipulations and should find broader application in synthesis and beyond.
This research program is funded by DST and CSIR. S.J.S.R. thanks CSIR for a doctoral fellowship. Thanks are also due to Mr Dipak Samanta, Mr Bijan Roy and Mr Vijith Shetty for their help with the X-ray crystallographic analysis.
Notes and references
- H. M. L. Davies and E. J. Sorensen, Chem. Soc. Rev., 2009, 38, 2981 RSC.
- For selected reviews, see: (a) E. García-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2011, 111, PR110 CrossRef PubMed; (b) T. Rovis, in New Frontiers in Asymmetric Catalysis, ed. K. Mikami and M. Lautens, Wiley, Hoboken, NJ, 2007, p. 275 Search PubMed; (c) M. C. Willis, J. Chem. Soc., Perkin Trans. 1, 1999, 1765 RSC.
- (a) L. S. Aitken, N. R. Arezki, A. Dell'Isola and A. J. A. Cobb, Synthesis, 2013, 2627 Search PubMed; (b) N. Ono, in The Nitro Group in Organic Synthesis, Wiley-VCH, Weinheim, 2001 CrossRef.
- R. Ballini, G. Bosica, D. Fiorini and A. Palmieri, Synthesis, 2004, 1938 CrossRef CAS PubMed.
- For non-asymmetric reactions involving 2-substituted 1,3-dinitropropanes, see: (a) L. Barboni, S. Gabrielli, A. Palmieri, C. Femoni and R. Ballini, Chem.–Eur. J., 2009, 15, 7867 CrossRef CAS PubMed; (b) D. Y. Park, K. Y. Lee, S. Gowrisankar and J. N. Kim, Bull. Korean Chem. Soc., 2008, 29, 701 CrossRef CAS; (c) D. Y. Park, K. Y. Lee and J. N. Kim, Tetrahedron Lett., 2007, 48, 1633 CrossRef CAS PubMed; (d) R. Ballini, L. Barboni, D. Fiorini, G. Giarlo and A. Palmieri, Chem. Commun., 2005, 2633 RSC.
- E. Reyes, H. Jiang, A. Milelli, P. Elsner, R. G. Hazell and K. A. Jørgensen, Angew. Chem., Int. Ed., 2007, 46, 9202 CrossRef CAS PubMed.
- (a) B. M. Trost and J.-P. Surivet, Angew. Chem., Int. Ed., 2000, 39, 3122 CrossRef CAS; (b) For a review, see: B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed.
- (a) X.-F. Yang, W.-H. Yu, C.-H. Ding, Q.-P. Ding, S.-L. Wan, X.-L. Hou, L.-X. Dai and P.-J. Wang, J. Org. Chem., 2013, 78, 6503 CrossRef CAS PubMed; (b) X.-F. Yang, C.-H. Ding, X.-H. Li, J.-Q. Huang, X.-L. Hou, L.-X. Dai and P.-J. Wang, J. Org. Chem., 2012, 77, 8980 CrossRef CAS PubMed.
- (a) G.-Y. Chen, F. Zhong and Y. Lu, Org. Lett., 2012, 14, 3955 CrossRef CAS PubMed; (b) G.-Y. Chen, F. Zhong and Y. Lu, Org. Lett., 2011, 13, 6070 CrossRef CAS PubMed . For non-asymmetric versions of this reaction, see: ; (c) D. Basavaiah and J. S. Rao, Tetrahedron Lett., 2004, 45, 1621 CrossRef CAS PubMed; (d) J. M. Kim, Y. J. Im, T. H. Kim and J. N. Kim, Bull. Korean Chem. Soc., 2002, 23, 657 CrossRef CAS . Also see: ; (e) S. W. Smith and G. C. Fu, J. Am. Chem. Soc., 2009, 131, 14231 CrossRef CAS PubMed.
- D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447 CrossRef CAS PubMed.
- For selected examples of allylic alkylations, see: (a) X. Companyó, A. Mazzanti, A. Moyano, A. Janecka and R. Rios, Chem. Commun., 2013, 49, 1184 RSC; (b) F. Zhong, J. Luo, G.-Y. Chen, X. Dou and Y. Lu, J. Am. Chem. Soc., 2012, 134, 10222 CrossRef CAS PubMed; (c) T. Furukawa, J. Kawazoe, W. Zhang, T. Nishimine, E. Tokunaga, T. Matsumoto, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2011, 50, 9684 CrossRef CAS PubMed; (d) H.-L. Cui, J.-R. Huang, J. Lei, Z.-F. Wang, S. Chen, L. Wu and Y.-C. Chen, Org. Lett., 2010, 12, 720 CrossRef CAS PubMed.
- For selected examples of annulations, see: (a) Q.-L. Wang, L. Peng, F.-Y. Wang, M.-L. Zhang, L.-N. Jia, F. Tian, X.-Y. Xu and L.-X. Wang, Chem. Commun., 2013, 49, 9422 RSC; (b) F. Zhong, G.-Y. Chen, X. Han, W. Yao and Y. Lu, Org. Lett., 2012, 14, 3764 CrossRef CAS PubMed; (c) F. Zhong, X. Han, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 7837 CrossRef CAS PubMed.
- For a recent review, see: T.-Y. Liu, M. Xie and Y.-C. Chen, Chem. Soc. Rev., 2012, 41, 4101 RSC.
- (a) L.-Q. Lu, X.-L. An, J.-R. Chen and W.-J. Xiao, Synlett, 2012, 490 Search PubMed; (b) J. Alemán, A. Parra, H. Jiang and K. A. Jørgensen, Chem.–Eur. J., 2011, 17, 6890 CrossRef PubMed; (c) W.-Y. Siau and J. Wang, Catal. Sci. Technol., 2011, 1, 1298 RSC; (d) Y. Takemoto, Chem. Pharm. Bull., 2010, 58, 593 CrossRef CAS; (e) S. J. Connon, Chem. Commun., 2008, 2499 RSC.
- See the ESI† for a more comprehensive optimization of catalyst and reaction conditions.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRef CAS PubMed.
- H.-L. Cui, X. Feng, J. Peng, J. Lei, K. Jiang and Y.-C. Chen, Angew. Chem., Int. Ed., 2009, 48, 5737 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all relevant compounds together with HPLC traces for all the products. CCDC 963860 (3ka) and 963861 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc47645f |
| This journal is © The Royal Society of Chemistry 2014 |