 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovering the pivotal role of carbonate in the formation of a bis-phenolate supported Co15 cluster†
Ross
McLellan
a,
Julien
Rezé
a,
Stephanie M.
Taylor
b,
Ruaraidh D.
McIntosh
*a,
Euan K.
Brechin
*b and
Scott J.
Dalgarno
*a
aInstitute of Chemical Sciences, Heriot – Watt University, Riccarton, Edinburgh, EH14 4AS, Scotland, UK. E-mail: R.McIntosh@hw.ac.uk; S.J.Dalgarno@hw.ac.uk; Fax: +44 (0)131 451 3180; Tel: +44 (0)131 451 8025
bEaStCHEM School of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, Scotland, UK. E-mail: ebrechin@staffmail.ed.ac.uk; Fax: +44 (0)131 650 6453; Tel: +44 (0)131 650 7545
First published on 16th January 2014
Abstract
Unexpected panelling by μ6-CO32− anions has been observed to support the formation of a new and unusual Co15 cluster. The arrangement is further stabilised by bis-phenolate ligands which assemble in a manner akin to tetrahomodioxacalix[4]arene. The reaction proceeds in very low yield as μ6-CO32− anions are formed in situ, but targeted synthesis by addition of carbonate improves synthetic yield and highlights the pivotal role of this anion in the overall assembly process.
Methylene-bridged calix[n]arenes have recently emerged as excellent platforms for the synthesis of new polynuclear metal clusters. We have investigated calix[4]arenes (C[4]s, Fig. 1A) thoroughly for this purpose with transition and lanthanide metal centres (TM and Ln respectively).1–9 In the course of these studies we discovered a range of novel cluster motifs including: (a) [MnIII2MnII2(TBC[4])2] Single-Molecule Magnets (SMMs);2,3 (b) [CuII9(TBC[4])3] clusters4 that are versatile anion binding materials (Fig. 1B); (c) [MnIII4LnIII4(C[4])4] clusters that are magnetic refrigerants or SMMs depending on the lanthanide employed;6,7 (d) [FeIII2LnIII2(TBC[4])2] clusters8 and (e) octahedral [LnIII6(TBC[4])2] clusters.9 We have been working towards a set of construction rules for C[4], and analysis of the coordination chemistry we have encountered thus far reveals that the lower-rim polyphenolic pocket preferentially binds TMIII ions for Mn or Fe, and TMII ions for Co or Cu.
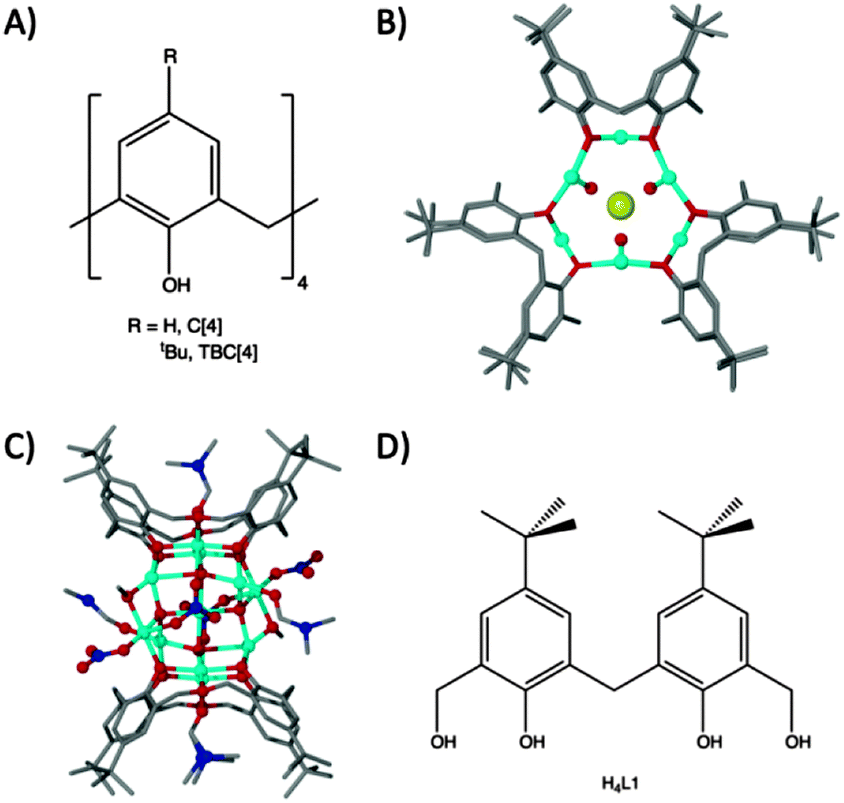 | ||
| Fig. 1 (A) General calix[4]arene framework. (B) Tri-capped trigonal prismatic [CuII9(TBC[4])3]Cl2 cluster, viewed down the face of the prism.4 (C) Cu11 cluster formed with tetrahomodioxacalix[4]arene.12 (D) Schematic of H4L1. | ||
We recently turned our attention to the oxacalixarenes which contain additional donor atoms in the form of ethereal bridges in place of the C[4] methylene links.10 This deviation from C[4]-supported cluster formation was undertaken with a view to exploring the effect that additional donor oxygen/s would have on cluster formation. We investigated their reaction with TMII salts under ambient conditions and (in this exploratory chemistry) found that reaction of homooxacalix[3]arene with MnII chloride in the presence of base affords a Mn10 supertetrahedron that has an unusual oxidation state distribution.11 We also found that reaction of two different oxacalix[4]arenes with CuII nitrate afforded Cu2, Cu4 and Cu11 clusters depending on the number of ethereal bridges included in the support (Cu2/Cu4 formed with one oxa-bridge present, and Cu11 with two), and on the order of reaction between the components employed (see Fig. 1C for Cu11); the Cu4 represents an ‘extended’ Cu2 cluster and forms by reacting a dmf solution of CuII nitrate with a MeOH solution of homooxacalix[3]arene, while the Cu2 cluster results from a one-pot reaction utilising both MeOH and dmf simultaneously.12
The molecule 6,6′-methylenebis(4-(tert-butyl)-2-(hydroxy-methyl)-phenol), H4L1 (Fig. 1D), is an intermediate employed in the synthesis of a number of oxacalixarenes.‡ To our surprise a search of the Cambridge Structural Database for metal complexes of H4L1 (in varying degrees of protonation) returned a small number of hits.13 As a consequence of this we began to explore this molecule as a support in cluster formation given that it (a) has calixarene character in the form of a methylene bridge between two p-tBu-phenols, giving the molecule distinct curvature, and (b) possesses two distinct types of OH group (aliphatic and phenolic) differing markedly in acidity. In our first report with H4L1 we detailed formation of linear lanthanide trimers supported by four ligands that displayed versatile degrees of deprotonation; there are eight phenols and eight methanol arms present which deprotonate appropriately to support a +9 charge from three LnIII centres.17 Here we show that a cluster of fifteen CoII centres, supported by L1 in differing degrees of deprotonation, is created concomitantly with the in situ formation of carbonate anions. Synthetic, structural and magnetic properties of this new assembly are presented. In addition we also discuss the pivotal role of carbonate anions in the assembly process.
Dissolution of H4L1 in dmf prior to mixing with a methanolic solution of CoII nitrate in the presence of base yielded a small quantity of red crystals upon prolonged standing and exposure to air; these were found to be of formula [CoII15(H1.33L1)6(μ6-CO3)6(μ3-OMe)2(μ-dmf)3(dmf)5(MeOH)0.5(H2O)2.5](MeOH)(dmf)5 (1, Fig. 2A). The asymmetric unit comprises the large central core of fifteen cobalt atoms which is supported by six μ6-CO32− anions in an approximate cubic arrangement (Fig. 2B). The cluster is further supported by six L1 ligands, varying in degrees of deprotonation, and two μ3-OMe− anions (Fig. 2). Additional coordination sites are occupied by ligated MeOH and dmf.
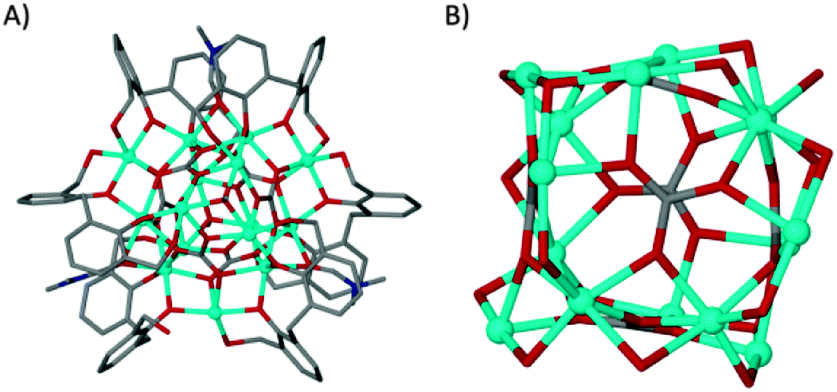 | ||
| Fig. 2 (A) Part of the asymmetric unit in the structure of 1. (B) Polymetallic skeleton in 1 showing the arrangement of the μ6-CO32− anions. | ||
The cobalt centres in 1 are all in the second oxidation state, evidenced by the Co–O bond lengths which all lie close to the typical range for Co(II)–O (2.1–2.2 Å, as opposed to 1.9–2.0 Å for Co(III)–O). They do not form a recognisable regular polyhedron (Fig. 2B) and as such the small unoccupied void within the cluster takes an unusual shape (Fig. S1, ESI†). The irregular cluster of Co atoms can be envisaged as a loosely equatorial enneanuclear belt, bi-capped by triangular clusters at each pole. The Co–Co distances are not uniform, resulting in the irregular nature of the polyhedron. One of the most striking features of the cluster is the face-to-face pairing of L1 ligands, which subsequently mimic C[4]s. Such structures are referred to as hemi-calixarenes and are implicated in the complicated, poorly defined chemistry which leads to the formation of C[n]s from oligomeric poly-phenols.18 The formation of calixarenes by joining linear units is postulated to be dependent on H-bonding, but this is unlikely to be responsible for the aforementioned arrangement of ligands in 1. Interestingly we observe that each L1 in the hemicalixarene is bound to a single metal and those metals are held together by the most strongly coordinating solvent present, dmf (Fig. 3A). We find a near identical coordination mode for tetrahomodioxacalix[4]arene (THDOC[4]) where two metal atoms bind within the cavity and are bridged by a coordinated dmf (Fig. 3B). The apparent preference to bind two metals is in contrast to that found for C4s, which reliably bind a single metal centre. A relationship clearly exists between H4L1 and THDOC[4] whereby these additional donor atoms dictate metal binding.
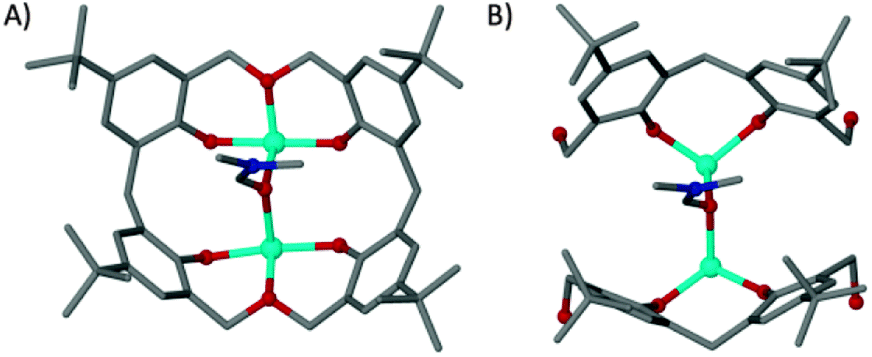 | ||
| Fig. 3 Binding pockets of tetrahomodioxacalix[4]arene (A)12 and a pair of L1 molecules (B). | ||
Each of the phenolic alcohols are deprotonated, as we might expect as these are the most acidic. The methanolic moieties are not completely deprotonated and therefore we see a mixture of alkoxides and OH lone pair donation. There is disorder present between coordinated solvent molecules and the aliphatic alcohols of L1, which is indicative of L-type ligand behaviour. This disorder goes hand in hand with the presence of proximal solvents of crystallisation (also disordered), which are located at positions consistent with hydrogen bonding interactions. Unfortunately disorder in the structure renders it impossible to unambiguously assign the location of all remaining protons.
The in situ formation of carbonate is not unprecedented and has previously been found to generate unexpected cluster geometries.19 Others have attributed formation of carbonate to arise from reactions including decomposition of dmf or TM–ligand complexes. The ambient conditions of our reaction, coupled with the limited carbonate formation suggested in situ generation via incorporation of atmospheric CO2 in the reaction mixture; the low concentration of dissolved CO2 at ambient pressure would thus be restricting the formation of carbonate, and is consistent with the extremely low yield observed. Enhancement of product yield was logically invoked through addition of sodium bicarbonate to the reaction mixture; the yield of 1 was subsequently raised from ∼2% to ∼10% by carbonate addition, demonstrating the pivotal role this anion plays in promoting formation of this new cluster motif.
Direct current magnetic susceptibility studies were performed on a polycrystalline sample of 1 in the 5–300 K range in an applied field of 0.1 T. The results are plotted as the χMT product versus T (where χM is the molar magnetic susceptibility and T the temperature) in Fig. S2 (ESI†). At 300 K the χMT value of ∼40 cm3 K mol−1 is well above the spin-only (g = 2.0) value of ∼28 cm3 K mol−1 expected for fifteen non-interacting high-spin CoII (3d7; S = 3/2) ions, reflecting the combined effects of spin–orbit coupling and the distortion of the octahedral crystal field, resulting in deviation from standard Curie law behaviour. The value of χMT decreases constantly with decreasing temperature reaching a maximum value of ∼11 cm3 K mol−1 at 5 K. This behaviour is therefore indicative of the presence of the magnetic anisotropy of the CoII ions and antiferromagnetic exchange between nearest neighbours, likely resulting in a small spin ground state. This is corroborated by magnetisation data taken in the 2–7 K and 0.5–7 T temperature and field ranges, which sees a near linear increase in magnetisation with increasing magnetic field strength, indicative of field-induced stabilisation of excited states with larger magnetic moments.
The bis-phenolate (H4L1) used for cluster formation in this study possesses significantly more flexibility with respect to coordination chemistry than the related methylene-bridged C[4]s. This increased flexibility is expected for the acyclic ligand, however this is coupled with the varied degrees of deprotonation which the ligand is found to exist in (C4s, unless protected, fully deprotonate). Despite the acyclic nature of the ligand we observe concerted coordination behaviour to mimic THDOC[4]. The Co15 cage that resulted from our exploratory cluster forming chemistry incorporated μ6-CO32− ions that were formed in situ. This process afforded very low yields of the cage, but targeted incorporation of CO32− into the reaction mixture caused a dramatic improvement. Anions are thus able to play a key role in cluster formation with this ligand, and the potential to have two sites of varied basicity opens up new avenues of coordination chemistry. Future work to understand assembly preferences for H4L1 is underway.
We thank the EPSRC for financial support of this work.
Notes and references
- C. Aronica, G. Chastanet, E. Zueva, S. A. Borshch, J. M. Clemente-Juan and D. Luneau, J. Am. Chem. Soc., 2008, 130, 2365 CrossRef CAS PubMed.
- G. Karotsis, S. J. Teat, W. Wernsdorfer, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Angew. Chem., Int. Ed., 2009, 48, 8285 CrossRef CAS PubMed.
- S. M. Taylor, G. Karotsis, R. D. McIntosh, S. Kennedy, S. J. Teat, C. M. Beavers, W. Wernsdorfer, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Chem.–Eur. J., 2011, 17, 7521 CrossRef CAS PubMed.
- G. Karotsis, S. Kennedy, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2010, 46, 3884 RSC.
- S. M. Taylor, R. D. McIntosh, C. M. Beavers, S. J. Teat, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2011, 47, 1440 RSC.
- G. Karotsis, M. Evangelisti, S. J. Dalgarno and E. K. Brechin, Angew. Chem., Int. Ed., 2009, 48, 9928 CrossRef CAS PubMed.
- G. Karotsis, S. Kenndy, S. J. Teat, C. M. Beavers, D. A. Fowler, J. J. Morales, M. Evangelisti, S. J. Dalgarno and E. K. Brechin, J. Am. Chem. Soc., 2010, 132, 12983 CrossRef CAS PubMed.
- S. Sanz, K. Ferreira, R. D. McIntosh, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2011, 47, 9042 RSC.
- S. Sanz, R. D. McIntosh, C. M. Beavers, S. J. Teat, M. Evangelisti, E. K. Brechin and S. J. Dalgarno, Chem. Commun., 2012, 48, 1449 RSC.
- Thiacalixarenes have also been used for this purpose, although they display different coordination chemistry. For example see: C. Desroches, G. Pilet, S. A. Borshch, S. Parola and D. Luneau, Inorg. Chem., 2005, 44, 9112 CrossRef CAS PubMed; T. Kajiwara, N. Iki and M. Yamashita, Coord. Chem. Rev., 2007, 251, 1734 CrossRef PubMed; Y. Bi, X.-T. Wang, W. Liao, X. Wang, X. Wang, H. Zhang and S. Gao, J. Am. Chem. Soc., 2009, 131, 11650 CrossRef PubMed; M. Liu, W. Liao, C. Hu, S. Du and H. Zhang, Angew. Chem., Int. Ed., 2012, 51, 1585 CrossRef PubMed.
- S. M. Taylor, R. D. McIntosh, J. Rezé, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2012, 48, 9263 RSC.
- R. E. Fairbairn, R. McLellan, R. D. McIntosh, M. A. Palacios, E. K. Brechin and S. J. Dalgarno, Dalton Trans. 10.1039/C3DT53088D.
- There are four entries for metal complexes of TBHOC[4] in the Cambridge Structural Database for complexes with U,14 Cs15 and Eu16.
- J. M. Harrowfield, M. I. Ogden and A. H. White, J. Chem. Soc., Dalton Trans., 1991, 979 RSC; B. Masci and P. Thuéry, Acta Crystallogr., Sect. E, 2007, 63, m59 Search PubMed.
- R. Assmus, V. Böhmer, J. M. Harrowfield, M. I. Ogden, W. R. Richmond, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1993, 2427 RSC.
- Z. Asfari, J. M. Harrowfield, M. I. Ogden, J. Vicens and A. H. White, Angew. Chem., Int. Ed. Engl., 1991, 30, 854 CrossRef.
- R. McLellan, M. A. Palacios, C. M. Beevers, S. J. Teat, E. K. Brechin and S. J. Dalgarno, Chem. Commun., 2013, 49, 9552 RSC.
- C. D. Gutsche, Calixarenes, Royal Society of Chemistry, 1989, ch. 2 and references therein Search PubMed.
- L. Wang, Y. Li, Y. Peng, Z. Liang, J. Yu and R. Xu, Dalton Trans., 2012, 41, 6242 RSC; K.-C. Xiong, F.-L. Jiang, Y.-L. Gai, D.-Q. Yuan, D. Han, J. Ma, S.-Q. Zhang and M.-C. Hong, Chem.–Eur. J., 2012, 18, 5536 CrossRef CAS PubMed.
- B. Masci, Calixarenes 2001, Kluwer Academic Press, Dordrecht, 2001, ch. 12 and references therein Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 977620 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc00167b |
‡ H4L1 was prepared according to literature procedure.20 Synthesis of 1: Co(NO3)2·6H2O (1.17 g, 4.00 mmol) and H4L1 (0.5 g, 1.35 mmol) were dissolved in a mixture of dmf (12 cm3) and MeOH (12 cm3) and stirred for 5 min and NEt3 (0.73 g, 7.2 mmol) was added. The deep red solution was stirred for an additional hour and filtered to remove traces of precipitate. Single crystals suitable for X-ray diffraction analysis were obtained by slow evaporation over several weeks in ca. 2% yield (based on H4L1). Crystallisation of 1 could be expedited by Et2O vapour diffusion into the mother liquor. Elemental analysis (%) calculated for dried crystalline 1, C196.5H284O61N13Co15: C, 49.03%; H, 6.27%; N, 3.99%. Found: C, 49.12%; H, 5.97%; N, 4.27%. Improved synthesis of 1: an analogous procedure also using sodium bicarbonate (0.6 g, 7.14 mmol) in the reaction mixture increased the yield of 1 to ca. 10%. Crystal data (CCDC 977620): C196.5H284O61N13Co15, M = 4688.47, red block, 0.40 × 0.30 × 0.25 mm3, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 20.8311(10), b = 22.9871(11), c = 29.1866(13) Å, α = 85.462(2), β = 71.615(2), γ = 64.348(2)°, V = 11928.4(10) Å3, Z = 2, 2θmax = 45.4°, 114 , a = 20.8311(10), b = 22.9871(11), c = 29.1866(13) Å, α = 85.462(2), β = 71.615(2), γ = 64.348(2)°, V = 11928.4(10) Å3, Z = 2, 2θmax = 45.4°, 114![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 209 reflections collected, 31699 unique (Rint = 0.0480). Final GooF = 1.494, R1 = 0.0895, wR2 = 0.2523, R indices based on 18732 reflections with I > 2σ(I) (refinement on F2). 209 reflections collected, 31699 unique (Rint = 0.0480). Final GooF = 1.494, R1 = 0.0895, wR2 = 0.2523, R indices based on 18732 reflections with I > 2σ(I) (refinement on F2). |
| This journal is © The Royal Society of Chemistry 2014 |