 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic azides: “energetic reagents” for the intermolecular amination of C–H bonds
Daniela
Intrieri
,
Paolo
Zardi
,
Alessandro
Caselli
and
Emma
Gallo
*
Università degli Studi di Milano - Chemistry Department, Via Golgi 19, 20136 Milano, Italy. E-mail: emma.gallo@unimi.it; Fax: +39 (0)2 50314405; Tel: +39 (0)2 50314374
First published on 27th May 2014
Abstract
This feature article provides an overview of the application of organic azides for the intermolecular amination of sp3 and sp2 C–H bonds. The catalytic activity of several metal complexes was reviewed underlining both synthetic and mechanistic aspects of the C–H amination. The majority of the aminated compounds reported in literature have been collected in this paper to provide a compendium of published procedures. In addition, the discussion of involved mechanisms has been included to assist the reader to envisage the future potential of organic azides in the synthesis of aza-derivatives.
 Daniela Intrieri | Daniela Intrieri was born in Italy in 1984, she obtained her master's degree in Chemistry in 2010 under the supervision of Prof. Sergio Cenini at the Chemistry Department of the University of Milano (Italy). In 2014 she got her PhD in Chemical Science at the University of Milano under the supervision of Prof. Emma Gallo. Her PhD thesis was on the synthesis of cobalt, ruthenium and iron porphyrins to be employed as homogeneous catalysts in C–C and C–N bond formation as well as on the study of reaction mechanisms. |
 Paolo Zardi | Paolo Zardi was born in Italy in 1987, he received his master's degree in Chemical Sciences in 2011 with an experimental thesis concerning the heterogenisation of copper complexes to use as catalysts for asymmetric cyclopropanation under the supervision of Dr Caselli in the Chemistry Department of the University of Milan. Currently he is a PhD Student at Milan University in Prof. Emma Gallo's group and his research is focused on the synthesis of ruthenium–porphyrin complexes and their employment as catalysts in C–N bond formation reactions. |
 Alessandro Caselli | Alessandro Caselli received his PhD under the supervision of Prof. Carlo Floriani at the University of Lausanne (CH) in 2000. He spent two years as post-doc at the University of Milano (Italy) under the supervision of Prof. Fulvia Orsini before joining Prof. Sergio Cenini's group. In 2003, he became assistant professor at the Chemistry Department of Milano University. His research has focused on: (i) the employment of metal complexes with macrocyclic N-donor ligands as homogeneous catalysts; (ii) the synthesis of asymmetric ligands; (iii) the mechanistic aspects of catalytic reactions and (iv) the heterogenisation of chiral catalysts. |
 Emma Gallo | Emma Gallo received her PhD under Prof. Carlo Floriani's guidance at the University of Lausanne (CH) in 1995. She spent one year in Floriani's Group as “Maitre assistant” before moving to Italy. After post-doctoral training alongside Prof. Sergio Cenini at the Chemistry Department of University of Milano (Italy), she became assistant professor and then associate professor at the same university. Her research is mainly devoted to the synthesis of fine chemicals by C–C or C–N bond formation, using metal porphyrin complexes as catalysts, as well as the study of catalytic reaction mechanisms. |
1. Introduction
Since the first preparation of organic azides (RN3) in 1864,1 this class of molecules has been involved in an increasing number of applications over the last 15 years, as documented by SciFinder (Fig. 1). Among all possible applications, organic azides are largely used as atom-efficient aminating agents due to the formation of eco-friendly molecular nitrogen as the only side product of the “NR” transfer reaction to an organic molecule.2,3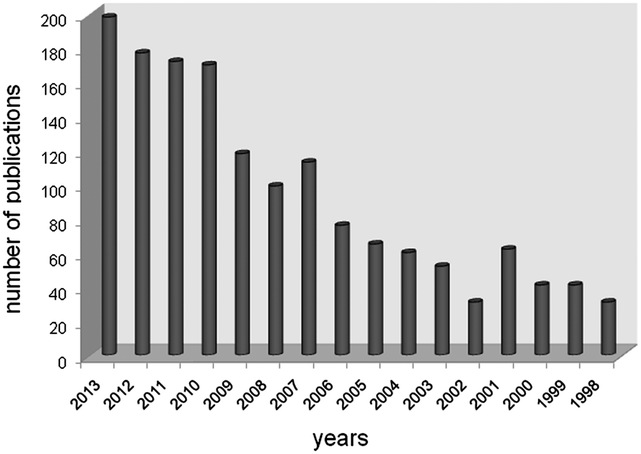 | ||
| Fig. 1 Manuscripts and patents on organic azides reported by SciFinder in the 1998–2013 period. | ||
The insertion of a nitrene moiety into an organic framework is efficiently catalysed by transition metal complexes and allows the synthesis of aza-compounds which often display important biological and pharmaceutical characteristics.4 As azides are active towards several classes of organic molecules, the amination reaction displays a great chemical versatility and it can be conducted using mild experimental conditions and no oxidant agent is required.
Considering the scientific interest to establish efficient methodologies to synthesise nitrogen containing molecules, several reviews on the use of organic azides as nitrogen sources have been published5–11 and great emphasis has been devoted to the amination of C–H bonds due to their ubiquity in almost all organic skeletons. Furthermore, the insertion of an aza-moiety into a hydrocarbon C–H bond allows the conversion of low cost reagents into high-added value aminated compounds.
This feature article provides an overview of the intermolecular C–H bond aminations by organic azides reported in Scheme 1, by underlining both synthetic and mechanistic aspects. The intramolecular amination was amply reviewed6,12–15 and it is not covered in this article.
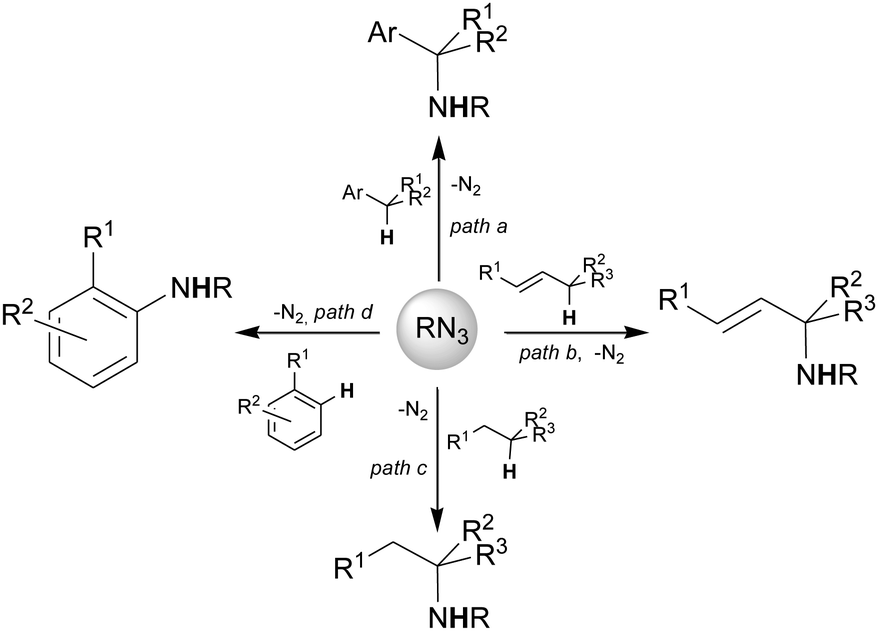 | ||
| Scheme 1 Intermolecular C–H aminations by organic azides discussed in this manuscript. | ||
We sincerely apologise if some important contributions to this topic have been unintentionally omitted.
2. Amination of activated sp3 C–H bonds
The insertion of a nitrene functionality into an activated C–H bond was initially studied by taking advantage of the thermal and photochemical instability of organic azides. Reactions were conducted without the assistance of a transition metal complex and consequentially drastic experimental conditions were required.16–19Later, a transition metal was employed to promote the cleavage of the Nα–Nβ bond (Scheme 2) and selectively insert the nitrene functionality into a specific position of the organic backbone.
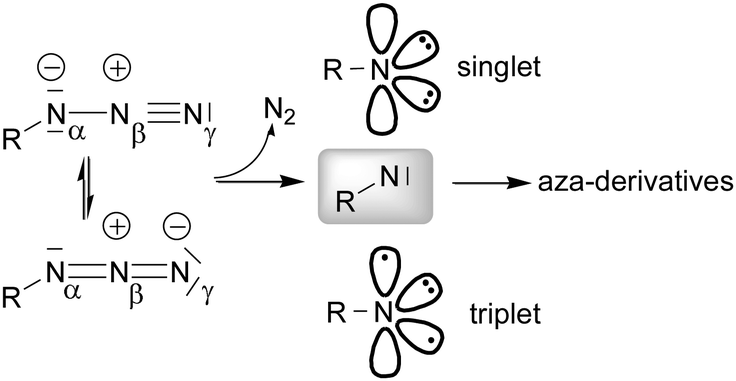 | ||
| Scheme 2 Nitrene formation by the cleavage of an azide Nα–Nβ bond. | ||
In 1967 Kwart and Khan20 reported on the catalytic activity of copper powder in the amination of cyclohexene and since then several transition metal catalysts have been developed to promote the amination of activated sp3 C–H bonds. The most representative classes of catalysts are reported in Fig. 2.
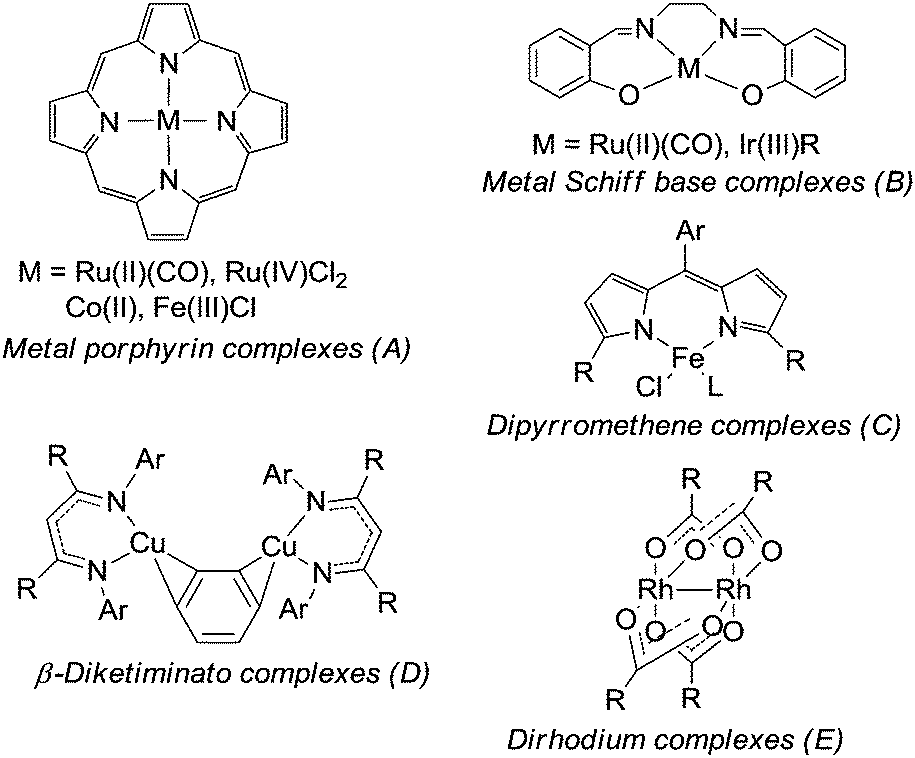 | ||
| Fig. 2 Catalysts for the amination of activated sp3 C–H bonds. | ||
2.1. Amination catalysed by metal porphyrin complexes (A)
Cenini et al. reported in 2000 that cobalt(II) porphyrin complexes are active catalysts that promote the amination of both allylic and benzylic C–H bonds by using aryl azides as nitrene sources.21–24 Aryl azides were chosen due to their stability–reactivity relationship that allows their use without any particular experimental caution.25 The benzylic amines were obtained together with the corresponding imines when the initially formed amine can be further oxidised by another ArN3 molecule. The stoichiometric by-product of this second step is the primary amine of the azide employed (Scheme 3).
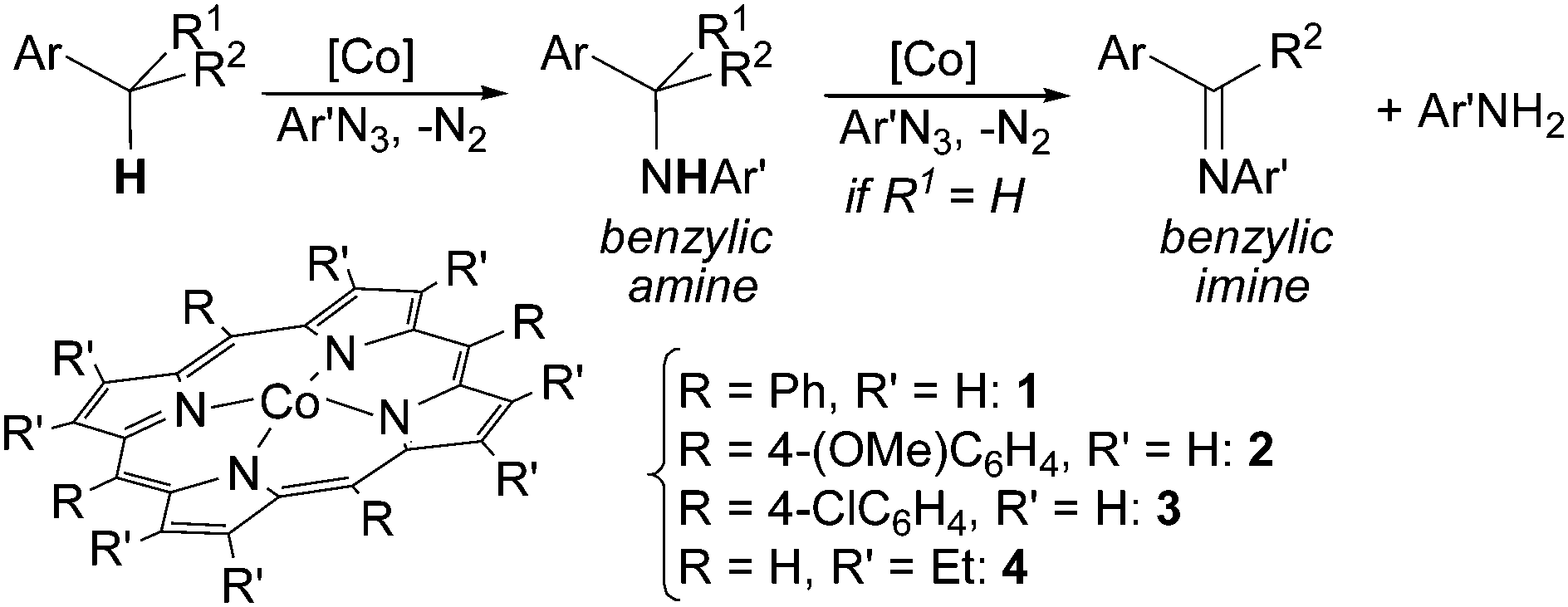 | ||
| Scheme 3 Synthesis of benzylic amines and imines catalysed by Co(II) porphyrin complexes. | ||
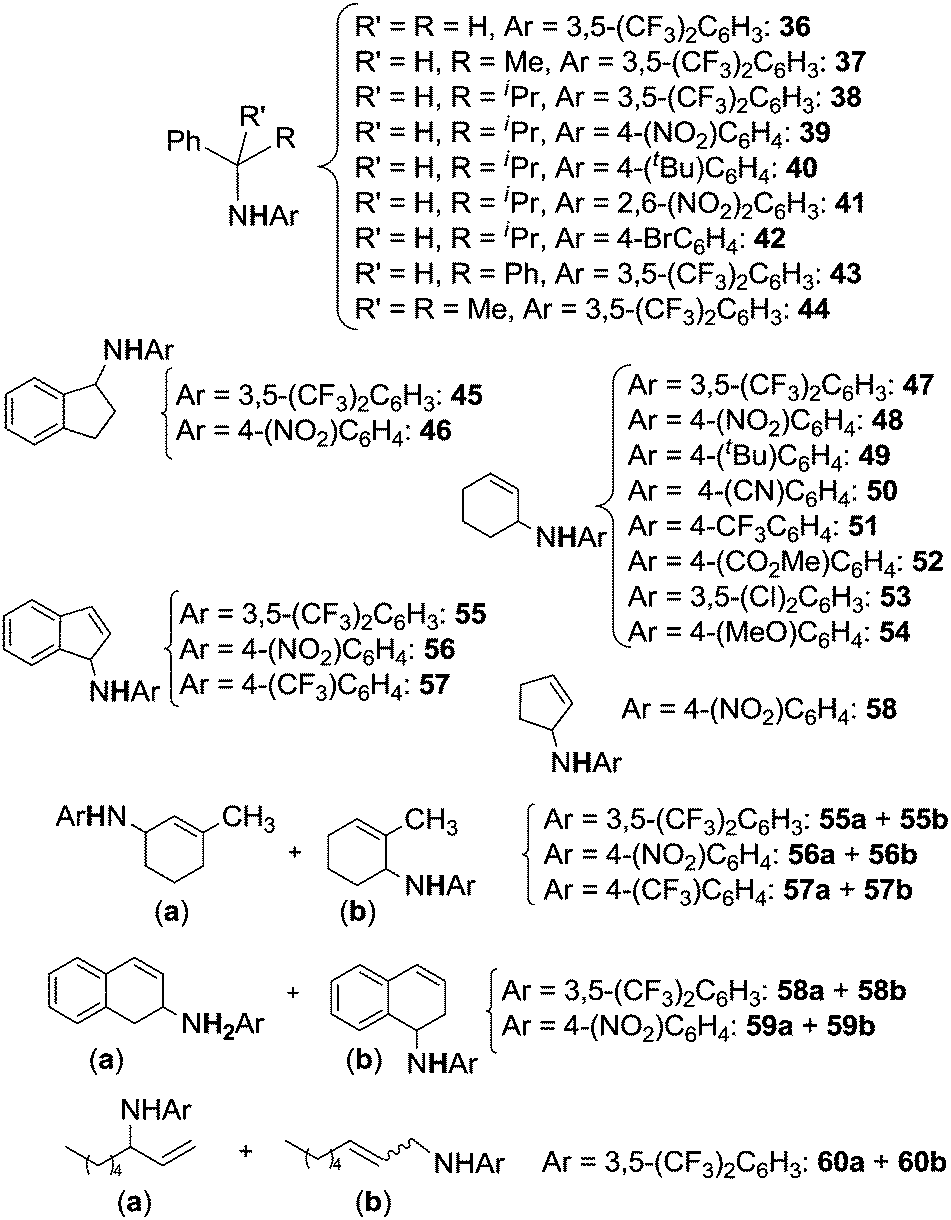
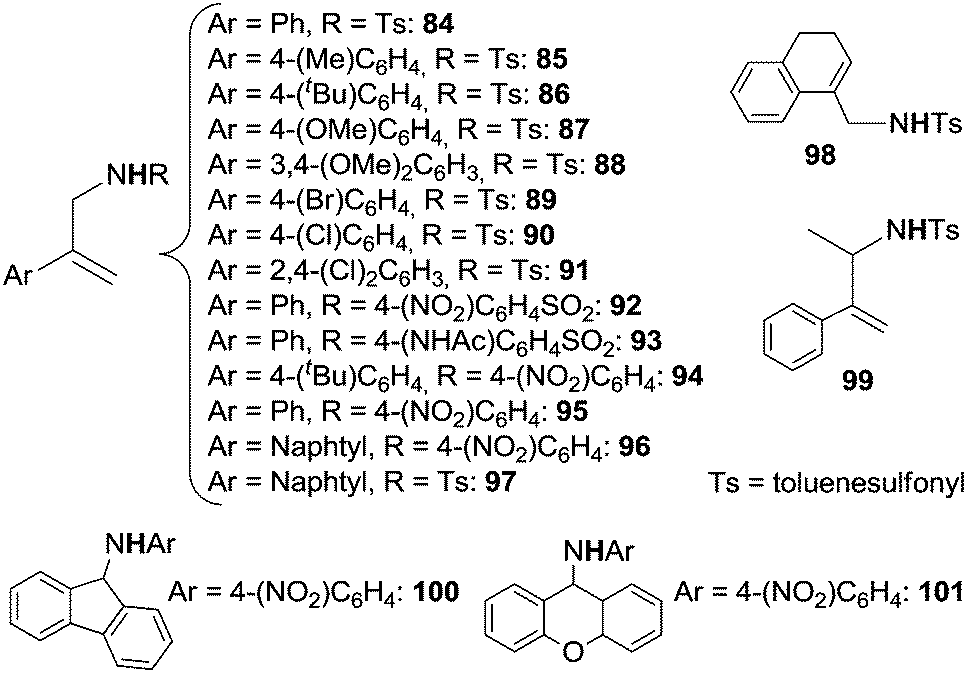
The scope of the reaction was investigated and the obtained products are reported in Chart 1.22
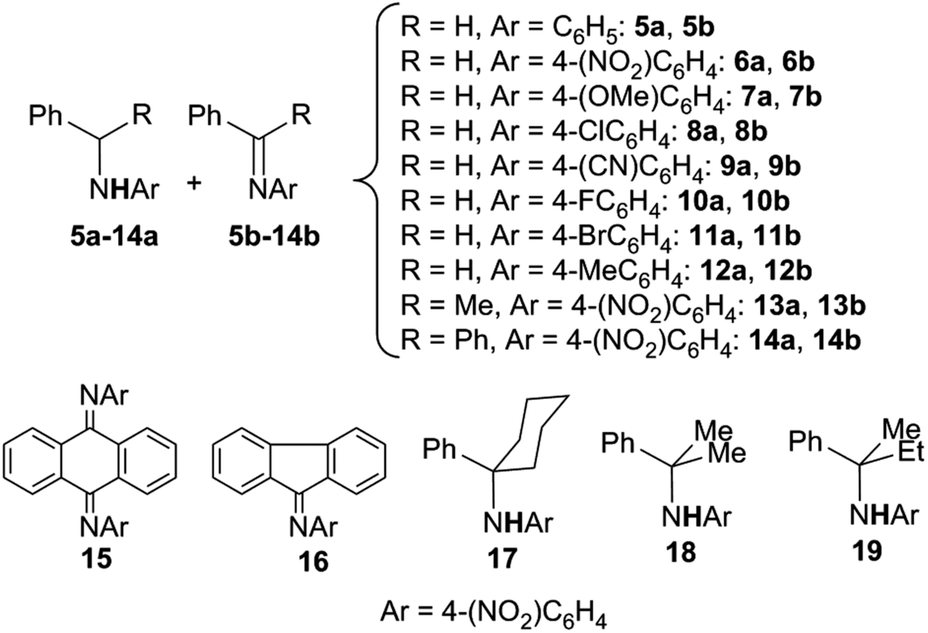 | ||
| Chart 1 Amination of benzylic substrates by aryl azides catalysed by Co(II)(porphyrins) leading to benzylic amines and imines. | ||
The reaction works well with azides bearing EWG on the aryl moiety indicating an electrophilic role of ArN3 in the catalysis.
The cobalt(II) porphyrin-catalysed amination of benzylic substrates was then performed by Zhang26 by using 2,2,2-trichloroethoxycarbonyl azide (TrocN3) as the nitrene source. The benzylic amines reported in Chart 2 were obtained without the contemporary formation of the corresponding imines. TrocN3 was demonstrated to be a better aminating agent than other azides such as sulfonyl, phosphoryl, and carbonyl azides confirming the electrophilic nature of azides in the catalytic process. It must be mentioned that the synthesis of compound 24, even though obtained in a low yield, represents the synthesis of an α-amino ester by the direct amination of the corresponding carboxylic ester.
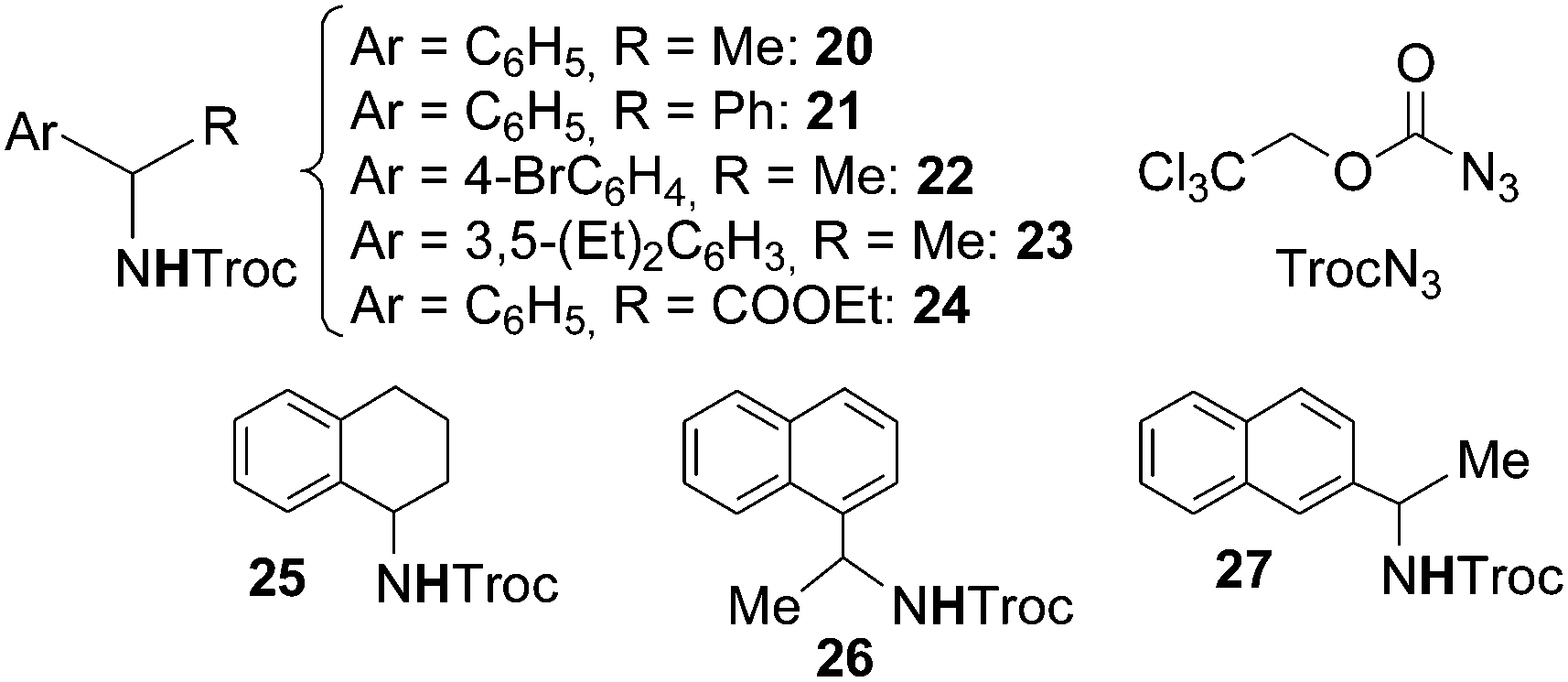 | ||
| Chart 2 Amination of benzylic substrates by TrocN3 catalysed by Co(II)(porphyrin) complexes leading to benzylic amines. | ||
Cobalt porphyrin complexes were also effective in aminating allylic C–H bonds (Scheme 1, path b) in moderate yields. It should be noted that the double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of endocyclic olefins, such as cyclohexene, did not react with the aryl azide to give the corresponding aziridine therefore indicating a good chemoselectivity towards the allylic amine formation.24
C bond of endocyclic olefins, such as cyclohexene, did not react with the aryl azide to give the corresponding aziridine therefore indicating a good chemoselectivity towards the allylic amine formation.24
Surprisingly, the allylic amination of dihydronaphthalene afforded the benzylic amine of tetrahydronaphthalene, instead of yielding the amine of dihydronaphthalene and neither the aziridination nor the benzylic amination of dihydronaphthalene were observed (Scheme 4) (see below for a mechanistic proposal of this reaction, ref. 39).
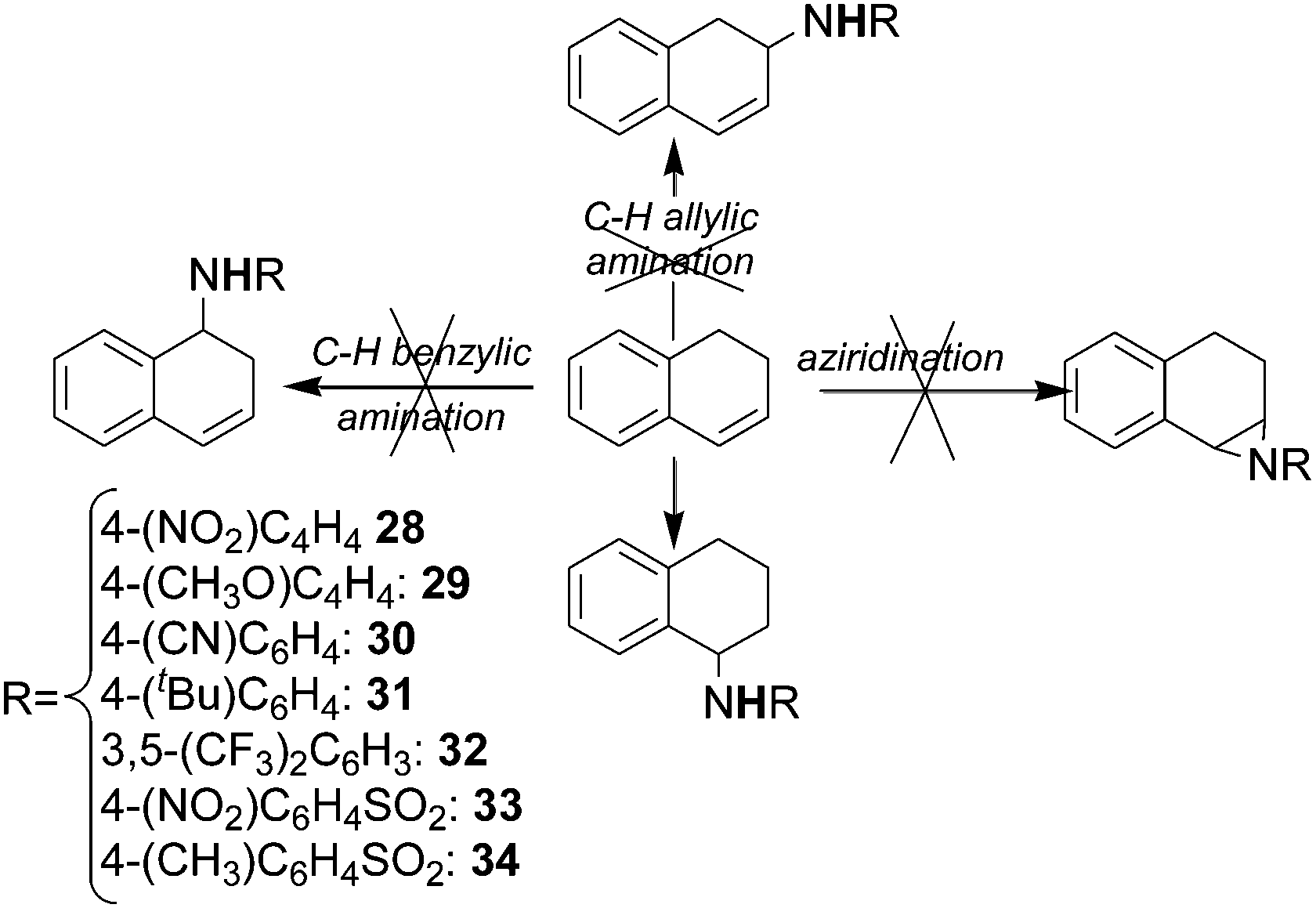 | ||
| Scheme 4 Reaction of dihydronaphthalene with aromatic and sulfonyl azides catalysed by cobalt(II) porphyrins. | ||
All data discussed up to now were collected employing achiral cobalt catalysts. Until very recently, to the best of our knowledge, only the chiral complex 35 (Fig. 3) was reported in literature for the intermolecular amination of C–H bonds in spite of the potential application of chiral amines as pharmaceuticals. Although the intermolecular aziridination reaction of styrenic double bonds performed in the presence of 35 occurred with some enantioselectivity, the allylic amination of cyclohexene afforded the racemate product, probably due to the presence of an atropoisomer mixture of the catalyst at the working temperature.23
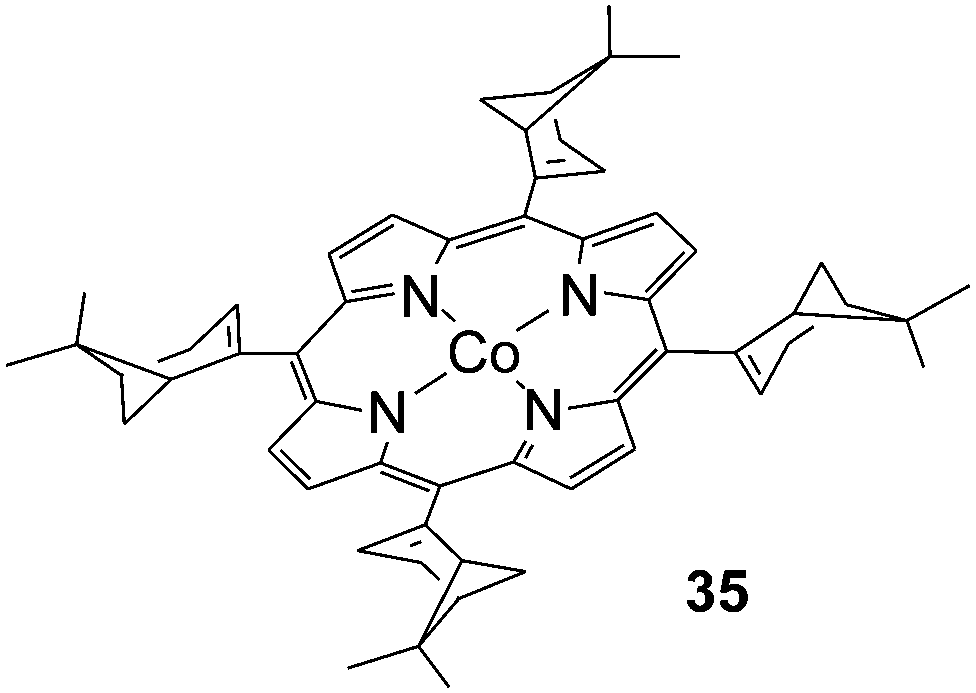 | ||
| Fig. 3 Structure of Co(II)(TmyrtP) (35) (TmyrtP = dianion of the meso-tetrakis[(1R)-apopinen-2-yl]porphyrin). | ||
Better synthetic results have been obtained by using ruthenium porphyrin complexes as catalytic species which are active in the amination of both benzylic and allylic C–H bonds.27–29 The commercially available Ru(II)(TPP)CO (TPP = dianion of tetraphenyl porphyrin) was a good catalyst to synthesise compounds 6a, 13a, 14a, 18 (Chart 1), 28, 32 (Scheme 4) and those reported in Chart 3. The best catalytic results were obtained by using aryl azides bearing EWG substituents on the aryl moiety and a high hydrocarbon excess.
 | ||
| Chart 3 Benzylic and allylic amines obtained by Ru(II)(TPP)CO-catalysed amination.27,28 | ||
While we were writing this review, Che and Lo reported the enhanced catalytic activity of bis(NHC)ruthenium(II) porphyrin complexes in nitrene insertion reactions into saturated C–H bonds (NHC = N-heterocyclic carbene ligands).30 Among the [Ru(porphyrin)(NHC)2] complexes tested, [Ru(4-F-TPP)(BIMe)2] (BIMe = 1,3-di-methyl-2,3-dihydro-1H-benzimidazol-2-ylidene) showed the highest activity and allowed the smooth insertion reaction of pentafluorophenyl azide into allylic (cyclohexene) and benzylic (toluene, ethyl benzene, 4-methoxy-ethyl benzene, 1-ethyl naphthalene, 1,2,3,4-tetrahydronaphthalene, and 2,3-dihydro-1H-indene) sp3 C–H bonds affording the corresponding amines in 88–96% isolated yields. The nitrene insertion reaction proceeded well also with the unactivated C–H bond of cyclohexane (90%). The authors proposed that the high catalytic activity is due to the strong donor strength of the axial NHC ligand in stabilising the trans M = NR moiety.
The use of chiral [Ru(D4-Por)(BIme)2] (D4 refers to the symmetry of the ligand) as the catalyst led to the formation of the allylic amine 13a (Ar = C6F5) in 91% yield and with 70% ee.30
Ru(porphyrin)CO complexes were also effective in synthesising α- and β-amino esters by amination of benzylic C–H bonds placed in the α or β position to an ester group. This synthesis, already explored by Zhang in the presence of cobalt porphyrins,26 occurred with yields up to 80% in the presence of ruthenium analogous, in spite of the poor reactivity of electron deficient benzylic positions toward electrophilic metallonitrene intermediates. The catalytic procedure is also effective in synthesising the two derivatives of methyl L-3-phenyllactate 73 and 74 in order to convert them into the corresponding β-lactam (Chart 4).31
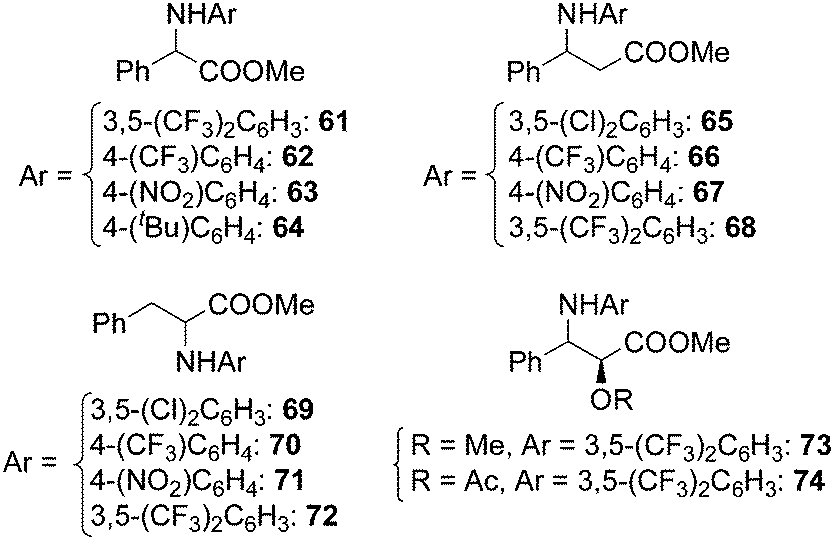 | ||
| Chart 4 α- and β-amino esters obtained by Ru(TPP)CO-catalysed amination of carboxylic esters. | ||
Very recently, Che et al.29 demonstrated that the amination of benzylic and allylic substrates by phosphoryl azides is also efficiently catalysed by ruthenium(IV) complexes. Among tested catalysts, Ru(F20-TPP)Cl2 performed the best (Chart 5).
 | ||
| Chart 5 Benzylic and allylic amines obtained by Ru(IV)(F20-TPP)Cl2-catalysed amination. | ||
A significant contribution to this topic was provided by Che et al. with the investigation of the catalytic activity of Fe(III)(F20-TPP)Cl.32 What makes this study important is the high sustainability of iron catalysts which are affordable, biocompatible and very reactive. Compounds 13a, 14a (Chart 1), 28 (Scheme 4) and those reported in Chart 6 were isolated in good yields.
 | ||
| Chart 6 Benzylic and allylic amines obtained by Fe(III)(F20-TPP)Cl-catalysed amination. | ||
The reported methodology displayed a very good chemoselectivity, in fact the allylic amination of α-methyl styrene derivatives occurred without the simultaneous formation of corresponding aziridines.
It is generally assumed that the active intermediate of the metal porphyrin-catalysed nitrene transfer reaction is an imido complex which is formed after the cleavage of the Nα–Nβ azide bond (Scheme 5).
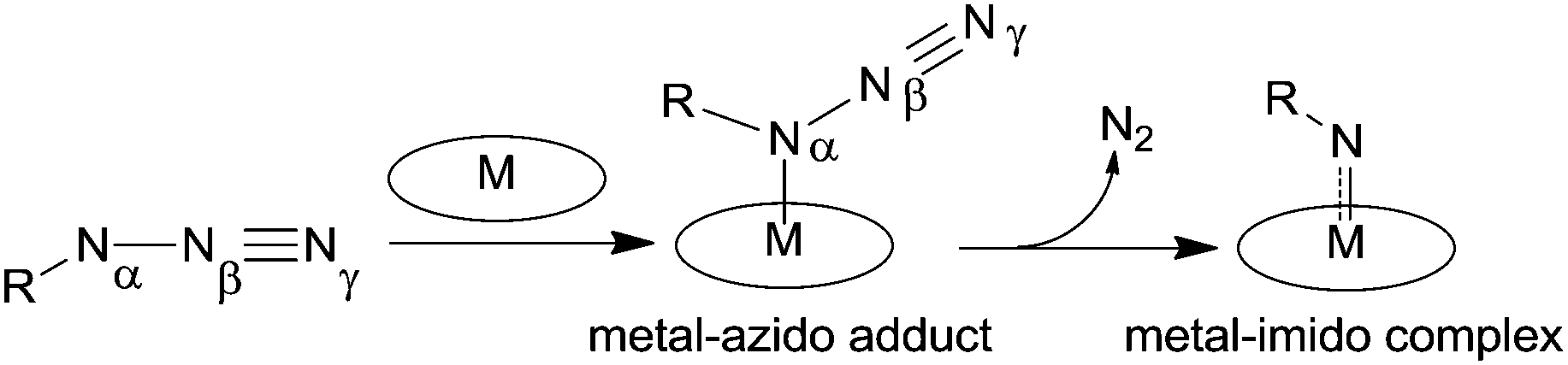 | ||
| Scheme 5 Metal–imido complex formation. | ||
The oxidation state of the metal in the imido complex depends on the oxidation state of the starting catalyst and it is crucial to define the reactivity of the imido intermediate towards the organic substrate.
When the amination reaction is catalysed by ruthenium(II) porphyrins the formation of a bis-imido ruthenium(VI) complex was generally proposed. Some years ago several ruthenium(VI) bis-imido complexes, of the general formula Ru(VI)(porphyrin)(NSO2R)2, were prepared by Che et al. by reacting ruthenium(II) precursors with imino iodinane compounds (PhI = NSO2R). All the complexes were fully characterised and the molecular structure of Ru(TMP)(NMs)2 (102) (Ms = 4-(MeO)C6H4SO2; TMP = dianion of 5,10,15,20 tetramesityl porphyrin) was solved by X-ray single crystal diffraction.33–35
Ru(VI)(porphyrin)(NSO2R)2 complexes were active in the stoichiometric nitrene transfer reaction to C–H bonds in the presence of pyrazole (Hpz) as an additional axial ligand. The suggested mechanism is depicted in Scheme 6.
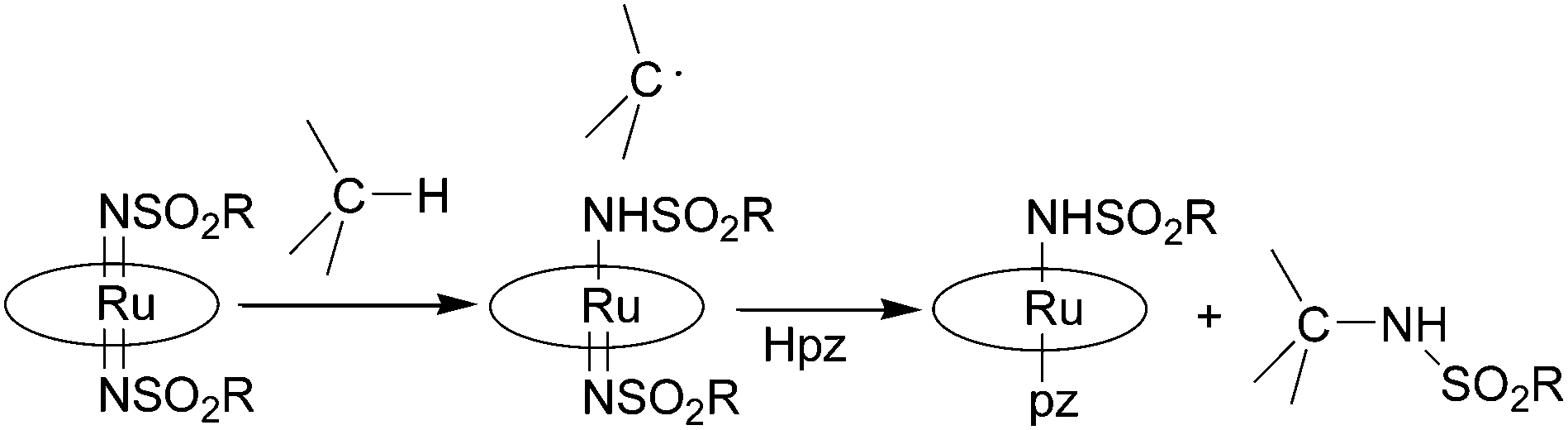 | ||
| Scheme 6 Suggested mechanism for the stoichiometric transfer of a nitrene moiety from a bis-imido ruthenium(VI) porphyrin to a C–H bond. | ||
The authors proposed that C–H cleavage is involved in the rate determining step of the reaction on the basis of the large isotopic effect registered for the amination of several substrates. In fact, the formation of a carboradical species by homolytic cleavage of the C–H bond is in accord with the observed kH/kD values of 6.1–12. Considering that the hydrogen atom of pyrazole is necessary for releasing the amine molecule, Hpz was always used in stoichiometric excess with respect to the hydrocarbon substrate.
The reaction mechanism illustrated in Scheme 6 was also studied by Density Functional Theory (DFT) calculations.35 Firstly, the molecular structure of 102 was optimised by DFT and achieved data were fitted with those collected by single crystal X-ray diffraction. Then, employed functionals indicated a singlet d2 ground state electronic configuration (1A) for Ru(TMP)(NMs)2 (102) that was found at a lower energy than the analogous triplet state. The energy profile of the ethyl benzene amination by 102 indicated that the singlet pathway is the most energetically favoured.
In Scheme 7 the proposed mechanism of the stoichiometric nitrene transfer from 102 to ethyl benzene is provided together with the relative potential energy surface. The last step of the mechanism illustrated in Scheme 6, which corresponds to the release of the amine and pyrazole coordination, has not been provided.
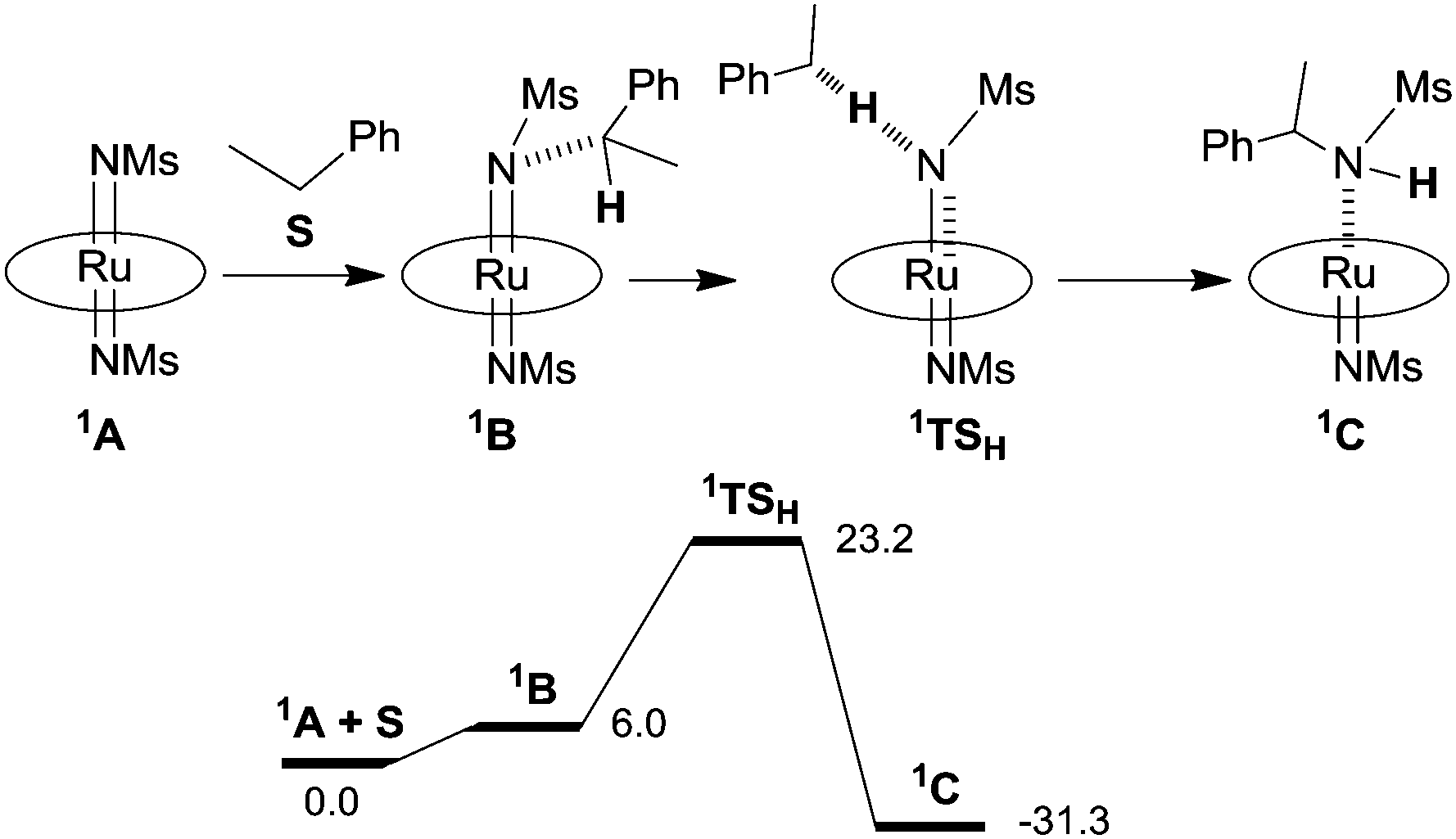 | ||
| Scheme 7 Mechanism of the ethyl benzene amination and relative potential energy surface (ΔG in kcal mol−1) based on DFT calculations. | ||
Another class of bis-imido ruthenium(VI) complexes have been synthesised by Gallo et al. reacting ruthenium(II) porphyrins with an aryl azide excess. The complex Ru(TPP)(NAr)2 (Ar = 3,5-(CF3)2C6H3) (103) was fully characterised and its molecular structure was determined by X-ray single crystal diffraction.28,36
Complexes 102 and 103 (Fig. 4) have similar RuN distances (1.79(3) Å vs. 1.808(4) Å) in the range of double bonds but the two imido complexes differ in the Ru–N–X (X = S for 102 and X = C for 103) imido angles. The angle of 162.5(3) of 102 indicates almost a linearity of the imido moiety whilst the values of 139.8(3)° and 143.7(4)° for Ru–N1–C and Ru–N2—C, respectively, of 103 indicate the existence of bent imido angles.
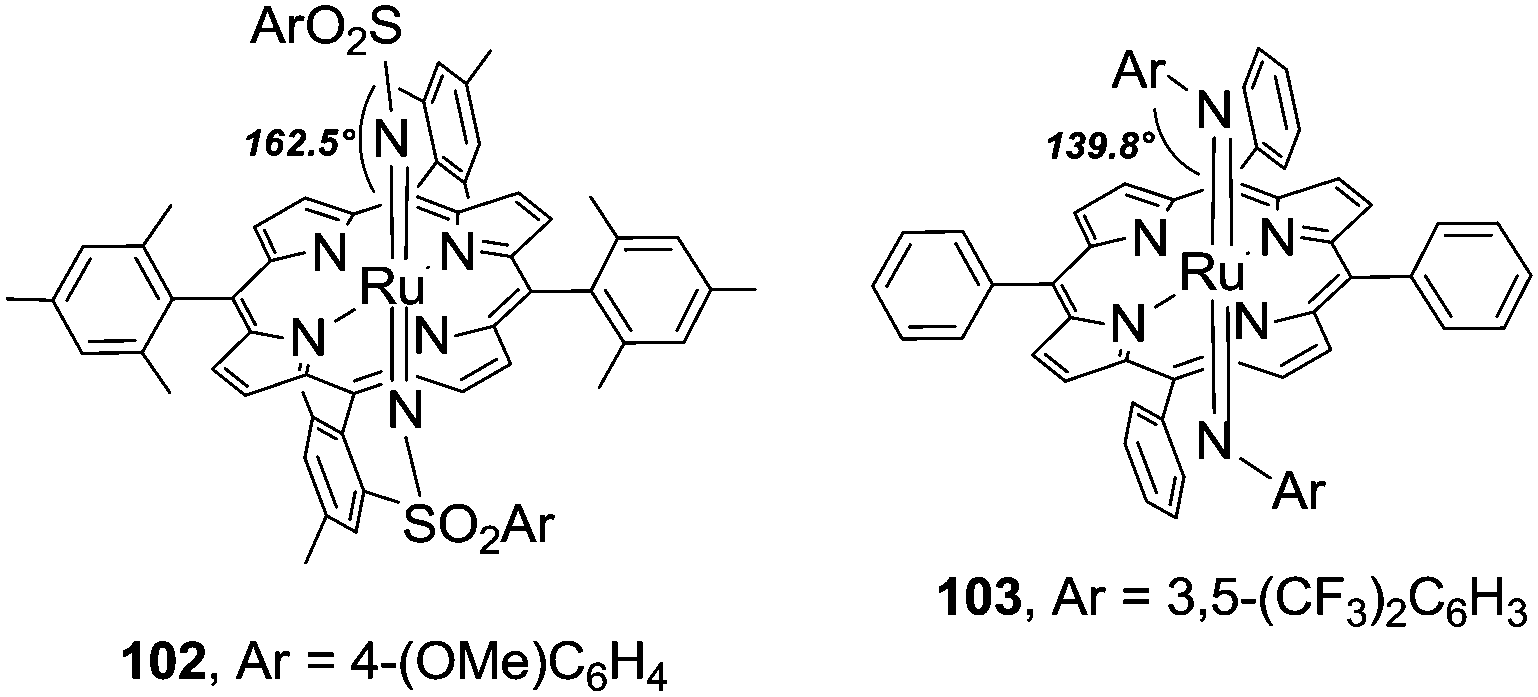 | ||
| Fig. 4 Structures of ruthenium(VI) imido complexes 102 and 103. | ||
Complex 103 displayed a good stability–reactivity relationship; it was stable for days in the solid state and decomposed when left standing in solution for a few hours. The bis-imido ruthenium(VI) complex 103 was active not only in stoichiometric nitrene transfer reaction from 103 to a C–H bond but was also an efficient catalyst of C–H amination. This last feature was essential to propose 103 as a possible key intermediate of the Ru(TPP)CO-catalysed amination reactions.
To shed some light on this reaction mechanism, a kinetic and theoretical investigation was undertaken. The kinetic investigation indicated the existence of another important catalytic intermediate which is formed by the reaction of Ru(TPP)CO with a single aryl azide molecule. This “elusive” Ru(IV)(TPP)(NAr)(CO) complex was suggested on the basis of kinetic data, however it was neither isolated nor spectroscopically observed. A DFT theoretical investigation of the allylic amination of cyclohexene confirmed kinetic results and indicated the existence of at least two different mechanisms in which, depending on experimental conditions, ruthenium(IV) (D) or ruthenium(VI) (E) complexes are involved.37 The release of the CO axial ligand of D yields a penta-coordinated mono-imido complex which forms E by reacting with another aryl azide molecule. The mechanistic proposal, based on both kinetic and DFT studies (Fig. 5), is reported in Scheme 8.
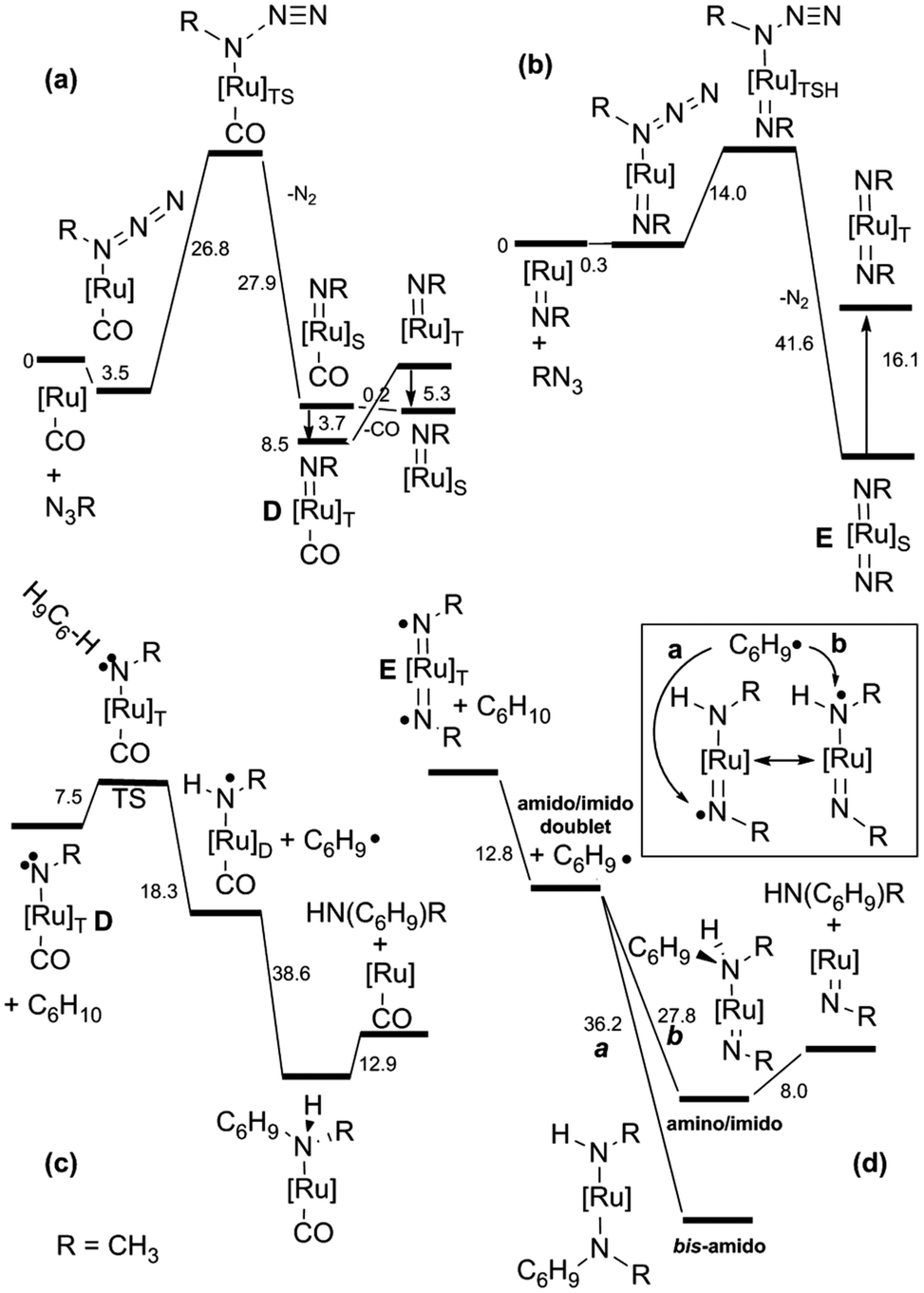 | ||
| Fig. 5 Energy profiles (ΔG in kcal mol−1) for (a) the D formation; (b) the E formation; (c) the D catalytic activity; (d) the E catalytic activity. | ||
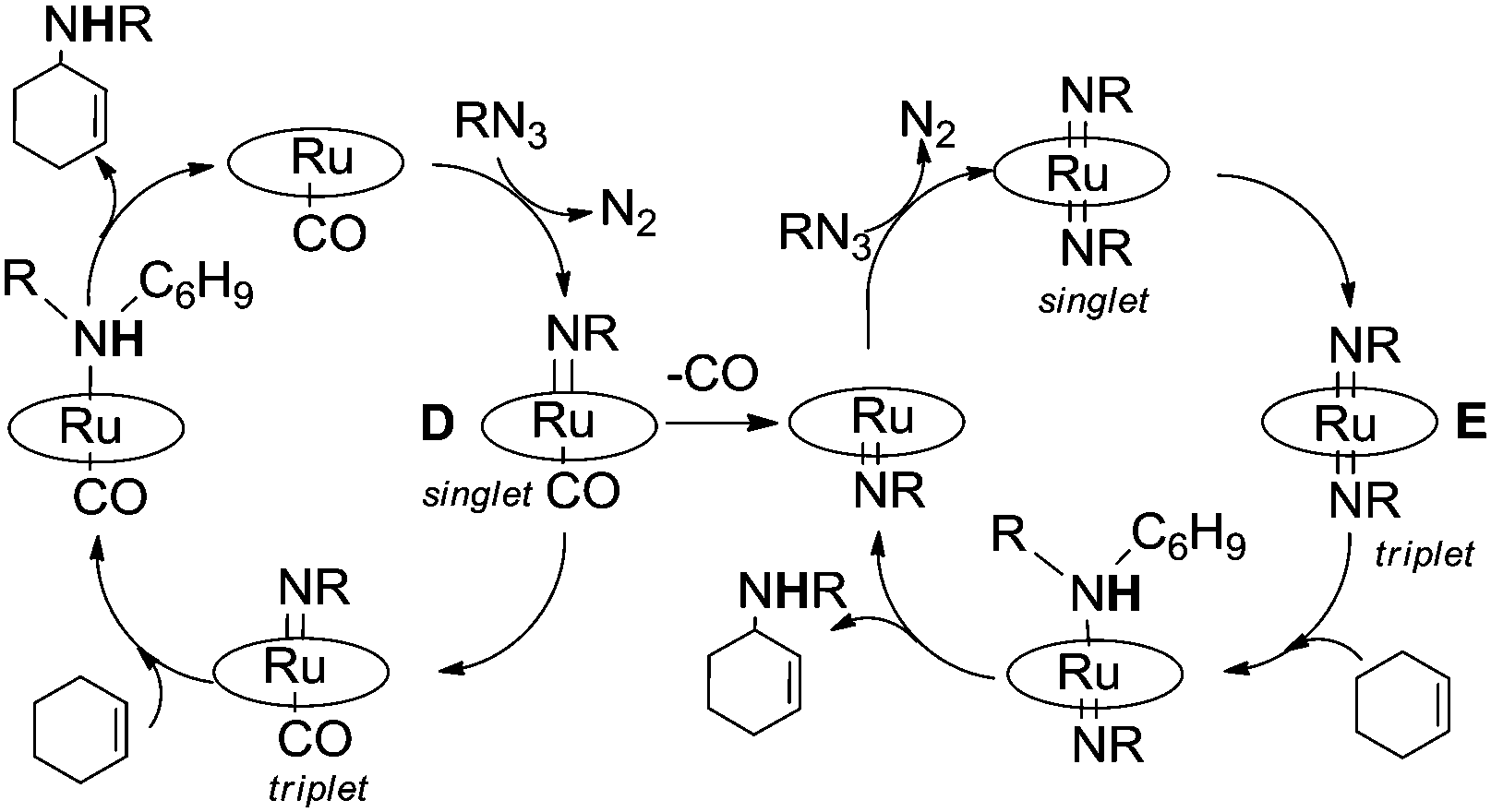 | ||
| Scheme 8 Mechanism suggested on the basis of kinetic and DFT studies. | ||
The computations indicated that both ruthenium(IV) mono-imido (D) and ruthenium(VI) bis-imido intermediates (E) are active in their triplet state which is necessary to accomplish the radical reaction, the associated intersystem crossing is generally feasible. The radical nature of the reaction was experimentally proven by performing reactions in the presence of radical traps such as TEMPO.
It is important to underline that a direct comparison between the two described DFT studies on the activity of complexes 102 and 103 may suggest a false analogy. In fact, the first study describes the stoichiometric transfer of a nitrene functionality from 102 to a C–H bond35 while the second analyses the catalytic activity of 103 in the amination of allylic C–H bonds.37 From these two different approaches a basic difference emerges, Che, Huang et al. interpreted the reactivity of the imido complex 102 based on the chemical evolution over a singlet ground state, while Manca, Gallo et al. underlined the critical importance of the triplet electronic configuration for the catalytic activity of 103.
A completely different approach was necessary to study the cobalt(II) porphyrin-catalysed amination of C–H bonds. This is due to the improbable formation of a diamagnetic cobalt(IV) imido intermediate from the reaction of cobalt(II) porphyrin with organic azide molecules. The first mechanistic proposal was provided in 2003 by Cenini et al. who, on the basis of a kinetic study,22 proposed that the first step of the benzylic amination by aromatic azides is a reversible coordination of the azide molecule to the cobalt(II) metal centre. This so-obtained cobalt(II) azido-adduct (F) can either react with the benzylic substrate forming the corresponding amine or lose molecular nitrogen in an unimolecular reaction to afford an imido intermediate (G) which could be responsible for the formation of by-products diazene and the primary amine corresponding to the azide employed (Scheme 9).
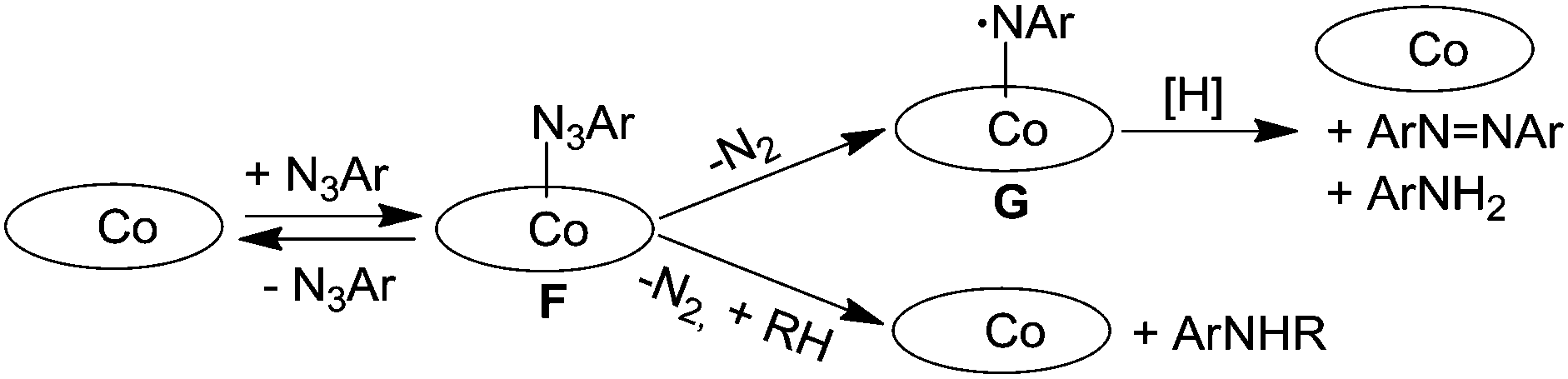 | ||
| Scheme 9 Suggested mechanism of the Co(porphyrin)-catalysed amination of benzylic substrates by aryl azides. | ||
When deuterated toluene was used instead of toluene as a hydrocarbon substrate, a large isotopic effect (kH/kD = 14) was observed indicating that the cleavage of the C–H bond is involved in the rate determining step of the catalytic cycle. A similar mechanism was also suggested for the allylic amination catalysed by cobalt(II) porphyrins.24
The mechanism of the cobalt porphyrin-catalysed amination of C–H bonds was further clarified by EPR and DFT studies recently performed by Zhang and de Bruin et al.38 In this mechanistic investigation it was proposed that the nitrene radical species G (Scheme 9) is not only involved in the formation of by-products but it is the key-intermediate of the catalysis.
An intermediate of type G was detected by EPR in the reaction of Co(TPP) with TrocN3 in the absence of a hydrocarbon substrate. The well resolved EPR spectrum was typical for nitrene radical ligand complexes and it fitted well with the simulated EPR spectrum. A DFT study of the reaction between methyl azidoformate, a model of TrocN3, and ethyl benzene was then performed and it also supported the formation of G as the catalytic active species. The mechanistic proposal and relative energy profiles are shown in Scheme 10 and Fig. 6.
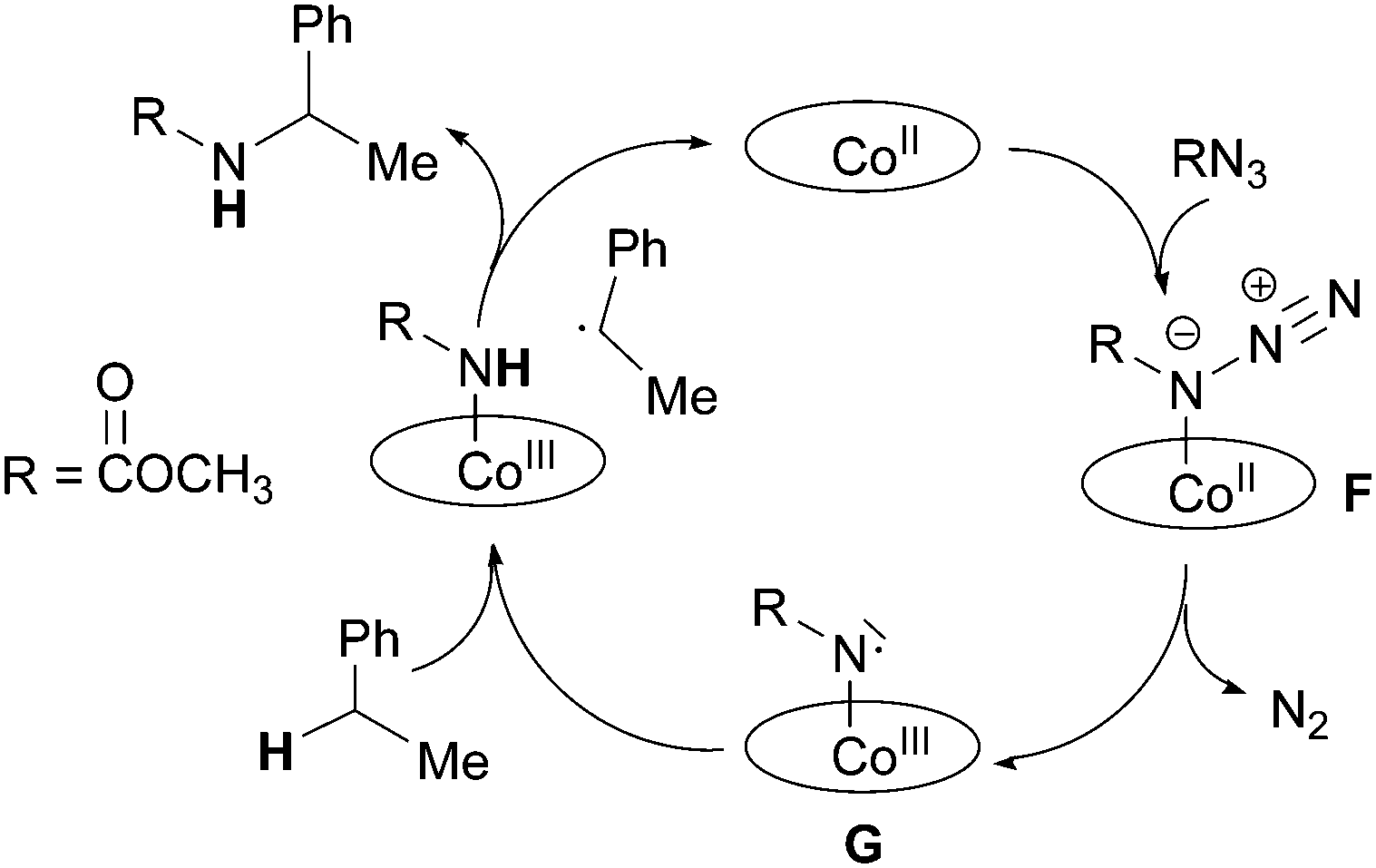 | ||
| Scheme 10 Proposed mechanism of the Co(porphyrin)-catalysed amination of ethyl benzene by methyl azidoformate. | ||
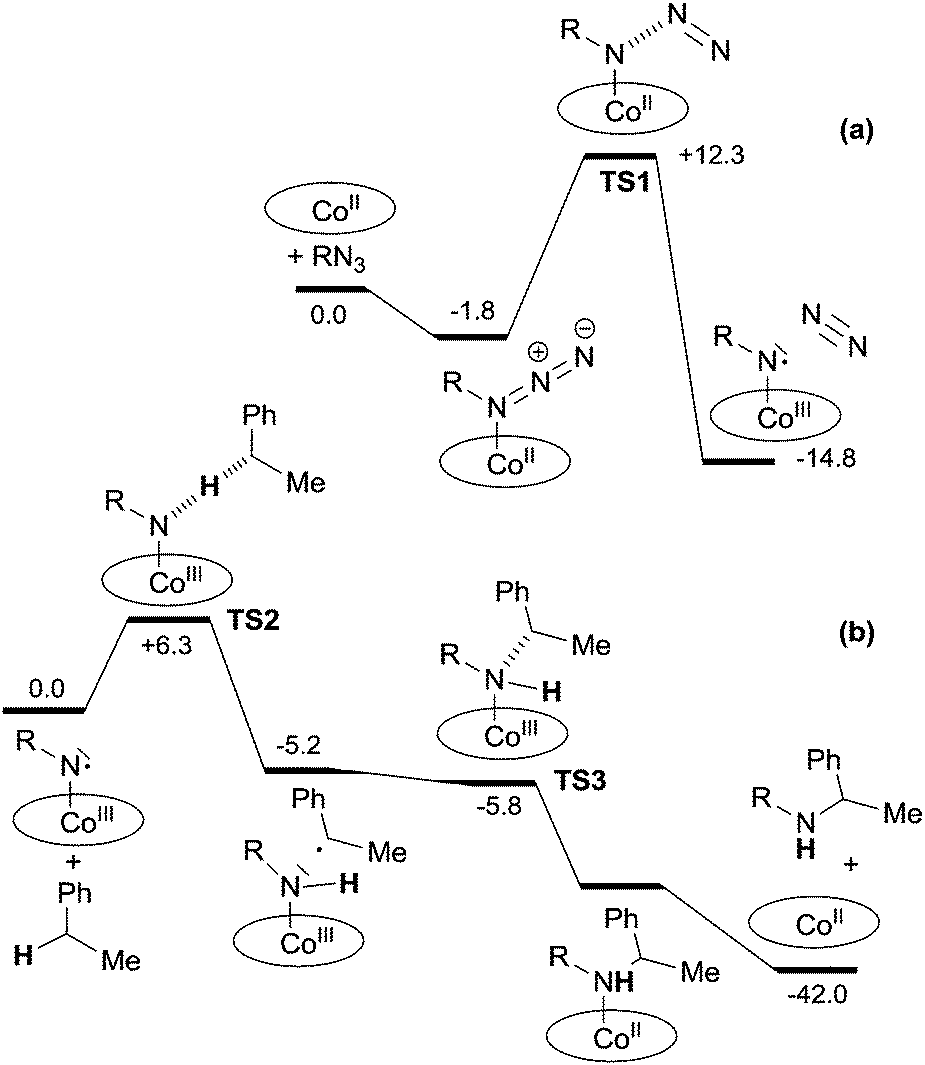 | ||
| Fig. 6 Reaction profile (ΔH in kcal mol−1) for the formation of (a) the nitrene radical species and (b) the benzylic amine. | ||
The formation of a cobalt(III) nitrene radical intermediate can also be proposed to understand the unusual formation of the amine of tetrahydronaphthalene from the reaction of dihydronaphthalene with an aryl azide (Scheme 11).39
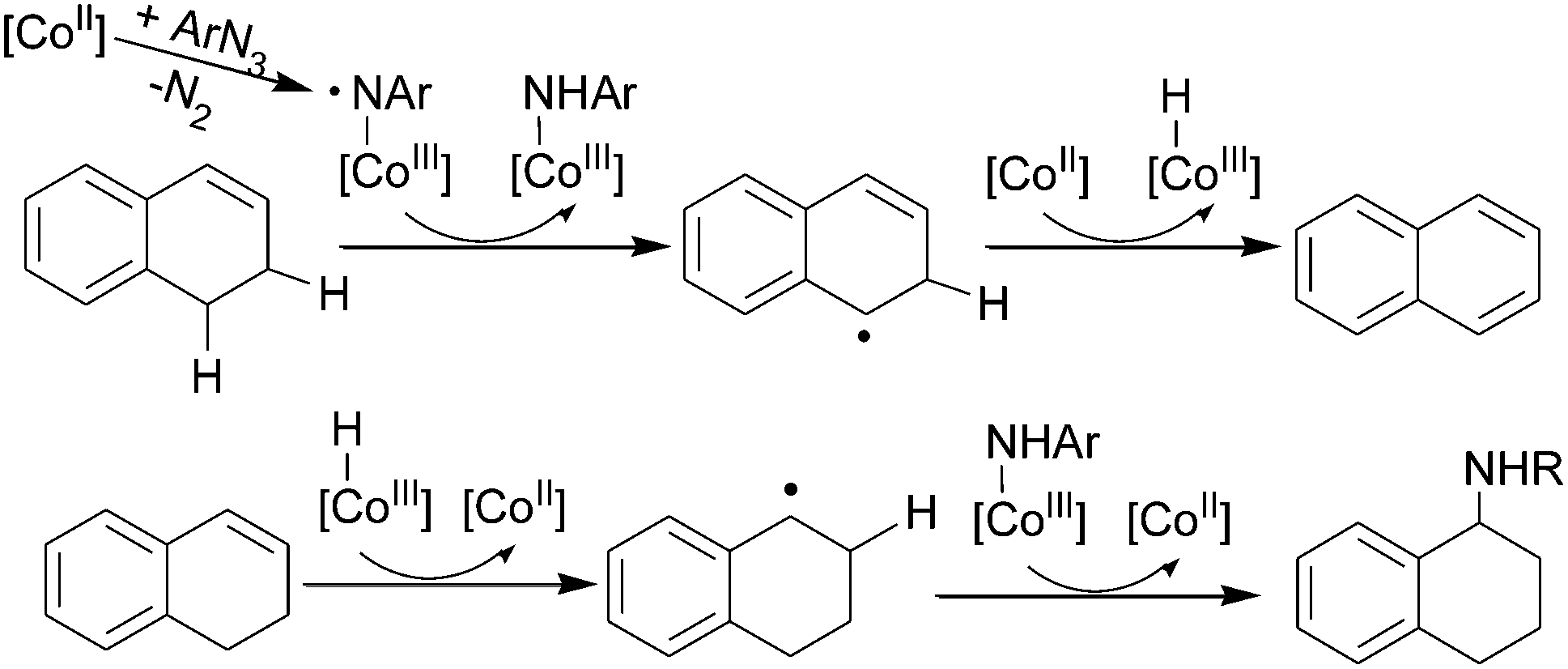 | ||
| Scheme 11 Reaction between tetrahydronaphthalene and ArN3. | ||
A mechanistic study on the iron(III) porphyrin-catalysed C–H amination has not been reported yet. It was suggested by Che et al. that the starting iron(III) complex can be reduced in situ to an iron(II) nitrene intermediate which evolves into the active iron(IV) imido derivatives.32 The latter is then responsible for the nitrene transfer reaction to the organic substrate.
2.2. Amination catalysed by metal Schiff base complexes (B)
Schiff base complexes are very active in intramolecular C–H amination and aziridination of olefins by organic azides5 but, to the best of our knowledge, only two papers by Katsuki et al. were published on the intermolecular amination of allylic and benzylic C–H bonds. The ruthenium(II) chiral catalysts employed (Fig. 7) promoted the enantioselective nitrene transfer from TsN3 (ref. 40) and SESN3 (SES = 2-(trimethylsilyl)ethanesulfonyl)41 to several hydrocarbons.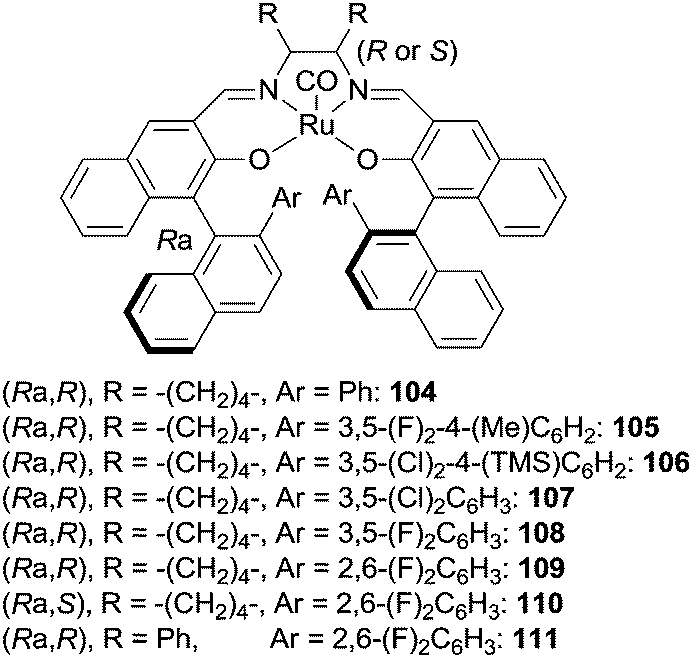 | ||
| Fig. 7 Ruthenium(II) Schiff base catalysts employed in the intermolecular amination of C–H bonds. | ||
Among all the complexes reported in Fig. 7, complexes 104 and 109 showed better catalytic efficiency by using TsN3 and SESN3 respectively. All the obtained aminated compounds are shown in Chart 7.
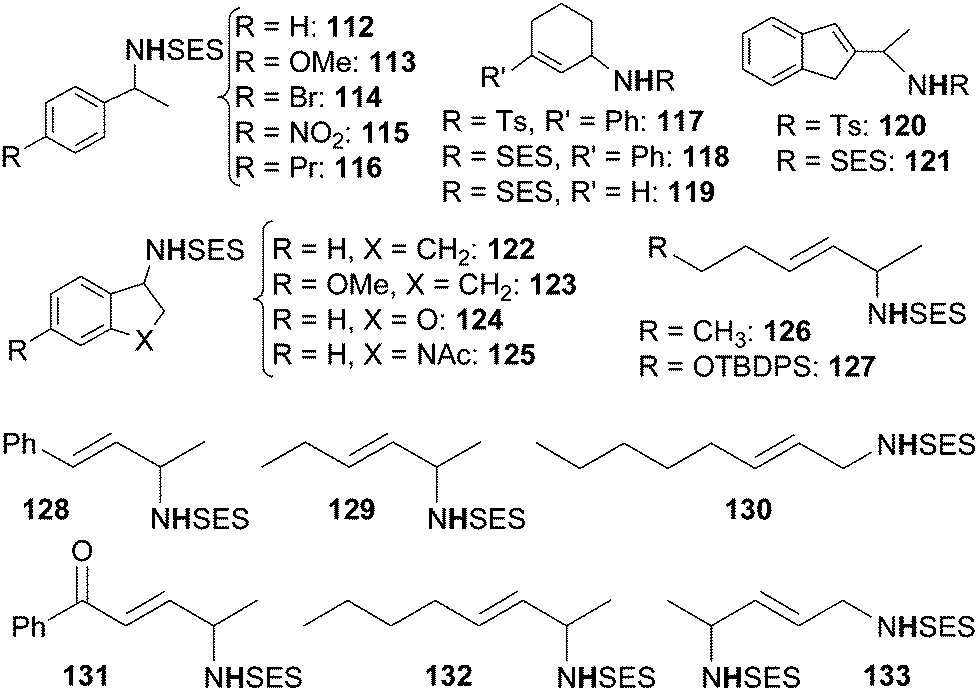 | ||
| Chart 7 Benzylic and allylic amines obtained by 104 and 109-catalysed amination (TBDPS = tert-butyldiphenyl silyl). | ||
The best catalytic results have been achieved by using SESN3 as the nitrene source and 109 as the catalyst. Yields and enantioselectivities up to 99% and 99% ee respectively were obtained. It is worth noting that also the regioselectivity of the amination reaction was extremely high and that this data represents the best results in terms of enantioselective C–H amination by organic azides.
Iridium(III) Schiff base complexes were also tested as catalysts in the amination of indane using SESN3 as the aminating agent; unfortunately the reaction occurred in low yields and modest enantioselectivities.41
2.3. Amination catalysed by dipyrromethene complexes (C)
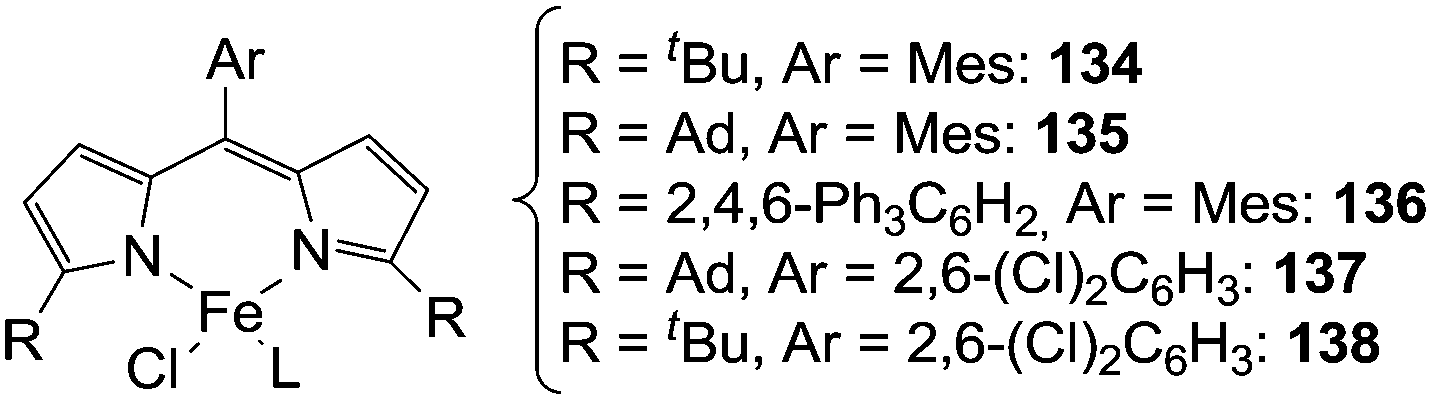 | ||
| Fig. 8 Iron(II) dipyrromethene catalysts employed in the intermolecular amination of C–H bonds (Mes = mesityl, Ad = adamanthyl). | ||
Initially, toluene was reacted with adamantyl azide (AdN3) in the presence of complex 135 as the catalyst to yield the corresponding benzylic amine.42 Then, in order to enlarge the reaction scope, other substrates were aminated in the presence of iron catalysts (Fig. 8).43 Complexes 137 and 138 showed the best catalytic efficiency to synthesize benzylic (139–143) and allylic (144–148) amines respectively (Chart 8).
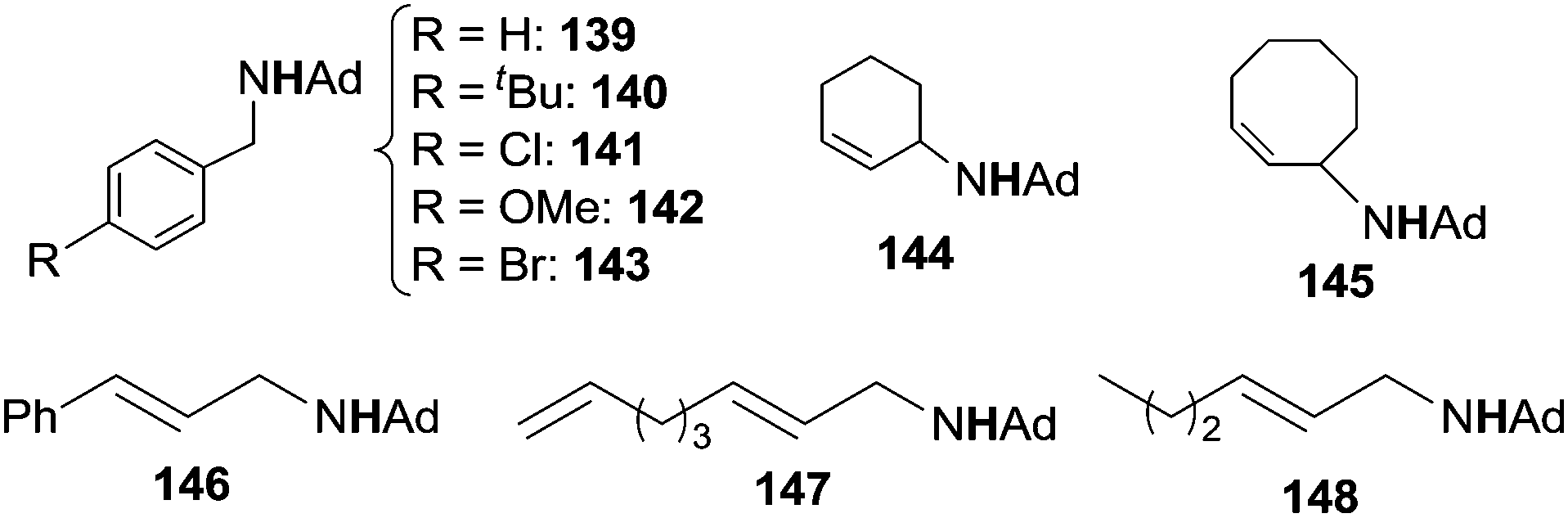 | ||
| Chart 8 Benzylic and allylic amines synthesised by 137 and 138 catalysed amination. | ||
When the reaction was run in an equimolar toluene/toluene-d8 mixture, an isotope effect kH/kD of 12.8 was observed indicating that the C–H bond cleavage was involved in the rate determining step of the cycle (Scheme 12).
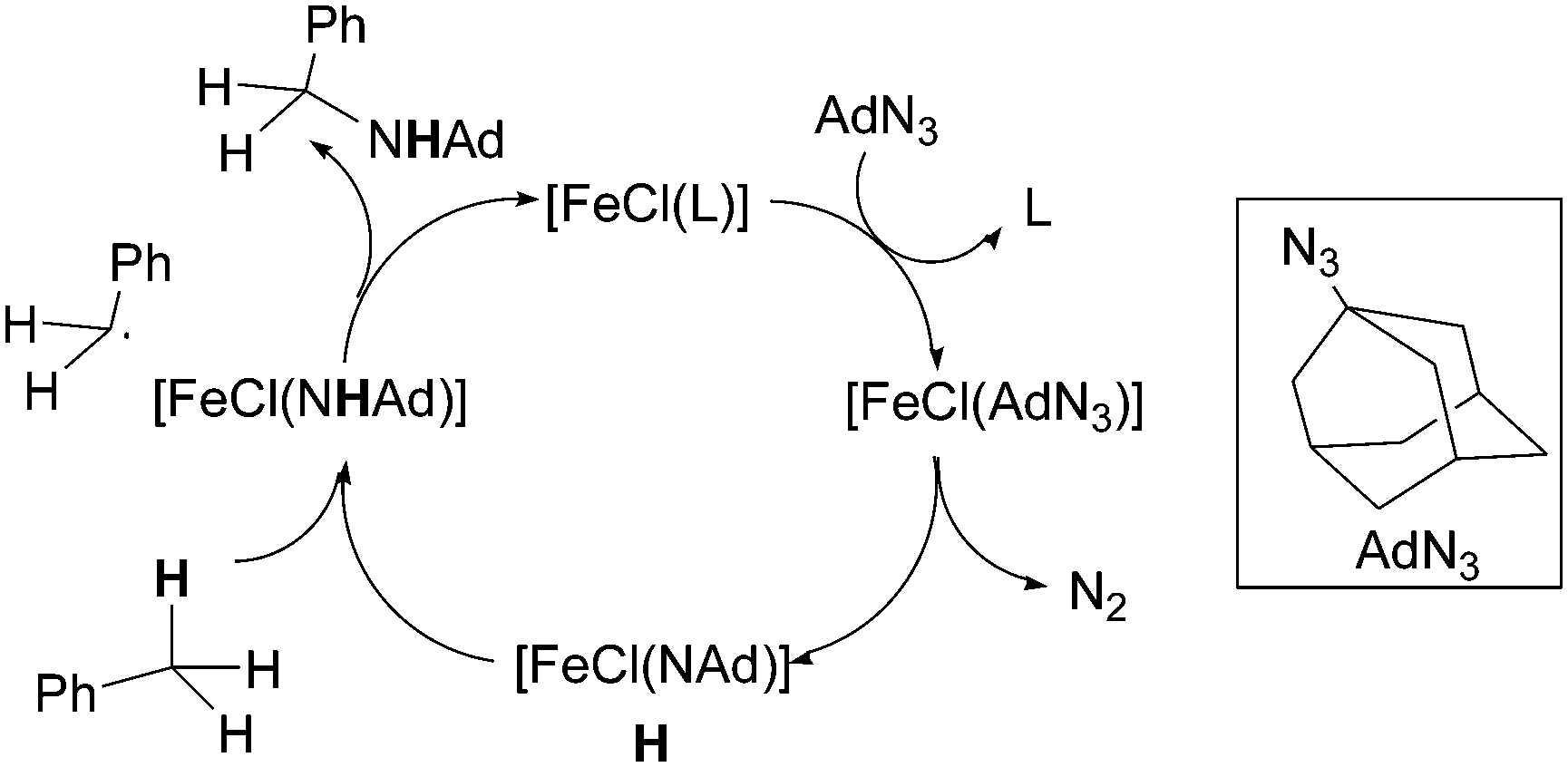 | ||
| Scheme 12 Proposed mechanism for the 135-catalysed toluene amination by AdN3. | ||
The authors suggested the formation of imido complex H which is responsible for hydrogen atom abstraction from the toluene substrate after which a radical rebound process finally yields the desired benzylic amine.
The formation of type H complex was supported by the isolation of terminal imido complex 149 from the stoichiometric reaction of p-tBuC6H4N3 with 136 (Fig. 9).
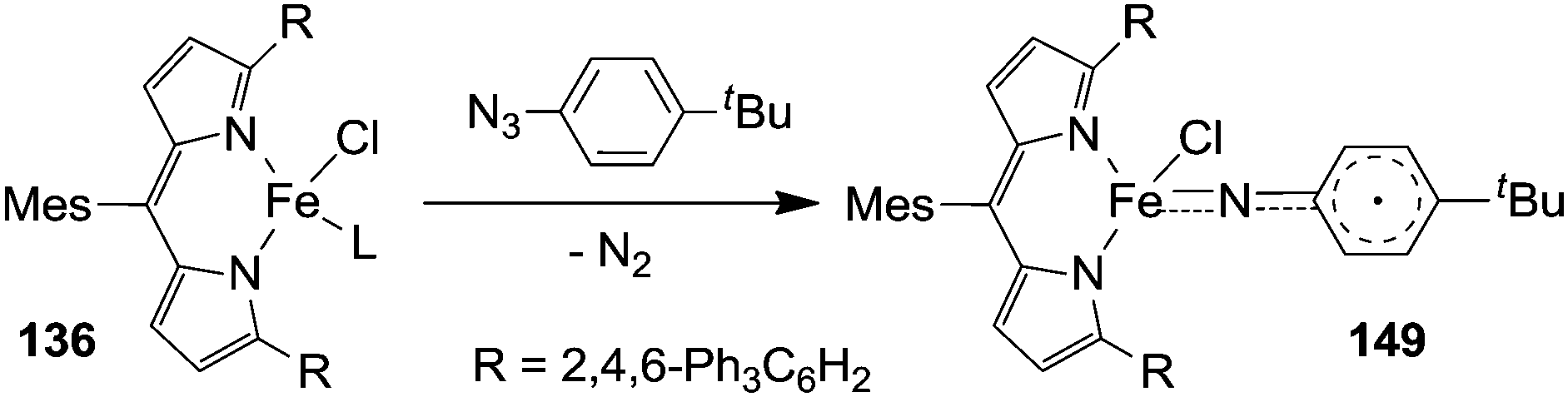 | ||
| Fig. 9 Synthesis of 149. | ||
The X-ray analysis of 149 disclosed the formation of a terminal iron-imido bond with a limited multiple-bond character.
This structural feature together with theoretical and magnetic data indicated the formation of a high-spin FeIII (d5, S = 5/2) centre antiferromagnetically coupled with an imido-based radical (S = 1/2). The resulting complex can be indicated as a high-spin (S = 2) iron(III) imido radical species.
It is noteworthy that complex 149 was able to deliver the nitrene moiety to C–H bonds to support its probable involvement in the catalytic cycle.
2.4. Amination catalysed by β-diketiminato complexes (D)
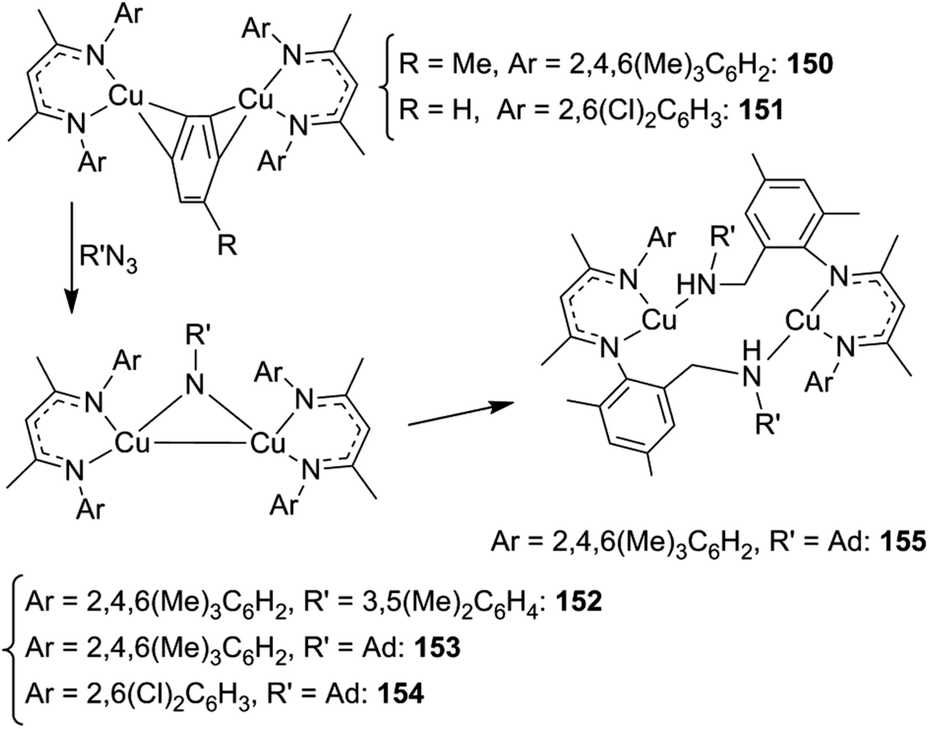 | ||
| Scheme 13 Synthesis of imido complexes 152–154. | ||
When the synthesis of 153 was conducted at room temperature for a long time, the intramolecular insertion of an adamantly nitrene functionality into a C–H bond of one β-diketiminate ortho-methyl group of 153 was observed yielding complex 155 (Scheme 13). This reaction demonstrated the capacity of this class of complexes to insert a nitrene functionality into a C–H bond.
In order to avoid the intramolecular side reaction and to promote intermolecular aminations, complex 151, which presents two chlorine atoms on the aryl ortho positions, was used as the starting material for the synthesis of imido complex 154. The intramolecular amination of the copper complex was not observed and 151 was a competent catalyst for the intermolecular amination of benzylic substrates. Compound 139 (Chart 8) together with compounds 156–158, reported in Chart 9, were obtained in good yields.
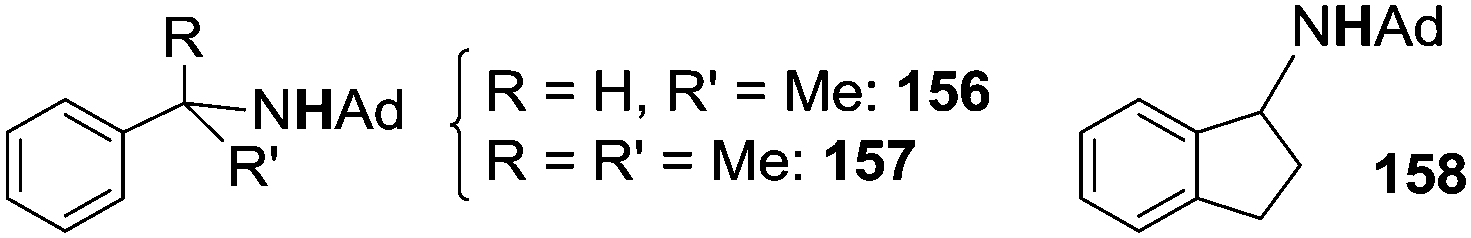 | ||
| Chart 9 Benzylic amines synthesised by 152 catalysed amination. | ||
The kinetic isotope effect kH/kD = 5.3, registered in the amination of ethyl benzene catalysed by 151, suggested that nitrene C–H insertion may be the rate limiting step of the catalysis. The mechanism reported in Scheme 14 was proposed.
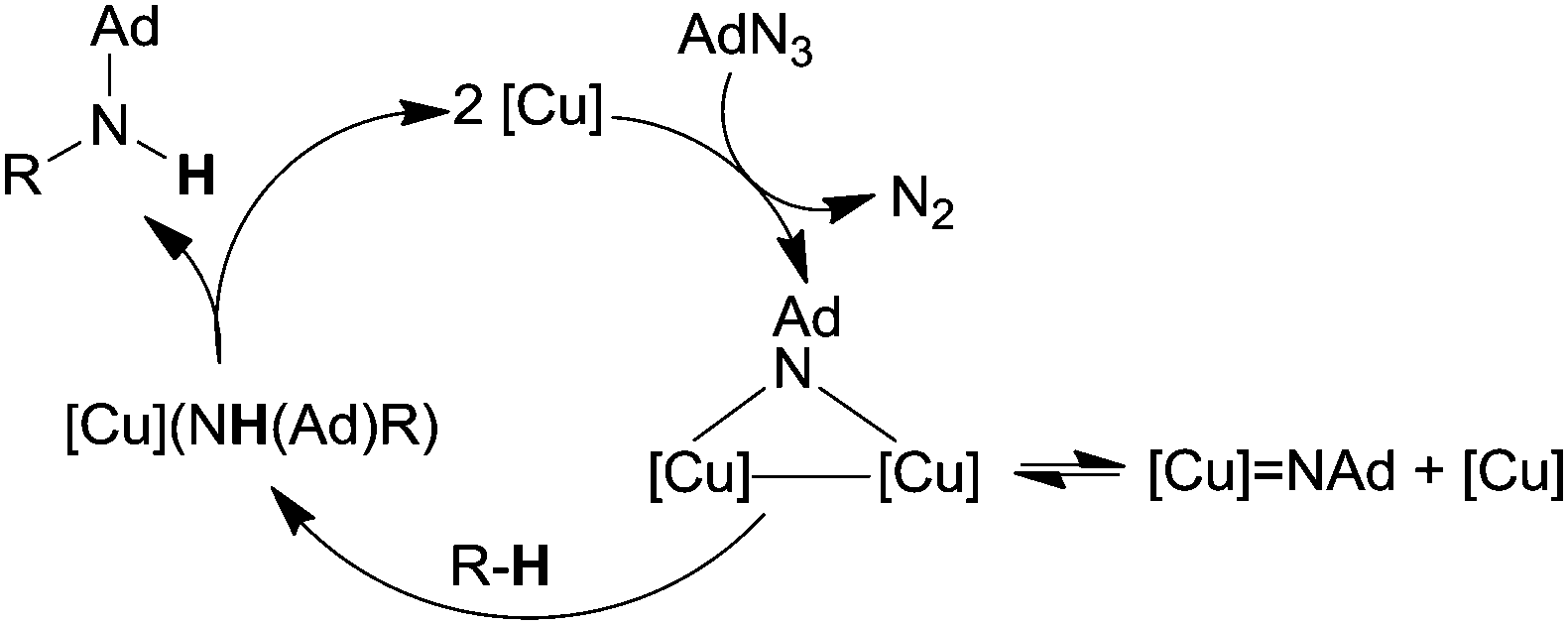 | ||
| Scheme 14 Suggested mechanism for a C–H amination catalysed by β-diketiminato copper(I) complexes. | ||
2.5. Amination catalysed by dirhodium complexes (E)
As supported by all data discussed up to now, best synthetic results were obtained by employing azides bearing EWG irrespective of the catalyst used. The positive effect of electrophilic azides has been recently underlined by the efficiency of nonafluorobutanesulfonyl azide (N3SO2(CF2)3CF3, NfN3) which aminated benzylic substrates in the presence of Rh2(OAc)4.47 No amination reaction occurred in the absence of the transition metal complex.The scope of the reaction was explored and the obtained aza-derivatives are reported in Chart 10.
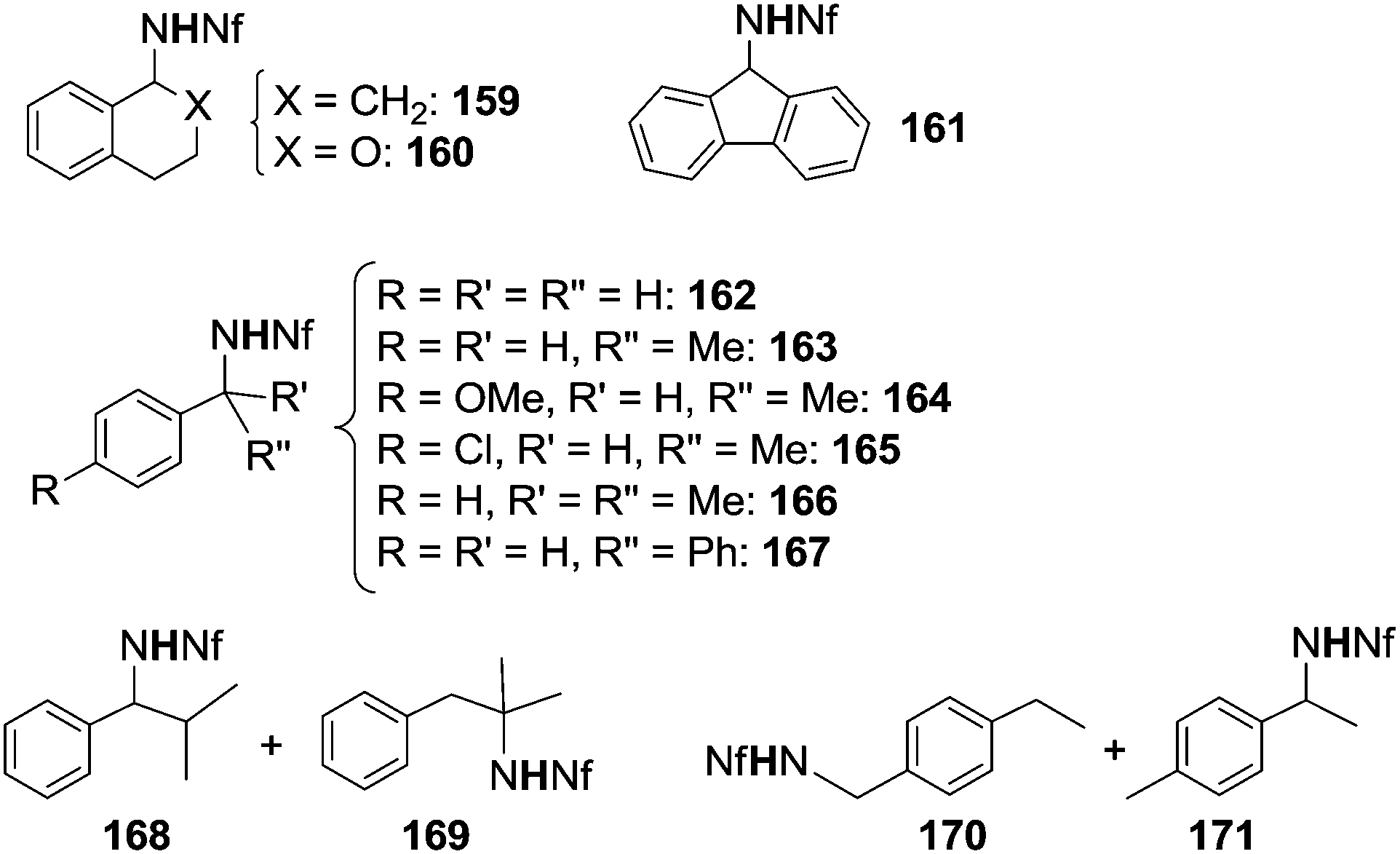 | ||
| Chart 10 Aza-compounds obtained by Rh2(OAc)4-catalysed reaction. | ||
The practical importance of this reaction is also due to the chemical versatility of polyfluoroalkanesulfonamides. The reported methodology can be used for the synthesis of a variety of secondary amines by a stepwise procedure. The N-alkylation reaction affords N,N-disubstituted nonafluorobutanesulfonamides which can then be transformed into the corresponding N-alkylated amines.
3. Amination of unactivated sp3 C–H bonds
Some of the catalysts mentioned up to now were also active in promoting the amination of compounds containing unactivated sp3 C–H bonds such as alkanes (Scheme 1, path c). These reactions confirm the high reactivity of some metallo-imido complexes or metallo-azido adducts (Scheme 5) in transferring the nitrene functionality into C–H bonds with high dissociation energy. Moreover, the amination of alkanes by organic azides constitutes a cost-effective strategy to obtain, in one pot, nitrogen containing molecules.The syntheses of aza-derivatives shown in Chart 11 were performed in the presence of Rh2(OAc)4,47 Cu(I) complex 151,45 Fe(III)(F20-TPP)Cl32 or Ru(IV)(F20-TPP)Cl2 (ref. 29) as the catalyst.
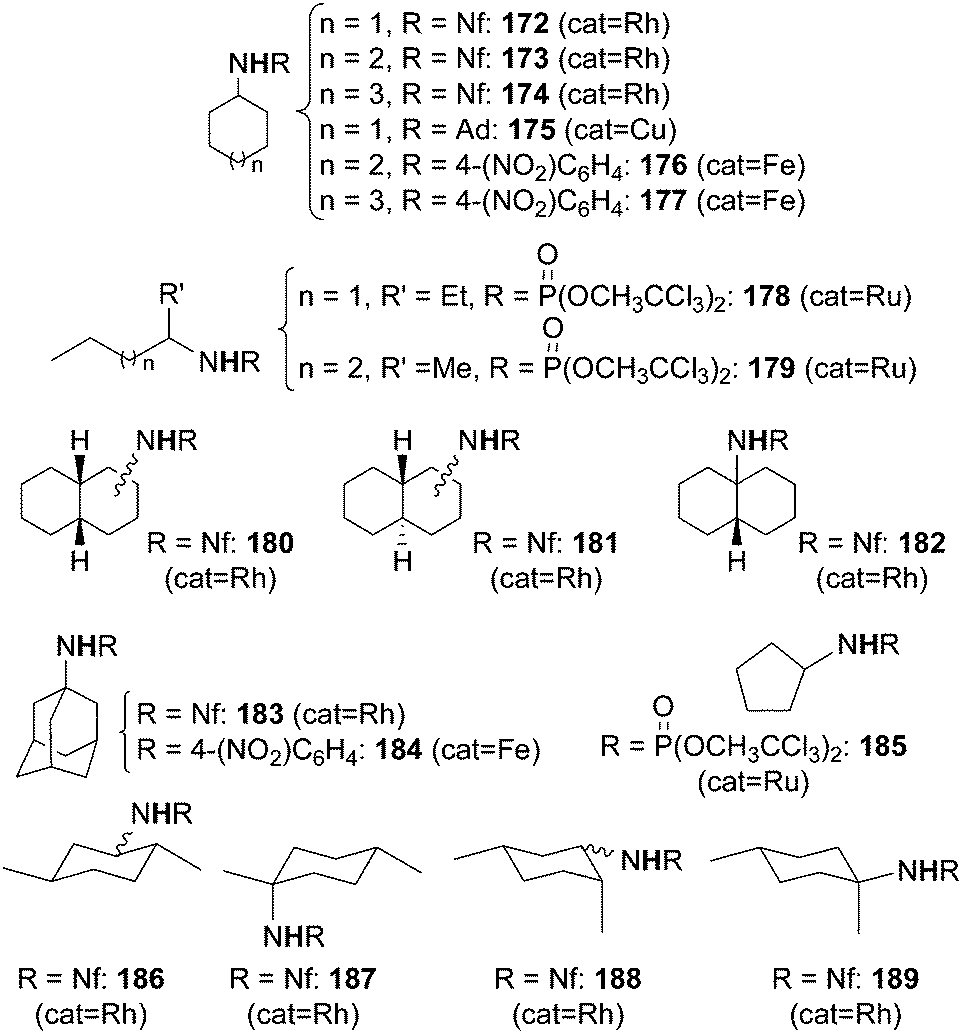 | ||
| Chart 11 Aminated compounds derived by the reaction of RN3 with alkanes. (Rh = Rh2(OAc)4, Cu = 151, Fe = Fe(III)(F20-TPP)Cl, Ru = Ru(IV)(F20-TPP)Cl2). | ||
It is difficult to compare the catalytic productivity of the different methodologies exploited for synthesising products of Chart 11 due to the diversity of employed experimental conditions. Generally speaking, to insert a nitrene functionality into a strong C–H bond of alkanes it is necessary to employ an azide which displays a strong electrophilic character. However, in the presence of the copper catalyst 151 (Scheme 13), also an alkyl azide such as adamathyl azide was reactive enough to aminate a cyclic alkane (Chart 11, product 175).
Very recently, Chang et al. reported on the catalytic activity of [IrCp*Cl2]2 (ref. 48) in the amination of ketoxime derivatives. The catalysis occurred in the presence of silver salt AgX which is necessary to form a cationic iridium complex as the catalytically active species (see paragraph 4.1.2. for the proposed mechanism). The reaction productivity was enhanced by adding acetate as an additive. The catalytic system [IrCp*Cl2]2/AgNTf2/AgOAc (Tf = triflate) was active in promoting the formation of a large class of compounds reported in Chart 12.
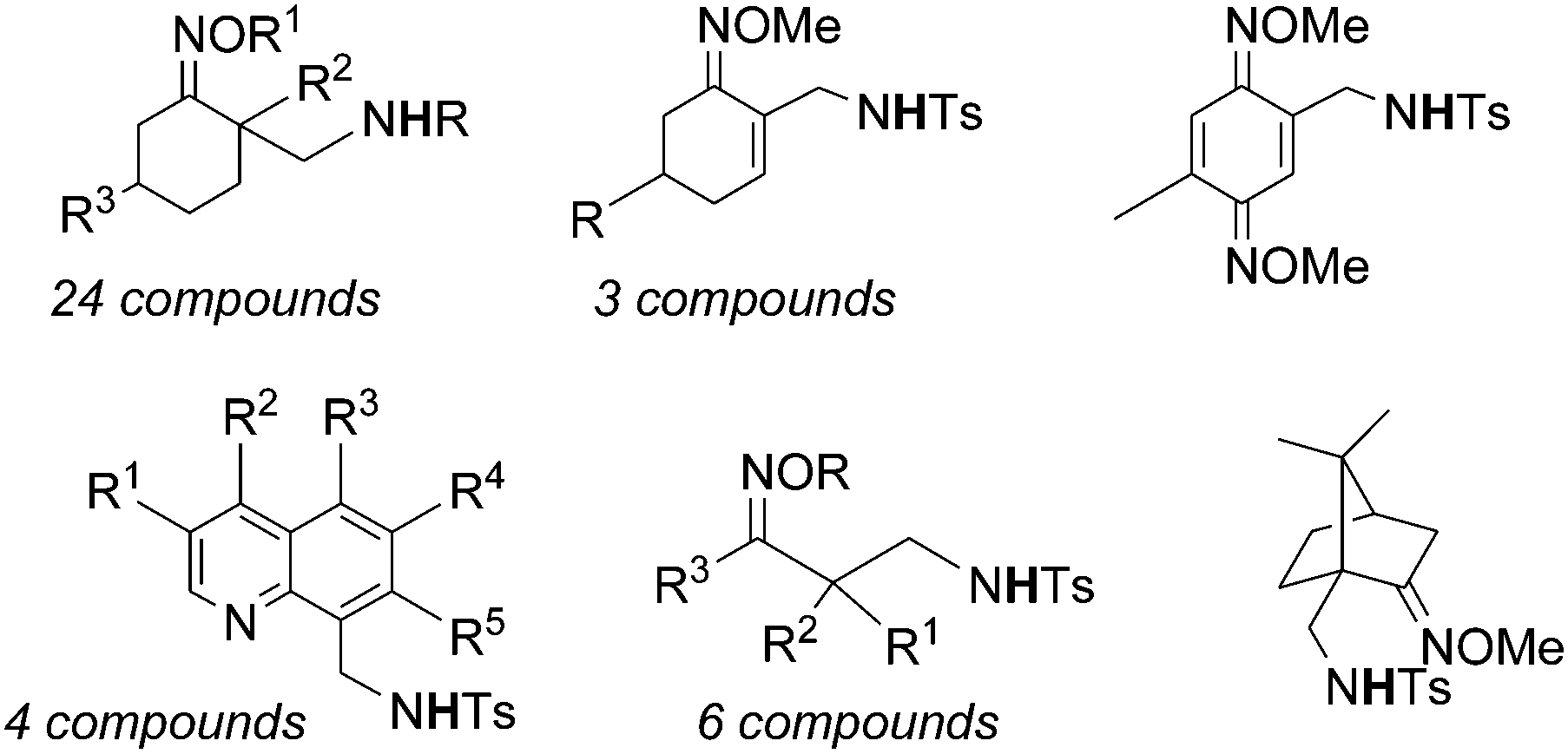 | ||
| Chart 12 Aminated compounds derived by the reaction of ketoximes with organic azides in the presence of the [IrCp*Cl2]2-based catalyst. | ||
The procedure reported above was also effective for late-stage C–H functionalisation of natural molecules with potential biological activities.
4. Amination of sp2 C–H bonds
4.1. Amination of aromatic sp2 C–H bonds
The amination of aromatic sp2 C–H bonds has been amply studied for more than ten years49–56 but only recently organic azides have been considered as direct aminating agents. The latter procedure allows the amination to proceed without the formation of waste products except for N2 (Scheme 1, path d). The presence of a directing group (DG) is always required (Scheme 15).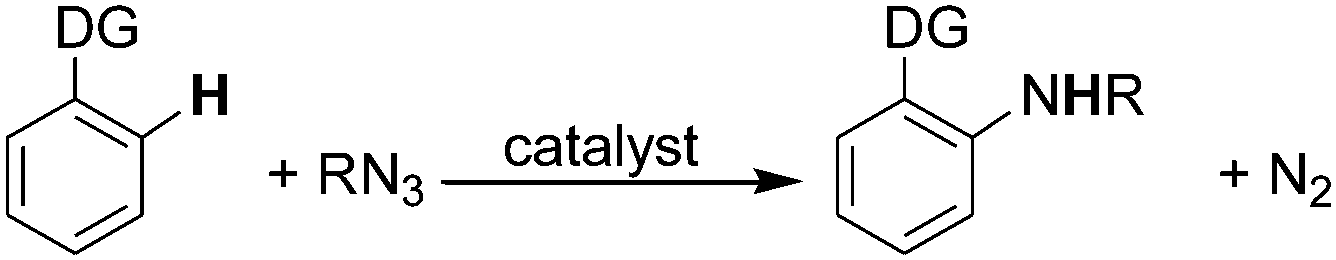 | ||
| Scheme 15 General scheme for the arylic amination by RN3. | ||
The catalysts employed to drive the reaction have a standard formula [M(L)Cl2]2 where M = Rh(III),57–63 Ru(II)63–65 and Ir(III).66,67 These catalysts were always employed in the presence of silver salt AgX which is able to generate the catalytic active cationic species [M(L)]X2 from the neutral precursors (see below for mechanistic studies).
In order to give the reader an overview of the synthetic potentiality of [RhCp*Cl2]2-based catalysis, several classes of aminated compounds are summarised in Chart 13. The chemical nature of all the substituents is not provided due to space constraints.
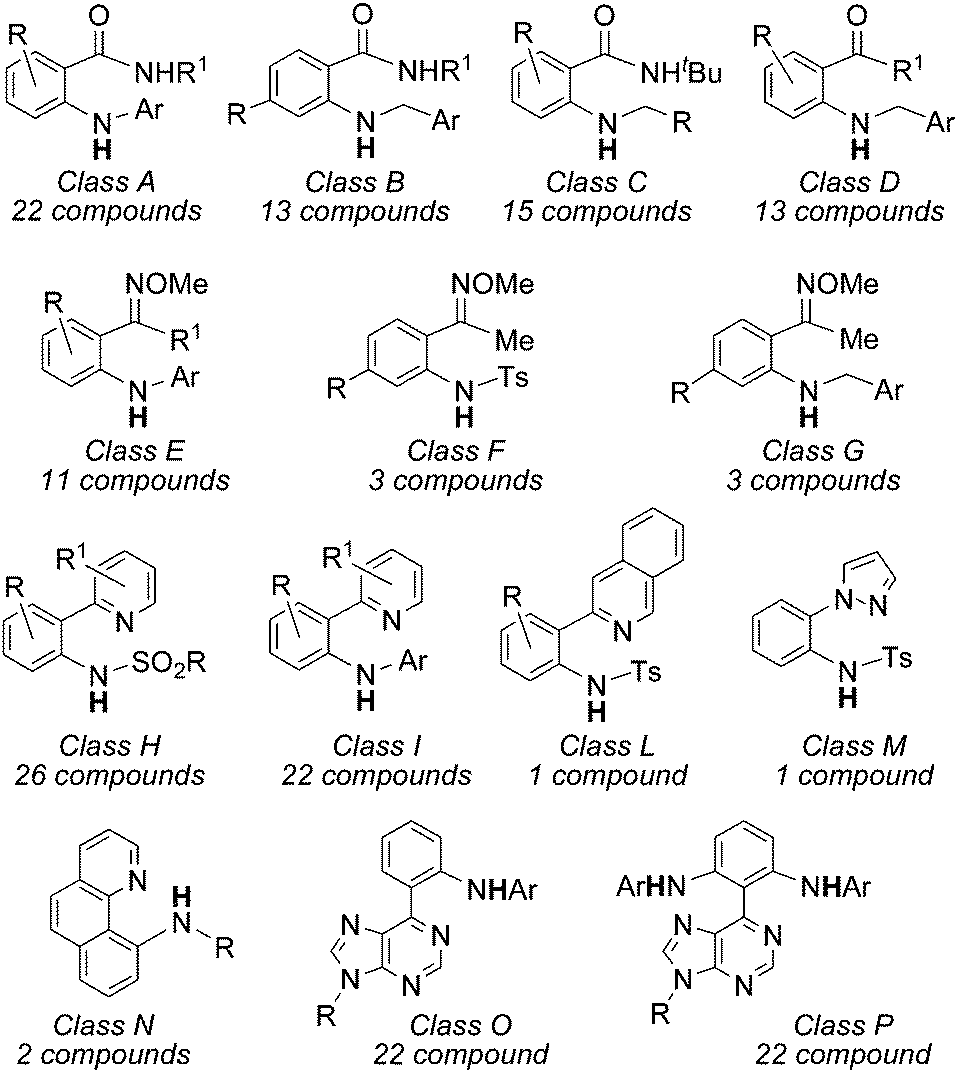 | ||
| Chart 13 Aza-compounds obtained by [RhCp*Cl2]2-catalysed reactions. | ||
Classes A, B and C have been obtained by reacting benzamide derivatives with aryl,57 benzyl60 and alkyl azides.60 Even if the reactivity of alkyl azides is generally modest in sp3 C–H aminations, all of these three classes of azides afforded high yields of desired compounds. The catalytic system also works well in the amination of aromatic ketones with satisfactory yields (class D).60
In view of positive synthetic results, Chang et al. turned their attention to the amination of aryl ketoximes and data indicated that the NOMe group is also very active as well to correctly drive the C–H amination (classes E, F and G).57,60
A large number of aza-derivatives were synthesised by using N-heterocyclic compounds as directing groups (DG) (classes H–N)58,59 and the best results were achieved by using azides bearing electron withdrawing groups (EWG). Interestingly, when purine was present on the aryl moiety as the DG,62 it was possible to isolate the mono and bis-aminated compounds depending on the experimental conditions employed (classes O and P).
Compounds belonging to classes A, D, H, M and F (Chart 13) were also obtained by using the [RuCl2(p-cymene)]2-based catalytic system. In 2013 two manuscripts by Chang et al.64 and Jiao et al.65 contemporarily appeared on this topic reporting almost identical results. The only difference between the two papers is the silver salt employed; AgNTf2 (Tf = triflate) was used by Chang et al. while Jiao et al. used AgSbF6. In both reported syntheses, sulfonyl azides were employed as aminating agents.
[RuCl2(p-cymene)]2/AgSbF6 combination in the presence of the acid additive 2-(NO2)C6H4COOH was also active in the amination of 2-phenyl piridines by using acyl azide as a nitrene source.68 Isolated compounds are reported in Chart 14.
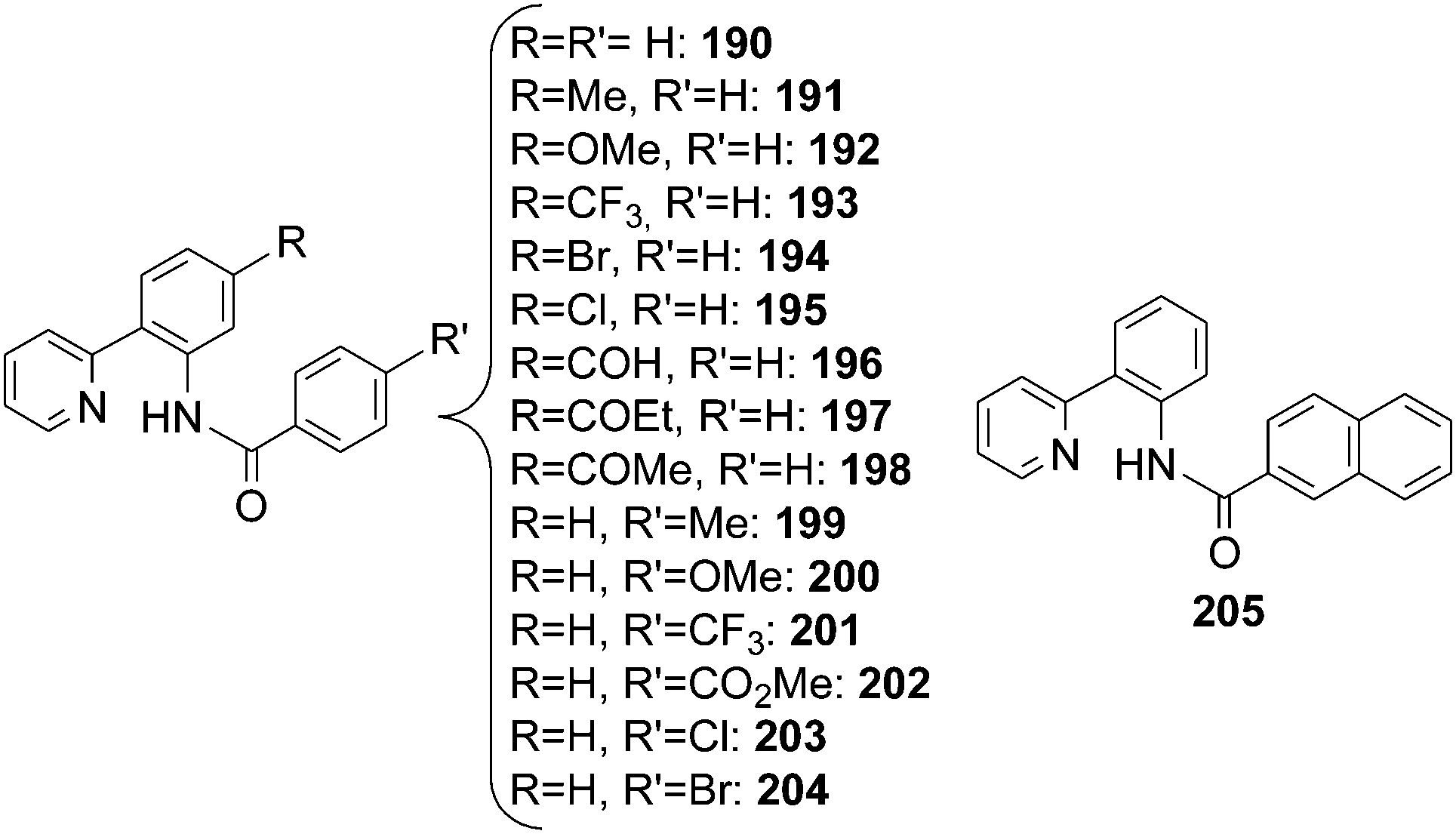 | ||
| Chart 14 Aza-compounds obtained by [RuCl2(p-cymene)]2 catalysed reactions. | ||
The synthetic strategy illustrated above for the amination of aromatic C–H bonds was further improved by employing milder experimental conditions in terms of temperatures and times of reactions and catalyst loading. This goal was reached by running aminations in the presence of [IrCp*Cl2]2/AgNTf2 catalytic systems.66,67 The amination of aryl benzamides by acyl azides was effective in obtaining twenty-five aza-derivatives and their general synthesis is reported in Scheme 16.66
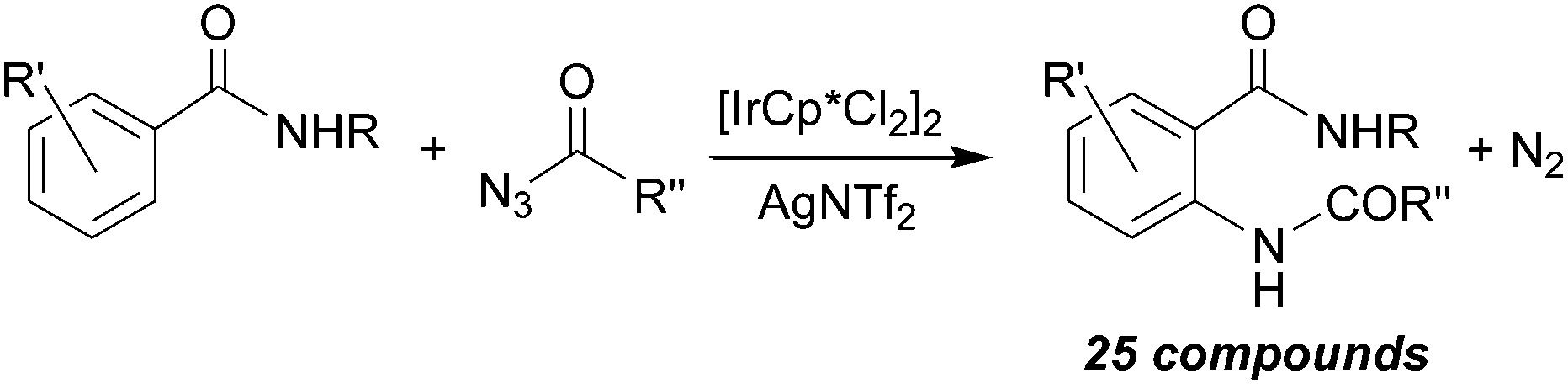 | ||
| Scheme 16 General reaction between aryl benzamides and acyl azides catalysed by the [IrCp*Cl2]2/AgNTf2 system. | ||
The authors also tested the applicability of the catalytic procedure to aminate aromatic compounds containing other directing groups, finding that this methodology displayed a general applicability and that it can also be exploited to synthesise natural molecules such as peptides.
To enlarge the reaction scope, the [IrCp*Cl2]2/AgNTf2 catalytic system was tested in amination reactions with sulfonyl or aryl azides as aminating agents. Several compounds belonging to almost all the classes reported in Chart 13 were obtained under mild experimental conditions, together with a new class of aza-derivatives achieved by aminating aryl carbamates with tosyl azide (Chart 15).
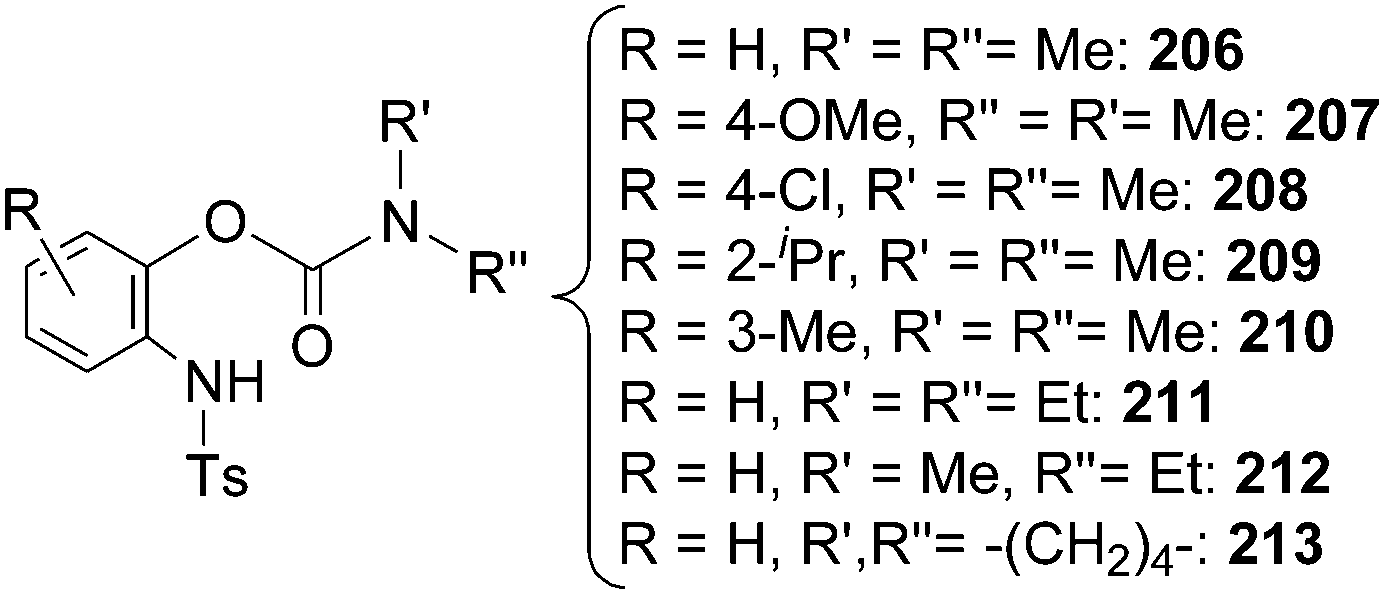 | ||
| Chart 15 Aza-derivatives obtained by the reaction of aryl carbamates with tosyl azide in the presence of the [IrCp*Cl2]2/AgNTf2 system. | ||
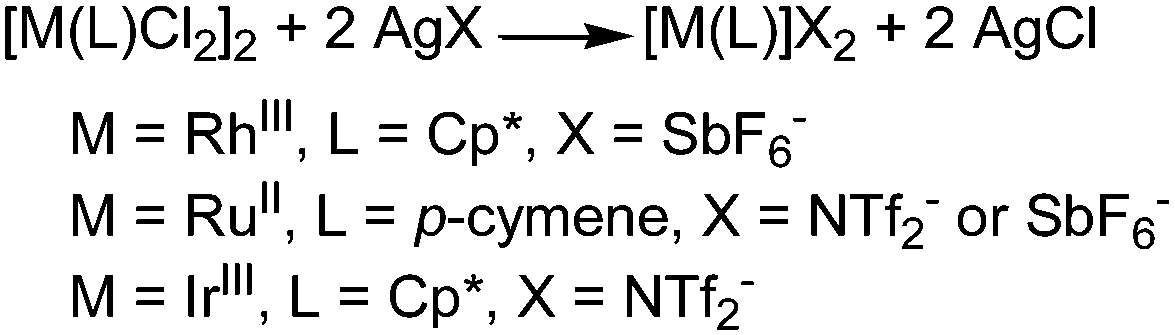 | ||
| Scheme 17 Formation of [M(L)]X2. | ||
For all catalytic systems presented in paragraph 4.1 it is possible to envisage a sequence of analogous mechanistic steps. First, the formed [M(L)]X2 complex can generate, by reaction with the aromatic substrate, a cationic metallacycle active species (I), which can coordinate the azide to promote the insertion of the nitrene unit into the aromatic C–H bond.
Scheme 18 provides a general mechanism which will be discussed in more depth for each catalytic system below.
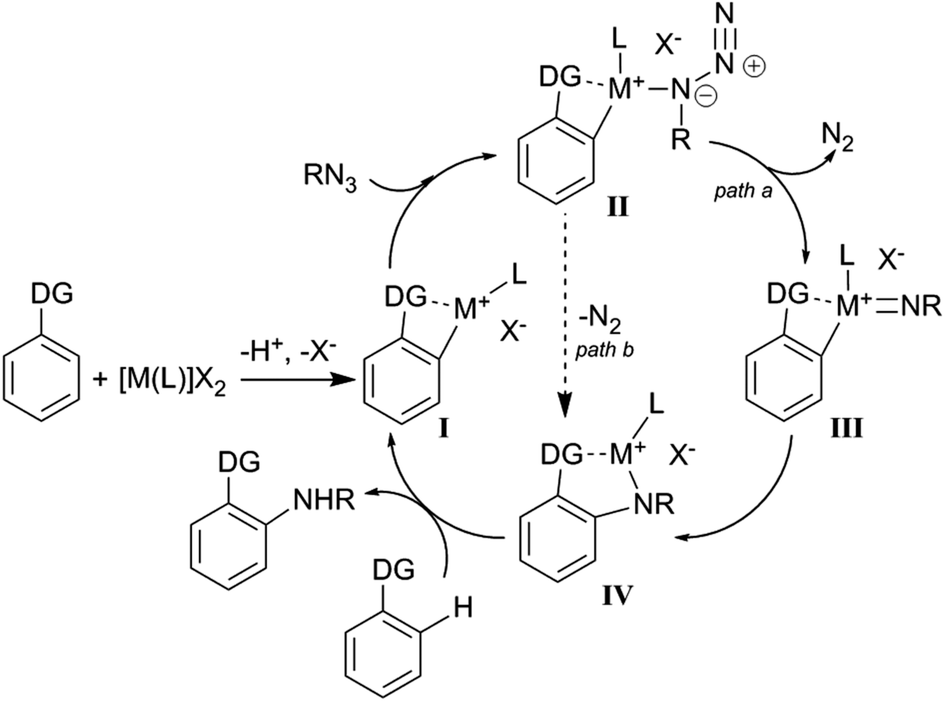 | ||
| Scheme 18 General mechanism for aminations catalysed by [M(L)Cl2]2/AgX. | ||
As shown in Scheme 18, the directing group (DG) plays a key role in the mechanism responsible for the formation of the metallacycle I. The unsaturated metallacycle I can coordinate the organic azide yielding IV either by formation of the metal-imido complex III (path a) or by directly releasing molecular nitrogen (path b). The reaction of IV with a proton source reforms the catalytic active species I with the contemporary formation of the aromatic amine.
The mechanism of the [RhCp*Cl2]2/AgSbF6 catalytic system was investigated by kinetic and theoretical studies and by isolating several intermediates.61 Complexes of type I and IV (Scheme 18) were isolated from the reaction of 2-phenyl pyridine with p-toluenesulfonazide (TsN3) and fully characterised also by X-ray diffraction analysis (Scheme 19).
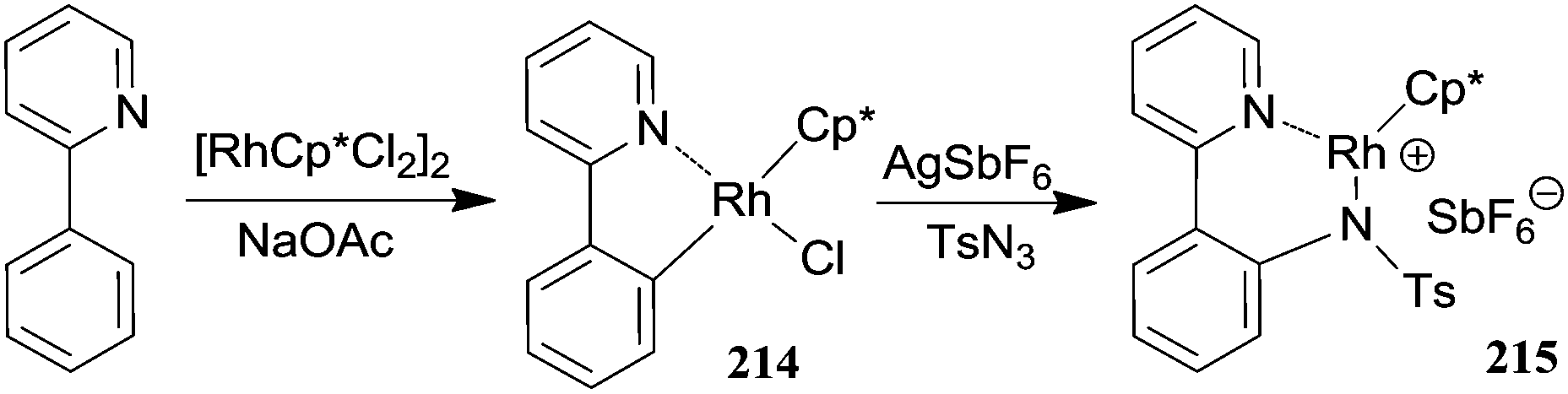
The involvement of complexes 214 and 215 in the catalytic cycle was supported by the catalytic activity of 214 in the amination of 2-phenyl pyridine by TsN3 and by the release of the aromatic amine from 215 when it was left reacting with a 2-phenyl pyridine excess as the proton source. This protodemetalation reaction was the last step of the cycle, being responsible for the regeneration of the catalyst I.
Considering that all the efforts to isolate or spectroscopically detect complexes of type II and III failed, Chang et al.61 performed a DFT study to shed some light on the catalytic mechanism. The theoretical study indicated rhodium(IV) imido complex formation as an energetically favourable process (path a, Scheme 18) and ruled out the direct conversion of I into IV by a concerted mechanism (path b, Scheme 18).
A kinetic isotope effect kH/kD (with values in the range 2–4) was observed in the amination of different substrates when they were tested in mixture with their deuterated form. These experiments indicated that the C–H bond cleavage was involved in the rate determining step of the cycle. An analogous mechanism was proposed by the same authors for the [IrIIICp*Cl2]2-catalysed reactions.66
Mechanistic studies conducted on [RhIIICp*Cl2]2-catalysed reactions laid the foundations to express an analogous mechanism for the aromatic amination catalysed by [RuIICl2(p-cymene)]2. The metallacycle 216 of type I (Scheme 18) was isolated and structurally characterised by Chang et al.64 from the reaction of 2-(p-tolyl)pyridine with [RuIICl2(p-cymene)]2. The treatment of 216 with sulfonyl azide gave the corresponding amido complex 217 which was detected by mass spectroscopy (Scheme 20).
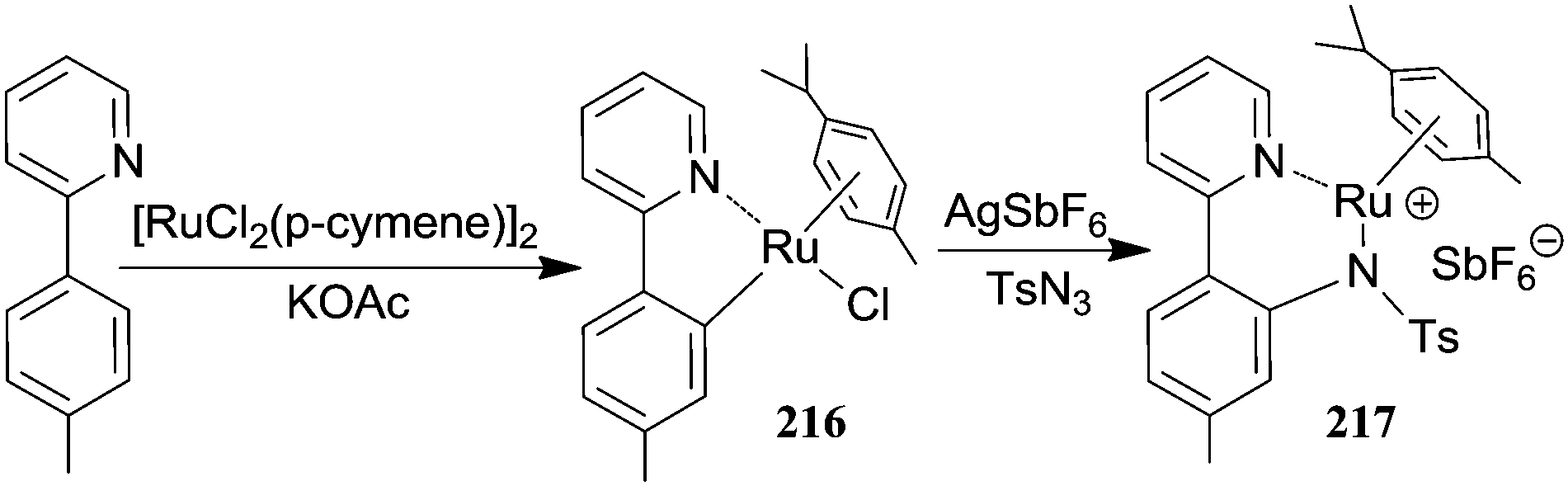 | ||
| Scheme 20 Synthesis of complexes 216 and 217. | ||
Also in this mechanistic investigation a kinetic isotope effect was registered indicating that the C–H cleavage is the rate limiting step of the process. The mechanistic analogies between ruthenium(II) and rhodium(III) catalysed processes allow the authors to assume equivalent catalytic cycles for both aminations of aromatic C–H bonds (Scheme 18).
4.2. Amination of sp2 C–H bonds of indoles and aldehydes
The reaction of indoles with organic azides afforded different compounds depending on the nature of the catalyst employed. When the [RhCp*Cl2]2/AgSbF6 catalytic system was used, the insertion of the nitrene moiety from an aryl azide was observed at the C2 position of the indole skeleton.57 The same position was also aminated by sulfonyl azides in the presence of [Cp*Rh(MeCN)3](SbF6)2 as the catalyst.69 In Chart 16 the obtained C2 aminated indoles are displayed.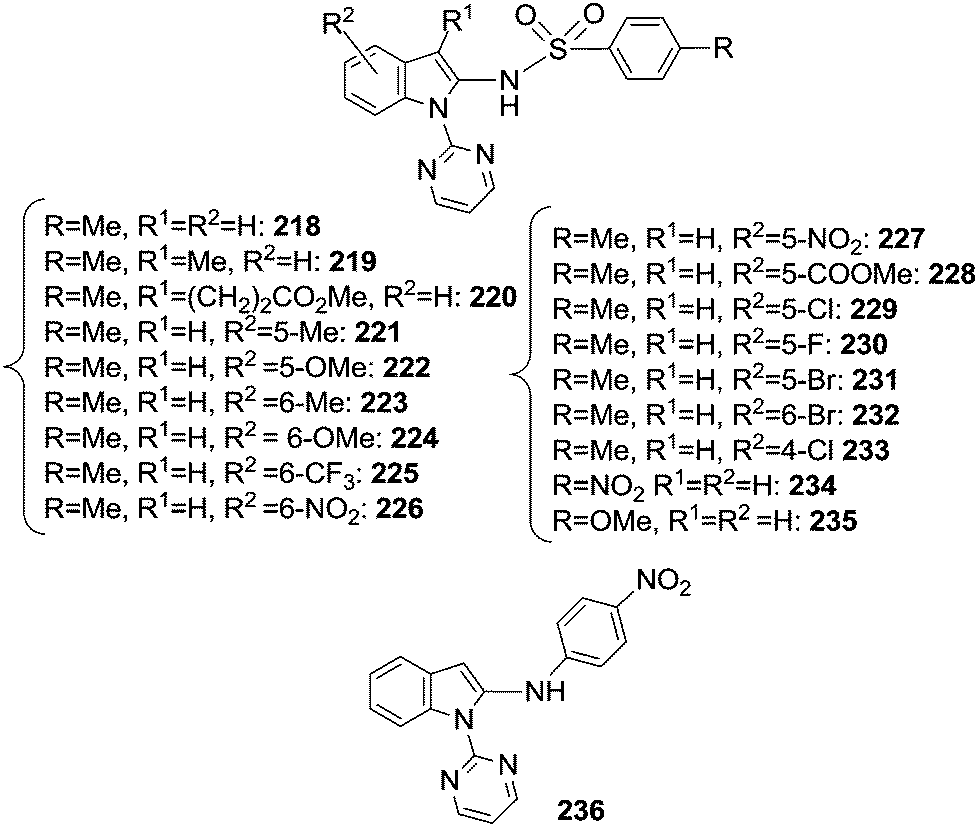 | ||
| Chart 16 C2 aminated indoles. | ||
It should be noted that the reaction of indoles with aromatic azides did not afford the corresponding aminated compounds when the catalyst [RhCp*Cl2]2/AgSbF6 was replaced by [Cp*Rh(MeCN)3](SbF6)2. In both cases, the DG pyrimidyl group can easily be removed yielding a wide class of 2-amino substituted indoles.
A different reactivity of aromatic azides towards indoles was observed by Che et al. by performing reactions in the presence of Ru(TPP)CO as the catalyst; the diimination of indoles was achieved instead of the formation of C2 aminated compounds.70 The study of the reaction scope allowed the synthesis of twenty differently substituted indoles (Chart 17).
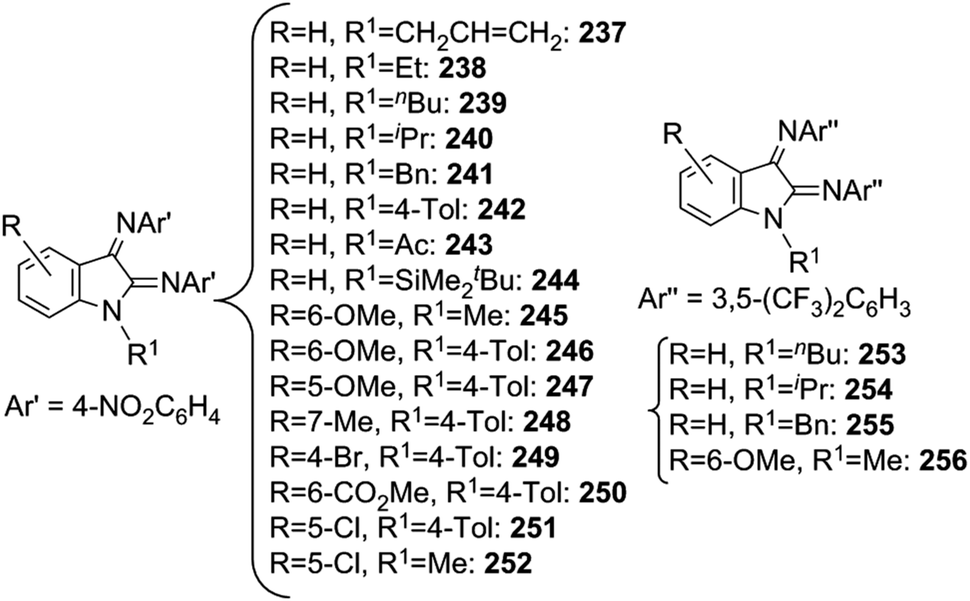 | ||
| Chart 17 Ru(TTP)CO-catalysed diimination of indoles. | ||
2,3-Diimination products were the only obtained aminated compounds and the monoiminated or aminated indole was neither obtained nor detected when the reaction was also performed by using an excess of indole.
A preliminary mechanistic study indicated that, when 3,5-(CF3)2C6H3N3 was used as the aromatic azide, the bis-imido ruthenium(VI) porphyrin 10328,36 can be suggested as the catalytic active intermediate.
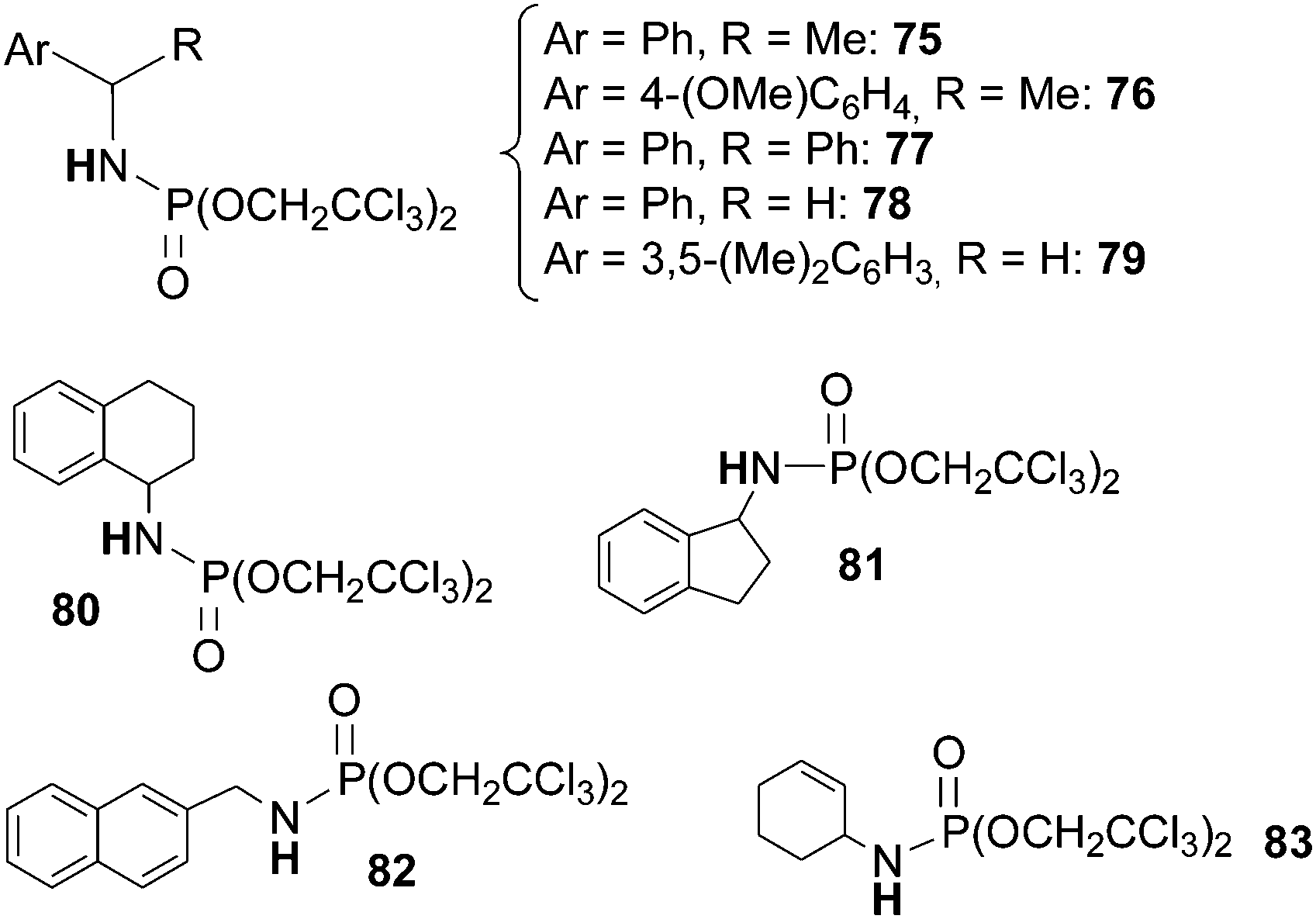
Organic azides are also competent aminating agents of sp2 C–H bonds of aldehydes. In 2008 Cundari and Warren et al.45 reported on the catalytic activity of complex 151 in the amidation of benzaldehyde with AdN3 (Scheme 21).
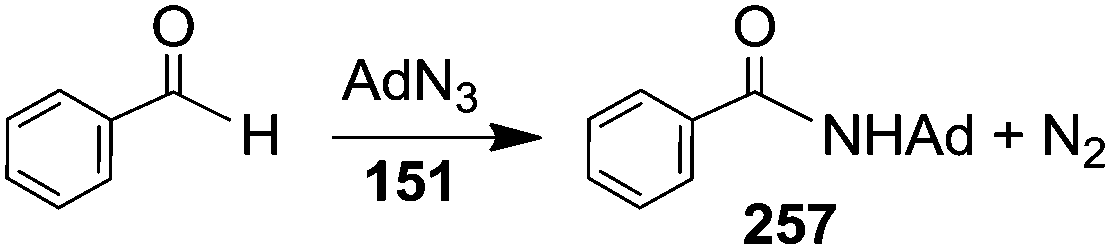 | ||
| Scheme 21 Amidation of benzaldehyde by AdN3 catalysed by 152. | ||
Some years later, Che et al.71 reported on the catalytic activity of the Ru(IV)(TPP)Cl2 complex in the same reaction by using phosphoryl azide as a nitrene source. The procedure was effective with several aldehydes and azides affording a large class of derivatives (Chart 18).
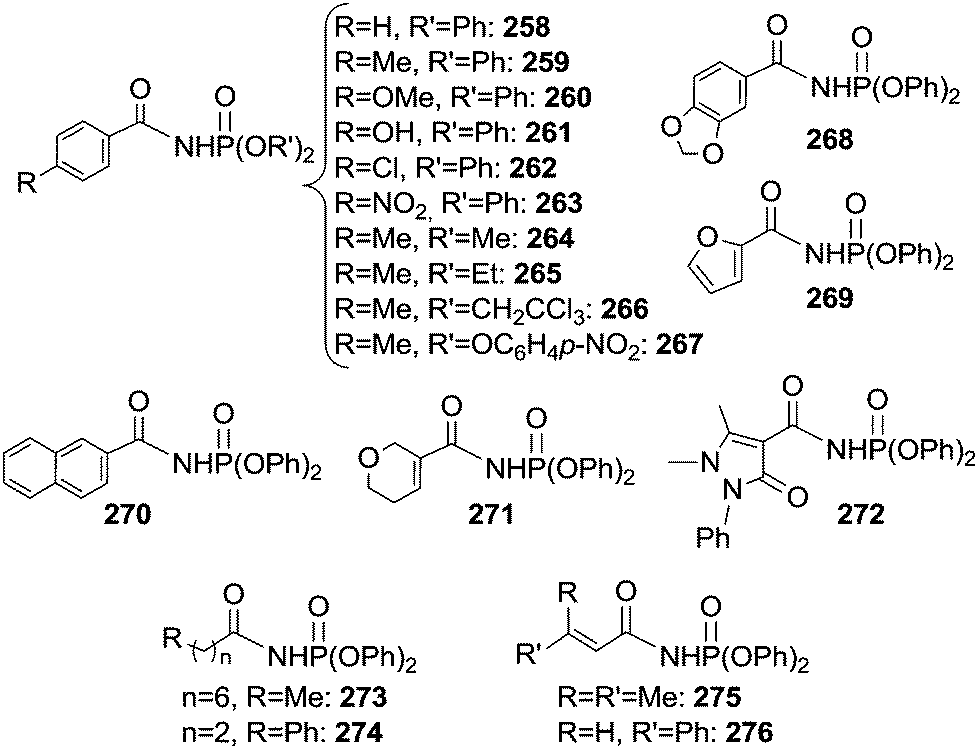 | ||
| Chart 18 Aza-compounds obtained by the reaction of aldehydes with phosphoryl azides in the presence of Ru(IV)(TPP)Cl2. | ||
The deuterium kinetic isotope effect for the reaction of benzaldehyde, benzaldehyde-d6 and N3P(O)(OCH2CCl3)2 gave a kH/kD value of 4.1 indicating that the C–H bond cleavage is the rate determining step of the catalysis.
5. Conclusions
The aim of this feature article is to present the state of the art on the use of organic azides as efficient reagents for intermolecular amination of C–H bonds. The incredible number of reported aza-compounds points out the great versatility of this class of aminating agents, which are currently receiving great interest from the scientific community.Several catalysts have been tested and in some cases complementary results have been reached in the presence of different catalytic systems. Therefore, it is possible to afford structurally dissimilar aza-derivatives starting from similar starting materials. Many efforts have been made to optimise catalytic protocols and to understand mechanisms with the belief that it is always necessary to have knowledge of catalytic pathways for planning more efficient synthetic procedures.
Considering the social request for eco-friendly chemical processes to synthesise fine chemicals, we strongly believe that organic azides are going to occupy a prominent role as aminating reagents in the near future. The production of benign N2 as the only by-product, coupled with the high atom efficiency and selectivity of nitrene transfer reactions, is an ideal starting point to design new and more sustainable synthetic procedures.
Notes and references
- P. Griess, Proc. R. Soc. London, 1864, 13, 375–384 CrossRef.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- Organic Azides Syntheses and Applications, ed. S. Bräse and K. Banert, John Wiley & Sons Ltd., 2010 Search PubMed.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- T. Uchida and T. Katsuki, Chem. Rec., 2014, 14, 117–129 CrossRef CAS PubMed.
- S. Chiba, Synlett, 2012, 21–44 CrossRef CAS PubMed.
- T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831–3846 CAS.
- M. Minozzi, D. Nanni and P. Spagnolo, Chem. – Eur. J., 2009, 15, 7830–7840 CrossRef CAS PubMed.
- T. Katsuki, Chem. Lett., 2005, 1304–1309 CrossRef CAS.
- S. Cenini, E. Gallo, A. Caselli, F. Ragaini, S. Fantauzzi and C. Piangiolino, Coord. Chem. Rev., 2006, 250, 1234–1253 CrossRef CAS PubMed.
- S. Fantauzzi, A. Caselli and E. Gallo, Dalton Trans., 2009, 5434–5443 RSC.
- T. G. Driver, Nat. Chem., 2013, 5, 736–738 CrossRef CAS PubMed.
- P. Panchaud, L. Chabaud, Y. Landais, C. Ollivier, P. Renaud and S. Zigmantas, Chem. – Eur. J., 2004, 10, 3606–3614 CrossRef CAS PubMed.
- S. Cenini, F. Ragaini, E. Gallo and A. Caselli, Curr. Org. Chem., 2011, 15, 1578–1592 CrossRef CAS.
- B. J. Stokes and T. G. Driver, Eur. J. Org. Chem., 2011, 4071–4088 CrossRef CAS.
- G. Smolinsky, J. Am. Chem. Soc., 1961, 83, 2489–2493 CrossRef CAS.
- K. R. Henery-Logan and R. A. Clark, Tetrahedron Lett., 1968, 801–806 CrossRef CAS.
- R. A. Abramovitch and E. P. Kyba, J. Am. Chem. Soc., 1974, 96, 480–488 CrossRef CAS.
- E. F. V. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297–368 CrossRef CAS.
- H. Kwart and A. A. Khan, J. Am. Chem. Soc., 1967, 89, 1951–1953 CrossRef CAS.
- S. Cenini, E. Gallo, A. Penoni, F. Ragaini and S. Tollari, Chem. Commun., 2000, 2265–2266 RSC.
- F. Ragaini, A. Penoni, E. Gallo, S. Tollari, C. Li Gotti, M. Lapadula, E. Mangioni and S. Cenini, Chem. – Eur. J., 2003, 9, 249–259 CrossRef CAS PubMed.
- A. Caselli, E. Gallo, F. Ragaini, F. Ricatto, G. Abbiati and S. Cenini, Inorg. Chim. Acta, 2006, 359, 2924–2932 CrossRef CAS PubMed.
- A. Caselli, E. Gallo, S. Fantauzzi, S. Morlacchi, F. Ragaini and S. Cenini, Eur. J. Inorg. Chem., 2008, 3009–3019 CrossRef CAS.
- M. Weber, G. Yilmaz and G. Wille, Chim. Oggi, 2011, 29, 8–10 CAS.
- H.-J. Lu, V. Subbarayan, J.-R. Tao and X. P. Zhang, Organometallics, 2010, 29, 389–393 CrossRef CAS.
- D. Intrieri, A. Caselli, F. Ragaini, S. Cenini and E. Gallo, J. Porphyrins Phthalocyanines, 2010, 14, 732–740 CrossRef CAS.
- D. Intrieri, A. Caselli, F. Ragaini, P. Macchi, N. Casati and E. Gallo, Eur. J. Inorg. Chem., 2012, 569–580 CrossRef CAS.
- W. Xiao, J. Wei, C.-Y. Zhou and C.-M. Che, Chem. Commun., 2013, 49, 4619–4621 RSC.
- K.-H. Chan, X. Guan, V. K.-Y. Lo and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 2982–2987 CrossRef CAS PubMed.
- P. Zardi, A. Caselli, P. Macchi, F. Ferretti and E. Gallo, Organometallics, 2014, 33, 2210–2218 CrossRef CAS.
- Y. Liu and C.-M. Che, Chem. – Eur. J., 2010, 16, 10494–10501 CrossRef CAS PubMed.
- S.-M. Au, J.-S. Huang, W.-Y. Yu, W.-H. Fung and C.-M. Che, J. Am. Chem. Soc., 1999, 121, 9120–9132 CrossRef CAS.
- S. K.-Y. Leung, W.-M. Tsui, J.-S. Huang, C.-M. Che, J.-L. Liang and N. Zhu, J. Am. Chem. Soc., 2005, 127, 16629–16640 CrossRef CAS PubMed.
- Z. Guo, X. Guan, J.-S. Huang, W.-M. Tsui, Z. Lin and C.-M. Che, Chem. – Eur. J., 2013, 19, 11320–11331 CrossRef CAS PubMed.
- S. Fantauzzi, E. Gallo, A. Caselli, F. Ragaini, N. Casati, P. Macchi and S. Cenini, Chem. Commun., 2009, 3952–3954 RSC.
- G. Manca, E. Gallo, D. Intrieri and C. Mealli, ACS Catal., 2014, 4, 823–832 CrossRef CAS.
- V. Lyaskovskyy, A. I. O. Suarez, H. Lu, H. Jiang, X. P. Zhang and B. de Bruin, J. Am. Chem. Soc., 2011, 133, 12264–12273 CrossRef CAS PubMed.
- P. Zardi, D. Intrieri, A. Caselli and E. Gallo, J. Organomet. Chem., 2012, 716, 269–274 CrossRef CAS PubMed.
- K. Omura, M. Murakami, T. Uchida, R. Irie and T. Katsuki, Chem. Lett., 2003, 354–355 CrossRef CAS.
- Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2013, 52, 1739–1742 CrossRef CAS PubMed.
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 CrossRef CAS PubMed.
- E. T. Hennessy, R. Y. Liu, D. A. Iovan, R. A. Duncan and T. A. Betley, Chem. Sci., 2014, 5, 1526–1532 RSC.
- Y. M. Badiei, A. Krishnaswamy, M. M. Melzer and T. H. Warren, J. Am. Chem. Soc., 2006, 128, 15056–15057 CrossRef CAS PubMed.
- Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961–9964 CrossRef CAS PubMed.
- T. R. Cundari, A. Dinescu and A. B. Kazi, Inorg. Chem., 2008, 47, 10067–10072 CrossRef CAS PubMed.
- J. R. Suarez and J. L. Chiara, Chem. Commun., 2013, 49, 9194–9196 RSC.
- T. Kang, Y. Kim, D. Lee, Z. Wang and S. Chang, J. Am. Chem. Soc., 2014, 136, 4141–4144 CrossRef CAS PubMed.
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed.
- F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969–5970 CrossRef CAS.
- A. S. Guram and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 7901–7902 CrossRef CAS.
- M. Kosugi, M. Kameyama and T. Migita, Chem. Lett., 1983, 927–928 CrossRef CAS.
- M. Kim and S. Chang, Org. Lett., 2010, 12, 1640–1643 CrossRef CAS PubMed.
- R. T. Gephart and T. H. Warren, Organometallics, 2012, 31, 7728–7752 CrossRef CAS.
- M. Zhang and A. Zhang, Synthesis, 2012, 1–14 CrossRef PubMed.
- J. E. R. Sadig and M. C. Willis, Synthesis, 2011, 1–22 CAS.
- J. Ryu, K. Shin, S. H. Park, J. Y. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 9904–9908 CrossRef CAS PubMed.
- J. Y. Kim, S. H. Park, J. Ryu, S. H. Cho, S. H. Kim and S. Chang, J. Am. Chem. Soc., 2012, 134, 9110–9113 CrossRef CAS PubMed.
- C. Tang, Y. Yuan, Y. Cui and N. Jiao, Eur. J. Org. Chem., 2013, 7480–7483 CrossRef CAS.
- K. Shin, Y. Baek and S. Chang, Angew. Chem., Int. Ed., 2013, 52, 8031–8036 CrossRef CAS PubMed.
- S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492–2502 CrossRef CAS PubMed.
- H. J. Kim, M. J. Ajitha, Y. Lee, J. Ryu, J. Kim, Y. Lee, Y. Jung and S. Chang, J. Am. Chem. Soc., 2014, 136, 1132–1140 CrossRef CAS PubMed.
- K. Shin, J. Ryu and S. Chang, Org. Lett., 2014, 16, 2022–2025 CrossRef CAS PubMed.
- J. Kim, J. Kim and S. Chang, Chem. – Eur. J., 2013, 19, 7328–7333 CrossRef CAS PubMed.
- Q.-Z. Zheng, Y.-F. Liang, C. Qin and N. Jiao, Chem. Commun., 2013, 49, 5654–5656 RSC.
- J. Ryu, J. Kwak, K. Shin, D. Lee and S. Chang, J. Am. Chem. Soc., 2013, 135, 12861–12868 CrossRef CAS PubMed.
- D. Lee, Y. Kim and S. Chang, J. Org. Chem., 2013, 78, 11102–11109 CrossRef CAS PubMed.
- K. Shin, J. Ryu and S. Chang, Org. Lett., 2014, 16, 2022–2025 CrossRef CAS PubMed.
- J. Shi, B. Zhou, Y. Yang and Y. Li, Org. Biomol. Chem., 2012, 10, 8953–8955 CAS.
- J. Wei, W. Xiao, C.-Y. Zhou and C.-M. Che, Chem. Commun., 2014, 50, 3373–3376 RSC.
- W. Xiao, C.-Y. Zhou and C.-M. Che, Chem. Commun., 2012, 48, 5871–5873 RSC.
| This journal is © The Royal Society of Chemistry 2014 |