 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
(Electro)catalytic C–C bond formation reaction with a redox-active cobalt complex†
Margarethe
van der Meer
a,
Yvonne
Rechkemmer
b,
Irina
Peremykin
b,
Stephan
Hohloch
a,
Joris
van Slageren
*b and
Biprajit
Sarkar
*a
aInstitut für Chemie und Biochemie, Anorganische Chemie, Freie Universität Berlin, Fabeckstraße 34-36, D-14195, Berlin, Germany. E-mail: biprajit.sarkar@fu-berlin.de
bInstitut für Physikalische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569, Stuttgart, Germany
First published on 29th May 2014
Abstract
Cooperativity between cobalt and non-innocent ligands in electron transfer processes has been utilized for (electro)catalytic C–C bond formation reactions.
Redox-active ligands have long fascinated coordination chemists because of the unusual and curious electronic structures of their metal complexes.1 More recently chemists have started developing novel catalysts based on metal complexes of non-innocent ligands.2 One feature of redox-active ligands which is often utilized in such catalytic processes is their active participation in various electron transfer steps. Quinones are an important class of redox-active ligands, and historically metal complexes of the all oxygen donating o-quinone ligand have been the most studied.1b More recently, metal complexes of o-iminoquinones have been thoroughly investigated.1c,3 Such [O] for [NR] isoelectronic substitutions often induce novel properties in the metal complexes; redox-tuning, and steric protection through the R groups being obvious gains of the approach.3do-Diiminoquinones where both the [O] groups have been replaced by [NR] have found rather limited use.4 Some such [NR] containing ligands used for synthesizing four-coordinate cobalt complexes have been listed in Fig. 1.4,5 We have been pursuing the development of new redox-active ligands where [O] donors are replaced by their isoelectronic [NR] counterpart. Such a substitution often turns out to be immensely helpful while using metal complexes of these ligands for various catalytic transformations.6 Herein we present the cobalt complex 1 with the ligand Q˙−. Geometric and electronic structure of this complex has been probed by a battery of methods, and its utility as an (electro)catalyst for C–C bond formation reactions has been investigated.
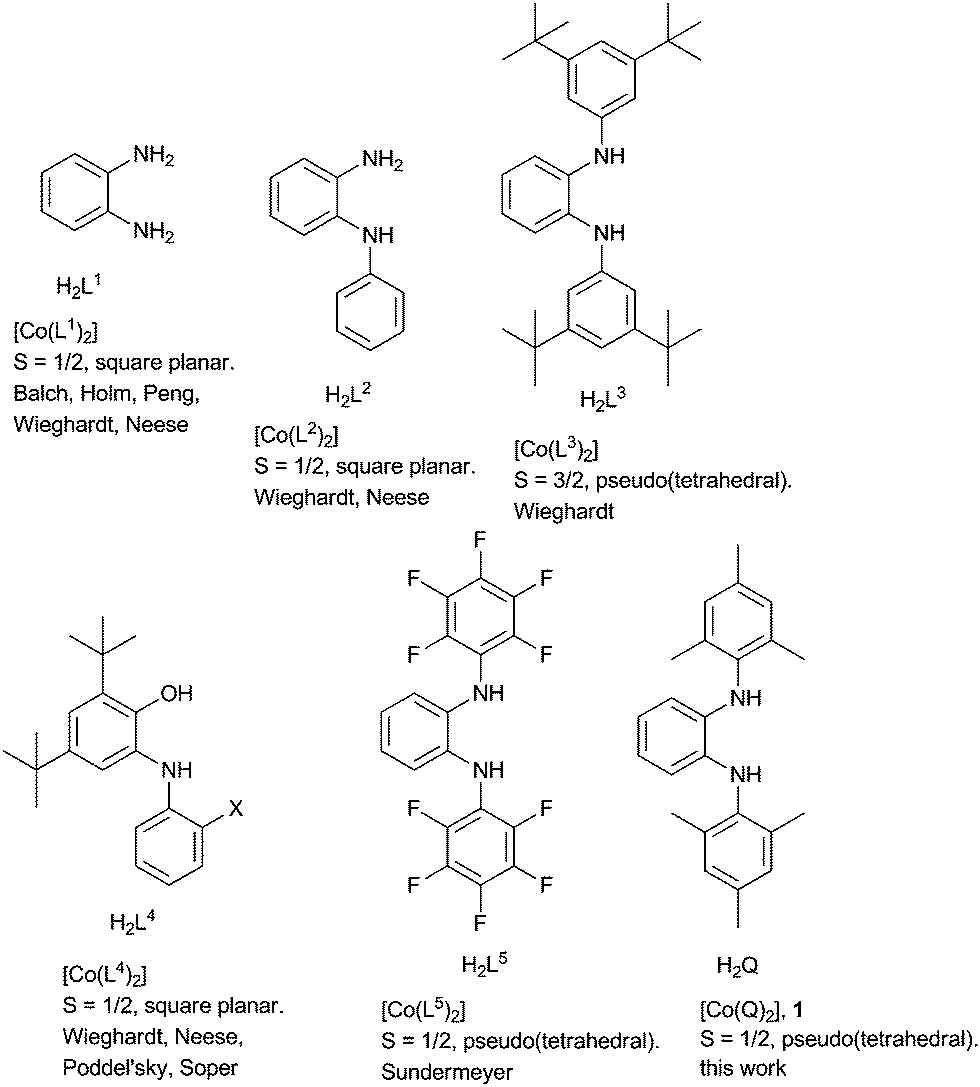 | ||
| Fig. 1 Selected redox-active ligands with which four-coordinate cobalt complexes have been reported together with the geometry and spin-states of those cobalt complexes. | ||
The complex 1 was synthesized by deprotonation of H2Q with n-BuLi, its reaction with CoCl2, and the subsequent oxidation of the reaction mixture with pure O2. Re-crystallization provided 1 in the pure form. The geometric structure of 1, which crystallizes as a CH2Cl2 solvate, was determined by single crystal X-ray diffraction studies. The cobalt center in 1 is in a (pseudo)tetrahedral environment; being coordinated by the nitrogen donor of two different ligands (Fig. 2).
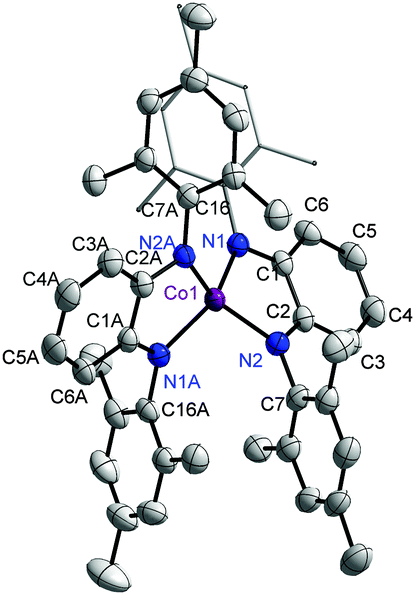 | ||
| Fig. 2 ORTEP plot of 1. Ellipsoids are drawn at 50% probability. Hydrogen atoms and solvent molecules have been left out for clarity. | ||
The τ value for this structure is 0.46. The dihedral angle between the two coordinating planes is 50°. The Co–N bond distances of about 1.92 Å points to a high spin (HS) Co(II) center. The intra-ring C–C bond display a quinoidal distortion with two short and four long bonds (Table S2, ESI†). The C–N bond lengths of 1.36 Å also points to the existence of these ligands as Q˙−, and hence neutral 1 can be formulated as [(Q˙−)CoII,HS(Q˙−)]. However, the difference in intra-ring bond lengths for the various redox forms of ligands with all nitrogen donors are known to be less compared to the various redox-forms of their [O,O] and [O,N] counterparts.7 Additionally, work by the groups of Wieghardt and Neese4c–e have shown the existence of valence ambiguity in the corresponding cobalt complex with [O,N] and [N,N] donor ligands (Fig. 1). With this background in mind, the temperature dependence of the magnetic susceptibility of 1 was investigated. The results resemble those of Sundermeyer et al.,4a indicating an S = 1/2 ground state due to strong antiferromagnetic exchange interactions between the central (HS) Co(II) ion and the surrounding radical ligands (Fig. S1, ESI†). The room-temperature effective magnetic moment was determined to be 2.47 μB. Furthermore, the strong g value anisotropy found in the 5 K EPR spectra of 1 both in the solid state and in frozen solution confirmed a cobalt-centered S = 1/2 spin. Best fits were obtained with g∥ = 4.0 ± 0.08, g⊥ = 1.78 ± 0.01 for the solid and g∥ = 3.75 ± 0.08, g⊥ = 1.80 ± 0.02 as well as a hyperfine coupling constant of A∥ = 850 ± 30 MHz for the frozen solution. (Fig. S2, ESI†). This combined approach delivered results that are best interpreted by considering the resonance form [(Q˙−)CoII,HS(Q˙−)]. Thus, it is seen that there are no simple correlations between ligand type, geometry at the metal center, and spin states for these classes of cobalt complexes (ca.Fig. 1).
Cyclic voltammogram of 1 in CH2Cl2/0.1 M Bu4NPF6 shows two reversible one-electron oxidation waves at −0.21 and 0.30 V and a reversible one-electron reduction wave at −1.47 V vs. Fc/Fc+ (Fig. S3, ESI†). A second reduction wave is also observed in CH2Cl2, which shows the onset of a catalytic current. This observation suggests that complex 1 is capable of activating the C–Cl bonds of CH2Cl2 catalytically. We note that most complexes listed in Fig. 1 show reversible reduction steps in dichloromethane, indicating that they do not react with CH2Cl2. Only the reduced form of complex [Co(L4)2] has been showed to perform C–Cl activation by Soper et al.5a Subtle substitution changes on the ligand backbone is thus shown to dramatically influence redox-induced chemical reactivity in these complexes. To prove the hypothesis of a catalytic reaction of complex 1 with CH2Cl2 following the second reduction step, we recorded the cyclic voltammogram of this complex in THF. Gratifyingly, both reduction waves are electrochemically reversible in THF (Fig. S4, ESI†). Reversibility was proven by comparing forward and reverse current heights (which is close to 1), and by plotting the square root of the scan rate versus the current peak. The latter plot is linear proving that reduction proceeds reversibly under diffusion control in THF (Fig. S5, ESI†). The oxidation waves become irreversible in THF. This is a possible indication of oxidation-induced binding of THF to the cobalt center. Such five-coordinated cobalt complexes have been previously isolated as oxidation products of the neutral four-coordinate cobalt complexes.4
Inspired by the catalytic wave observed in CH2Cl2 after the second reduction of 1, we wanted to investigate the utility of 1 as an (electro)catalyst for C–C bond formation reactions. To test this, aliquots of benzyl bromide was added to a solution of 1 in THF. Each additional amount of benzyl bromide resulted in further increase in the catalytic current (Fig. 3), proving a catalytic reaction of 1 with benzyl bromide after the second reduction wave.
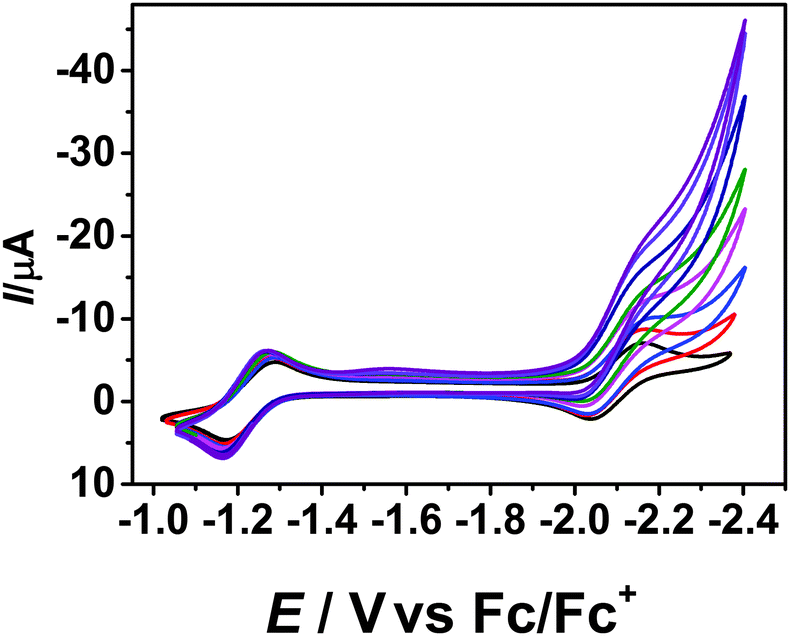 | ||
| Fig. 3 Cyclic voltammogram of 1 with the addition of benzyl bromide. THF/0.1 M Bu4NPF6, 298 K, 100 mV s−1. | ||
It should be noted, that the addition of benzyl bromide does not lead to a change in the intensity or position of the first reduction wave. The catalytic current was determined for each wave after the addition of benzyl bromide. A plot of icat/ipversus benzyl bromide concentration delivers a linear fit (Fig. S6, ESI†). Additionally, the plot of icat/ipversus the square root of the concentration of benzyl bromide is linear as well, proving that the reaction is first order in benzyl bromide concentration (Fig. S7, ESI†). Using the icat/ip ratio, we have calculated the observed rate constant (kobs) at various scan rates. kobs is seen to be dependent on scan rate, with a stronger dependence observed at lower scan rates (Fig. S8, ESI†). This observation is consistent with the formation of the catalytically active species after the second reduction process. At a scan rate of 100 mV s−1, the value of kobs is approximately 10 s−1 displaying relatively fast reaction rates. We also tested benzyl chloride as a substrate for this reaction. The trends observed are similar to the reaction with benzyl bromide (Fig. S10–S12, ESI†). However, reaction rates with benzyl chloride are slower. For example, at a scan rate of 100 mV s−1, the value of kobs for the reaction with benzyl chloride is about 2.8 s−1. This observation is consistent with the breaking of the C–halide bond as the possible rate determining step. Control reactions were also performed with Bu4NBr to rule out the involvement of species derived from Br− in catalysis. Such a reaction did not display any catalytic current (Fig. S9 and S13, ESI†).
We then turned our attention to the possible mechanism of this (electro)catalytic reaction. A mixture of in situ generated [Co(Q2−)2]2− and benzyl chloride were stirred together, and a ESI mass spectrum of the mixture was recorded. The main product observed from this mixture was the five-fold coordinated cobalt complex [Co(Q˙−)2(CH2Ph)]− (Fig. S15, ESI†). Additionally, a chromatographic work-up of the reaction mixture, and 1H NMR characterization of the organic phase delivered dibenzyl as the exclusive product (Fig. S16, ESI†). Control experiments showed that CoCl2 without the ligand does not deliver any product (Fig. S17, ESI†). Taking these observations into consideration, a mechanism shown in Scheme 1 can be postulated. The complex 12− activates the C–X bonds of the substrates which lead to a release of X− and the formation of the aforementioned five-fold coordinated species. The formation of the C–C coupled dibenzyl product leads to a release of 1−, the reduction of which regenerates the active catalyst 12−. This cycle ensures that the 1/1− redox couple of complex 1 remains unchanged during the catalytic cycle, as has been experimentally observed (Fig. 3).
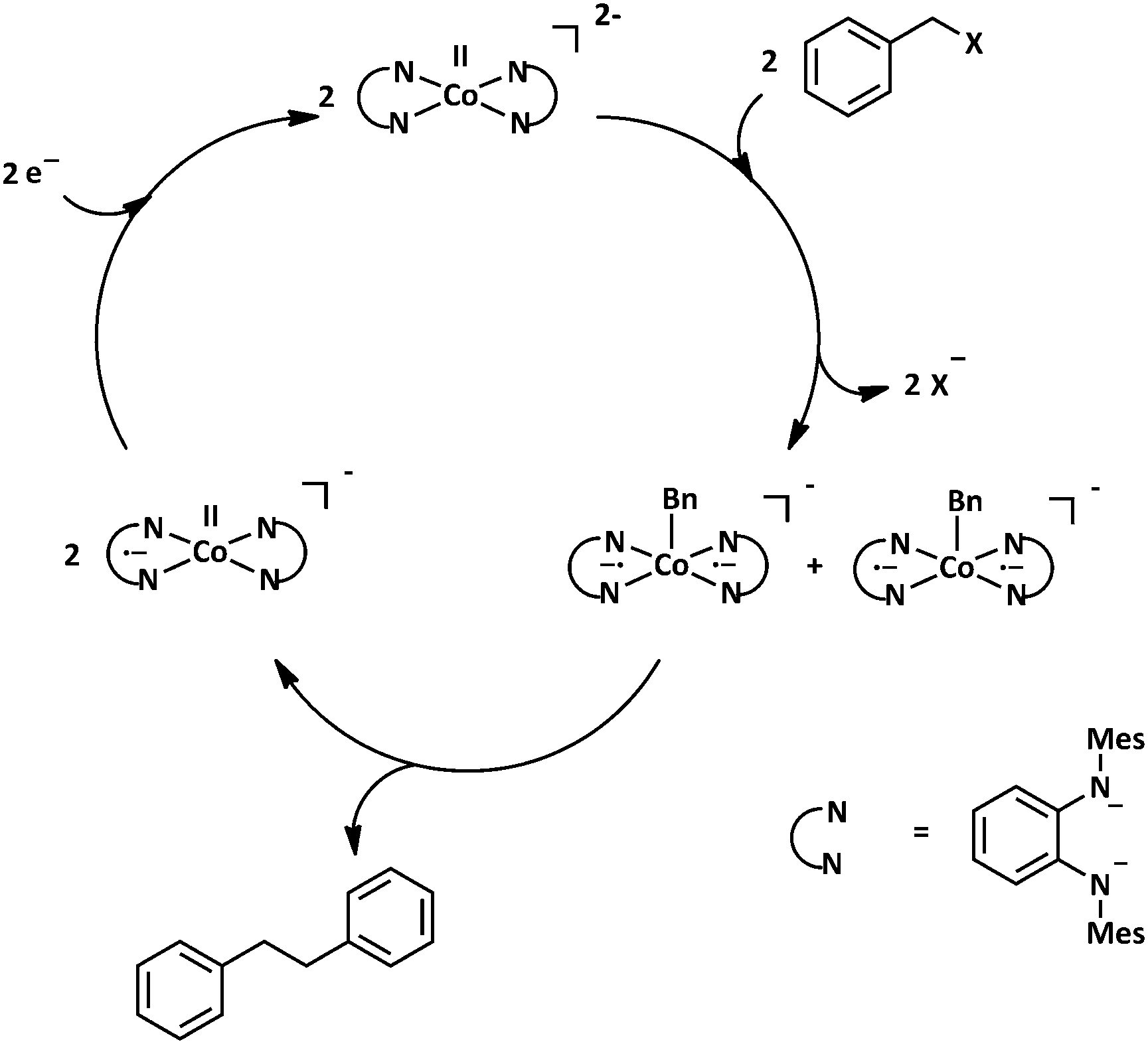 | ||
| Scheme 1 Proposed catalytic cycle for the (electro)catalytic C–C bond formation reaction with 1. | ||
Summarizing, we have presented here a four-coordinate cobalt complex with a redox-active ligand. The geometric and electronic structure of this complex has been probed by a variety of methods, and the complex 1 can be best described as [(Q˙−)CoII,HS(Q˙−)]. Despite the apparent similarity of this complex with several reported cobalt complexes, unique catalytic reactivities have been observed with it. Quantification of the catalytic results and a possible mechanistic picture has been presented. It is the subtle cooperative interplay between cobalt and the redox-active ligands that make such catalytic bond formation possible. The results presented here display the catalytic utility of redox-active metal complexes in C–C bond formation reactions, and provide impetus for carrying out studies with a systematic variation of the ligand backbone. Such investigations are likely to shed more light on the catalytic activity of such metal complexes.
Deutsche Forschungsgemeinschaft (DFG, SA 1580/5-1, SL 104/2-1) is kindly acknowledged for financial support of this project. We thank Prof. Dr M. Dressel for access to the SQUID facilities of the 1. Physikalisches Institut, Universität Stuttgart.
Notes and references
- (a) M. D. Ward and J. A. McCleverty, J. Chem. Soc., Dalton Trans., 2002, 275 RSC; (b) C. G. Pierpont, Coord. Chem. Rev., 2001, 216–217, 95 Search PubMed; (c) P. Chaudhuri, C. N. Verani, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 2213 CrossRef CAS PubMed.
- For selected examples see: (a) B. de Bruin, D. G. H. Hetterscheid, A. J. J. Koekkoek and H.-J. Grützmacher, Prog. Inorg. Chem., 2007, 55, 247 CrossRef CAS; (b) P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794 CrossRef CAS PubMed; (c) V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., 2012, 124, 10374 CrossRef; (d) M. K. Tsai, J. Rochford, D. E. Polyansky, T. Wada, K. Tanaka and J. T. Muckerman, Inorg. Chem., 2009, 48, 4372 CrossRef CAS PubMed; (e) Forum Issue on Redox-Active Ligands, Inorg. Chem., 2011, 50, 9737–9914 Search PubMed; (f) Cluster Issue, Cooperative and Redox Non-Innocent Ligands in Directing Organometallic Reactivity, Eur. J. Inorg. Chem., 2012, 340–580 Search PubMed; (g) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270 CrossRef CAS.
- For selected examples see: (a) A. I. Poddel'sky, V. K. Cherkasov and G. A. Abakumov, Coord. Chem. Rev., 2009, 253, 291 CrossRef CAS PubMed; (b) Z. Sun, H. Chun, K. Hildebrandt, E. Boethe, T. Weyhermüller, F. Neese and K. Wieghardt, Inorg. Chem., 2002, 41, 4295 CrossRef PubMed; (c) P. Ghosh, A. Begum, D. Herebian, E. Boethe, K. Hildebrand, T. Weyhermüller and K. Wieghardt, Angew. Chem., Int. Ed., 2003, 42, 563 CrossRef CAS PubMed; (d) M. R. Ringenberg, S. L. Kokatam, Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2008, 130, 788 CrossRef CAS PubMed; (e) D. Das, H. Agarwala, A. Dutta Chowdhury, T. Patra, S. M. Mobin, B. Sarkar, W. Kaim and G. K. Lahiri, Chem. – Eur. J., 2013, 19, 7384 CrossRef CAS PubMed.
- (a) M. M. Khusniyarov, K. Harms, O. Burghaus, J. Sundermeyer, B. Sarkar, W. Kaim, J. van Slageren, C. Duboc and J. Fiedler, Dalton Trans., 2008, 1355 RSC; (b) A. L. Balch and R. H. Holm, J. Am. Chem. Soc., 1966, 88, 5201 CrossRef CAS; (c) D. Herebian, K. Wieghardt and F. Neese, J. Am. Chem. Soc., 2003, 125, 10997 CrossRef CAS PubMed; (d) E. Bill, E. Bothe, P. Chaudhuri, K. Chlopek, D. Herebian, S. Kokatam, K. Ray, T. Weyhermüller, F. Neese and K. Wieghardt, Chem. – Eur. J., 2005, 11, 204 CrossRef PubMed; (e) K. Chlopek, E. Bothe, F. Nesse, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2006, 45, 6298 CrossRef CAS PubMed.
- (a) A. L. Smith, K. I. Hardcastle and J. D. Soper, J. Am. Chem. Soc., 2010, 132, 14358 CrossRef CAS PubMed; (b) W. I. Dzik, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Angew. Chem., Int. Ed., 2011, 50, 3356 CrossRef CAS PubMed.
- (a) N. Deibel, D. Schweinfurth, S. Hohloch, J. Fiedler and B. Sarkar, Chem. Commun., 2012, 48, 2388 RSC; (b) N. Deibel, D. Schweinfurth, S. Hohloch, M. Delor, I. V. Sazanovich, M. Towrie, J. Weinstein and B. Sarkar, Inorg. Chem., 2014, 53, 1021 CrossRef CAS PubMed; (c) S. Hohloch, P. Braunstein and B. Sarkar, Eur. J. Inorg. Chem., 2012, 546 CrossRef CAS.
- S. Bhattacharya, P. Gupta, F. Basuli and C. G. Pierpont, Inorg. Chem., 2002, 41, 5810 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, crystallography, magnetic measurements, EPR spectroscopy, electrochemistry and mechanistic studies. CCDC 902892. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc03309d |
| This journal is © The Royal Society of Chemistry 2014 |