
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the interaction of copper(II) with disulfiram†
David J.
Lewis‡
ab,
Parikshit
Deshmukh
a,
Aleksander A.
Tedstone
a,
Floriana
Tuna
c and
Paul
O'Brien§
*ab
aSchool of Chemistry, University of Manchester, Oxford Road, M13 9PL, UK. E-mail: Paul.O'Brien@manchester.ac.uk; Fax: +44 (0)161 275 4616; Tel: +44 (0)161 275 4653
bSchool of Materials, University of Manchester, Oxford Road, M13 9PL, UK
cEPSRC EPR National Research Facility and Service, Photon Science Institute, University of Manchester, Oxford Road, M13 9PL, UK
First published on 11th September 2014
Abstract
In combination with copper(II) ions, disulfiram (DSF) has been reported to be a potentially potent anticancer agent based on in vitro results. The interaction of DSF with copper(II) chloride in solution has been studied using a range of spectroscopic techniques. There is strong evidence for the rapid formation of the bis(N,N-diethyl dithiocarbamato)copper(II) complex in situ. Kinetic experiments were used to determine rate laws for the reaction that give insight into the mechanism of the process which may help to explain the observed in vitro cytotoxicity.
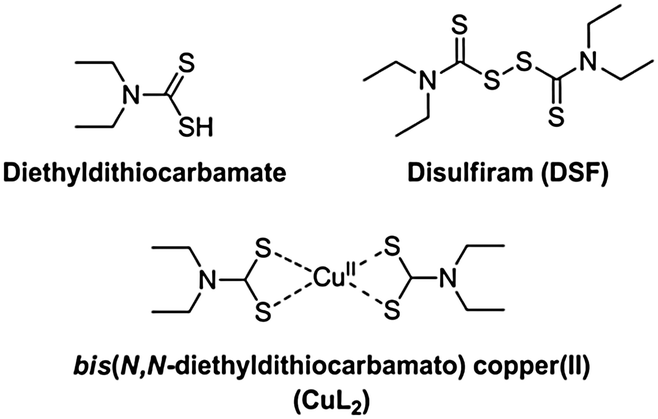
Disulfiram (tetraethylthiuram disulfide, DSF), the two-electron oxidised dimer of diethyl dithiocarbamate (Scheme 1), is a useful pharmaceutical agent in the treatment of chronic alcoholism.1 Marketed under the name Antabuse, the drug acts primarily by irreversible inhibition of the enzyme acetaldehyde dehydrogenase, leading to unpleasant physical feelings accompanied by nausea or vomiting when ethanol is consumed. Recently, there has been renewed interest in DSF, primarily for its use in adjuvant therapies (e.g. clinical trials gov. identifier NCT00312819). DSF is also rapidly becoming considered as a potentially convincing anticancer drug. Reports have emerged recently of the induction of apoptosis in vitro in cancer cell lines generally thought to be chemotherapy-resistant, such as glioblastoma multiforme, by DSF either alone or in synergy with an anticancer agent (gemcitabine).2,3 However, the cytotoxic activity observed in both cases was reliant on the presence of copper(II) salts and the active species responsible for the remarkable anticancer activity was not identified. Cen et al. reported a similar copper dependency in the induction of apoptosis in human melanoma cells by DSF/Cu2+.4 Conticello et al. reported the effective use of DSF or DSF–copper(II) against human haematological malignancies in vitro.5 It is known that DSF can also be used with other transition metals to effect anti-cancer activity,6,7 though copper(II) is interesting due to its natural presence in vivo, requiring, in theory, only treatment of a patient with DSF alone to establish a prototype chemotherapy.
 | ||
| Scheme 1 | ||
Dithiocarbamates are a class of sulfur-containing bidentate chelating ligands. The ability of the diethyldithiocarbamate ligands to complex a range of transition metal ions including Cu2+ is well-known.8 We have previously been interested in using metal–dithiocarbamate and metal–diselenocarbamate complexes as single-source precursors9 for various semiconductors for photovoltaic applications10–16 as well as for nanocrystalline semiconducting quantum dots.17,18 Bifunctional dithiocarbamates have been used to functionalise the surface of noble metal nanoparticles with transition metal or lanthanide(III) complexes.19,20
The electronic properties of bis(N,N-diethyldithiocarbamato)copper(II) (CuL2, Scheme 1) have been investigated by electron paramagnetic resonance (EPR) spectroscopy, giving insights into its self-association.21 Similarly, the photochemistry of CuL2 has been explored, with the complex displaying an intense ligand-to-metal charge transfer band (LMCT), with maximum absorbance at around 430 nm, a signature of its formation.22,23
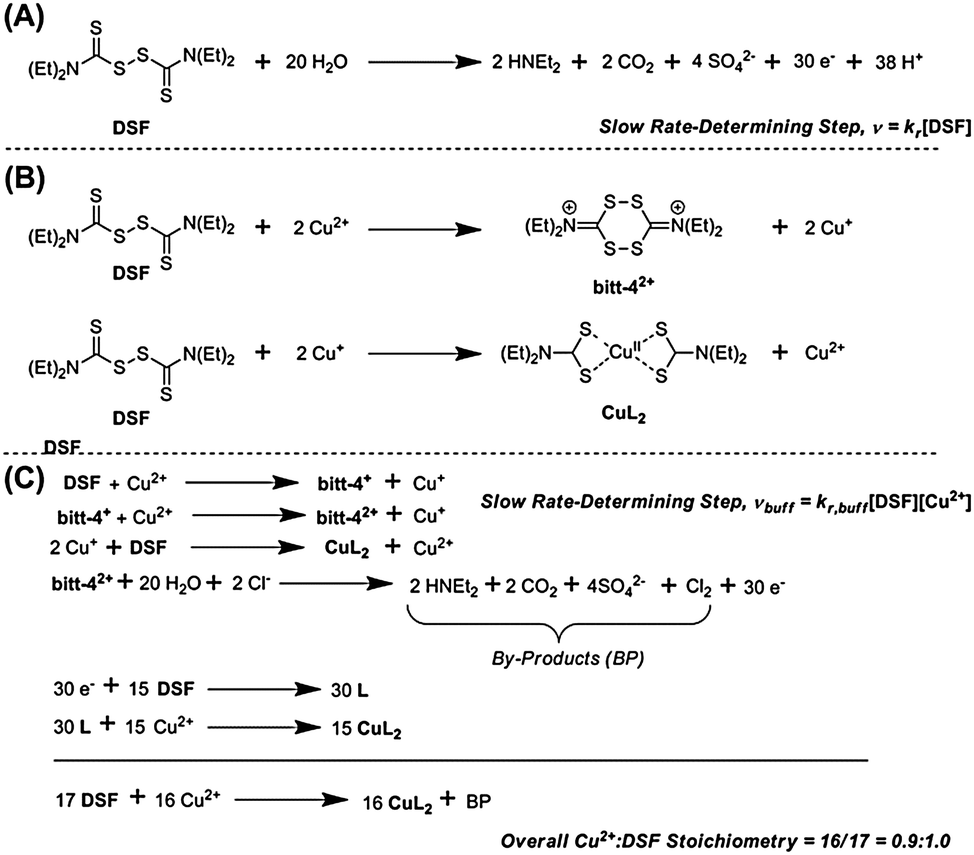
It has been claimed that DSF forms CuL2 species in solution upon addition of copper(II) salts. The chelating ligand, diethyldithiocarbamate, is the product of the two-electron reduction of DSF. Chen et al. noted a “dramatic colour change” on addition of copper(II) salts to DSF, probably due to the aforementioned LMCT transition in CuL2 formed in situ.24 Farmer and co-workers reported that the mechanism of formation of CuL2 from a mixture of Cu(II) and DSF proceeded through the spontaneous decomposition of a small fraction of DSF in the presence of water to produce 30 electrons (Scheme 2, A) which then proceed to reduce 15 molecules of DSF to form the diethyldithiocarbamate, L, which then complexes copper(II) in situ to form CuL2 in a theoretical 93% yield.4
 | ||
| Scheme 2 | ||
The main evidence presented for such a mechanism was an assay based on UV-Vis absorbance spectroscopy. The work was performed in biphasic systems (soluble Cu2+ and solid DSF) formed on the addition of DSF to aqueous CuCl2, requiring extraction of reaction products into chloroform prior to analysis, as well as long reaction times (e.g. 24 h). Here we report the direct observation of CuL2 species and study the EPR properties compared to isolated CuL2 The reaction kinetics of the interaction of DSF with Cu2+ ions are studied in both unbuffered and buffered (HEPES, pH 7.4) mixed-solvent solutions.
A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of DSF and CuCl2 was analysed by positive-mode electrospray ionisation mass spectrometry in order to observe directly if the dithiocarbamate–copper(II) complex, CuL2, is formed. We were able to identify the product in solution: a peak manifold was observed at m/z 359, consistent with CuL2 (ESI†). The isotope pattern of the peak observed at m/z 359 confirmed the incorporation of a single copper centre as-compared to the theoretical pattern expected for protonated CuL2. Accurate mass determination of the [M+H]+ peak by high-resolution mass spectrometry (HRMS, ES+) gave a value that corresponded exactly to the theoretical mass of protonated CuL2 (calc. for C10H21N2S4Cu, [M+H]+: 359.9878. Found: 359.9878), thus confirming that the species analysed by this method in situ is indeed protonated CuL2.
:1 molar ratio of DSF and CuCl2 was analysed by positive-mode electrospray ionisation mass spectrometry in order to observe directly if the dithiocarbamate–copper(II) complex, CuL2, is formed. We were able to identify the product in solution: a peak manifold was observed at m/z 359, consistent with CuL2 (ESI†). The isotope pattern of the peak observed at m/z 359 confirmed the incorporation of a single copper centre as-compared to the theoretical pattern expected for protonated CuL2. Accurate mass determination of the [M+H]+ peak by high-resolution mass spectrometry (HRMS, ES+) gave a value that corresponded exactly to the theoretical mass of protonated CuL2 (calc. for C10H21N2S4Cu, [M+H]+: 359.9878. Found: 359.9878), thus confirming that the species analysed by this method in situ is indeed protonated CuL2.
EPR spectroscopy was used to probe structural similarities between a solution of laboratory-synthesised CuL2 (synthesised by the method of Jeliazkova et al.,22 characterisation data in ESI†) and a mixture of DSF and CuCl2 in 1:1 molar ratio in a 95:5 THF:water glass at 80 K (Fig. 1) in order to compare the structure of the species formed in situ when stoichiometric amounts of DSF and CuCl2 are mixed together in a solution. We also simulated the EPR spectrum of CuL2 to compare both solutions to theory. The EPR spectra of both mixtures and the simulated spectrum of CuL2 revealed striking similarities, thus suggesting that the species formed when DSF is mixed with a stoichiometric amount of Cu2+ is CuL2. The EPR parameters obtained from experiment are in good agreement with the published EPR data of CuL2.21
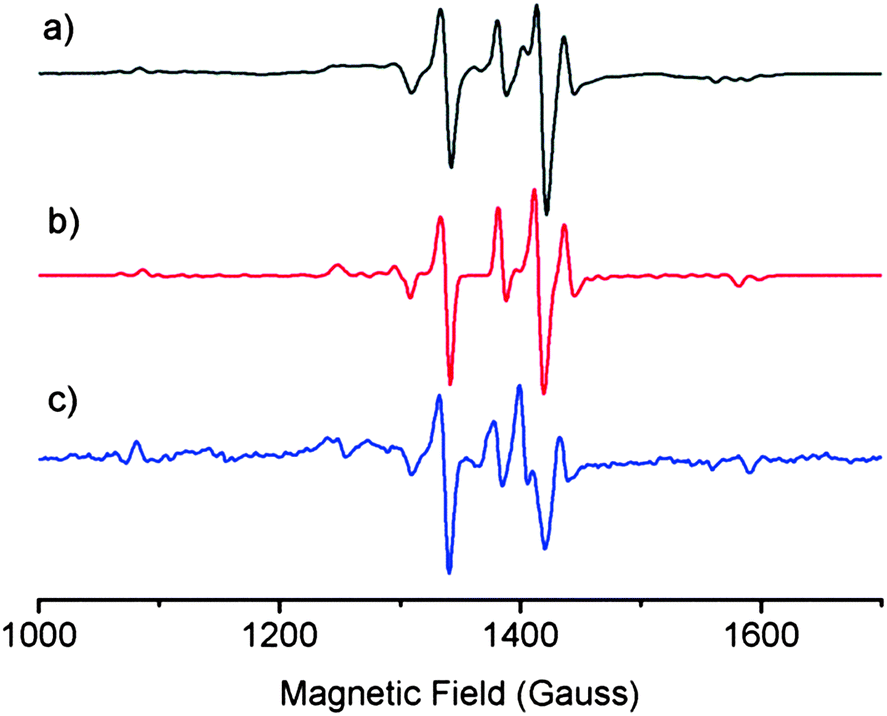 | ||
| Fig. 1 S-band (3.8 GHz) EPR spectra at 80 K of 95:5 THF:water glasses of (a) isolated CuL2 and (c) a mixture of DSF and CuCl2 in 1:1 molar ratio. The red line (b) represents the calculated EPR spectrum of CuL2, using the parameters g|| = 2.071; g⊥ = 2.002; A|| = 165 G and A⊥ = 43 G. | ||
UV-Vis absorption spectroscopy in a mixed solvent system (aqueous THF, 0.7% v/v) was used to probe complex formation under steady-state and kinetic conditions. The LMCT transition of CuL2, which has an absorbance maximum at ca. 425 nm can be used as a spectroscopic handle to confirm the formation of CuL2 species in situ when CuCl2 and DSF are mixed in stoichiometric amounts. Titration of a solution of DSF in THF into a solution of copper(II) chloride in water demonstrated the steady growth of the LMCT absorption band, with peak maxima at 425 nm, characteristic of CuL2 (ESI†). It is important that the solution remained homogeneous throughout the titration, ensuring that all the species formed were analysed. Kinetic studies of the using a 1:1 ratio of DSF:CuCl2 exhibited a marked increase in the LMCT absorption at 425 nm to a plateau in around 150 min, indicating that complexation was fully complete only after this time (Fig. 2). Isosbestic points in absorbance spectra give important information regarding the relationship between two species in solution. The presence of three well-defined isosbestic points at 250 nm, 290 nm and 325 nm demonstrate that the copper(II)-bound and unbound ligand (i.e. Cu(II)–DSF and DSF) species are, most likely, related linearly by stoichiometry as expected.
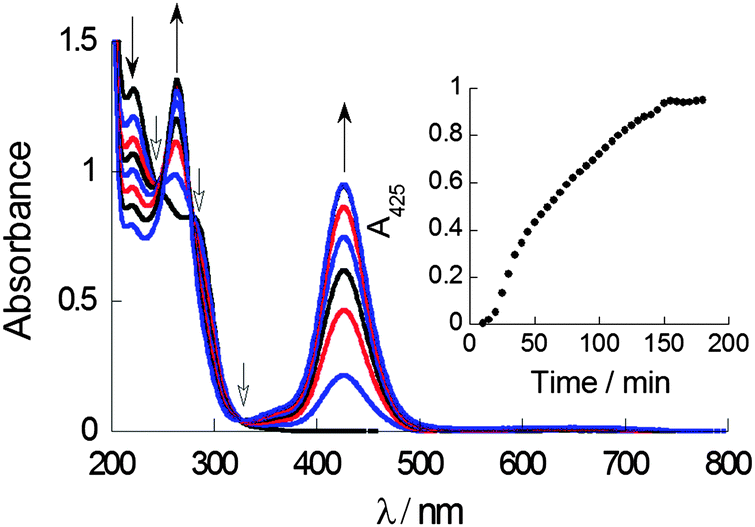 | ||
| Fig. 2 UV-Vis absorption time-course plot for a 1:1 ratio of Cu2+ and DSF in THF/water. Solid-headed arrows show the either the hyperchromic (upward arrows) or hypochromic (downward arrow) nature of the electronic transitions during the study. The three isosbestic points at 250, 290 and 325 nm are marked with hollow-headed arrows. Inset: the growth of the LMCT electronic transition over time, with an interval of 5 min between points. | ||
Continuous variation plots using the LMCT electronic transition at 425 nm were used to further probe the stoichiometry of the complex formed between CuCl2 and DSF in situ. A continuous variation plot (the method of Job25) of the of mole fraction of DSF, x, (such that x = [DSF]/([Cu2+] + [DSF])) plotted vs. absorption at 425 nm (A425) in HEPES buffer revealed a value of x to be 0.61 ± 0.07, interpolated via the most appropriate linear fits to both halves of the plot (Fig. 3 and ESI† for calculation of intersect and calculation of random error in linear regression). This means therefore that the overall stoichiometry of the system is in slight excess of DSF; a value of x = 0.50 corresponds to CuL2, thus the slight shift to x > 0.5 indicates a slight stoichiometric excess of DSF in the system. This overall stoichiometry is fully consistent with the stoichiometric equations which were subsequently derived from kinetic experiments in buffered solution (vide infra and Scheme 2, C).
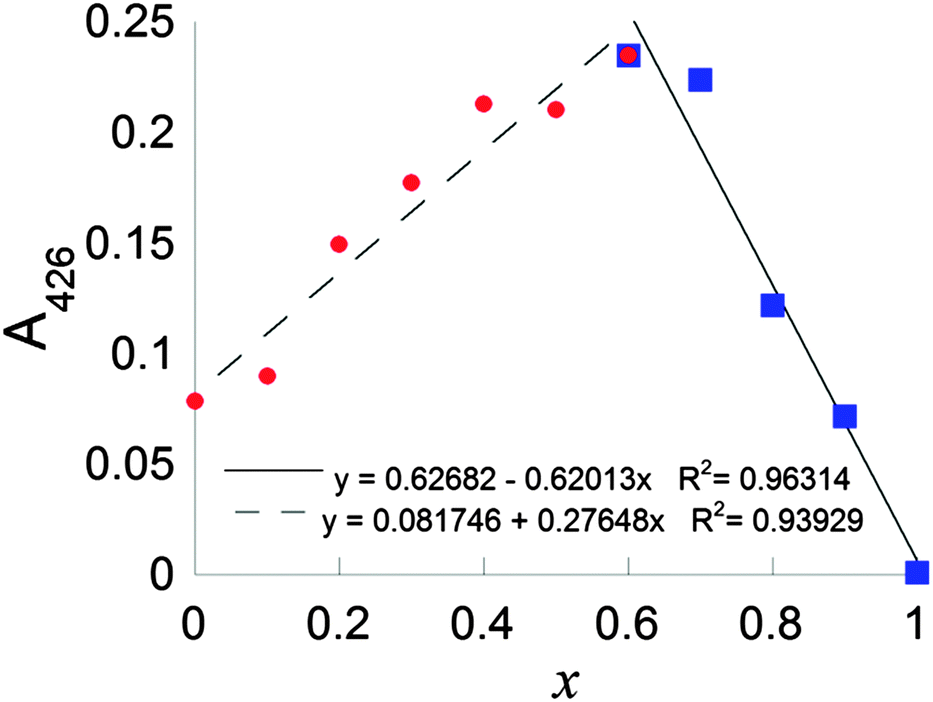 | ||
| Fig. 3 Continuous variation plot of mole fraction of DSF, x, (such that x = [DSF]/([Cu2+] + [DSF])) vs. absorption at 425 nm (A425). | ||
UV-Vis absorption spectroscopy allowed the determination of a rate law for the reaction in unbuffered solution using the method of initial rates.26 In this method, initial rates for the reaction being studied (v0) are measured for reactants isolated by concentration and plots of log v0vs. log[R] (where [R] is the concentration of the reactant being studied) give straight lines with slope equal to the order of the reaction with respect to reactant R, and y-intercepts equivalent to logk, where k is the observed rate constant with respect to the isolated reactant. The rate of formation of CuL2-type species can thus be quantified by UV-Vis spectroscopy using the absorbance of the LMCT transition, and the orders of reaction determined for both DSF and Cu. Initial rates of reaction (v0; t ≤ 5 min) were measured for a range of concentrations of DSF and Cu2+ in a mixed solvent system (aqueous THF, 0.4–14% v/v see ESI† for full details). Plots of log v0vs. log[DSF] or logv0vs. log[Cu2+] for isolated reactants (N = 5 at every point with full analysis of systematic and random error for each point; ESI†) gave linear relationships with slopes approximately equal to the order of reaction for each individual reactant; the rate is first-order with respect to DSF, but zero-order with respect to copper i.e. v = kr[DSF][Cu2+]0 or simply v = kr[DSF], with the observed rate constant for the DSF isolated reaction equal to 1.653 × 10−2 s−1. The rate law derived here gives great credence to the mechanism proposed by Farmer and co-workers (vide supra):4 disproportionation of DSF in water (i.e. pseudo first-order kinetics) to release the electrons required for reduction, followed by complexation of copper(II) and acidic runaway would indeed be pseudo first-order with respect to DSF, and probably rate-limiting, assuming that the complexation step is relatively rapid in comparison.
In HEPES-buffered solution an interesting change in the rate law to the form vbuff = kr,buff[DSF][Cu2+] is observed (ESI†) i.e. the reaction becomes first-order with respect to copper(II) whilst remaining first-order with respect to DSF. The observed rate constants determined are k = 1.854 × 10−4 mol−1 dm3 s−1 with respect to isolated DSF and k = 4.851 × 10−5 mol−1 dm3 s−1 with respect to isolated copper. We propose that the mechanism in buffered solutions occurs via reduction of Cu2+ to Cu+ by the initial oxidation of DSF to the bitt-42+ intermediate4 which is followed by a rapid two-electron reduction of another DSF molecule by two Cu+ ions (Scheme 2, B) to form CuL2 and regenerate a single Cu2+ ion. The rapid disproportionation of bitt-42+ intermediate to release 30 electrons plus by-products then facilitates the formation of further amounts of CuL2via the two-electron reduction of DSF (Scheme 2, C). Pathways to complexation via the bitt-42+ intermediate have been suggested by Farmer and co-workers.4 The overall stoichiometry of the reaction with respect to Cu2+:DSF ratio is ca. 0.9:1.0, as observed in the Job plot (vide supra). It is likely that this may be the mechanism for the reaction of DSF with copper(II) ions under biological conditions. It is also likely that the release of copper(I) ions during the formation of CuL2 and the catastrophic decomposition of bitt-42+ could lead to massive oxidative stress on cells in vitro assays and result in the apoptosis observed.
In conclusion, the interaction of DSF with copper(II) ions in solution has been comprehensively studied. Mass spectrometry and EPR suggest the species formed in solution is CuL2 where L = (S2CNEt2). UV-Vis absorption spectroscopy titrations at steady-state demonstrate the growth of the characteristic LMCT absorbance for this species. Kinetic studies using UV-Vis absorption spectroscopy to monitor the LMCT absorption band demonstrate that the rate law is first-order with respect to DSF in unbuffered solution, which suggests a rate-limiting step reliant only on the concentration of DSF within the mixture. The rate law changes to a first order dependence on both DSF and copper(II) ions in buffered solutions, with a mechanism for the formation of CuL2 involving an oxidised ligand intermediary, bitt-42+, and copper(I) ions. In this sense, DSF may behave non-innocently. We have, therefore, confirmed the first step of the mechanism proposed previously to this study,4 which can involve a pH runaway, as well as proposing a mechanism for the altered kinetics observed in buffered solutions.
These observations potentially have serious implications regarding the potential use of DSF as a therapeutic agent in vivo. It is likely that in in vitro assays, on addition of the copper(II) ions to the media, the cells are exposed to a rapid decomposition of DSF to bitt-42+ and copper(I) ions with a catastrophic release of reactive oxygen species such as H2O2 arising from Fenton chemistry, the latter which are known to cause apoptosis.27 The role of reactive oxygen species in apoptosis of cancer cells treated with DSF and DSF–copper(II) cocktails has not escaped the attention of researchers though a potential explanation for this has, until now, been elusive.5 The oxidation reactions suggested here, as we have seen, are likely to be relatively rapid and thus may be highly cytotoxic (produces a greater dose of H2O2 over time).28 Therefore, the induction of apoptosis in tumour cells by a copper(II) DSF cocktail in vivo is difficult to envisage as it is probably not caused by a discrete copper–DSF complex but rather is due to a reaction.
Obviously, if the product of this reaction were a therapeutic molecule there would be a candidate drug that could be delivered in vivo but this does not seem to be the case. The situation is rather similar to the recent case of cisplatin solvated in DMSO, where reactivity of the solvent medium may have produced misleading results from in vitro assays.29 Care must therefore be taken when it is suggested that the anticancer activity of DSF in combination with copper(II) observed in vitro can be translated in vivo.
The authors would like to acknowledge the EPSRC National EPR Research Facility (NS/A000014/1).
Notes and references
- R. K. Fuller, L. Branchey, D. R. Brightwell, R. M. Derman, C. D. Emrick, F. L. Iber, K. E. James, R. B. Lacoursiere, K. K. Lee, I. Lowenstam, I. Maany, D. Neiderhiser, J. J. Nocks and S. Shaw, J. Am. Med. Assoc., 1986, 256, 1449–1455 CrossRef CAS.
- X. Guo, B. Xu, S. Pandey, E. Goessl, J. Brown, A. L. Armesilla, J. L. Darling and W. Wang, Cancer Lett., 2010, 290, 104–113 CrossRef CAS PubMed.
- P. Liu, S. Brown, T. Goktug, P. Channathodiyil, V. Kannappan, J. P. Hugnot, P. O. Guichet, X. Bian, A. L. Armesilla, J. L. Darling and W. Wang, Br. J. Cancer, 2012, 107, 1488–1497 CrossRef CAS PubMed.
- D. Z. Cen, D. Brayton, B. Shahandeh, F. L. Meyskens and P. J. Farmer, J. Med. Chem., 2004, 47, 6914–6920 CrossRef CAS PubMed.
- C. Conticello, D. Martinetti, L. Adamo, S. Buccheri, R. Giuffrida, N. Parrinello, L. Lombardo, G. Anastasi, G. Amato, M. Cavalli, A. Chiarenza, R. De Maria, R. Giustolisi, M. Gulisano and F. Di Raimondo, Int. J. Cancer, 2012, 131, 2197–2203 CrossRef CAS PubMed.
- E. M. Nagy, L. Ronconi, C. Nardon and D. Fregona, Mini-Rev. Med. Chem., 2012, 12, 1216–1229 CrossRef CAS.
- D. Buac, S. Schmitt, G. Ventro, F. R. Kona and Q. P. Dou, Mini-Rev. Med. Chem., 2012, 12, 1193–1201 CrossRef CAS.
- A. Hulanicki, Talanta, 1967, 14, 1371–1392 CrossRef CAS.
- M. A. Malik, M. Afzaal and P. O'Brien, Chem. Rev., 2010, 110, 4417–4446 CrossRef CAS PubMed.
- K. Ramasamy, M. A. Malik and P. O'Brien, Chem. Sci., 2011, 2, 1170–1172 RSC.
- K. Ramasamy, V. L. Kuznetsov, K. Gopal, M. A. Malik, J. Raftery, P. P. Edwards and P. O'Brien, Chem. Mater., 2013, 25, 266–276 CrossRef CAS.
- M. Afzaal, K. Ellwood, N. L. Pickett, P. O'Brien, J. Raftery and J. Waters, J. Mater. Chem., 2004, 14, 1310–1315 RSC.
- M. B. Hursthouse, M. A. Malik, M. Motevalli and P. O'Brien, Organometallics, 1991, 10, 730–732 CrossRef CAS.
- M. B. Hursthouse, M. A. Malik, M. Motevalli and P. O'Brien, Polyhedron, 1992, 11, 45–48 CrossRef CAS.
- M. Lazell, P. O'Brien, D. J. Otway and J.-H. Park, J. Chem. Soc., Dalton Trans., 2000, 4479–4486 RSC.
- P. O'Brien, J. R. Walsh, I. M. Watson, L. Hart and S. R. P. Silva, J. Cryst. Growth, 1996, 167, 133–142 CrossRef.
- B. Ludolph, M. A. Malik, P. O'Brien and N. Revaprasadu, Chem. Commun., 1998, 1849–1850 RSC.
- M. A. Malik, N. Revaprasadu and P. O'Brien, Chem. Mater., 2001, 13, 913–920 CrossRef CAS.
- E. R. Knight, A. R. Cowley, G. Hogarth and J. D. E. T. Wilton-Ely, Dalton Trans., 2009, 607–609 RSC.
- S. Sung, H. Holmes, L. Wainwright, A. Toscani, G. J. Stasiuk, A. J. P. White, J. D. Bell and J. D. E. T. Wilton-Ely, Inorg. Chem., 2014, 53, 1989–2005 CrossRef CAS PubMed.
- N. D. Yordanov and D. Shopov, J. Chem. Soc., Dalton Trans., 1976, 883–886 RSC.
- B. G. Jeliazkova and G. C. Sarova, J. Photochem. Photobiol., A, 1996, 97, 5–9 CrossRef CAS.
- V. F. Plyusnin, A. V. Kolomeets, V. P. Grivin, S. V. Larionov and H. Lemmetyinen, J. Phys. Chem. A, 2011, 115, 1763–1773 CrossRef CAS PubMed.
- D. Chen, Q. C. Cui, H. Yang and Q. P. Dou, Cancer Res., 2006, 66, 10425–10433 CrossRef CAS PubMed.
- J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., Int. Ed., 2013, 52, 11998–12013 CrossRef CAS PubMed.
- P. W. Atkins and J. de Paula, The Elements of Physical Chemistry, W. H. Freeman, 5th edn, 2009 Search PubMed.
- K. D. Held, F. C. Sylvester, K. L. Hopcia and J. E. Biaglow, Radiat. Res., 1996, 145, 542–553 CrossRef CAS.
- K. D. Held and J. E. Biaglow, Radiat. Res., 1994, 139, 15–23 CrossRef CAS.
- M. D. Hall, K. A. Telma, K.-E. Chang, T. D. Lee, J. P. Madigan, J. R. Lloyd, I. S. Goldlust, J. D. Hoeschele and M. M. Gottesman, Cancer Res., 2014, 74, 3913–3922 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental and instrumental details, synthetic procedures, electrospray mass spectrum, and initial rate plots. See DOI: 10.1039/c4cc04767b |
| ‡ Parts of this communication were written by DJL whilst a visiting Scholar at the University of Illinois at Urbana-Champaign, The United States of America. DJL and POB would like to thank Professors Shen J. Dillon, Pascal Bellon and Robert Averback for their kind invitation. |
| § We wish to thank warmly Dr. K. A. F. O’Brien for turning our attention to the copper(II)–DSF question. |
| This journal is © The Royal Society of Chemistry 2014 |