
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The use of the sterically demanding IPr* and related ligands in catalysis
Frédéric
Izquierdo
,
Simone
Manzini
and
Steven P.
Nolan
*
EaStCHEM School of Chemistry, University of St Andrews, St Andrews, KY16 9ST UK. E-mail: snolan@st-andrews.ac.uk
First published on 26th August 2014
Abstract
This account highlights the synthesis and applications of one of the very bulky NHC ligands, IPr* (1,3-bis(2,6-bis(diphenylmethyl)-4-methylphenyl)imidazo-2-ylidene). This ligand and some of its derivatives have been found very effective in several catalytic applications and have enabled the isolation of highly reactive organometallic complexes. More specifically, applications of this ligand in Pd and Ni chemistry have permitted challenging transformations under mild reaction conditions and low catalyst loadings. We report the successes as well as the limitations encountered using transition-metal systems bearing this ligand-type. This report will hopefully serve as a guide to synthetic chemists, providing insights as to when the very sterically demanding IPr* ligand (and its congeners) and in a broader context, very bulky NHC ligands, should be used.
Introduction
Since the discovery of N-heterocyclic carbenes (NHC) and their use as ligands in the coordination environment of metals, several variations of the basic structural motif have been reported.1–5 Specifically, varying the steric hindrance (and also the flexibility) of the NHCs has been shown to have a significant influence on catalysis.6,7 A bulkier ligand can possibly stabilize the active species during the catalytic reaction, disfavoring bimolecular decomposition pathways or C–H activation and cyclometallation.8–11 However, a much larger ligand can disfavor the substrate approach to the metal and thereby reduce catalytic activity. Therefore a careful balance of the two aspects proves critical.8,12 In order to reduce catalyst deactivation, one of the first sterically demanding NHCs, 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) and its saturated analogue 1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazol-2-ylidene (SIPr), exhibited positive effects on catalysis compared to their smaller congeners. Because of their straightforward synthesis and their commercial availability,13 they are two of the most commonly used ligands in LTM–NHC-based (LTM = Late Transition Metal) catalysis.5 Several other hindered NHCs have been reported, in order to further reduce decomposition pathways, while maintaining high catalytic activity (Fig. 1). From this portfolio of sterically demanding ligands, three classes can be highlighted: IBiox,14 CAAC (Cyclic Alkyl Amino Carbenes)15 and 1,3-bis(aryl)imidazolylidenes such as IPent, IPr* and SICyoctNap16–20 (Fig. 1).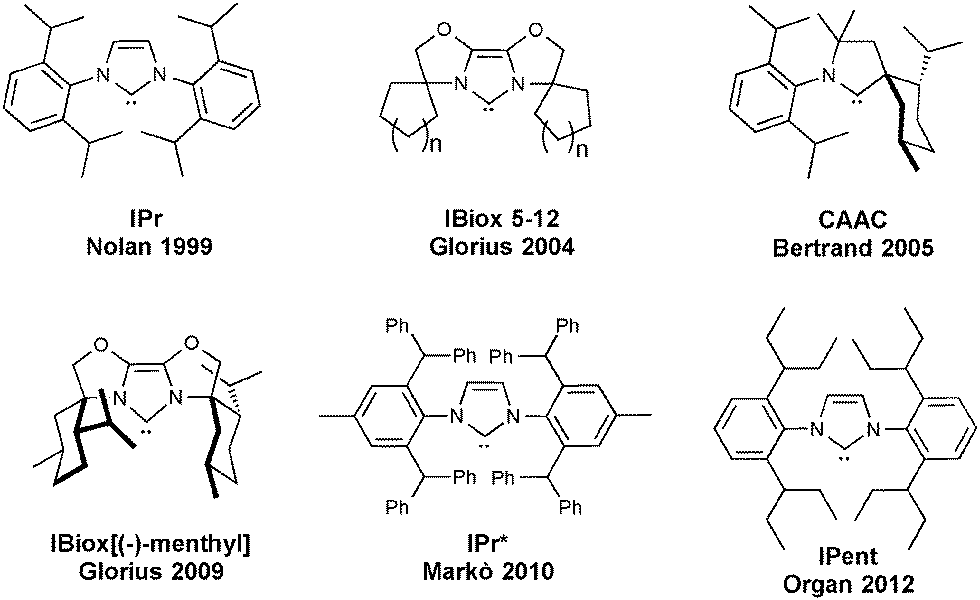 | ||
| Fig. 1 Bulky N-heterocyclic carbenes. | ||
The IBiox ligands are prepared from imidazolium salts, which are easily obtained from the related bioxazolines.14 The ligand bears a tricyclic backbone that confers rigidity to the system. The facile synthesis of IBiox-type carbenes allows access to ligands with different properties, including chirality.21 The IBiox series has mainly been used by Glorius7 and Lautens,21 particularly in palladium and rhodium catalysis, but their use is far from general and has only been examined for a limited number of applications.22,23 Another class of bulky NHCs is represented by the Cyclic Alkyl Amino Carbenes (CAAC), developed by Bertrand.15 These carbenes are based on a pyrrolidine ring bearing two quaternary carbon centers, making it one of the most electron-donating types of NHCs. CAACs have been used as ligands in several metal-catalyzed reactions,24 particularly with palladium,15 gold25–27 and ruthenium.28
Due to their facile synthesis, 1,3-bis(aryl)imidazolylidene carbenes are some of the most commonly used NHCs and several variations have been reported. From this type of NHC, IPr and SIPr are the most widely used with a variety of metals in catalysis. Increasing their steric hindrance, three ligand subsets display interesting properties: the ITent (Tent for tentacular), IPr* and SINap series. Based on the IPr structure, the ITent family bears symmetrical n-alkyl chains at the 2- and 6-positions of the N-aryl substituent, conferring increased hindrance on the metal center, but at the same time, a high level of flexibility. The IPent ligand, first reported by Organ and co-workers, has mainly been used on palladium.18,29 Dorta and co-workers have prepared substituted 1,3-bis(naphthyl)imidazolylidene carbenes and amongst them SICyoctNap has showed great activity in palladium-catalyzed coupling reactions.20,30,31 Keeping this background in mind, in this account, we will focus on the use and reactivity of one of the largest members of these families, namely the IPr* ligand and its uses in catalysis.
Synthesis of the IPr* ligand and its derivatives
Among highly hindered imidazol-2-ylidene ligands, IPr* possesses remarkable properties. First synthesized by Markò and co-workers, IPr*·HCl (Scheme 1, 1a) can be accessed in a straightforward manner. The synthesis begins with a solvent-free condensation of p-toluidine and benzhydrol in the presence of HClconc. and ZnCl2. Then, the generated aniline was transformed into the diimine under acidic conditions using glyoxal and MgSO4 as a water abstractor. Subsequently, the cyclization towards the formation of the imidazolium salt was achieved with moderate yields by the use of p-formaldehyde under acidic conditions, assisted by the presence of ZnCl2 as a templating agent.19 By varying the aniline starting material, IPr*Tol·HCl (1b) and IPr*OMe·HCl (1c) have been obtained.32,33 Using a 4,4′-benzhydrol substituted with octa-tert-butyl derivative in the first step, IPr**·HCl (1d) is achieved.34 Finally, Markò and co-workers prepared IPr*(2-Np)·HCl (1e, Np = naphthyl) starting from the aniline used to generate IPr* and di(naphthalen-2-yl)methanol.35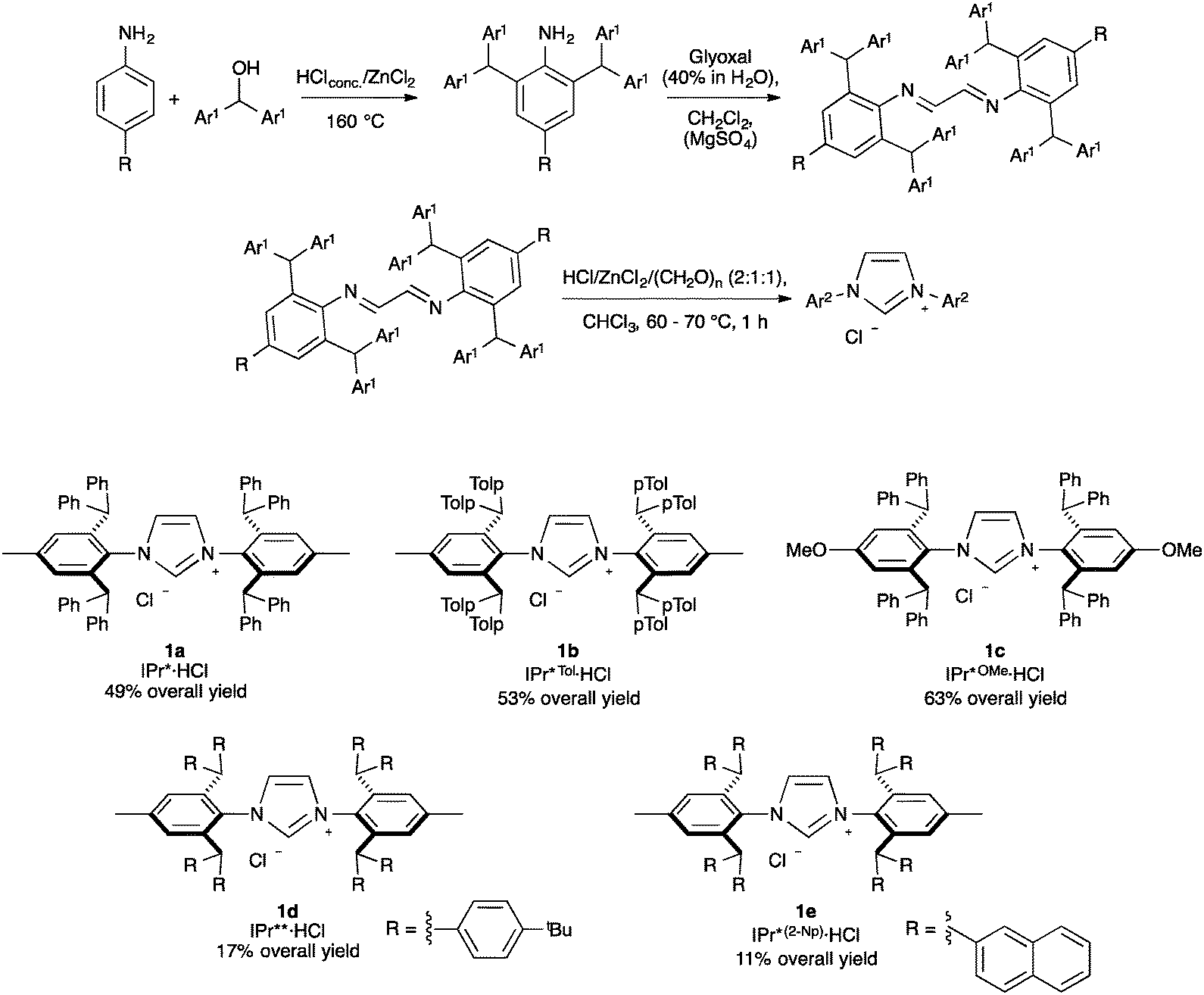 | ||
| Scheme 1 Synthesis of IPr*·HCl and its derivatives. | ||
Nickel, palladium and platinum chemistry with IPr*
In nickel chemistry, the use of IPr* was first explored by Hillhouse and co-workers who isolated a series of Ni–IPr* complexes, including an exceptionally low coordinate nickel imido complex, (IPr*)Ni![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(dmp) (Scheme 2, 2c, dmp = 2,6-dimesitylphenyl). X-ray studies of 2c showed that the C–Ni–N atoms were in a linear arrangement with a short Ni–N distance and multiple-bond character for the C–Ni–N core. When the activity of this imido nickel complex was evaluated, 2c permitted the formation of 2d by a nitrene-group transfer and C–H insertion was observed for the reaction with ethene.36
N(dmp) (Scheme 2, 2c, dmp = 2,6-dimesitylphenyl). X-ray studies of 2c showed that the C–Ni–N atoms were in a linear arrangement with a short Ni–N distance and multiple-bond character for the C–Ni–N core. When the activity of this imido nickel complex was evaluated, 2c permitted the formation of 2d by a nitrene-group transfer and C–H insertion was observed for the reaction with ethene.36
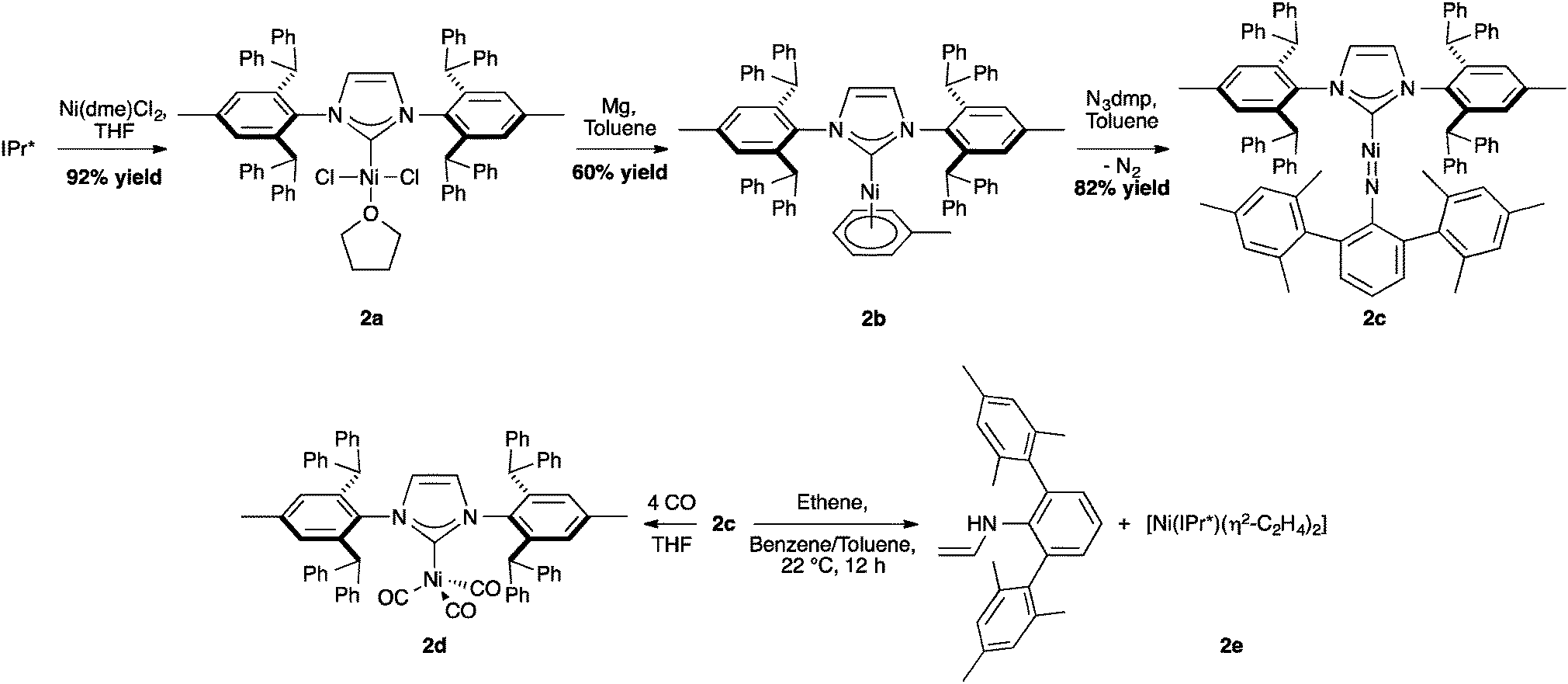 | ||
| Scheme 2 Synthesis of [Ni(IPr*)(η6-C7H8)] and other Ni-complexes. | ||
Nolan and co-workers reported a highly efficient method to form C–N bonds using [Ni(NHC)(η3-allyl)Cl] complexes bearing bulky NHCs such as IPr* (Scheme 3, 3a, %VBur = 42.4) or IPr*OMe (3b, %VBur = 42.7).37 Complex 3b shows outstanding reactivity and outperformed the previous state-of-the-art procedure published by Nicasio and co-workers, using [Ni(IPr)(allyl)Cl] (3c, %VBur = 36.9).38 As reported with [Ni(NHC)(Cp)Cl] (Cp = cyclopentadienyl), steric hindrance plays an important role for the catalytic activity of the system, presumably due to the stabilization of the active species during the reaction, reducing possible bimolecular deactivation.39
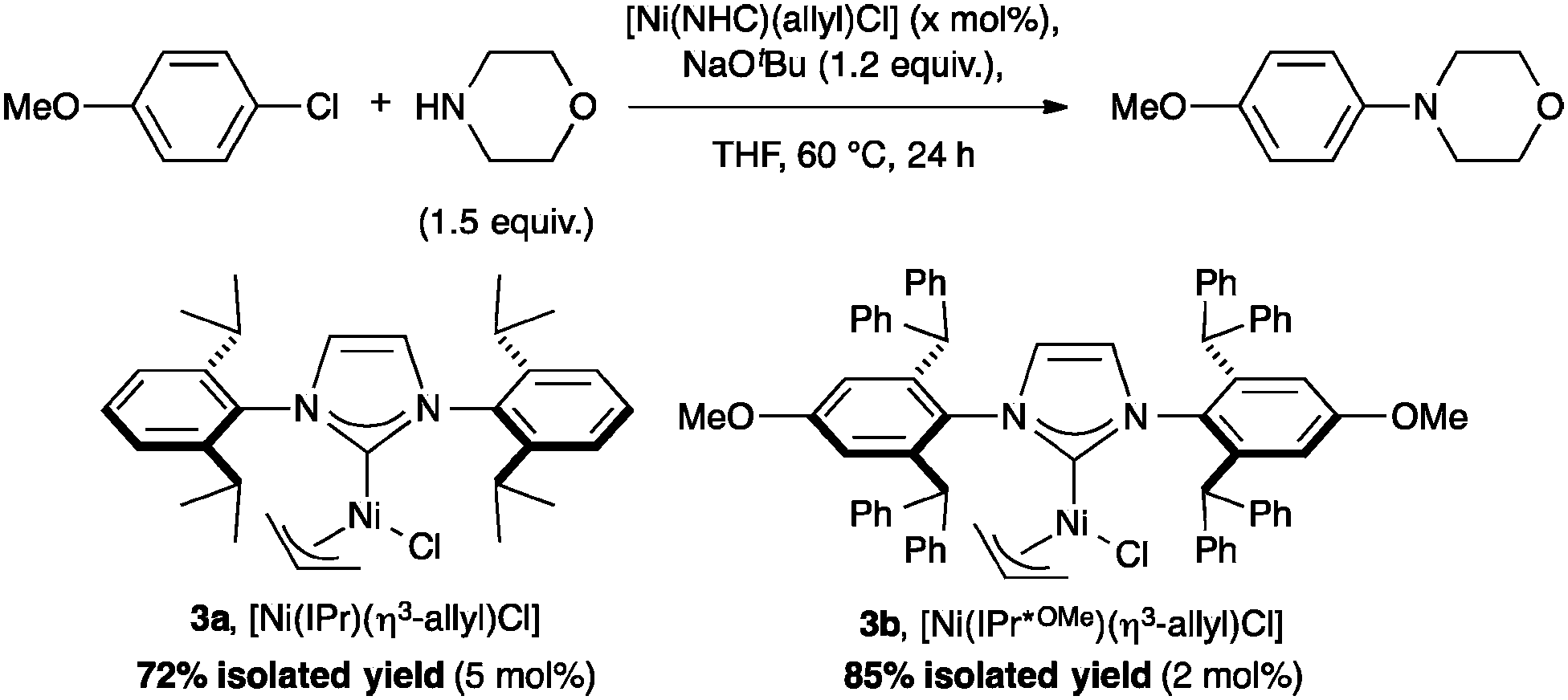 | ||
| Scheme 3 Efficiency of nickel pre-catalysts in the Buchwald–Hartwig reaction. | ||
In addition, the same pre-catalyst was used under similar conditions to perform C–S cross-couplings at very low catalyst loadings.37,38
The highly beneficial effects of the IPr* ligand in nickel catalysis was further demonstrated in the coupling of organoboron reagents with CO2 with [Ni(IPr*)(allyl)Cl] to produce carboxylic acid. As shown in Scheme 4, varying the NHC ligand in the well-defined Ni complexes from IPr* to the smaller IPr dramatically reduces the reactivity.40
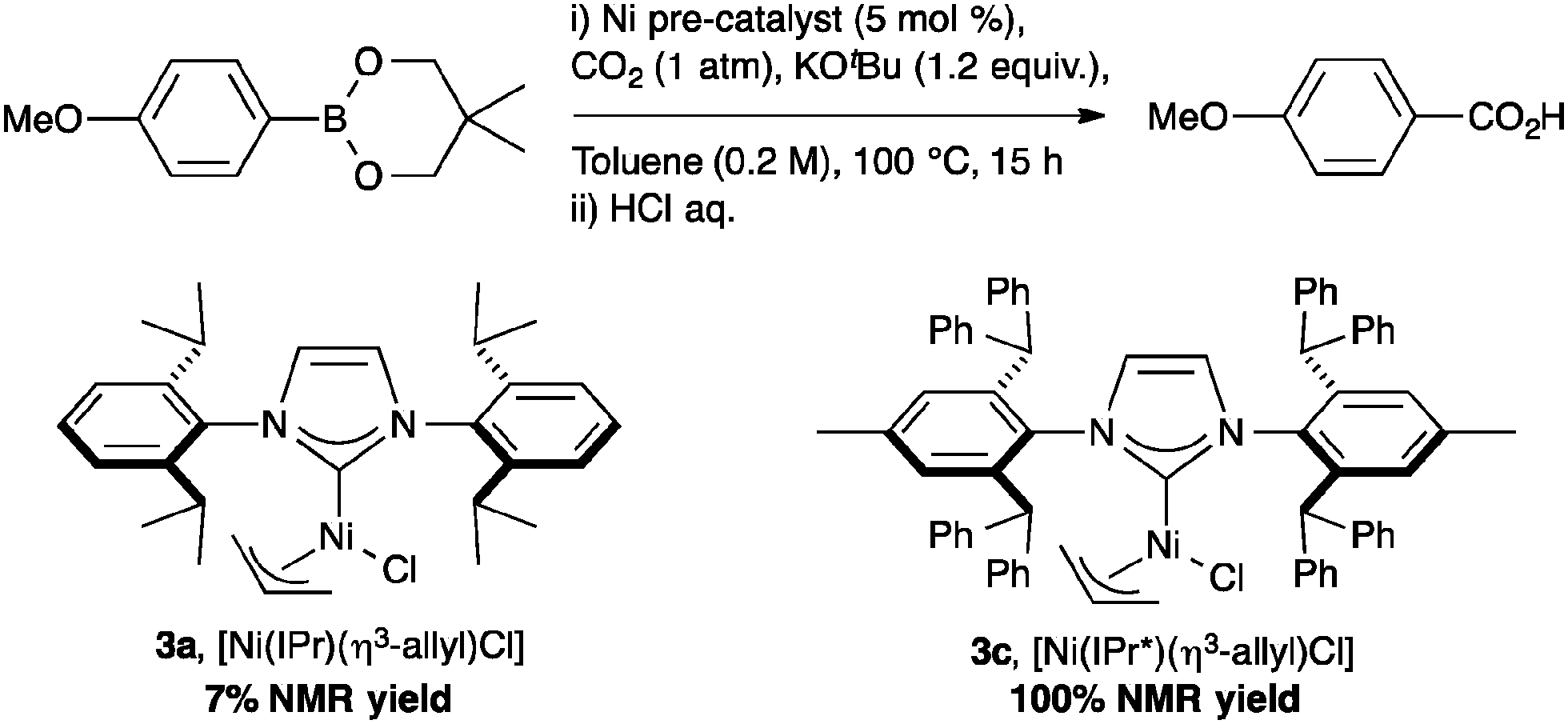 | ||
| Scheme 4 Nickel-catalyzed carboxylation. | ||
Under optimized conditions, a broad scope of aryl- and heteroarylboronates as well as alkenylboronates could be reacted with CO2 (Scheme 5) to produce the corresponding carboxylic acids. As seen from p-methoxy- and p-trifluoromethyl-substituted compounds, the reaction is not influenced by the electronic properties of the substituents on the arylboronates. In addition, this nickel-catalyzed carboxylation tolerated a variety of functional groups such as silyl ethers and esters.40
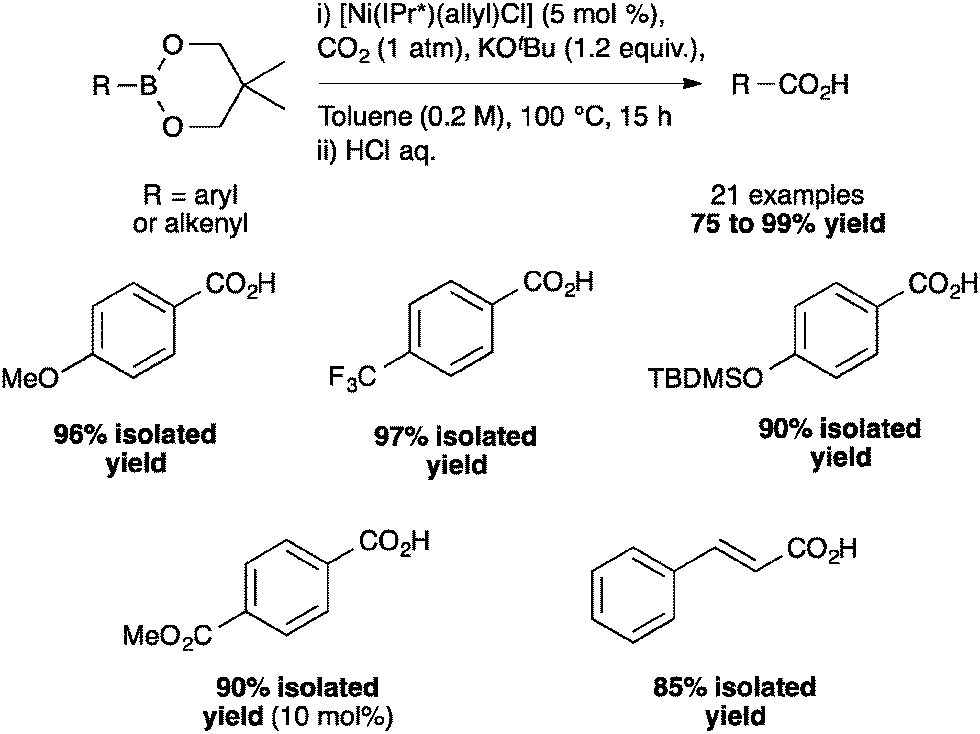 | ||
| Scheme 5 Substrate scope of the nickel-catalyzed carboxylation of arylboronates. | ||
As palladium has proven to be an efficient metal in numerous coupling reactions, Nolan and co-workers reported the synthesis of several IPr* Pd-complexes, bearing IPr* and variations of this core motif: [Pd(IPr*)(cin)Cl] (Scheme 6, 4a cin = cinnamyl) with a %Vbur of 44.6,41 [Pd(IPr*)(acac)Cl] (4c) with 42.2,42 Pd–PEPPSI–IPr* (4e) with 43.1;43 and more recently [Pd(IPr*OMe)(acac)Cl] (4d) with 39.333 and [Pd(IPr*OMe)(cin)Cl] (4b). Markò and co-workers reported [Pd(IPr*(2-Np))(cin)Cl] (4f).35 Interestingly 4a is the bulkiest palladium complex reported to date, while the presence of OMe substituents, as in 4d, improved in certain case the catalytic activity of the complexes.33
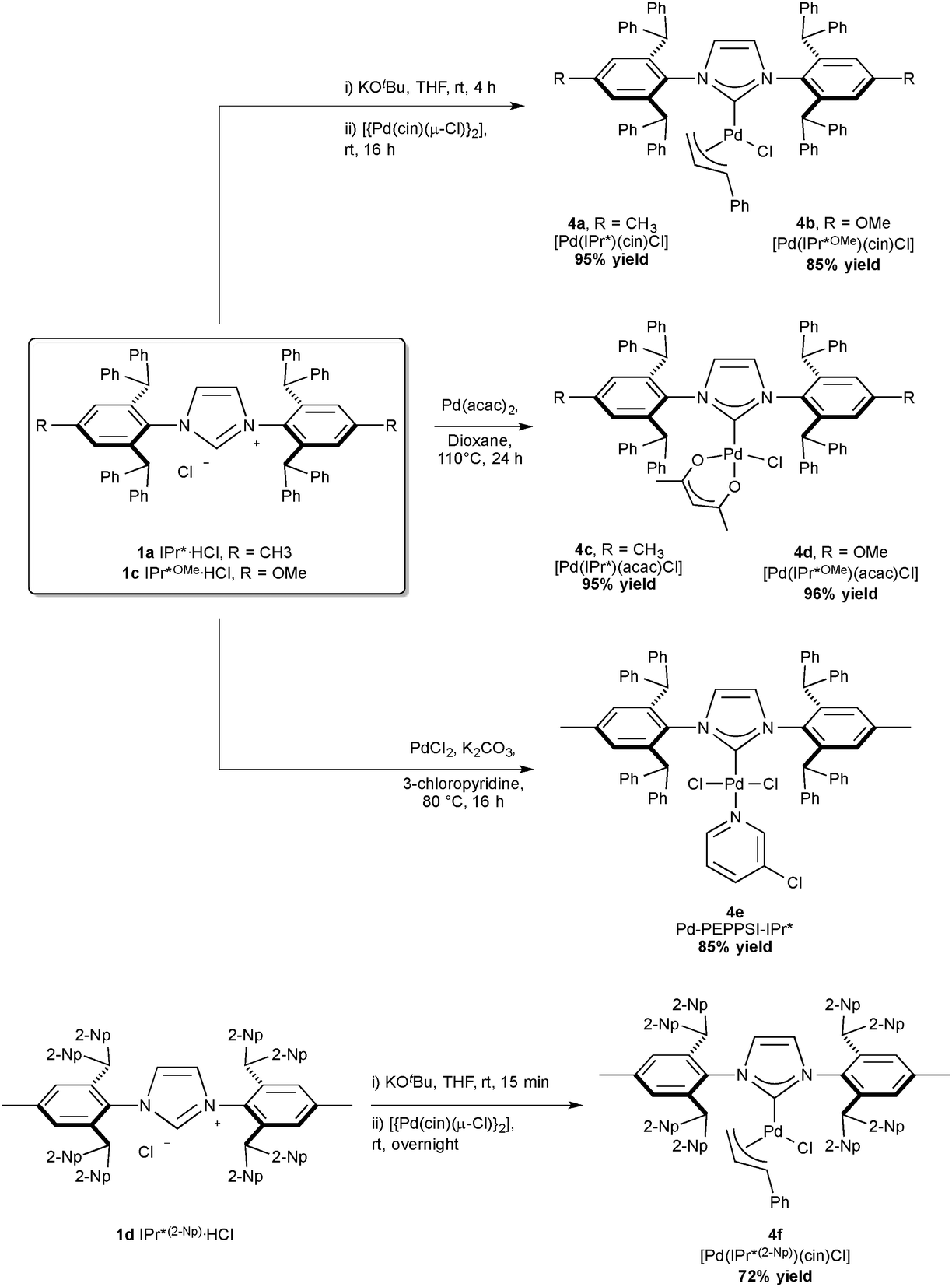 | ||
| Scheme 6 Synthesis of IPr*-based palladium complexes. | ||
These complexes have been used successfully in several reactions. In Buchwald–Hartwig amination, complexes 4a and 4e exhibited improved efficiency at low catalyst loading when compared with the less bulky [Pd(IPr)(acac)Cl] complexes (Table 1).42,44,45
|
|
||||
|---|---|---|---|---|
| Entry | Pd cat. | Cat. loading (mol%) | Conditions | Yieldb (%) |
| a Reaction conditions: (A) aryl chloride (1 mmol), amine (1.1 equiv.), KOtBu (1.1 equiv.), in DME (1 M) at 50 °C. (B) Aryl chloride (1 mmol), amine (1.1 equiv.), KOtAm (1.1 equiv.), in toluene (1 M) at 110 °C for 2 h. (C) Aryl chloride (0.5 mmol), amine (1.1 equiv.), LiHMDS (1.1 equiv.), in dioxane (1 M) at 110 °C for 3 h. (D) Aryl chloride (1 mmol), amine (1.1 equiv.), KOtBu (1.1 equiv.), in toluene (1 M) at 110 °C for 1.5 h. b Isolated yield after chromatography on silica gel. | ||||
| 1 | [Pd(IPr)(acac)Cl] | 1 | A | 97 |
| 2 | [Pd(IPr*)(cin)Cl] (4a) | 0.025 | B | 95 |
| 3 | [Pd(IPr*)(acac)Cl] (4c) | 0.4 | C | 78 |
| 4 | Pd–PEPPSI–IPr* (4e) | 0.025 | D | 96 |
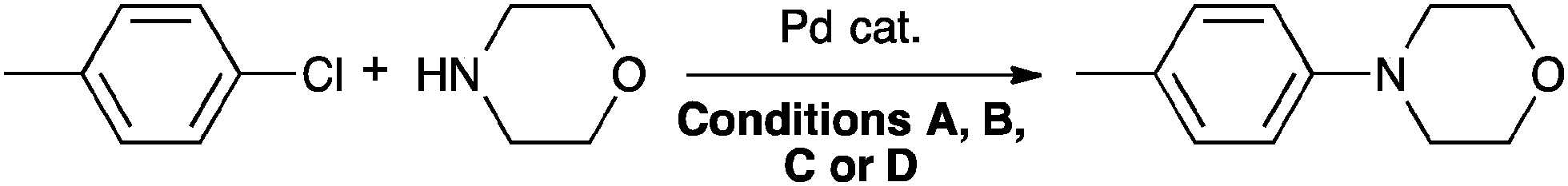
Pre-catalyst [Pd(IPr*)(cin)Cl] permitted the coupling of a wide range of amines with non-activated, deactivated and hindered aryl chlorides. The system was also efficient in the more challenging couplings involving unactivated chlorides with N-methylaniline (Scheme 7).45
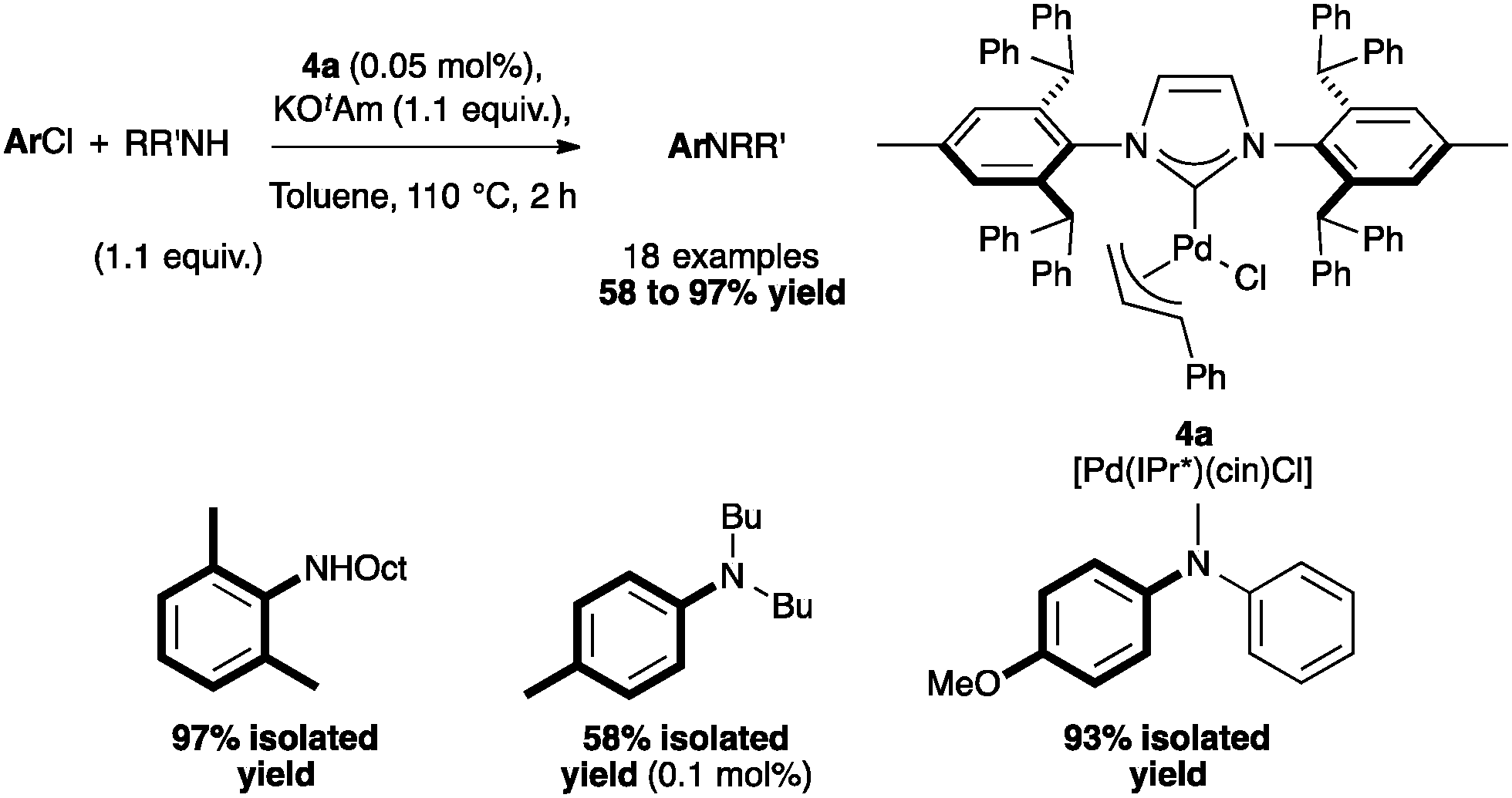 | ||
| Scheme 7 Aryl amination with [Pd(IPr*)(cin)Cl]. | ||
This methodology for aryl amination with 4a has also been extended by Nolan and co-workers to a solvent-free protocol, using 1 mol% catalyst loading, which permitted the coupling with unactivated arylchlorides in excellent yields (83–99%). The catalytic activity of several palladium complexes has been studied (Table 2). Among the pre-catalysts tested, [Pd(IPr*)(cin)Cl] again showed the highest efficiency.46
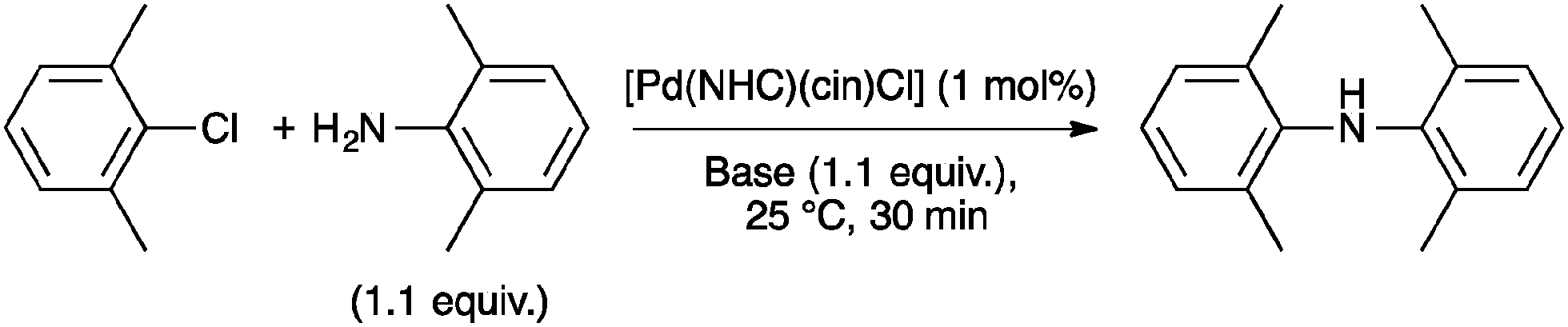
The reactivity of 4a was assessed and a selection of examples are depicted in Scheme 8. The complex appeared to be active with primary amines, especially bulky ones in short reaction times. The coupling with electron-rich chlorides required longer reaction times. Of note, the reaction proceeded in excellent yields with non-aromatic primary and secondary amines.46 In an effort to develop this reaction for flow systems, Guido and co-workers designed a process of C–N bond formation in a continuous flow microreactor using 4a.47
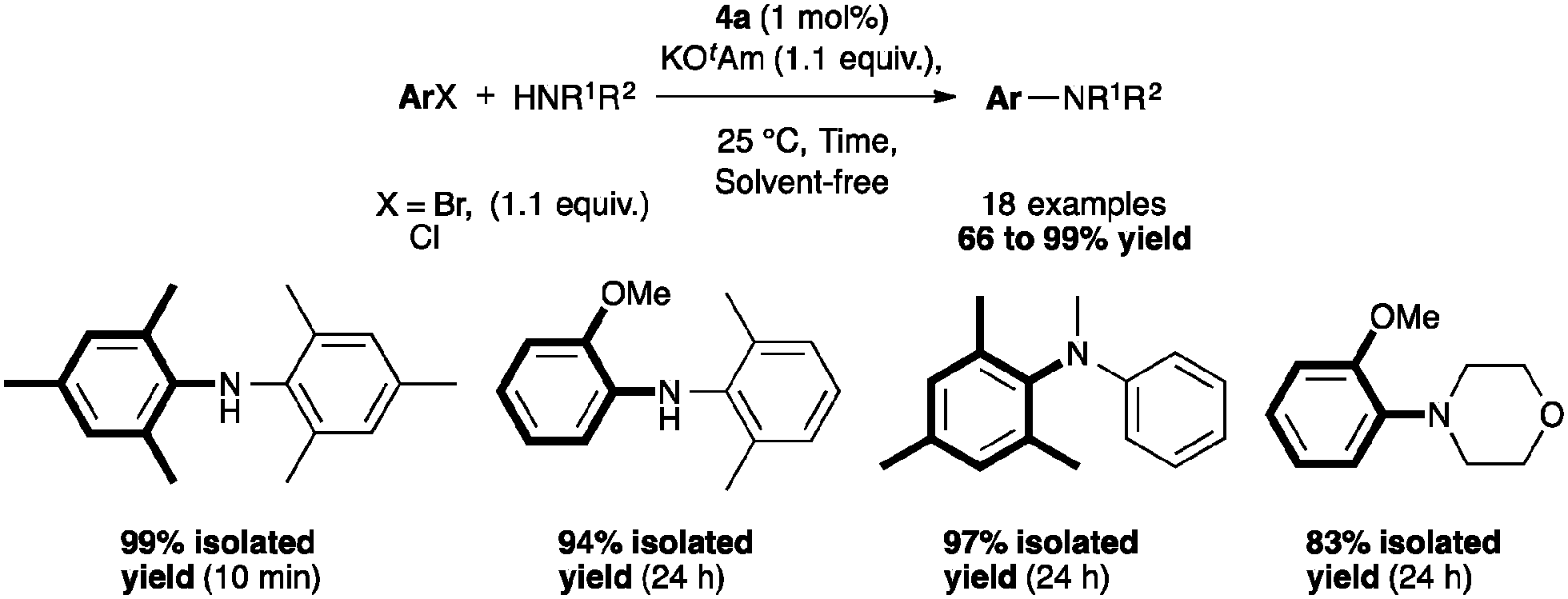 | ||
| Scheme 8 Selected examples for solvent-free amination. | ||
Aryl amination has also been achieved with [Pd(IPr*OMe)(cin)Cl] (4b)48 and [Pd(IPr*OMe)(acac)Cl] (4d).33 Complex 4d was compared to [Pd(IPr*)(acac)Cl] (4c), and showed higher activity, which might be explained by a slightly stronger σ-donor character compared to IPr*. Complex 4d allowed the coupling of secondary biaryl anilines with highly deactivated aryl chlorides (Scheme 9).33
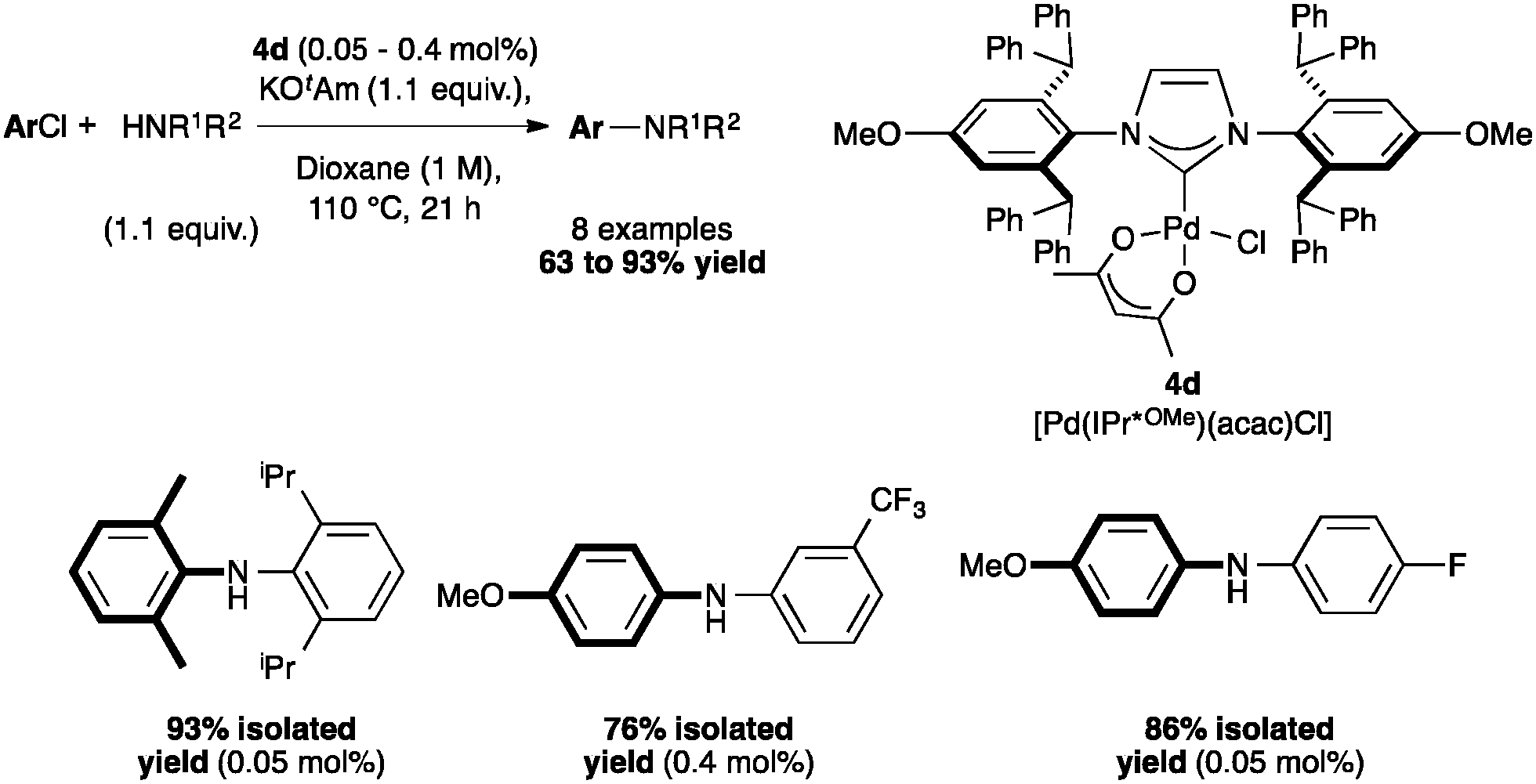 | ||
| Scheme 9 Selected examples of arylamination with [Pd(IPr*OMe)(acac)Cl]. | ||
Moreover, Navarro and co-workers were able to show that such coupling reactions (10 examples, 90 to 99%) can be performed under air using their complex [Pd(IPr*)(TEA)Cl] (TEA: triethylamine) at 1 mol%.49
While the efficiency of numerous NHC systems has been shown in routine C–C cross-coupling reactions, only a few of these studies involve the IPr*-ligand.16,50–55 NHCs have shown comparable reactivity compared to phosphine ligands state-of-the-art, in the depicted cross-couplings.
Among NHC complexes, the [Pd(IPr*)(cin)Cl] catalytic system showed high activity in Suzuki–Miyaura reactions forming several tetra-ortho-substituted biaryl compounds at very low catalyst loadings and under mild conditions (Scheme 10).41,48 These examples show a superior performance than previously reported results described by Dorta and Organ.20,56
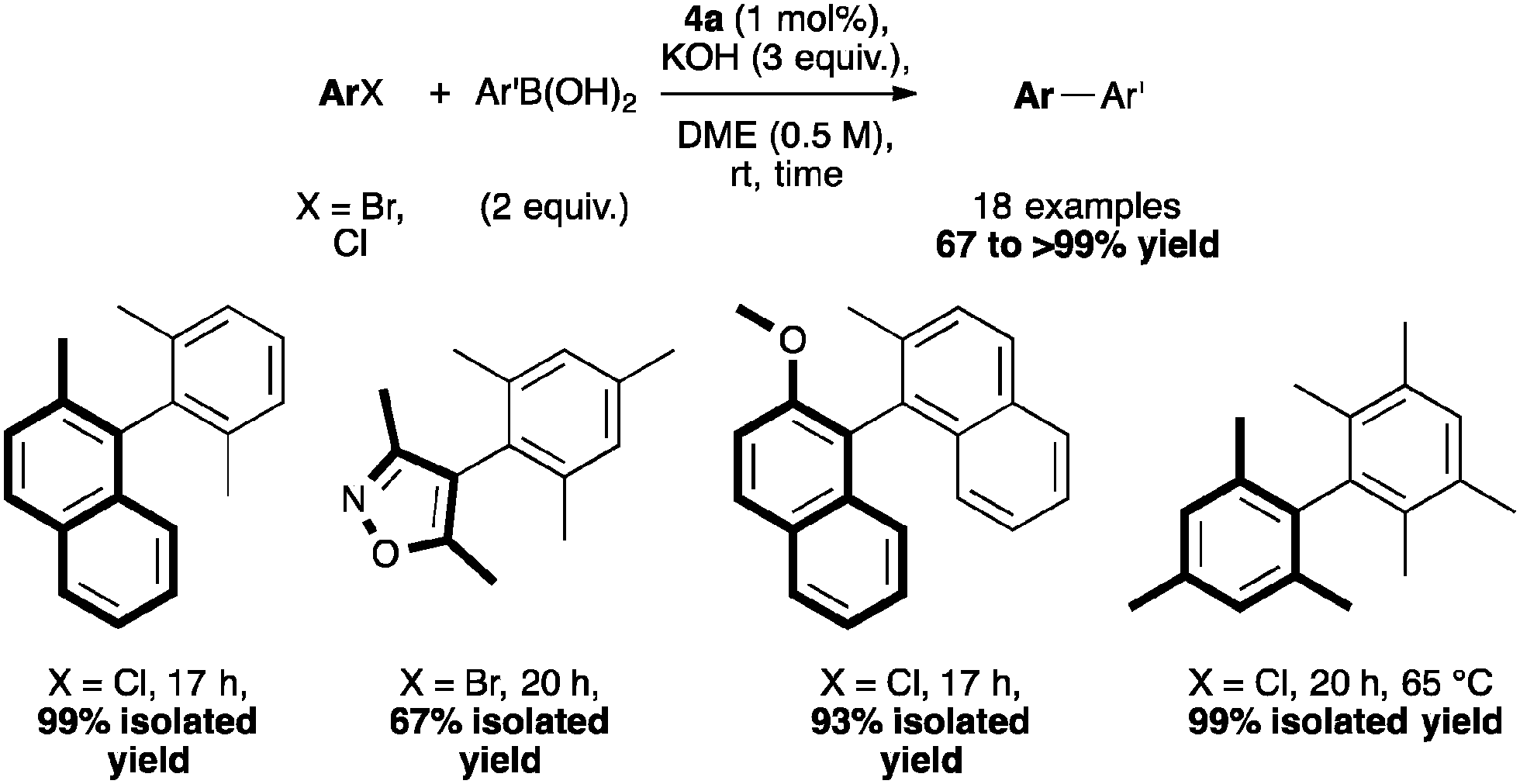 | ||
| Scheme 10 Suzuki–Miyaura cross-coupling using [Pd(IPr*)(cin)Cl] on hindered substrates. | ||
The Kumada–Tamao–Corriu reaction has been investigated by Cazin and co-workers,57 using the palladium dimer: [Pd(IPr*)(μ-Cl)Cl]2 (Scheme 11, 5) which can be accessed easily by treating the cinnamyl complex 4a with ethereal HCl. As shown in Scheme 11, this method allows to form hindered biaryl compounds from both bromides and chlorides at 60 °C with reasonable catalyst loading.57
 | ||
| Scheme 11 Kumada–Tamao–Corriu cross-coupling using [Pd(IPr*)(μ-Cl)Cl]2 on hindered substrates. | ||
The methoxy-derivative [Pd(IPr*OMe)(cin)Cl] (4b) showed remarkable activity in C–S cross coupling reactions. Hindered and deactivated aryl halides reacted with both aryl thiols (Scheme 12) and particularly challenging alkyl thiols.58
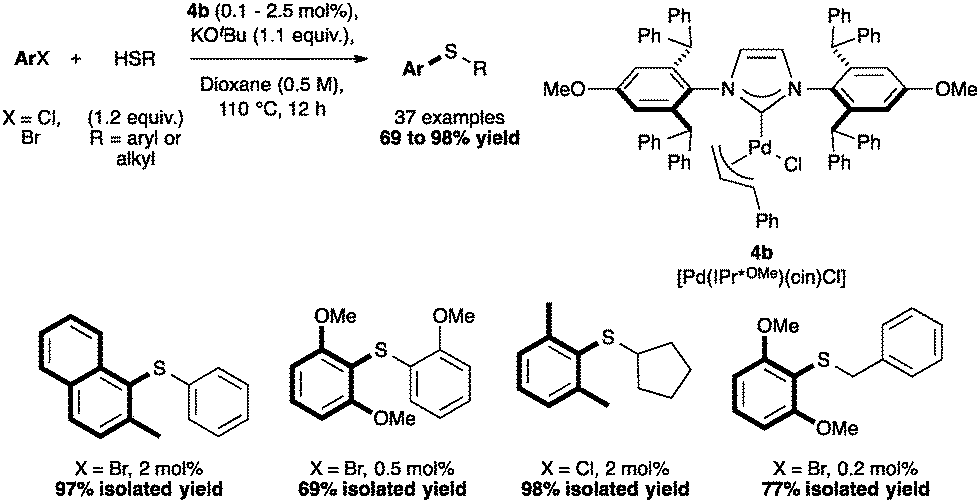 | ||
| Scheme 12 Selected examples of C–S couplings. | ||
Complex 4a also showed interesting reactivity in S-arylation reactions.59 Poli and Madec explored such cross-couplings for the preparation of bis-arylated sulfoxides.60–62 The latter were formed from β-sulfinyl esters60 or allyl sulfoxides61 using Pd2(dba)3 and Xantphos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene). Recently, in a protocol developed in our laboratory, [Pd(IPr*)(cin)Cl] (4a) alone allowed access to the direct S-arylation starting from aryl methyl sulfoxides by cleavage of the CH3–S bond (Scheme 13).
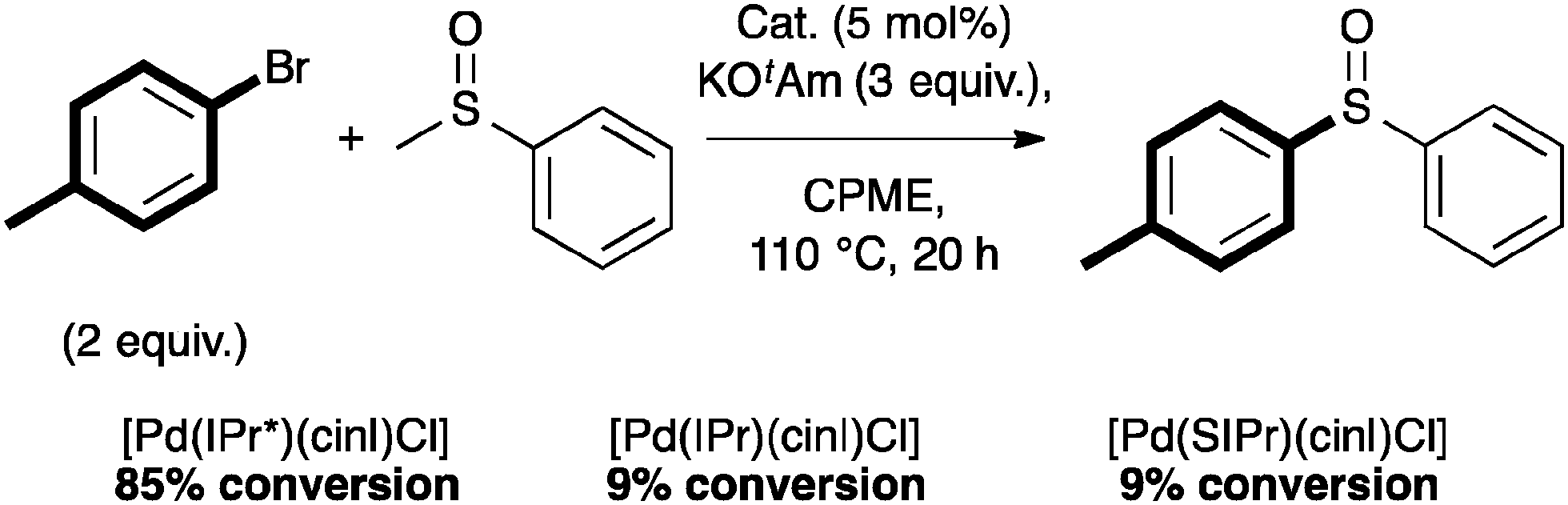 | ||
| Scheme 13 Pre-catalyst screening in S-arylation. | ||
Using 5 mol% of 4a, non-activated and activated sulfoxides were coupled quite efficiently with p-bromo- or p-chlorotoluene (Scheme 14). However, slight modifications of the aryl halide such as m-chlorotoluene, significantly reduced the reactivity.59
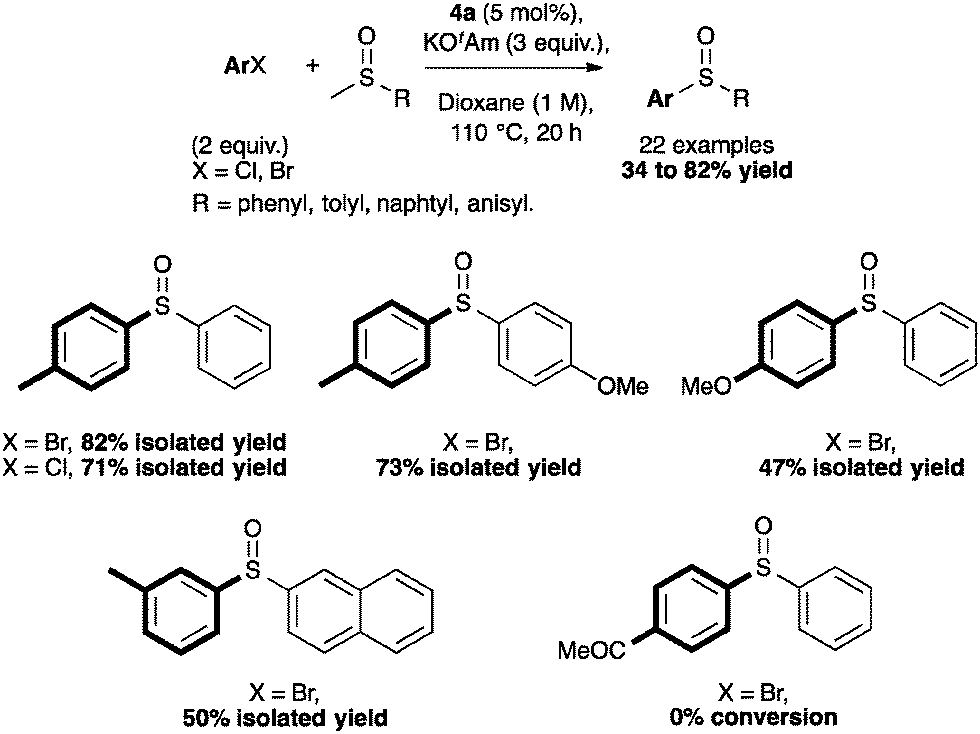 | ||
| Scheme 14 Selected examples for the S-arylation reaction. | ||
Although there have been no applications of IPr* complexes of platinum to date, Cundari and co-workers, through the use of different models including one based on IPr*, were able to study in silico mechanisms permitting methane C–H bond activation with cationic and neutral PtII–NHC. In the case of the IPr* model, computational results highlight the impact of the steric bulk of the NHC ligand on the reaction.63
Copper, silver and gold chemistry with IPr*
With the exclusion of palladium, the synthetic chemistry of the IPr*-ligand family has been mostly explored with gold and copper, while only two complexes with silver has been reported in the literature, namely [Ag(IPr*)Cl] (70% yield, %Vbur = 53.5) and the analogous complex of the larger congener, IPr*(2-Np) ([Ag(IPr*(2-Np))Cl], 91% yield, %Vbur = 57.4).19,35In 2011, Nolan and Cazin reported the synthesis of [Cu(IPr*)Cl] (Scheme 15, 6a, %VBur = 50.1). They developed and improved a general synthetic route to [Cu(NHC)(X)] complexes, using CuCl and an excess of K2CO3, in acetone at reflux. Using this method, the desired product was obtained under mild and environmentally friendly conditions.64 Very recently, Markò and co-workers prepared a Cu-complex of IPr*(2-Np) (6b, %VBur = 57.1).35
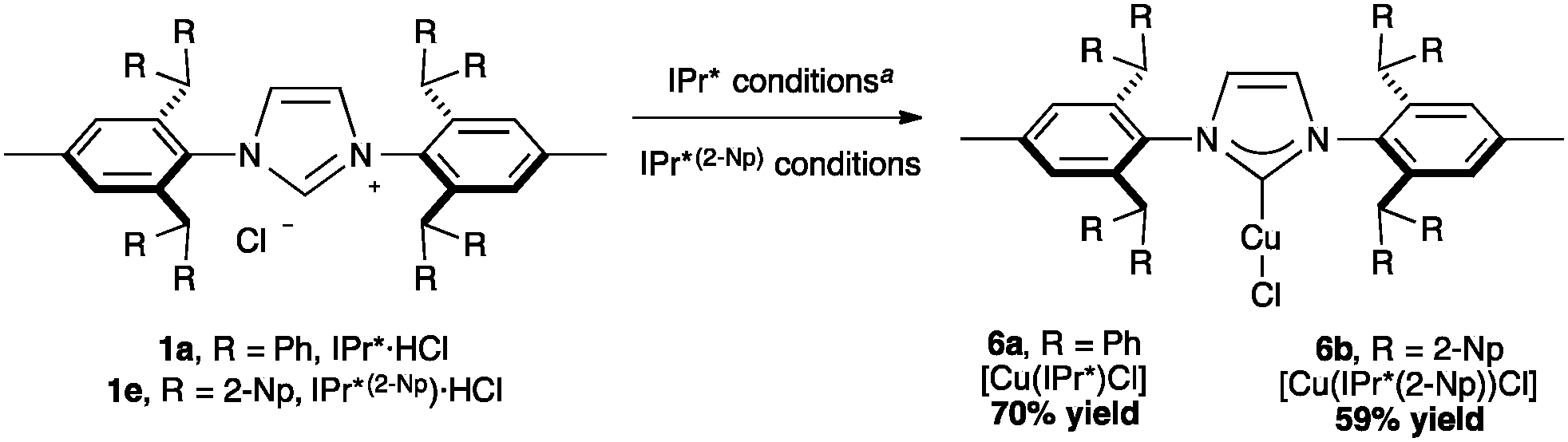 | ||
| Scheme 15 Synthesis of [Cu(IPr*)(Cl)] and [Cu(IPr*(2-Np))(Cl)]. aIPr* conditions: IPr*·HCl (0.10 mmol), CuCl (1 equiv.), K2CO3 (2 equiv.) in acetone (1 M) at reflux for 24 h. IPr*(2-Np) conditions: IPr*(2-Np)·HCl (0.125 mmol), CuCl (1 equiv.), KOtBu (1.5 equiv.) in DCM (0.02 M) at room temperature, overnight. | ||
Interesting variations of the copper IPr* system have been explored by Rasika Dias and co-workers. Under CO atmosphere, they were able to prepare the dicarbonyl complex [Cu(IPr*)(CO)2][SbF6] from (IPr*)CuBr and AgSbF6 (Scheme 16, A), which showed high moisture sensitivity and quickly incorporated a molecule of water, forming [Cu(IPr*)(H2O)(CO)][SbF6]. Interestingly, under reduced pressure it is possible to remove the second coordinated CO, giving [Cu(IPr*)(H2O)][SbF6] (Scheme 16, B).65
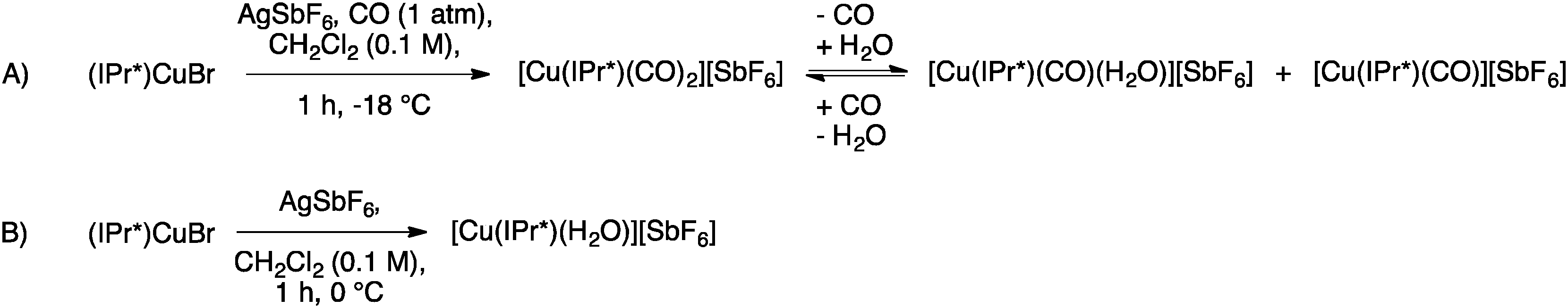 | ||
| Scheme 16 Synthesis of [Cu(IPr*)(CO)2][SbF6] and [Cu(IPr*)(H2O)][SbF6]. | ||
The first IPr*–gold complex described in the literature is [Au(IPr*)Cl] (Scheme 17, %VBur = 50.4), which was easily obtained either from [Au(DMS)Cl] and the free NHC, or through the novel route using the corresponding imidazolium salt and potassium carbonate in acetone. Using the same procedure the analogous, [Au(IPr*Tol)Cl] could be prepared. Interestingly, the gold complex cannot be obtained through transmetallation from [Cu(IPr*)Cl] or [Ag(IPr*)Cl].64,66
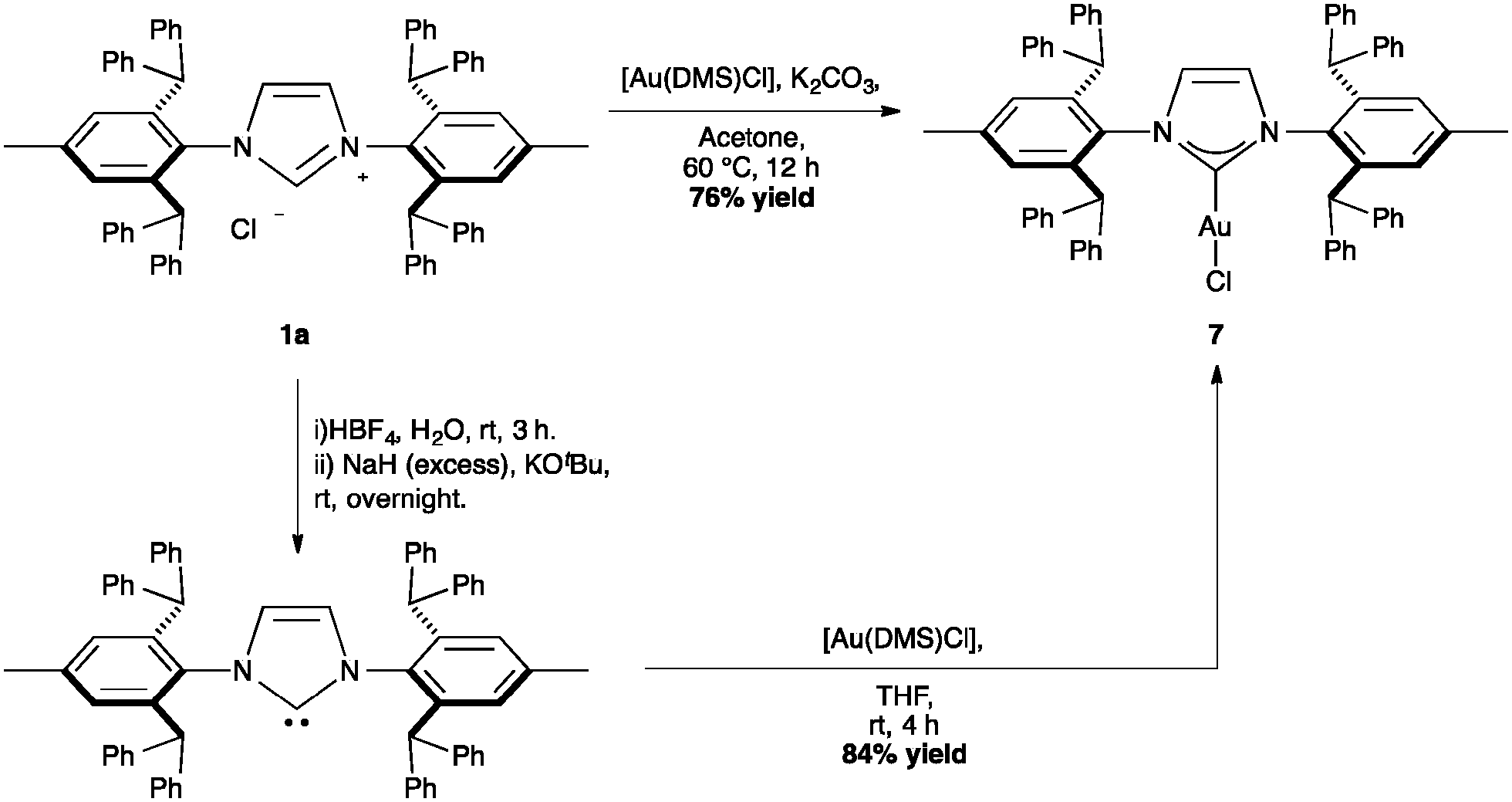 | ||
| Scheme 17 Synthesis of [Au(IPr*)Cl]. | ||
NTf2 gold species have been synthesized, in particular [Au(NHC)(NTf2)] and the digold species [{Au(NHC)}2(μ-OH)][BF4].64,67 Complex 7 has been used to prepare the corresponding cationic and digold derivatives, respectively 8c, 8a and 8b (Scheme 18), which were then tested in a number of catalytic reactions (Schemes 19–21). The IPr*·Au-complex 8a proved to be less active in these catalytic reactions depicted in Schemes 19–21; probably due to the steric hindrance and showing the limits in this chemistry of the larger is more efficient concept.67
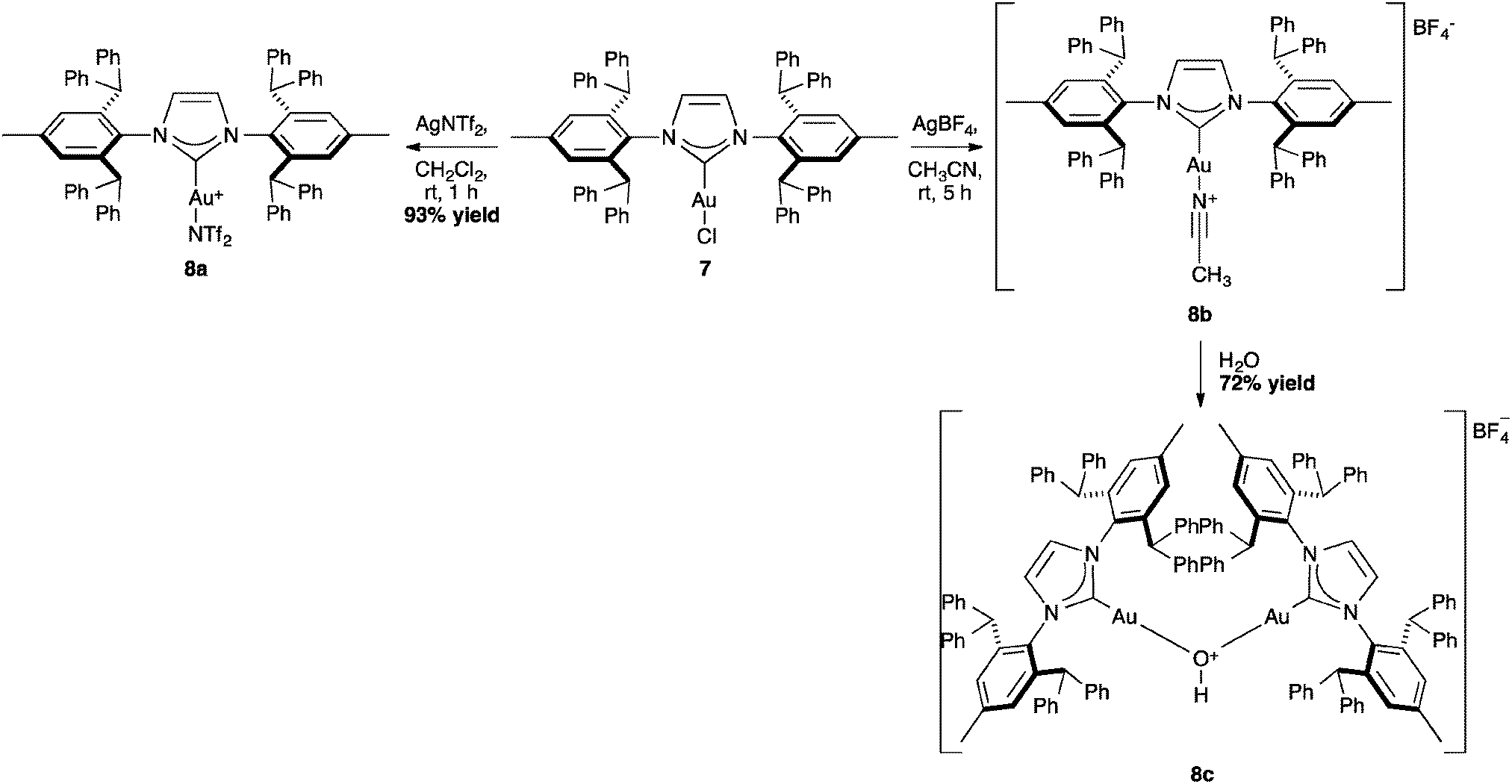 | ||
| Scheme 18 Synthesis of other gold complexes from [Au(IPr*)Cl]. | ||
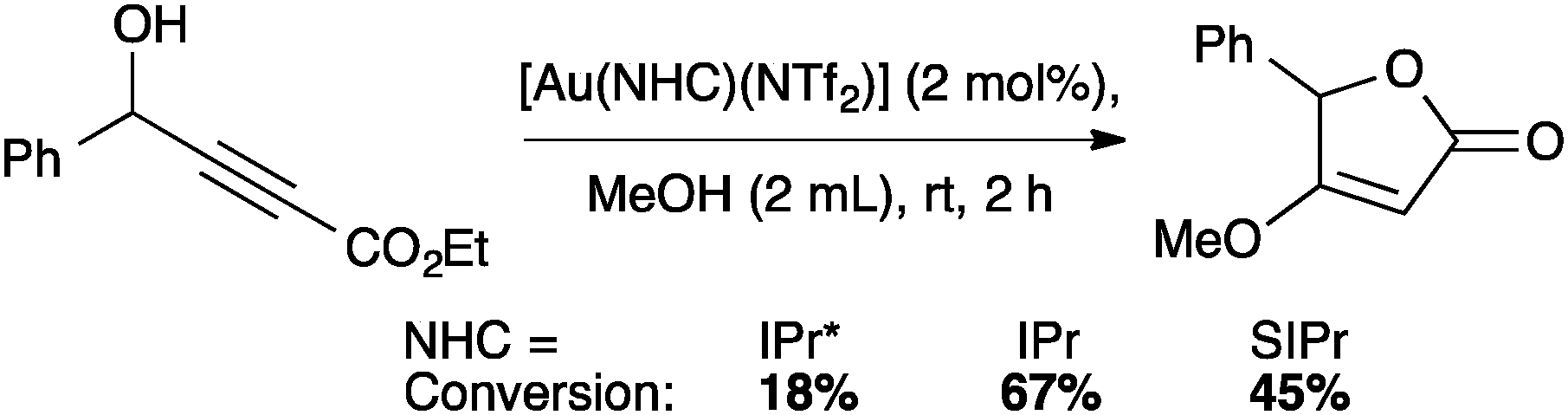 | ||
| Scheme 19 Catalyst screening for the synthesis of furanone. | ||
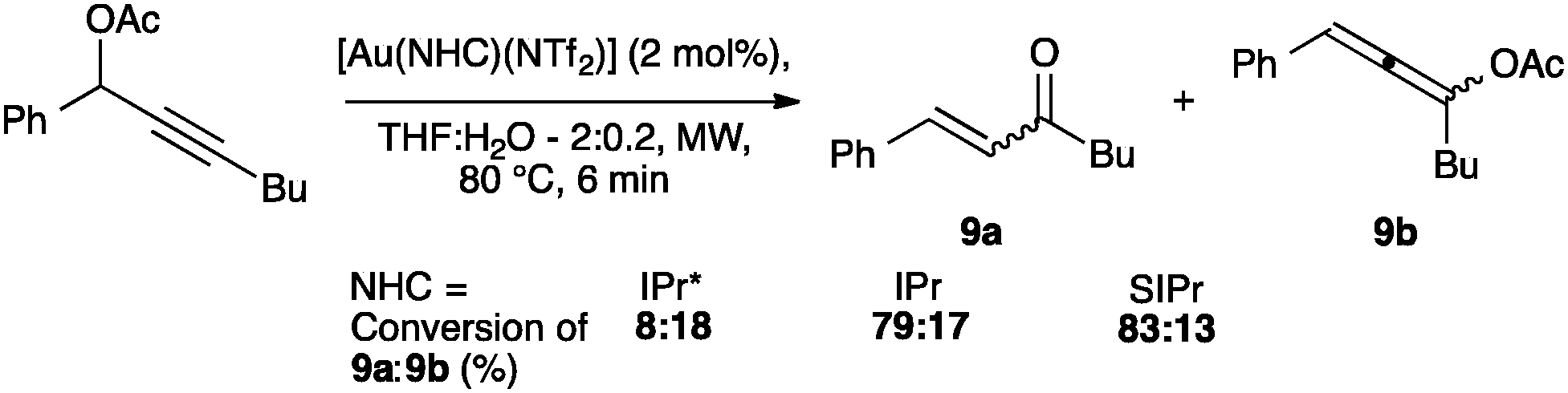 | ||
| Scheme 20 Catalyst screening of the isomerization of propargylic acetate. | ||
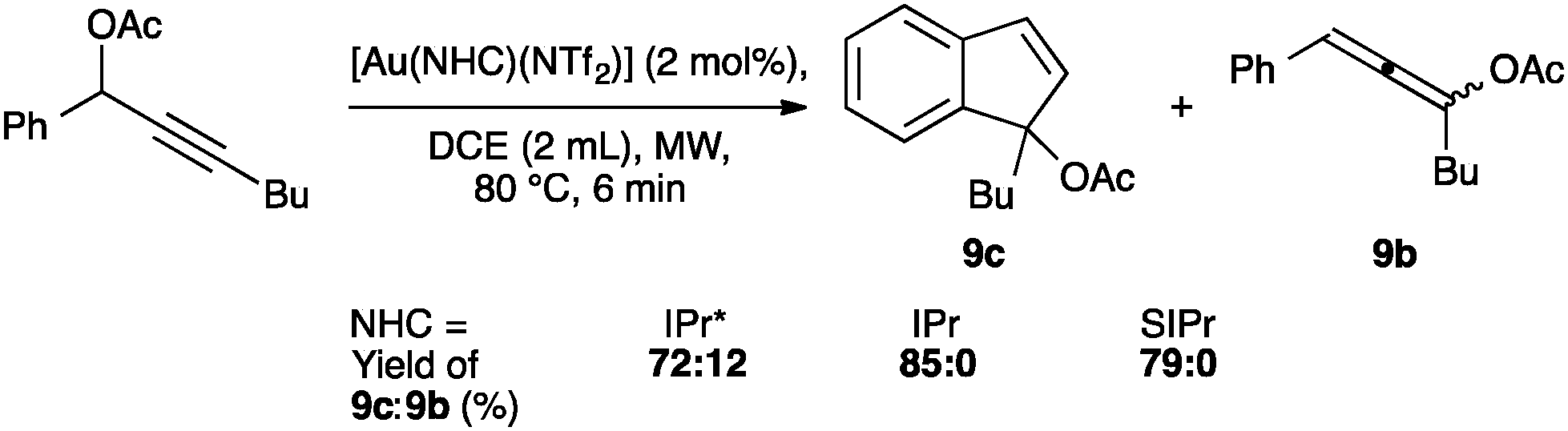 | ||
| Scheme 21 Catalyst screening for the synthesis of substituted indenes. | ||
Interestingly, the activity of IPr* based complexes showed a significant dependence on the nature of the solvent (Scheme 22). Changing the reaction solvent from tetrahydrofuran to the less coordinating dichloroethane resulted in an improvement of the reactivity.64
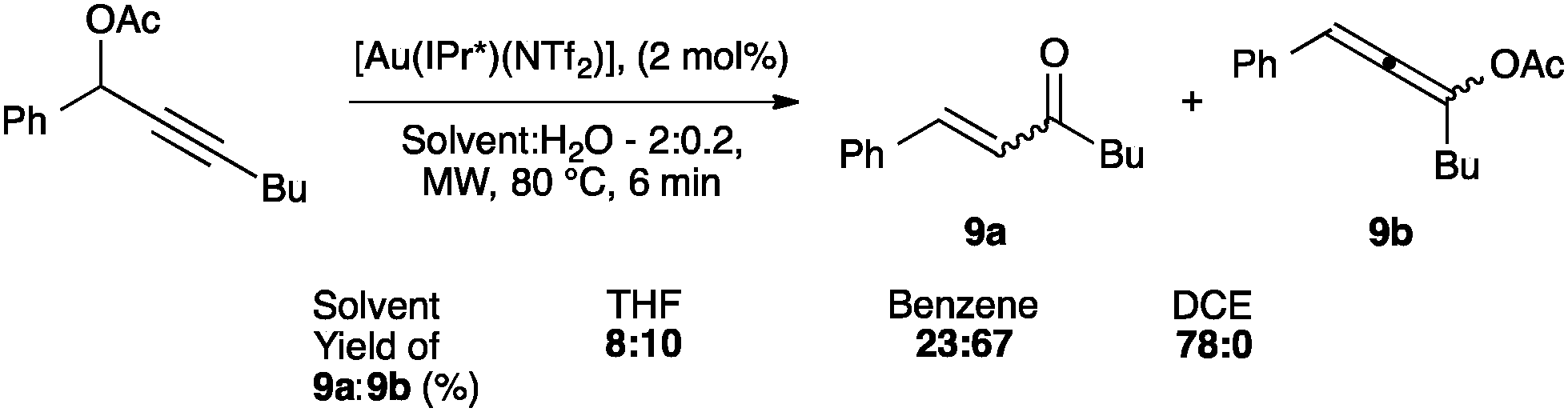 | ||
| Scheme 22 Solvent dependency of IPr*-based catalyst. | ||
The IPr* digold hydroxide species [{Au(IPr*)}2(μ-OH)][BF4] shows a similar trend in activity (Schemes 23–25), confirming that NHC ligands with excessive bulk can be detrimental to activity in some instances.67
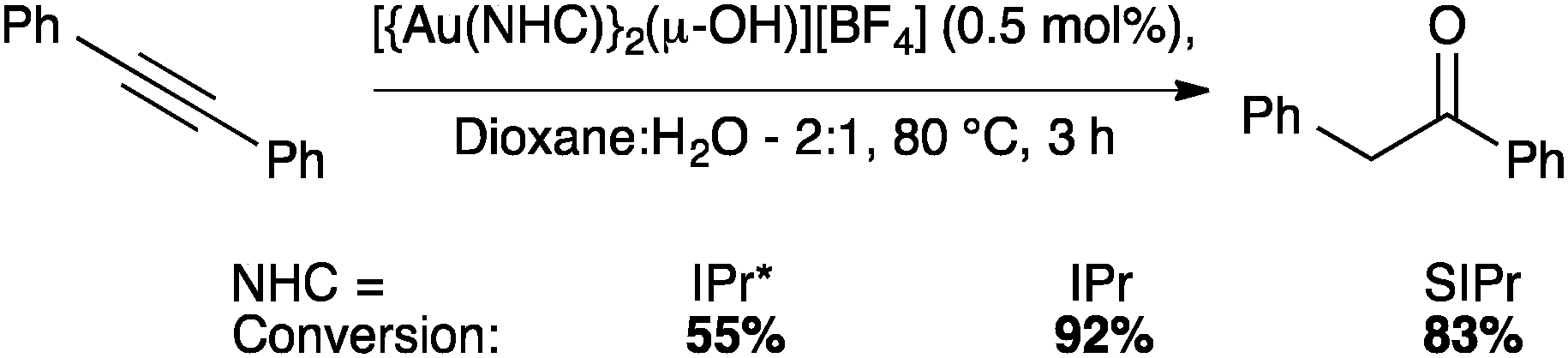 | ||
| Scheme 23 Gold-catalyzed alkyne hydration. | ||
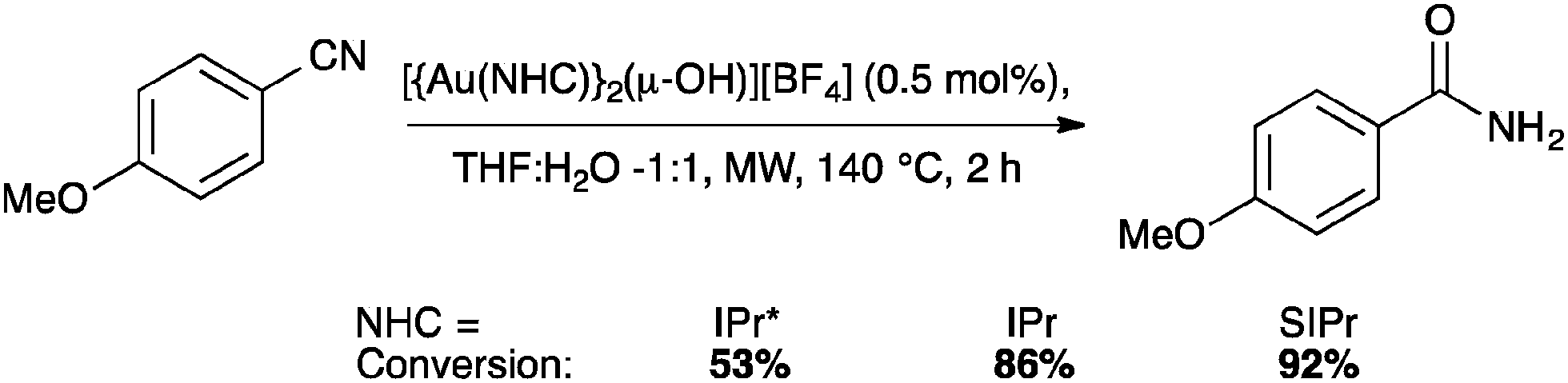 | ||
| Scheme 24 Gold-catalyzed nitrile hydration. | ||
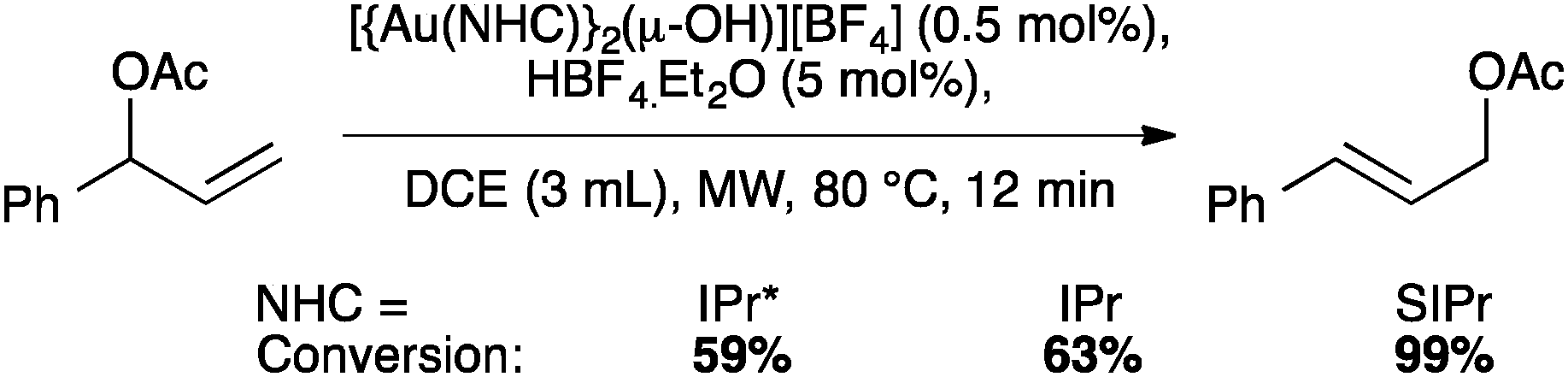 | ||
| Scheme 25 Gold-catalyzed allylic acetate rearrangement. | ||
The bulkiest variation of IPr*, the IPr** ligand introduced by Straub and co-workers has been investigated in the isolation of reactive cationic intermediates in gold catalysis. The synthesis of cationic [Au(NHC)][SbF6] proceeded through the chloride abstraction in complex 10a (%Vbur = 55.4) with silver hexafluoroantimonate. In fact, the increased steric hindrance of the ligand stabilized the formation of the mesomeric gold–silver species 10d, which was characterized by X-ray analysis. In this complex, the AgCl salt is coordinated to the [Au(IPr**)] moiety through a σ-bond interaction (Scheme 26).68
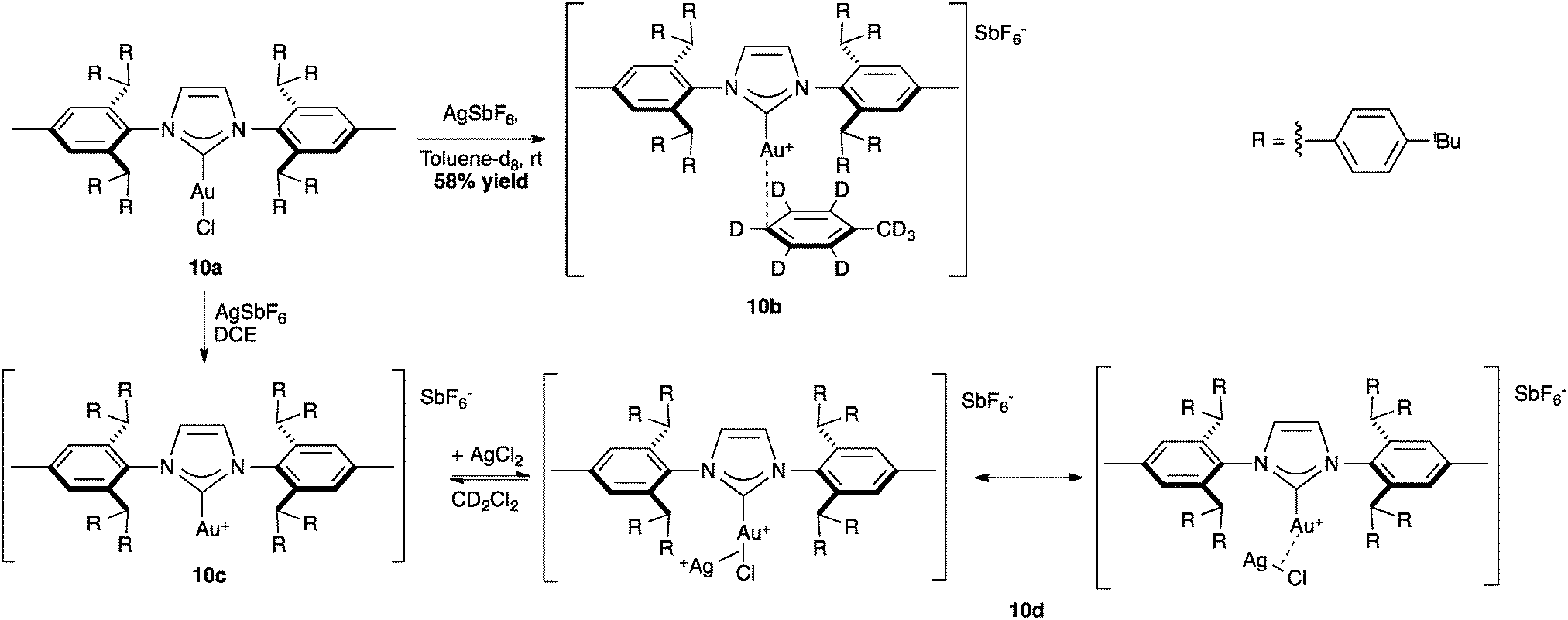 | ||
| Scheme 26 Gold synthesis of cationic [Au(NHC)][SbF6]. | ||
The positive effect of the shielding of the IPr** ligand also allowed for the isolation of intermediates, such as complex 11a (Scheme 27). Interestingly, the crystal data show a linear structure with C2-symmetry, in which the two hindered N-aryl substituents protect the gold center from bimolecular decomposition.69
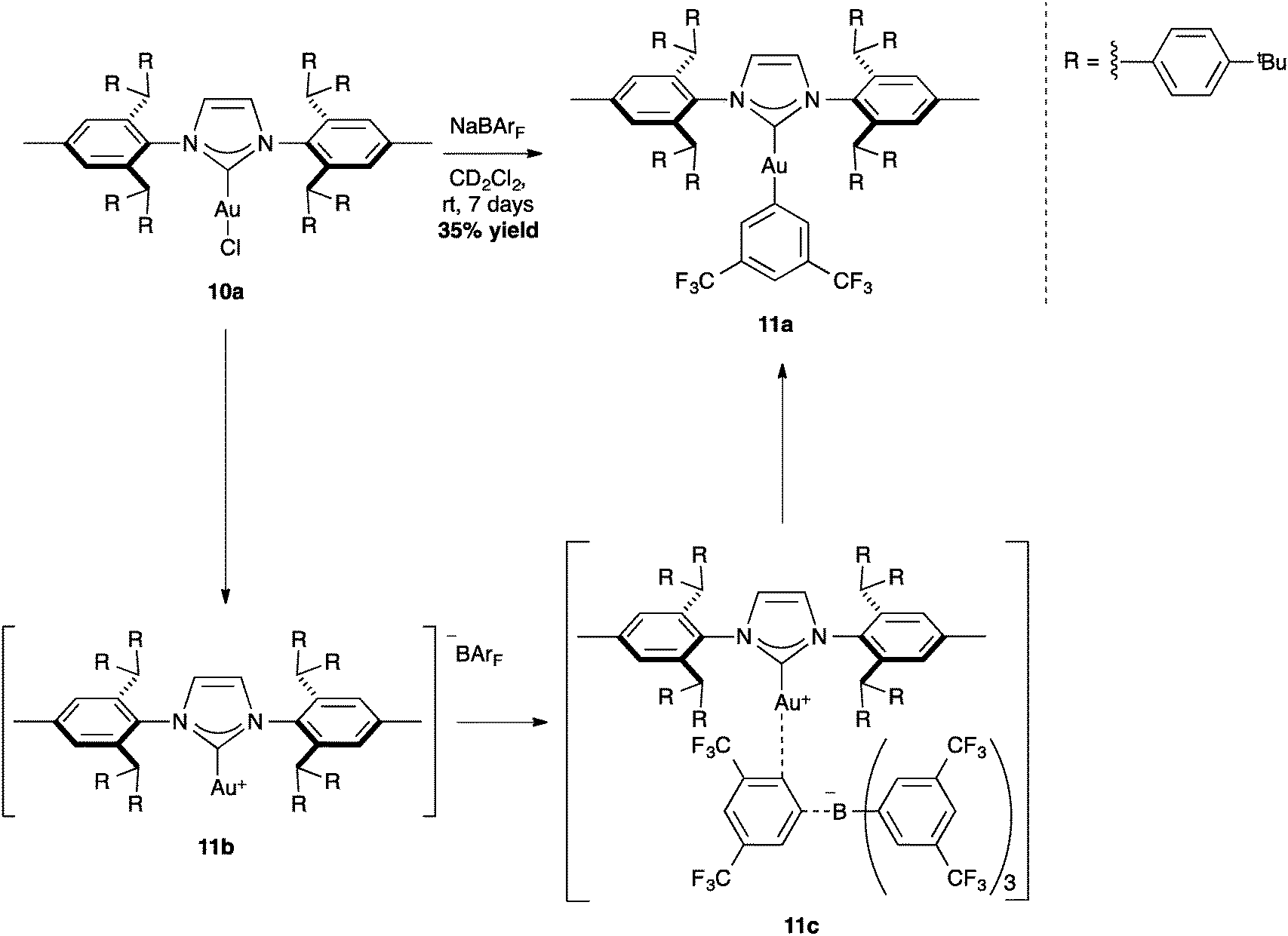 | ||
| Scheme 27 Synthesis of the Au-complex 11a. | ||
Using the positive effect of the shielding of the IPr** ligand, Straub and co-workers successfully isolated crystals of non-heteroatom-stabilized gold–carbene complex (Scheme 28, 12b).70 This example shows again the importance of the steric bulk to avoid the decomposition of sensitive complexes.
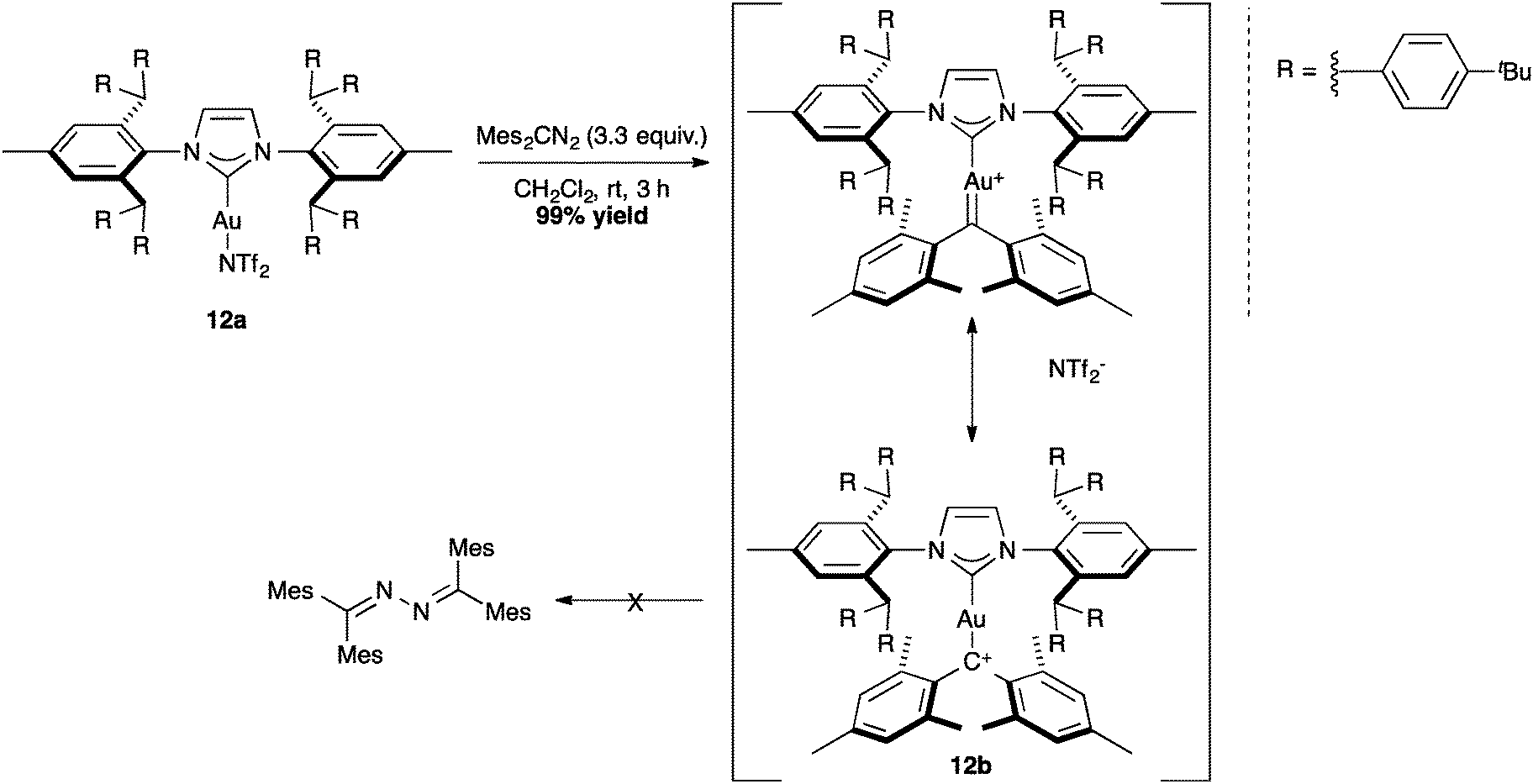 | ||
| Scheme 28 Non-heteroatom-stabilized Au–carbene complex 12b. | ||
Ruthenium complexes: synthesis and activity
The use of IPr* in ruthenium catalyzed reaction has been explored in olefin metathesis and in alcohol racemization.71,72 Concerning olefin metathesis, it has been stated that hindered N-aryl NHCs prevent bimolecular deactivation and ortho-metalation, increasing the lifespan of the propagating species during the reaction.11 In addition, bulky NHCs have shown beneficial effects in catalysis; the introduction of IPr and SIPr in ruthenium indenylidene complexes, improved the catalytic activity for certain substrates, promoting ring-closing metathesis of unhindered olefins at very low catalyst loadings (Scheme 29).73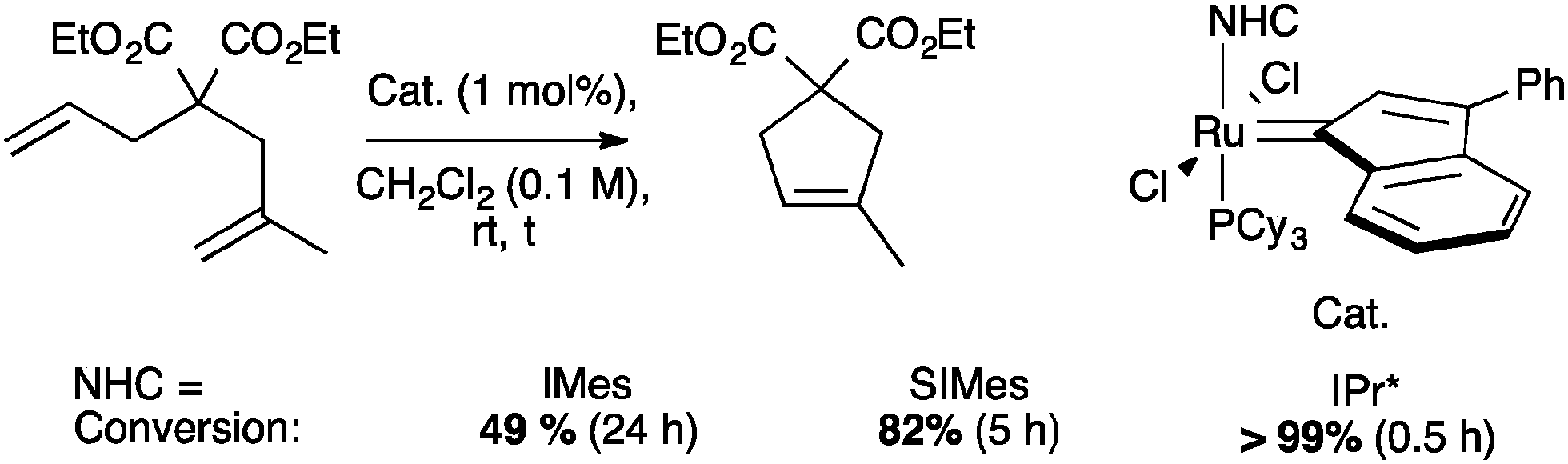 | ||
| Scheme 29 Ring-closing metathesis of unhindered olefins using ruthenium indenylidene complexes. | ||
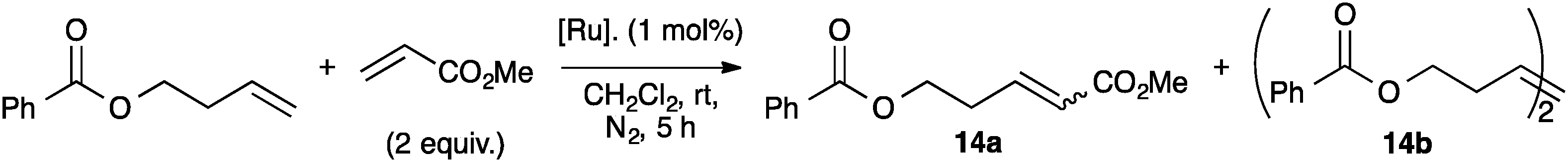
However, in order to understand how much steric demand can be tolerated without adversely affecting the catalytic activity, IPr* was considered. Using the general procedure to synthesize second and third generation indenylidene-type pre-catalysts, reported by Nolan and co-workers,74 complexes 13a and 13c were prepared in modest yields (43% and 73% respectively, Scheme 30).71
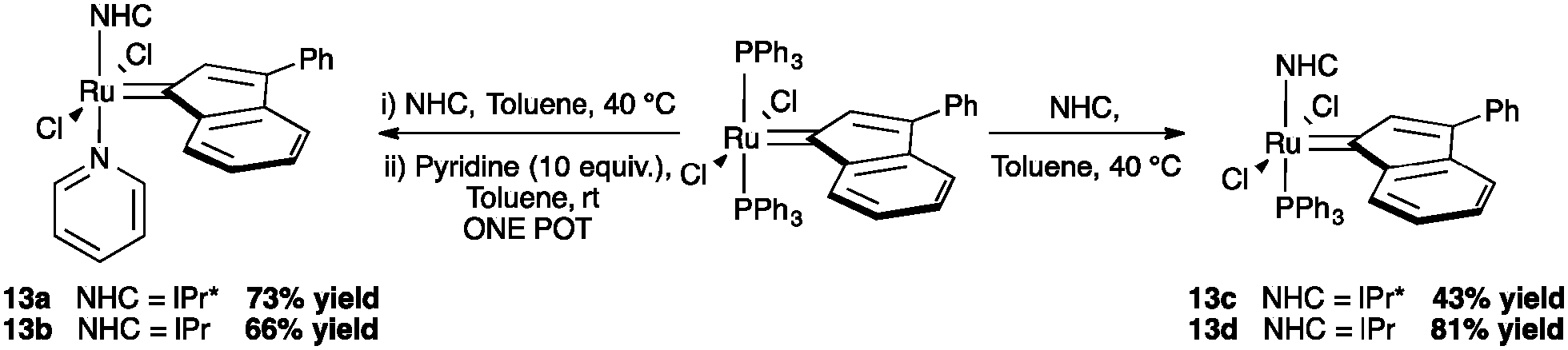 | ||
| Scheme 30 Synthesis of NHC ruthenium indenylidene complexes. | ||
In order to understand the reactivity of these complexes, 13a and 13c were evaluated via DFT calculations using ethylene as a substrate. In particular, even though the two pre-catalysts showed the same %Vbur in the solid state; the increased bulkiness of IPr* had a detrimental effect during the ligand dissociation and olefin binding steps of the metathesis process.12 The calculations were in accordance with the experimental results (Tables 3 and 4), showing that for complexes 13a and 13c, longer reaction times were usually required to achieve full conversions than with 13b and 13d (Table 3).71
| Entry | Substrate | Product | Cat. | Time (h) | Conversionb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: substrate (0.25 mmol), Cat. (1 mol%), CH2Cl2 (0.1 M), N2, RT. b Conversions determined by 1H NMR. | |||||
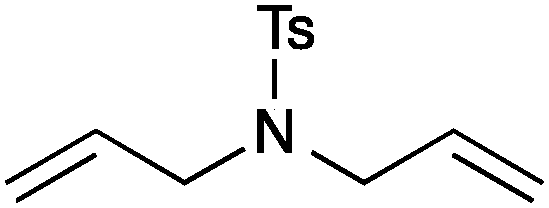
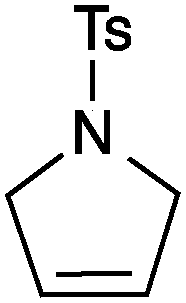
| 1 |
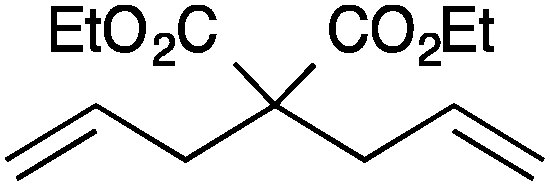
|
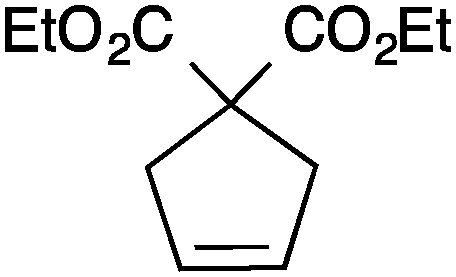
|
13a | 2 | 99 |
| 2 | 13c | 3 | 95 | ||
| 3 | 13b | 1 | 99 | ||
| 4 | 13d | 0.25 | 99 | ||
| 5 |
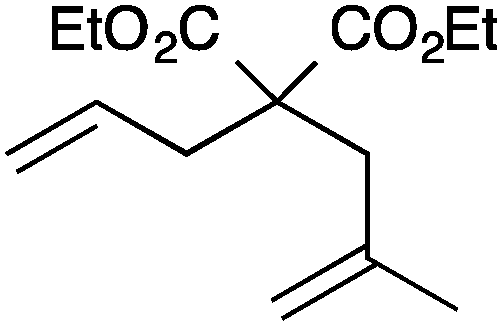
|
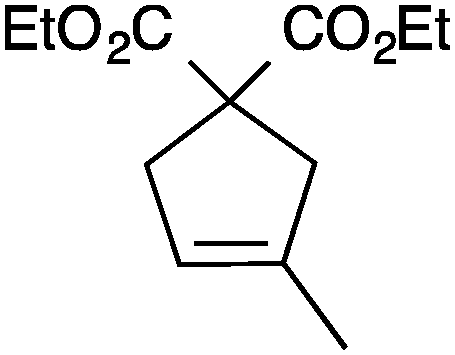
|
13a | 3 | 75 |
| 6 | 13c | 3 | 94 | ||
| 7 | 13b | 3 | 99 | ||
| 8 | 13d | 3 | 90 | ||
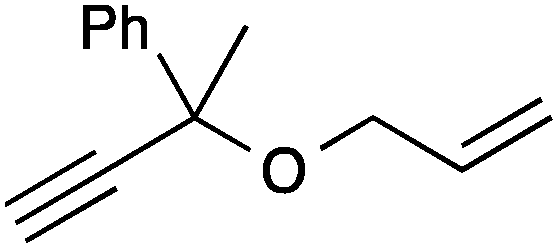
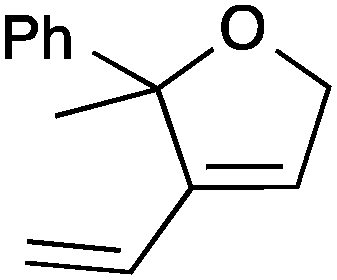
| 9 |
|
|
13a | 3 | >99 |
| 10 | 13c | 3 | 96 | ||
| 11 | 13b | 0.5 | >99 | ||
| 12 | 13d | 0.25 | >99 | ||
| 13 |
|
|
13a | 3 | 42 |
| 14 | 13c | 3 | >99 | ||
| 15 | 13b | 3 | 87 | ||
| 16 | 13d | 3 | 95 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:Regarding the racemization of chiral alcohols, Nolan and co-workers investigated [Ru(NHC)Cp*Cl] (Cp* = 1,2,3,4,5-pentamethylcyclopentadienyl), as catalyst motif, which was known to be active in this transformation. The steric hindrance of IPr* seemed to be detrimental as the desired complex (15) was obtained in very low yields (Scheme 31). This complex presented a %VBur of 31.1.72
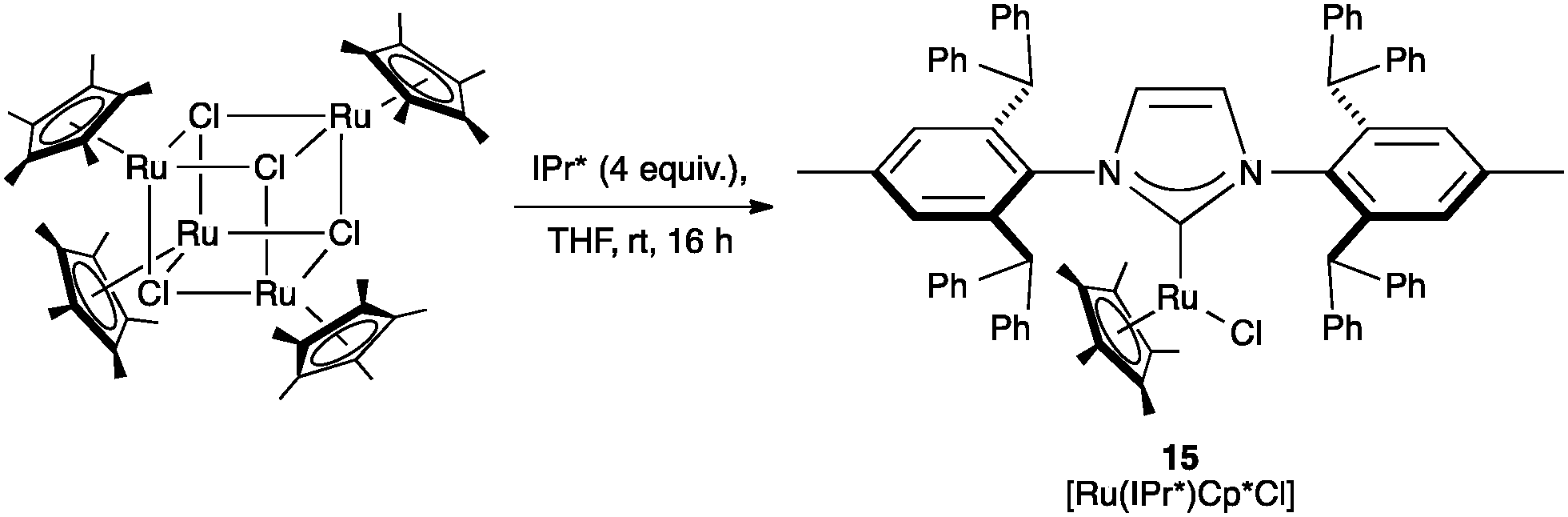 | ||
| Scheme 31 Synthesis of [Ru(IPr*)Cp*Cl]. | ||
The catalytic performance of [Ru(IPr*)Cp*Cl] (15) was similar to that of the corresponding IMes derivative, which is comparable in terms of buried volume (Table 5). However, in a more in depth analysis of the activity of the catalysts in relation to the steric bulk, no relevant trend was found, suggesting that the electronic contributions play a more important role in this transformation.72
|
|
|||
|---|---|---|---|
| Entry | Cat. | %VBur | racb (%) |
| a Reaction conditions: substrate (0.25 mmol), [Ru(NHC)Cp*Cl] (2 mol%), NaOtBu (5 mol%), toluene (2 mL). N2, RT. b Conversions determined by chiral HPLC, rac = (1 − ee) × 100. | |||
| 1 | [Ru(IMes)Cp*Cl] | 31.4 | 13 |
| 2 | [Ru(IAd)Cp*Cl] | 34.0 | 29 |
| 3 | [Ru(IPr)Cp*Cl] | 31.2 | 34 |
| 4 | [Ru(ICy)Cp*Cl] | 25.6 | >99 |
| 5 | [Ru(IPr*)Cp*Cl] | 31.1 | 12 |
Conclusions
Sterically demanding NHCs have been shown to play an important role in several aspects of organometallic chemistry. Within this family, IPr* and its derivatives have been shown to be helpful for the stabilization of catalytically active species in which bimolecular decomposition pathways can be a most significant drawback. Furthermore, these ligands provide beneficial effects on the catalytic activity of certain processes. In contrast, IPr* showed lower catalytic activities when transformations require specific electronic properties and/or the generation of a vacant site. As previously seen, there does not appear to be a universal NHC that shows highest catalytic activity throughout the catalytic landscape. The IPr* ligand does show superior activity in some transformations but forms complexes that are likely too sterically crowded to activate in an efficient manner in others. The search continues but these caveats should be kept in mind when deciding on the ligand of choice for a given catalytic transformation.Acknowledgements
We gratefully acknowledge the EC for funding through the seventh framework program SYNFLOW, the ERC (Advanced Investigator Award ‘FUNCAT’ to SPN) and the EPSRC. SPN is a Royal Society Wolfson Research Merit Award holder. We also would like to thank Dr David Nelson, Dr Fady Nahra, Dr Jose Fernandez Salas, Dr Marcel Brill, Dr Martin Corpet, Mr Scott R. Patrick and Mr Byron Truscott for helpful discussions.Notes and references
- N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, 2006 Search PubMed.
- N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, 2007, vol. 21, p. 231.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- S. Würtz and F. Glorius, Acc. Chem. Res., 2008, 41, 1523–1533 CrossRef PubMed.
- T. Szilvási and T. Veszprémi, ACS Catal., 2013, 1984–1991 CrossRef.
- R. A. Widenhoefer and S. L. Buchwald, Organometallics, 1996, 15, 2755–2763 CrossRef CAS.
- A. Poater and L. Cavallo, Theor. Chem. Acc., 2012, 131, 1–6 CAS.
- A. Poater, N. Bahri-Laleh and L. Cavallo, Chem. Commun., 2011, 47, 6674–6676 RSC.
- A. Poater, L. Falivene, C. A. Urbina-Blanco, S. Manzini, S. P. Nolan and L. Cavallo, Dalton Trans., 2013, 42, 7433–7439 RSC.
- X. Bantreil and S. P. Nolan, Nat. Protoc., 2011, 6, 69–77 CrossRef CAS PubMed.
- G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195–15201 CrossRef CAS PubMed.
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed.
- J. Huang and S. P. Nolan, J. Am. Chem. Soc., 1999, 121, 9889–9890 CrossRef CAS.
- A. J. Arduengo III, R. Krafczyk, R. Schmutzler, H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Tetrahedron, 1999, 55, 14523–14534 CrossRef.
- M. G. Organ, S. Çalimsiz, M. Sayah, K. H. Hoi and A. J. Lough, Angew. Chem., Int. Ed., 2009, 48, 2383–2387 CrossRef CAS PubMed.
- G. Berthon-Gelloz, M. A. Siegler, A. L. Spek, B. Tinant, J. N. H. Reek and I. E. Marko, Dalton Trans., 2010, 39, 1444–1446 RSC.
- L. Wu, E. Drinkel, F. Gaggia, S. Capolicchio, A. Linden, L. Falivene, L. Cavallo and R. Dorta, Chem. – Eur. J., 2011, 17, 12886–12890 CrossRef CAS PubMed.
- J. Bexrud and M. Lautens, Org. Lett., 2010, 12, 3160–3163 CrossRef CAS PubMed.
- A. B. Chaplin, Organometallics, 2014, 33, 3069–3077 CrossRef CAS.
- A. B. Chaplin, Organometallics, 2014, 33, 624–626 CrossRef CAS.
- V. Lavallo, A. El-Batta, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2011, 50, 268–271 CrossRef CAS PubMed.
- X. Zeng, M. Soleilhavoup and G. Bertrand, Org. Lett., 2009, 11, 3166–3169 CrossRef CAS PubMed.
- X. Zeng, R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 942–945 CrossRef CAS PubMed.
- D. S. Weinberger, M. Melaimi, C. E. Moore, A. L. Rheingold, G. Frenking, P. Jerabek and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 8964–8967 CrossRef CAS PubMed.
- D. R. Anderson, V. Lavallo, D. J. O'Leary, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2007, 46, 7262–7265 CrossRef CAS PubMed.
- C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed.
- X. Luan, R. Mariz, M. Gatti, C. Costabile, A. Poater, L. Cavallo, A. Linden and R. Dorta, J. Am. Chem. Soc., 2008, 130, 6848–6858 CrossRef CAS PubMed.
- L. Vieille-Petit, X. Luan, R. Mariz, S. Blumentritt, A. Linden and R. Dorta, Eur. J. Inorg. Chem., 2009, 1861–1870 CrossRef CAS PubMed.
- A. R. Martin, A. Chartoire, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2012, 8, 1637–1643 CrossRef CAS PubMed.
- S. Meiries, K. Speck, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2012, 32, 330–339 CrossRef.
- S. G. Weber, C. Loos, F. Rominger and B. F. Straub, ARKIVOC, 2012, 2012, 226–242 CrossRef.
- S. Dierick, D. F. Dewez and I. E. Markó, Organometallics, 2014, 33, 677–683 CrossRef CAS.
- C. A. Laskowski, A. J. M. Miller, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2010, 133, 771–773 CrossRef PubMed.
- A. R. Martin, D. J. Nelson, S. Meiries, A. M. Z. Slawin and S. P. Nolan, Eur. J. Org. Chem., 2014, 3127–3131 CrossRef CAS PubMed.
- M. J. Iglesias, A. Prieto and M. C. Nicasio, Adv. Synth. Catal., 2010, 352, 1949–1954 CrossRef CAS PubMed.
- A. R. Martin, Y. Makida, S. Meiries, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 6265–6270 CrossRef CAS.
- Y. Makida, E. Marelli, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2014, 50, 8010–8013 RSC.
- A. Chartoire, M. Lesieur, L. Falivene, A. M. Z. Slawin, L. Cavallo, C. S. J. Cazin and S. P. Nolan, Chem. – Eur. J., 2012, 18, 4517–4521 CrossRef CAS PubMed.
- S. Meiries, A. Chartoire, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2012, 31, 3402–3409 CrossRef CAS.
- A. Chartoire, X. Frogneux, A. Boreux, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2012, 31, 6947–6951 CrossRef CAS.
- O. Navarro, N. Marion, N. M. Scott, J. González, D. Amoroso, A. Bell and S. P. Nolan, Tetrahedron, 2005, 61, 9716–9722 CrossRef CAS PubMed.
- A. Chartoire, X. Frogneux and S. P. Nolan, Adv. Synth. Catal., 2012, 354, 1897–1901 CrossRef CAS PubMed.
- A. Chartoire, A. Boreux, A. R. Martin and S. P. Nolan, RSC Adv., 2013, 3, 3840–3843 RSC.
- A. Pommella, G. Tomaiuolo, A. Chartoire, S. Caserta, G. Toscano, S. P. Nolan and S. Guido, Chem. Eng. J., 2013, 223, 578–583 CrossRef CAS PubMed.
- G. Bastug and S. P. Nolan, Organometallics, 2014, 33, 1253–1258 CrossRef CAS.
- D. Guest, M.-T. Chen, G. J. Tizzard, S. J. Coles, M. L. Turner and O. Navarro, Eur. J. Inorg. Chem., 2014, 2200–2203 CrossRef CAS PubMed.
- E. A. B. Kantchev, G.-R. Peh, C. Zhang and J. Y. Ying, Org. Lett., 2008, 10, 3949–3952 CrossRef CAS PubMed.
- N. Hadei, E. A. B. Kantchev, C. J. O'Brie and M. G. Organ, Org. Lett., 2005, 7, 3805–3807 CrossRef CAS PubMed.
- C. Zhang, J. Huang, M. L. Trudell and S. P. Nolan, J. Org. Chem., 1999, 64, 3804–3805 CrossRef CAS.
- T. Weskamp, V. P. W. Böhm and W. A. Herrmann, J. Organomet. Chem., 1999, 585, 348–352 CrossRef CAS.
- H. M. Lee and S. P. Nolan, Org. Lett., 2000, 2, 2053–2055 CrossRef CAS PubMed.
- J. A. Loch, M. Albrecht, E. Peris, J. Mata, J. W. Faller and R. H. Crabtree, Organometallics, 2002, 21, 700–706 CrossRef CAS.
- M. G. Organ, S. Çalimsiz, M. Sayah, K. H. Hoi and A. J. Lough, Angew. Chem., 2009, 121, 2419–2423 CrossRef PubMed.
- M. Lesieur, A. M. Z. Slawin and C. S. J. Cazin, Org. Biomol. Chem., 2014, 12, 5586–5589 CAS.
- G. Bastug and S. P. Nolan, J. Org. Chem., 2013, 78, 9303–9308 CrossRef CAS PubMed.
- F. Izquierdo, A. Chartoire and S. P. Nolan, ACS Catal., 2013, 3, 2190–2193 CrossRef CAS.
- G. Maitro, S. Vogel, G. Prestat, D. Madec and G. Poli, Org. Lett., 2006, 8, 5951–5954 CrossRef CAS PubMed.
- E. Bernoud, G. t. Le Duc, X. Bantreil, G. Prestat, D. Madec and G. Poli, Org. Lett., 2010, 12, 320–323 CrossRef CAS PubMed.
- G. Maitro, G. Prestat, D. Madec and G. Poli, Tetrahedron: Asymmetry, 2010, 21, 1075–1084 CrossRef CAS PubMed.
- B. M. Prince and T. R. Cundari, Organometallics, 2012, 31, 1042–1048 CrossRef CAS.
- A. Gómez-Suárez, R. S. Ramón, O. Songis, A. M. Z. Slawin, C. S. J. Cazin and S. P. Nolan, Organometallics, 2011, 30, 5463–5470 CrossRef.
- C. Dash, A. Das, M. Yousufuddin and H. V. R. Dias, Inorg. Chem., 2013, 52, 1584–1590 CrossRef CAS PubMed.
- A. Collado, A. Gomez-Suarez, A. R. Martin, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2013, 49, 5541–5543 RSC.
- A. Gómez-Suárez, Y. Oonishi, S. Meiries and S. P. Nolan, Organometallics, 2013, 32, 1106–1111 CrossRef.
- S. G. Weber, F. Rominger and B. F. Straub, Eur. J. Inorg. Chem., 2012, 2863–2867 CrossRef CAS PubMed.
- S. G. Weber, D. Zahner, F. Rominger and B. F. Straub, Chem. Commun., 2012, 48, 11325–11327 RSC.
- M. W. Hussong, F. Rominger, P. Krämer and B. F. Straub, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201404032.
- S. Manzini, C. A. Urbina Blanco, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2012, 31, 6514–6517 CrossRef CAS.
- J. Balogh, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2012, 31, 3259–3263 CrossRef CAS.
- C. A. Urbina-Blanco, A. Leitgeb, C. Slugovc, X. Bantreil, H. Clavier, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2011, 17, 5044–5052 Search PubMed.
- C. A. Urbina-Blanco, S. Manzini, J. P. Gomes, A. Doppiu and S. P. Nolan, Chem. Commun., 2011, 47, 5022–5024 RSC.
| This journal is © The Royal Society of Chemistry 2014 |