
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly regioselective hydride transfer, oxidative dehydrogenation, and hydrogen-atom abstraction in the thermal gas-phase chemistry of [Zn(OH)]+/C3H8†‡
Xiao-Nan
Wu
,
Hai-Tao
Zhao
 ,
Jilai
Li
,
Maria
Schlangen
* and
Helmut
Schwarz
*
,
Jilai
Li
,
Maria
Schlangen
* and
Helmut
Schwarz
*
Institut für Chemie, Technische Universität Berlin, Straße des 17. Juni 135, 10623, Berlin. E-mail: Maria.Schlangen@mail.chem.tu-berlin.de; Helmut.Schwarz@tu-berlin.de
First published on 12th September 2014
Abstract
The thermal reactions of [Zn(OH)]+ with C3H8 have been studied by means of gas-phase experiments and computational investigation. Two types of C–H bond activation are observed in the experiment, and pertinent mechanistic features include inter alia: (i) the metal center of [Zn(OH)]+ serves as active site in the hydride transfer to generate [i-C3H7]+ as major product, (ii) generally, a high regioselectivity is accompanied by remarkable chemoselectivity: for example, the activation of a methyl C–H bond results mainly in the formation of water and [Zn(C3,H7)]+. According to computational work, this ionic product corresponds to [HZn(CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)]+. Attack of the zinc center at a secondary C–H bond leads preferentially to hydride transfer, thus giving rise to the generation of [i-C3H7]+; (iii) upon oxidative dehydrogenation (ODH), liberation of CH3CH2CH2 occurs to produce [HZn(H2O)]+. Both, ODH as well as H2O loss proceed through the same intermediate which is characterized by the fact that a methylene hydrogen atom from the substrate is transferred to the zinc and one hydrogen atom from the methyl group to the OH group of [Zn(OH)]+. The combined experimental/computational gas-phase study of C–H bond activation by zinc hydroxide provides mechanistic insight into related zinc-catalyzed large-scale processes and identifies the crucial role that the Lewis-acid character of zinc plays.
CH2)]+. Attack of the zinc center at a secondary C–H bond leads preferentially to hydride transfer, thus giving rise to the generation of [i-C3H7]+; (iii) upon oxidative dehydrogenation (ODH), liberation of CH3CH2CH2 occurs to produce [HZn(H2O)]+. Both, ODH as well as H2O loss proceed through the same intermediate which is characterized by the fact that a methylene hydrogen atom from the substrate is transferred to the zinc and one hydrogen atom from the methyl group to the OH group of [Zn(OH)]+. The combined experimental/computational gas-phase study of C–H bond activation by zinc hydroxide provides mechanistic insight into related zinc-catalyzed large-scale processes and identifies the crucial role that the Lewis-acid character of zinc plays.
1. Introduction
Enhancing the efficiency for the selective activation of carbon–hydrogen bonds is linked to the success in generating new or improving existing catalysts.1–3 To this end, great efforts have been undertaken to reveal the mechanisms of bond activation processes at a molecular level.3–8 Among the various catalysts so far applied in industry, quite a few employ transition metals. Zinc-based catalysts are also in use, for example in the oxidative conversion of CH4, C2H6 and C3H8.9 Further, zinc-doped zeolites are known to be effective catalysts for promoting dehydrogenation and aromatization of light alkanes, and Zn species including [Zn(OH)]+ are believed to play a key role in these mechanistically rather complex transformations.10–12 Also, pure or Li doped zinc oxides act as catalyst for the C–H bond activation of light alkanes, e.g. in the oxidative coupling of methane or the oxidative dehydrogenation of ethane and propane.13 While in all these reactions, Zn species are considered as the active ingredients, the reaction mechanisms as well as the precise structure and exact composition of the active sites of the catalysts are still under debate.12,14,15 In this respect, gas-phase experiments have proven useful because they provide in an unperturbed way rather detailed insight into the elementary steps of numerous transformations mediated by zinc-containing catalysts.16–18There have been several experimental and theoretical studies on the gas-phase reactions with various zinc species.19–23 As shown by Georgiadis and Armentrout, C–C bond cleavage of alkanes can be achieved by atomic [Zn]+.24 Further, the interaction of neutral [ZnO] with CH4 has been investigated by theoretical methods, and possible pathways yielding syngas, CH2O, and CH3OH, respectively, have been identified.25 Kretschmer et al. reported N–H bond activation of NH3 by [Zn(OH)]+,17 and CO2 activation has been brought about in the reaction of [LnZn(OH)]+ (L = imidazole and pyridine; n = 1, 2) in analogy with the Lipscomb mechanism for carbonic anhydrase.18 However, the activation of C–H bonds of light alkanes with zinc hydroxide is much less investigated. This is rather surprising given the fact that well-designed gas-phase processes of transition-metal fragments using advanced mass-spectrometric techniques in conjunction with theoretical studies have greatly helped in uncovering mechanistic aspects underlying C–H bond activation.3,5,7,8,26–33
Herein we present a combined experimental/theoretical investigation of the gas-phase reactions of cationic zinc hydroxide with alkanes. While [Zn(OH)]+ does not activate methane and ethane, mechanistically rather remarkable processes with propane are observed. As will be shown, studying the mechanistic aspects of C–H bond activation by zinc species proves helpful to understand the role of zinc in catalysis in a broader context.
2. Methods
Experiments were performed with a VG BIO-Q mass spectrometer of QHQ configuration (Q: quadrupole, H: hexapole) equipped with an ESI source, as described previously in detail.34 To this end, Q(1) is used for mass-selection of the ion of interest and then, in the rf-only hexapole, the ion/molecule reactions are conducted. Ionic products are analyzed by scanning Q(2). Further, for ESI, millimolar solutions of Zn(NO3) in pure methanol were introduced through a fused-silica capillary to the ESI source by a syringe pump (ca. 4 μL min−1) to produce the [Zn(OH)]+ cations. Nitrogen was used as a nebulizing and drying gas at a source temperature of 80 °C. Maximal yields of the desired complexes were achieved by adjusting the cone voltage (Uc) to 80 V; Uc determines the degree of collisional activation of the incident ions in the transfer from the ESI source to the mass spectrometer. The identity of the ions was confirmed by comparison with the calculated isotope patterns, which also assisted in the choice of the adequate precursor ion to avoid coincidental mass overlaps of isobaric species in the mass-selected ion beam.35 Here, we selected [64Zn(OH)]+ as the reactant ions by means of Q(1). In the hexapole, the ion/molecule reactions with CH4, C2H6, C2D6, C3H8, C3D8, CD3CH2CD3, and CH3CD2CH3 were probed at a collision energy (Elab) set to nominally 0 eV; this, in conjunction with the kinetic energy width of about 0.4 eV of the parent ion at peak half-height, allowed the investigation of quasi-thermal reactions, as demonstrated previously.18Since absolute rate constants cannot readily be determined by using the experimental setup of the VG BIO-Q mass spectrometer, the rate constant and the branching ratios of the reaction of [Zn(OH)]+ with C3H8 have been determined by using a Spectrospin CMS 47X Fourier Transform Ion Cyclotron Resonance (FT-ICR) mass spectrometer; details of the instrument have been described previously.36,37 Atomic [Zn]+ ions were generated by laser ablation of pure Zn metal disks using a Nd:YAG laser operating at 1064 nm in the presence of helium carrier gas. The [64Zn]+ isotope was isolated and allowed to react with a mixture of N2O and H2O (ca. 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give [64Zn(OH)]+. The so-formed product ions are subsequently quenched by collisional thermalization with the buffer gas (argon, ca. 2 × 10−8 mbar). After collisional thermalization, the [64Zn(OH)]+ species were mass-selected and exposed to react with C3H8 by introducing the substrate through a leak-valve. For the thermalized ions a temperature of 298 K was assumed.36,37 The branching ratios have been determined by extrapolating the ratios at different reaction times to t = 0 s. Note, that somehow different branching ratios are obtained by using the two types of mass spectrometers applied in this study, cf.Fig. 1a (branching ratios of reactions (a), (b) and (c) are 78%, 12% and 10%) and eqn (a)–(c); these differences may reflect the lack of proper collisional thermalization in the experiments using the VG BIO-Q mass spectrometer.
:1) to give [64Zn(OH)]+. The so-formed product ions are subsequently quenched by collisional thermalization with the buffer gas (argon, ca. 2 × 10−8 mbar). After collisional thermalization, the [64Zn(OH)]+ species were mass-selected and exposed to react with C3H8 by introducing the substrate through a leak-valve. For the thermalized ions a temperature of 298 K was assumed.36,37 The branching ratios have been determined by extrapolating the ratios at different reaction times to t = 0 s. Note, that somehow different branching ratios are obtained by using the two types of mass spectrometers applied in this study, cf.Fig. 1a (branching ratios of reactions (a), (b) and (c) are 78%, 12% and 10%) and eqn (a)–(c); these differences may reflect the lack of proper collisional thermalization in the experiments using the VG BIO-Q mass spectrometer.
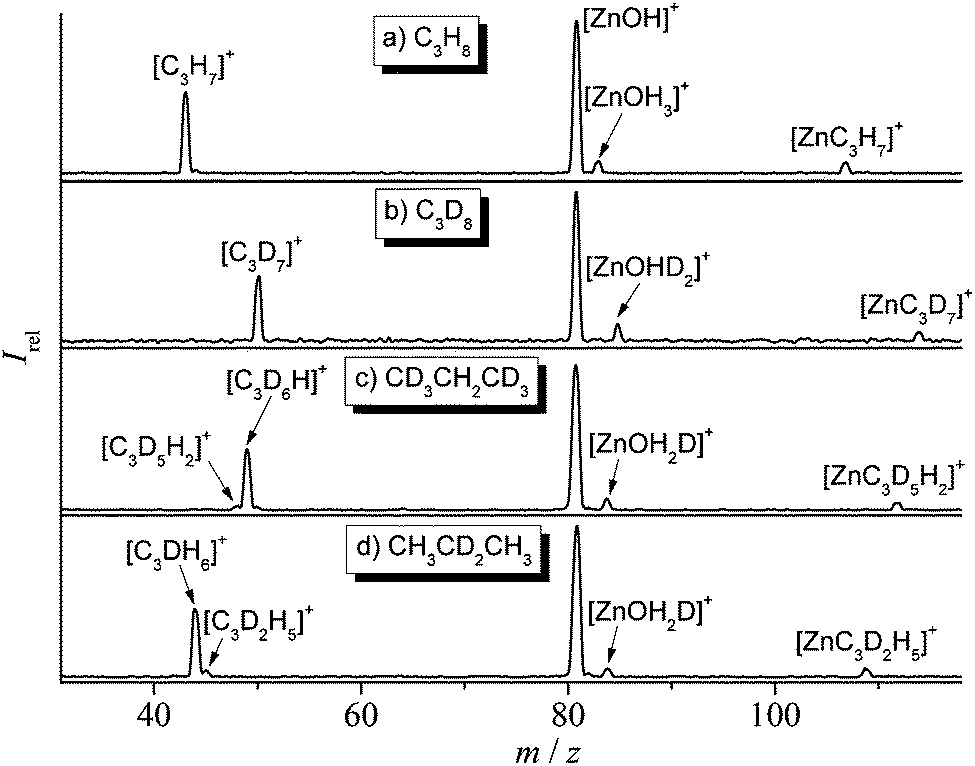 | ||
| Fig. 1 Mass spectra showing the ion/molecule reactions of mass-selected [64Zn(OH)]+ with C3H8 (a), C3D8 (b), CD3CH2CD3 (c), and CH3CD2CH3 (d) at a pressure of 1.0 × 10−3 mbar in the VG BIO-Q mass spectrometer. | ||
Calculations were carried out by using the Gaussian 09 program suite.38 Potential energy surfaces (PESs) are calculated by using the Møller–Plesset second-order perturbation MP2 method39,40 employing a triple-zeta level basis set with diffuse and polarization functions 6-311++G(2d,2p) for all atoms.41 To obtain even more accurate energies of the relevant structures, the coupled-cluster CCSD(T) method42,43 with single, double, and perturbative treatment of triple excitations in conjunction with the correlation-consistent polarized valence triple-zeta basis sets cc-pVTZ was used.44,45 The MP2/6-311++G(2d,2p) optimized geometries were employed for the single-point coupled cluster calculations without reoptimization at the CCSD(T)/cc-pVTZ levels, and the results are in line with the MP2 calculations (Table 1). All geometries were fully optimized without symmetry constraints. Vibrational frequency calculations were performed to identify the nature of reaction intermediates, transition states (TSs) and products. To corroborate which minima are linked by the considered transition states, normal coordinate analyses were performed on these TS structures by intrinsic reaction coordinate (IRC) routes in both reactant and product directions.46–48 Additional geometry optimizations starting from the last IRC structures were carried out when the IRC calculations did not converge. Unscaled vibrational frequencies were used to calculate zero-point energy (ZPE) corrections. To demonstrate the applicability of the MP2 method selected for this study, test calculations were performed at the MP2/6-311++G(2d,2p) level of theory (Table S1, ESI‡); the results are in agreement, within 0.36 eV, of experimental values.21,22,49
| Path 1 | I1 | TS1/2 | I2 | P1 | I3 | TS3/4 | I4 | P2 |
|---|---|---|---|---|---|---|---|---|
| MP2 | −1.70 | −0.38 | −0.39 | −0.20 | −1.70 | −0.40 | −1.78 | −1.12 |
| CCSD(T) | −1.65 | −0.31 | −0.32 | −0.13 | −1.65 | −0.38 | −1.62 | −1.19 |
| TS4/5 | I5 | P3 | Path 2 | I6 | P4 | I7 | TS7/8 | I8 |
|---|---|---|---|---|---|---|---|---|
| −1.70 | −2.22 | −1.23 | MP2 | −1.64 | 0.06 | −1.82 | −0.12 | −2.61 |
| −1.75 | −2.23 | −1.12 | CCSD(T) | −1.58 | 0.19 | −1.80 | −0.17 | −2.51 |
| P5 | Path 3 | I9 | TS9/10 | I10 | P6 | TS1 | TS2 |
|---|---|---|---|---|---|---|---|
| −1.07 | MP2 | −0.17 | 0.16 | −0.16 | 0.06 | 0.16 | 0.30 |
| −0.89 | CCSD(T) | −0.17 | 0.06 | −0.07 | 0.08 | 0.18 | 0.20 |
The relative energies of P1 and TS3/4 in Fig. 2 have also been calculated by DFT using different functionals50–59 and the 6-311++G(2d,2p) basis set, as well as by single-point energy calculations using the CCSD(T) method; the results are listed in Table S2 (ESI‡). MP2 calculated relative energies of high-spin and low-spin products of the reaction of [Zn(OH)]+ with C3H8, C2H6, and CH4, respectively, are shown in Tables S3–S5 (ESI‡) with respect to the particular ground state separated reactant pair.
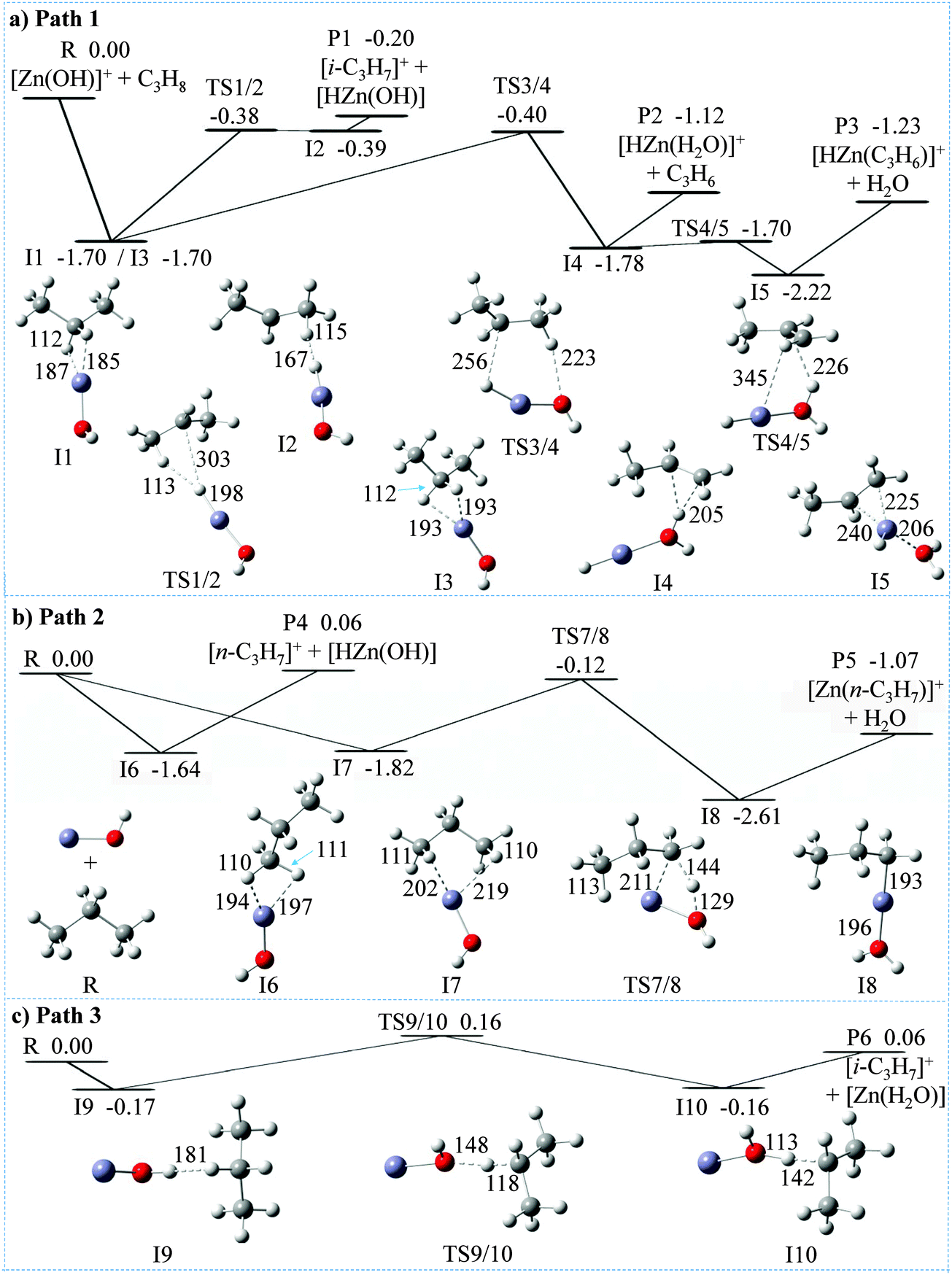 | ||
| Fig. 2 MP2-calculated potential-energy profiles for the reactions of [Zn(OH)]+ with C3H8. (a) Path 1 and (b) path 2 show the channels starting with the initial coordination of a secondary and primary C–H bond of C3H8, to the Zn site; (c) path 3 depicts the channel starting with C–H bond activation by the OH ligand. Color code: blue Zn, red O, gray C, and white H. Selected bond lengths are given in pm; relative ΔH0K energies (in eV) are given with reference to the separated reactant pair. | ||
3. Results and discussion
In the thermal reaction of [Zn(OH)]+ with propane (Fig. 1a), the generation of [C3H7]+ by hydride transfer60–64 corresponds to the main process (reaction (a)); in competition, one observes oxidative dehydrogenation, (reaction (b)) as well as the generation of [Zn(C3,H7)]+ accompanied by the elimination of water (reaction (c)). The reaction pathways are confirmed in labeling experiments in which C3D8, CD3CH2CD3, and CH3CD2CH3 have been employed as substrates (Fig. 1b–d). The structural assignments of the neutral and/or cationic products described in eqn (a) to (c) are based on theoretical results (see below). The labeling experiments are quite instructive regarding the reaction mechanisms: (i) in the hydride transfer reaction, a secondary C–H bond is preferentially activated. A best fit with the data obtained in Fig. 1 is obtained by assuming an average kinetic isotope effect (KIE) of 1.1 and a specificity of 1:45 in favor of the activation of a secondary C–H bond of propane.64 In contrast, it is the primary C–H bond of C3H8 which exclusively ends up as water in reaction (c). At the detection limit, only [Zn(C3D5H2)]+ and [Zn(C3H5D2)]+ are formed in the ion/molecule reactions with CD3CH2CD3 and CH3CD2CH3, respectively. Thus, both hydrogen-transfer processes, i.e. reactions (a) and (c), do not share a common intermediate like [Zn(H2O)(C3H7)]+ as might be anticipated. Also, based on the labeling experiments, ODH proceeds via a specific transfer of HD in the reactions of [Zn(OH)]+ with CD3CH2CD3 and CH3CD2CH3, respectively. Finally, the branching ratios given in eqn (a)–(c) as well as the rate constant k([Zn(OH)]+/C3H8) of 3.2 × 10−8 cm3 s−1 molecule−1 have been measured by using the FT-ICR mass spectrometer; the rate constant corresponds to an efficiency of 30%, relative to the collision rate.65,66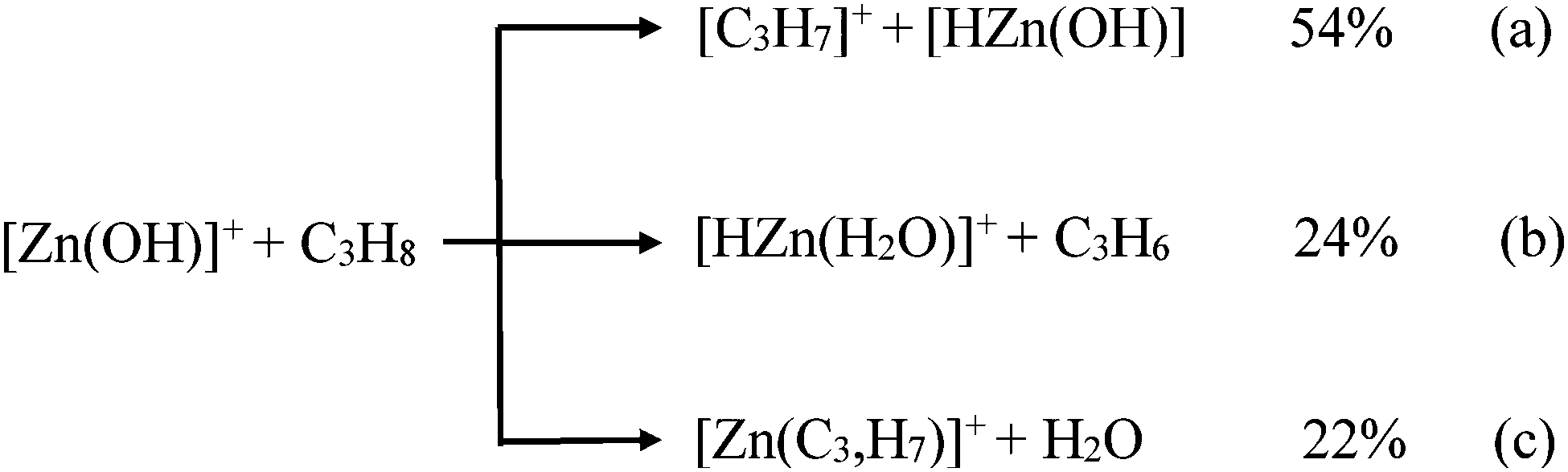
To obtain additional insight in the mechanisms of the reactions of [Zn(OH)]+ with C3H8, MP2/6-311++G(2d,2p) calculations have been performed, and the corresponding PESs are shown in Fig. 2 and Fig. S1 (ESI‡). Overall, these reactions are controlled by the Lewis-acid character of the metal center interacting with the electron donating C–H bonds of propane. Some pertinent details of the most favorable PESs are shown in Fig. 2a and b; possible pathways which involve the OH moiety interacting with C–H bonds of propane have also been tested (Fig. 2c) but turned out to be higher in energy. Four different encounter complexes have been located on the PES for the initial interaction of the metal center with C–H bonds of C3H8. In the iso-energetic I1 and I3, the metal interacts with two hydrogen atoms of the secondary position, and H–Zn bond lengths amount to 187/185 pm for I1 and 193 pm for I2, respectively. In I6 two hydrogens from one methyl group participate while in I7, one C–H bond of each of the two methyl groups is involved. All four intermediates form remarkably stable ion/molecule complexes [Zn(OH)(C3H8)]+, and these complexes profit from the electron donation from the C–H bonds into the empty 4s–4p hybrid orbital of zinc (see also below). These interactions are indicated by the fact that the coordinating C–H bonds are slightly elongated (from 109 pm in free propane to 112 pm in I1 and I3, and to 110–111 pm in I6 and in I7, respectively). Regarding the hydride transfer from a methyl group in intermediate I6 to the Zn atom, no barrier has been located in this step I6 → P4 ([n-C3H7]+/[HZn(OH)]); in contrast, the hydride transfer from I1 to P1 ([i-C3H7]+/[HZn(OH)]) proceeds via transition structure TS1/2 and intermediate I2. However, TS1/2 and I2 are almost iso-energetic, i.e. this pathway proceeds in a quasi barrier-free process. Notably, while the formation of product P1 is exothermic (−0.20 eV) and thus accessible under thermal conditions, product P4 is much higher in energy (0.06 eV); this is in line with the labeling experiments clearly favoring the former reaction (see above). As shown in Fig. S1a (ESI‡), formation of [i-C3H7]+ is also accessible via the more complex pathway R → I1′ → TS1′/2′ → I2′ → P1. In the formation of [i-C3H7]+ (Fig. 2a and Fig. S1a, ESI‡), neutral HZn(OH) is co-generated; the alternative to produce a neutral water complex Zn(H2O) is less favorable both kinetically and thermodynamically (product P6 of path 3, Fig. 2c). Likewise, the combined formation of Zn(H2O) and [n-C3H7]+ is more endothermic than generating HZn(OH) and [n-C3H7]+; the former product pair is 0.30 eV above the entrance channel (P8, Fig. S1c, ESI‡). Thus, the interaction of the OH group of [Zn(OH)]+ to C–H bonds of C3H8 (Fig. 2c and Fig. S1c, ESI,‡ respectively) to form neutral Zn(H2O) cannot compete with the initial coordination of the metal center to secondary or primary C–H bonds of propane, respectively (Fig. 2a and b). These findings are also confirmed in single point energy calculations using the CCSD(T) method.
For the regiospecific ODH process, the sequence I3 → TS3/4 → I4 → P2 (Fig. 2a) constitutes the energetically most favorable pathway. Formation of propene takes place via TS3/4, in which neutral HZn(OH) interacts with [i-C3H7]+ resulting in intermediate I4; the NBO atomic charge of the HZn(OH) moiety in TS3/4 corresponds to 0.06 |e|, and the associated IRC paths are shown in Fig. S2 (ESI‡). The weakly bound propene ligand in I4 can easily be liberated to yield the experimentally observed ODH product [HZn(H2O)]+ (P2). In competition, I5 is generated in which the Zn atom is coordinated to the CC double bond of C3H6. The weakly-bound H2O group can be eliminated yielding the cationic product [HZn(CH3CHCH2)]+ (P3), which is experimentally observed in reaction (c). A kinetically less favorable pathway for the elimination of water and formation of [Zn(n-C3H7)]+ of reaction (c) is shown in Fig. 2b (see below). The energetic requirement to produce P2 and P3 are comparable (−1.12 versus −1.23 eV); this is consistent with similar branching ratio of reactions (b) and (c) as observed experimentally. As to the energetics of the competitive productions of P1 versus P2 and P3, the relative energy of P1 (−0.20 eV) is higher than that of TS3/4 (−0.40 eV). The same order of relative energies of P1 and TS3/4 are also obtained by DFT calculations using different functionals as well as single point energy calculations using the CCSD(T) method (see Table S2, ESI‡). Taking into account the errors as well as previous work,67–69 our calculations are in agreement with the branching ratio of the production of [i-C3H7]+/[HZn(OH)] (reaction (a)) versus reactions (b) and (c) (ca. 54%:46% by using the FT-ICR mass spectrometer). In addition, the direct dissociation I1 → P1 is kinetically favored over a more complex rearrangement/dissociation path proceeding via the tight transition state TS3/4.
For the alternative water formation pathway of reaction (c), as shown in Fig. 2b, the generation of [Zn(n-C3H7)]+ occurs along the route I7 → TS7/8 → I8 → P5 with the intermediate formation of a propyl–water complex I8, [Zn(C3H7)(H2O)]+; mechanistically, this is a σ bond metathesis reaction. The relative energy of TS7/8 amounts to −0.12 eV, which is higher in energy as compared to TS3/4 (−0.40 eV), and thus unlikely to compete efficiently under thermal conditions. A brief comparison of structural features of the transition states for the σ-metathesis reaction, i.e. TS7/8 versus TS1 (Fig. S1b, ESI‡) is indicated. In TS7/8, the metal center is not only a constituent of the four-membered ring which is essential for a σ-bond metathesis but is also coordinated by a C–H bond of the distal methyl group of propane; this results in a slight but clearly discernible elongation of the C–H bond from 109 pm to 113 pm. Quite likely, this agostic interaction is beneficial for the stabilization of TS7/8 versus TS1; the latter one lacks this stabilization.
Finally, with regard to the C–H bond activation in [Zn(OH)]+, the 4s orbital hybridizes with a 4p orbital of Zn leading to the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), respectively (Fig. 3a and b). While the former is used to bind to the OH ligand, the latter can accept two electrons from a C–H bond of propane resulting in hydride transfer and carbocation formation; this view is supported by the similarity of the HOMO of neutral [HZn(OH)] with the LUMO of [Zn(OH)]+ (Fig. 3b and c).
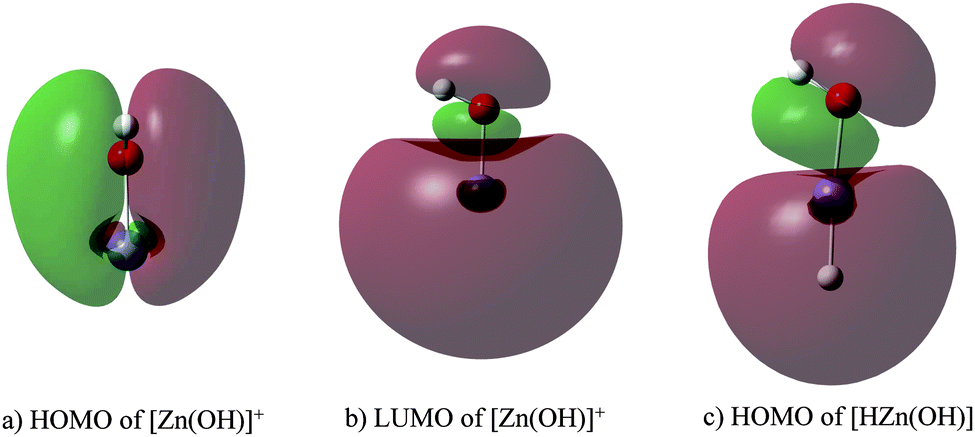 | ||
| Fig. 3 HOMO (a) and LUMO (b) of [Zn(OH)]+, and HOMO (c) of neutral [HZn(OH)]. | ||
On the triplet PES, all reaction channels described above for the [Zn(OH)]+–C3H8 system have been calculated to be endothermic (Table S3, ESI‡); therefore, this spin state has not been considered in further calculations.70,71
As mentioned above, the reactions of [Zn(OH)]+ with ethane and methane have also been studied in the experiments; here, only adduct formations are observed for [Zn(OH)]+–C2H6 (Fig. S3, ESI‡). In agreement with these findings, all reactions involving C–H bond activation for the [Zn(OH)]+–C2H6 and [Zn(OH)]+–CH4 systems are endothermic according to MP2 calculations (see Tables S4 and S5, ESI‡).
With regard to catalysis, [Zn(OH)]+ species have been conjectured to play a role in Zn/Na-ZSM5 catalysts for the conversion of propane to propene and aromatic compounds.14,72–75 As shown in this study, the metal Zn and the resulting strong Lewis-acid character in [Zn(OH)]+ are of crucial importance for the hydride transfer from propane to generate both [i-C3H7]+ and propene.72,74 Thus, the identification of the active sites of ZnOH species helps to unravel part of the enigma associated with the conversion of alkane by zinc catalysts.72–75
4. Conclusion
Here, we report and analyze the thermal gas-phase reactions of [Zn(OH)]+ with C3H8 by using experimental and theoretical methods. The reactivity of cationic zinc hydroxide [Zn(OH)]+ toward C3H8 is characterized by C–H bond activation; the main reaction channel corresponds to a hydride transfer from the hydrocarbon to the Lewis acid metal center resulting in the generation of [HZn(OH)]/[i-C3H7]+. Homolytic C–H bond activation give rise to an ODH channel (generation of propene) as well as the competitive formation of [HZn(CH3CHCH2)]+/H2O. Our study may prove helpful to further understand the industrially relevant, catalytic conversion of small alkanes by Zn species.
Acknowledgements
This work is supported by the Fonds der Chemischen Industrie, the Deutsche Forschungsgemeinschaft (DFG), and the Cluster of Excellence “Unifying Concepts in Catalysis” (coordinated by the Technische Universität Berlin and funded by the DFG). For computational resources, the Institut für Mathematik at the Technische Universität Berlin is acknowledged. Dr Xiaonan Wu is grateful to the Alexander von Humboldt-Stiftung for a postdoctoral fellowship. We thank Dr Robert Kretschmer, Dr Patricio A. González-Navarrete, Dr Shiya Tang, Dr Shaodong Zhou, and Dr Nicole Rijs for helpful suggestions and discussions. Andrea Beck is to be thanked for technical assistance, and the Reviewer for thoughtful comments.References
- C. Coperet, Chem. Rev., 2010, 110, 656 CrossRef CAS PubMed.
- A. Sattler and G. Parkin, Nature, 2010, 463, 523 CrossRef CAS PubMed.
- J. Roithová and D. Schröder, Chem. Rev., 2010, 110, 1170 CrossRef PubMed.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS PubMed.
- K. Eller and H. Schwarz, Chem. Rev., 1991, 91, 1121 CrossRef CAS.
- A. D. Ryabov, Chem. Rev., 1990, 90, 403 CrossRef CAS.
- A. W. Castleman, Jr., Catal. Lett., 2011, 141, 1243 CrossRef.
- X.-L. Ding, X.-N. Wu, Y.-X. Zhao and S.-G. He, Acc. Chem. Res., 2012, 45, 382 CrossRef CAS PubMed.
- S. Arndt, B. Uysal, A. Berthold, T. Otrebma, Y. Aksu, M. Driess and R. Schomäcker, J. Nat. Gas Chem., 2012, 21, 581 CrossRef CAS.
- E. A. Pidko and R. A. van Santen, J. Phys. Chem. C, 2007, 111, 2643 CAS.
- M. V. Frash and R. A. van Santen, Phys. Chem. Chem. Phys., 2000, 2, 1085 RSC.
- S. M. Almutairi, B. Mezari, P. C. Magusin, E. A. Pidko and E. J. Hensen, ACS Catal., 2011, 2, 71 CrossRef.
- S. Arndt, Y. Aksu, M. Driess and R. Schomacker, Catal. Lett., 2009, 131, 258 CrossRef CAS PubMed.
- H. Berndt, G. Lietz and J. Volter, Appl. Catal., A, 1996, 146, 365 CrossRef CAS.
- J. Heemsoth, E. Tegeler, F. Roessner and A. Hagen, Microporous Mesoporous Mater., 2001, 46, 185 CrossRef CAS.
- D. Schröder, H. Schwarz, S. Polarz and M. Driess, Phys. Chem. Chem. Phys., 2005, 7, 1049 RSC.
- R. Kretschmer, M. Schlangen and H. Schwarz, ChemPlusChem, 2013, 78, 952 CrossRef CAS.
- D. Schröder, H. Schwarz, S. Schenk and E. Anders, Angew. Chem., Int. Ed., 2003, 42, 5087 CrossRef PubMed.
- C. Bergquist, T. Fillebeen, M. M. Morlok and G. Parkin, J. Am. Chem. Soc., 2003, 125, 6189 CrossRef CAS PubMed.
- C. R. A. Catlow, S. T. Bromley, S. Hamad, M. Mora-Fonz, A. A. Sokol and S. M. Woodley, Phys. Chem. Chem. Phys., 2010, 12, 786 RSC.
- L. N. Zack, M. Sun, M. P. Bucchino, D. J. Clouthier and L. M. Ziurys, J. Phys. Chem. A, 2012, 116, 1542 CrossRef CAS PubMed.
- I. Iordanov, K. D. D. Gunaratne, C. L. Harmon, J. O. Sofo and A. W. Castleman, Jr., J. Chem. Phys., 2012, 136, 214314 CrossRef CAS PubMed.
- M. A. Flory, A. J. Apponi, L. N. Zack and L. M. Ziurys, J. Am. Chem. Soc., 2010, 132, 17186 CrossRef CAS PubMed.
- R. Georgiadis and P. Armentrout, J. Am. Chem. Soc., 1986, 108, 2119 CrossRef CAS PubMed.
- Z. Su, S. Qin, D. Tang, H. Yang and C. Hu, J. Mol. Struct., 2006, 778, 41 CrossRef CAS PubMed.
- H. Schwarz, Acc. Chem. Res., 1989, 22, 282 CrossRef CAS.
- Z.-C. Wang, N. Dietl, R. Kretschmer, J.-B. Ma, T. Weiske, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2012, 51, 3703 CrossRef CAS PubMed.
- N. Dietl, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2012, 51, 5544 CrossRef CAS PubMed.
- X. N. Wu, X. N. Li, X. L. Ding and S. G. He, Angew. Chem., Int. Ed., 2013, 125, 2504 CrossRef.
- J. B. Ma, B. Xu, J. H. Meng, X. N. Wu, X. L. Ding, X. N. Li and S. G. He, J. Am. Chem. Soc., 2013, 135, 2991 CrossRef CAS PubMed.
- G. E. Johnson, E. C. Tyo and A. W. Castleman, Jr., Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18108 CrossRef CAS PubMed.
- G. E. Johnson, R. Mitric, M. Nossler, E. C. Tyo, V. Bonacic-Koutecky and A. W. Castleman, Jr., J. Am. Chem. Soc., 2009, 131, 5460 CrossRef CAS PubMed.
- A. Bozovic and D. K. Bohme, Phys. Chem. Chem. Phys., 2009, 11, 5940 RSC.
- C. Trage, D. Schröder and H. Schwarz, Chem. – Eur. J., 2005, 11, 619 CrossRef CAS PubMed.
- D. Schröder and H. Schwarz, Can. J. Chem., 2005, 83, 1936 CrossRef.
- K. Eller and H. Schwarz, Int. J. Mass Spectrom. Ion Processes, 1989, 93, 243 CrossRef CAS.
- D. Schröder, H. Schwarz, D. E. Clemmer, Y. Chen, P. Armentrout, V. I. Baranov and D. K. Böhme, Int. J. Mass Spectrom. Ion Processes, 1997, 161, 175 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1 asasas, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- H. B. Schlegel, J. Chem. Phys., 1986, 84, 4530 CrossRef CAS PubMed.
- J.-L. Li, C.-Y. Geng, X.-R. Huang and C.-C. Sun, J. Chem. Theory Comput., 2006, 2, 1551 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS PubMed.
- J. A. Pople, M. Headgordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968 CrossRef CAS PubMed.
- G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910 CrossRef CAS PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS.
- G. A. Natanson, B. C. Garrett, T. N. Truong, T. Joseph and D. G. Truhlar, J. Chem. Phys., 1991, 94, 7875 CrossRef CAS PubMed.
- D. E. Clemmer, N. F. Dalleska and P. B. Armentrout, J. Chem. Phys., 1991, 95, 7263 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- T. Schwabe and S. Grimme, Phys. Chem. Chem. Phys., 2006, 8, 4398 RSC.
- X. Cai, Y. Li, E. R. O'Grady and J. A. Farrar, Int. J. Mass. Spectrom., 2005, 241, 271 CrossRef CAS PubMed.
- S. Feyel, D. Schröder and H. Schwarz, J. Phys. Chem. A, 2006, 110, 2647 CrossRef CAS PubMed.
- N. Dietl, M. Engeser and H. Schwarz, Chem. – Eur. J., 2009, 15, 11100 CrossRef CAS PubMed.
- D. Schröder, H. Florencio, W. Zummack and H. Schwarz, Helv. Chim. Acta, 1992, 75, 1792 CrossRef.
- M. Schlangen, D. Schröder and H. Schwarz, Chem. – Eur. J., 2007, 13, 6810 CrossRef CAS PubMed.
- T. Su and M. Bowers, Int. J. Mass Spectrom. Ion Phys., 1973, 12, 347 CrossRef CAS.
- R. Wesendrup, D. Schröder and H. Schwarz, Angew. Chem., Int. Ed., 1994, 33, 1174 CrossRef.
- D. Schröder and J. Roithova, Angew. Chem., Int. Ed., 2006, 45, 5705 CrossRef PubMed.
- N. Dietl, C. van der Linde, M. Schlangen, M. K. Beyer and H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 4966 CrossRef CAS PubMed.
- D. J. McAdoo, Mass. Spectrom. Rev., 1988, 7, 363 CrossRef CAS.
- J.-L. Li, C.-Y. Geng, X.-R. Huang and C.-C. Sun, Theor. Chem. Acc., 2007, 117, 417 CrossRef CAS PubMed.
- D. Schröder, S. Shaik and H. Schwarz, Acc. Chem. Res., 2000, 33, 139 CrossRef PubMed.
- X.-L. Sun, X.-R. Huang, J.-L. Li, R.-P. Huo and C.-C. Sun, J. Phys. Chem. A, 2012, 116, 1475 CrossRef CAS PubMed.
- J. A. Biscardi and E. Iglesia, Phys. Chem. Chem. Phys., 1999, 1, 5753 RSC.
- H. A. Aleksandrov and G. N. Vayssilov, Catal. Today, 2010, 152, 78 CrossRef CAS PubMed.
- H. A. Aleksandrov, E. A. I. Shor, A. M. Shor, V. A. Nasluzov, G. N. Vayssilov and N. Rosch, Soft Mater., 2012, 10, 216 CrossRef CAS.
Footnotes |
| † Dedicated to Professor A. W. Castleman, Jr., in recognition of his inspiring work on gas-phase catalysis. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp02139h |
| This journal is © the Owner Societies 2014 |