 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctional interlocked systems
Stijn F. M.
van Dongen
,
Seda
Cantekin
,
Johannes A. A. W.
Elemans
,
Alan E.
Rowan
and
Roeland J. M.
Nolte
*
Radboud University Nijmegen, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: R.Nolte@science.ru.nl
First published on 26th September 2013
Abstract
With the advent of supramolecular chemistry and later nanotechnology a great deal of research has been focused on new types of molecular structures, which are not held together by covalent bonds but by non-covalent mechanical interactions. Examples include the catenane, rotaxane, and knot interlocked structures. The design and synthesis of these architectures is an art by itself and as such is worth being reviewed. In this tutorial review we will focus, however, on the functional aspects of interlocked molecules and discuss how these can find applications, e.g. as artificial muscles, as molecular valves, as components of electronic devices, and as catalysts.
 From left to right: Johannes A. A. W. Elemans, Alan E. Rowan, Roeland J. M. Nolte, Seda Cantekin, Stijn F. M. van Dongen | After finishing his MSc studies (cum laude) at the ETH Zürich (Switzerland), Stijn van Dongen obtained his PhD in 2010 with Profs. Roeland Nolte and Jan van Hest at the Radboud University Nijmegen (The Netherlands), working on self-assembled polymersome nanoparticles for catalysis and chemical biology. He then moved to Paris (France) for a stay at the École Normale Supérieure and the Institut Curie. Working from a personal grant, he developed dynamic surface coatings for cell culture with Dr Christophe Tribet and Dr Matthieu Piel. In 2012, Stijn returned to the group of Prof. Nolte to explore protein-based rotaxanes. |
Seda Cantekin received her MSc (chemistry) from the Middle East Technical University with Prof. M. Balci. In 2008 she joined the Molecular Science and Technology group at the Institute for Complex Molecular Systems (Eindhoven University of Technology, Netherlands) as a PhD student under the supervision of Prof. E. W. Meijer and Dr A. R. A. Palmans. During this research she investigated the limits of supramolecular chirality in synthetic supramolecular polymers, mainly focusing on the combination of chemical reactions with noncovalent interactions. Currently she works as a postdoctoral research associate with prof. R. J. M. Nolte to explore the kinetics of polymer threading through flexible porphyrin macrocycles. |
Hans Elemans completed his PhD in 2001 with Prof. Roeland Nolte at the Radboud University Nijmegen (The Netherlands). After that he initiated and developed a collaboration between the physics and chemistry departments of the Radboud University, with the aim to study self-assembly and catalytic processes at the single molecule level using Scanning Probe Microscopy. In 2008 he stayed in the group of Prof. Steven De Feyter at K. U. Leuven (Belgium), after which he returned to Nijmegen in 2009 to start his independent research group. In 2010 he was awarded an ERC Starting Grant to further study catalysis at the nanoscale. |
Professor Dr Alan Rowan studied at the University of Liverpool, England, where he obtained a BSc 1st Honours in chemistry and then a PhD in physical organic chemistry. In 1992 he moved to New Zealand where he completed a postdoctoral study in the field of supramolecular chemistry with Prof. C. Hunter. In 1994 he moved to Nijmegen (the Netherlands) as a Marie Curie Fellow, and then as Assistant and Associate Professor with Prof. R. J. M. Nolte. In 2005, he set up a new department of Molecular Materials in Nijmegen. His interests are in the relationship between molecular architecture and function, in self-assembling and macromolecular (bio)organic and magnetic materials. |
Roeland J. M. Nolte is professor of organic chemistry at the Radboud University Nijmegen, where he was director of the Institute for Molecules and Materials. He is a member of the Royal Netherlands Academy of Science and holds a special Royal Academy of Science Chair in chemistry. His research interests span many topics at the interfaces of supramolecular, macromolecular and biomimetic chemistry, in which he focuses on the design of catalysts and materials. His contributions to science have been recognized with numerous awards and prizes including the Izatt–Christensen award for excellence in macrocyclic chemistry, the first Royal Netherlands Academy of Science chair in chemistry, and a knighthood in 2003. He has served on the editorial boards of many scientific journals, including the journals ‘Science’ and ‘Chemical Communications’ (as Chairman). |
Key learning points˙ Topologically linked architectures enable unique interactions and motions.˙ The mechanical interlocking of components allows important motions to be repeatable. ˙ Real mechanical contact enables the actual ‘touching’ of other molecules. ˙ The position of a rotaxane's ring on specific ‘stations’ found in its axle can be sufficiently stable for a rotaxane to maintain its supramolecular configuration, or to change it if the ‘stations’ change. ˙ Mechanically linking a catalyst to its substrate enhances that catalyst's performance. |
1. Introduction
The complex molecular machineries that are routinely employed by nature to drive processes essential to life are a major source of inspiration for research in today's nanoscience. Stimulated by the efficient operating mechanisms of these machines, much effort is put into the development of simplified artificial mimics. A fascinating approach to create synthetic machines in which controlled motion plays a dominant role involves the organisation of molecules into mechanically interlocked (or topologically linked) architectures. A mechanical link entails separate molecules that are linked together not through a covalent bond, but through their shape. Chain links in an iron chain, for example, are not covalently linked, yet they are clearly and strongly stuck together as a result of their shape and their spatial configuration. A molecular analogy of two chain links featuring two macrocyclic rings that are similarly interlinked is called a catenane. Rotaxanes are closely related, but here a macrocycle is stuck on a linear axle – the name ‘rotaxane’ is based on the Latin roots for ‘wheel’ (rota) and ‘axle’ (axis). A system where such a ring can still slide off of its axle is dubbed a pseudorotaxane. Should the axle feature bulky moieties that the macrocycle cannot pass over, then the macrocycle is trapped and the system is a true rotaxane.Since the 1980s this research field of interlocked molecules, which was pioneered by the groups of Stoddart1 and Sauvage,2 has been booming and it has become one of the contemporary pillars of supramolecular chemistry. A wealth of appealing interlocked structures, including rotaxanes, catenanes, knots and ‘Borromean rings’, have spiced up the literature. What these systems have in common is that they contain macrocyclic components, which are held together via mechanical, non-covalent bonds. While over the years a variety of efficient synthetic procedures have been established to optimise the yield of these complex structures opening new avenues to even more sophisticated systems, an important question still remains: will they ever be of any practical use?
Some interesting future applications were already proposed for the earliest interlocked molecules, most of them based on the idea that different ‘stations’ can be incorporated on the thread-component of a rotaxane or catenane. The ring-component can, in principle, shuttle between these stations in a repeatable fashion, and control over the shuttling process might be obtained by the application of chemical, electrochemical, or photochemical stimuli. Such complexes were coined as switchable components in molecular electronics, as they would be able to exhibit tuneable on/off behaviour. Once it becomes possible to induce and control directionality in the motion of molecular shuttles, and make many of them operate in tandem, movement of objects on a macroscopic scale might be achieved, thereby delivering work. Section 2 of this review deals with such defined motion in interlocked structures, and the behaviour of miniature devices with illustrious names like ‘artificial muscles’, ‘tape readers’ and ‘elevators’ will be discussed, as well as the first successful efforts to translate microscopic into macroscopic motion.
Mechanically interlocked architectures can also be employed to regulate access to molecules, or parts of them, and this will be the subject of Section 3. Rotaxanes can serve as ‘molecular valves’ that are capable of opening or closing compartments that host other molecules, which can then be either released or contained. In addition, regulated access can be useful if a particular station of a rotaxane needs protection, for example because it has a certain function which requires controlled expression.
In Section 4, we will discuss another originally foreseen application of interlocked systems, namely as components of molecular electronic devices. Because of their earlier mentioned ability to shuttle macrocyclic components between stations on a thread, rotaxanes can in principle function as excellent miniature switches and logic gates.
Finally, in Section 5 we will introduce and elaborate on a relatively new application of interlocked architectures, namely as catalysts and in particular as mimics of processive enzymes. In nature such enzymes play an essential role in DNA synthesis and degradation, with fascinating examples being DNA polymerase and λ-exonuclease. They operate by threading the biopolymer through a hole in their toroidal structure. After threading, several rounds of catalysis (e.g., replication or degradation) take place before the enzyme dissociates from the biopolymer, giving rise to a process that occurs with high efficiency and fidelity. We will discuss the role of mechanically interlocked species in the first artificial systems that mimic these processes.
This paper is meant to be read as a tutorial review and does not completely cover the literature published on interlocked systems. Emphasis is on applications and the most important examples from the last years are presented.
2. Mechanical forces
As mechanically interlocked species, the nature of rotaxanes is in large part defined by forces. It is force that keeps the macrocyclic ring locked around its axle, and the application of sufficient force to the ring can cause it to move away from any preferential binding sites it may have. But due to this tendency of the rings to always slide towards certain areas of their axis, the inverse can also be true: rotaxane-based molecular muscles can exert force, e.g. if they are attached to a surface. This chapter deals with precisely that type of situation: use of a rotaxane to exert forces on an environment. Where the typical strong man in a circus may bend thick iron bars, this field has yet to bend anything larger than a micron-sized cantilever. But given the fact that the muscles involved in the bending force are merely individual molecules, this is an impressive feat. Next to muscles, we will also discuss the exploration of what type of force is exerted by rotaxanes, and what macroscopic effects have already been realised.2.1 Artificial muscles
Mechanically interlocked systems have often been used for the construction of molecular motors or machines.3 Therefore, it is fitting that one of the earliest achievements of rotaxanes as model systems was their ability to imitate the action of biological muscles. The omnipresence and importance of muscles in vertebrates makes them greatly interesting structures, and as such there is a certain demand for muscle mimics. In nature, skeletal muscles do work by having their individual proteins make stepwise contractions along a filament. The sarcomere, which is the basic unit of a muscle, consists of a thick myosin-containing filament which moves unidirectionally along a thin actin filament to induce contraction. Movement in the other direction results in stretching of the muscle, relaxing it again.The group of Sauvage was amongst the first to recognise the ability of a double rotaxane to emulate this behaviour.4Fig. 1 shows a doubly threaded bimolecular setup which forms a single interlocked system. Systems such as this are referred to as daisy chains, and an integer in square brackets denotes how many ‘daisies’ are in the chain. The schematic representation in Fig. 1A thus represents a [2]daisy chain, and it gives a clear insight into the intended action of this mimic: analogous to actual muscles, the two ‘filaments’ of this double rotaxane could glide along one another. It is the interlocked nature of the double rotaxane that keeps all elements together. Movement was directed by the metal coordinating capabilities of the macrocycles, which contained a phenanthroline-derived bidentate ligand. The axles offered a complementary choice between either a similar bidentate or a tridentate terpyridine ligand, so that the entire system could accommodate either four- or five-coordination, depending on the position of the macrocycle. Indeed, when complemented by the bidentate in the axle, four-coordinate Cu(I) could be chelated in a pseudo-tetrahedral complex, locking the ‘muscle’ in its extended position. To switch to the contracted position, the copper was removed using KCN. Subsequent remetalation with Zn(II) drove the macrocycles towards the tridentate moieties of the axles to enable formation of the thermodynamically more favourable five-coordinating complex, as required for Zn(II) complexation. This locked the ‘muscle’ in its contracted position (see Fig. 1B).
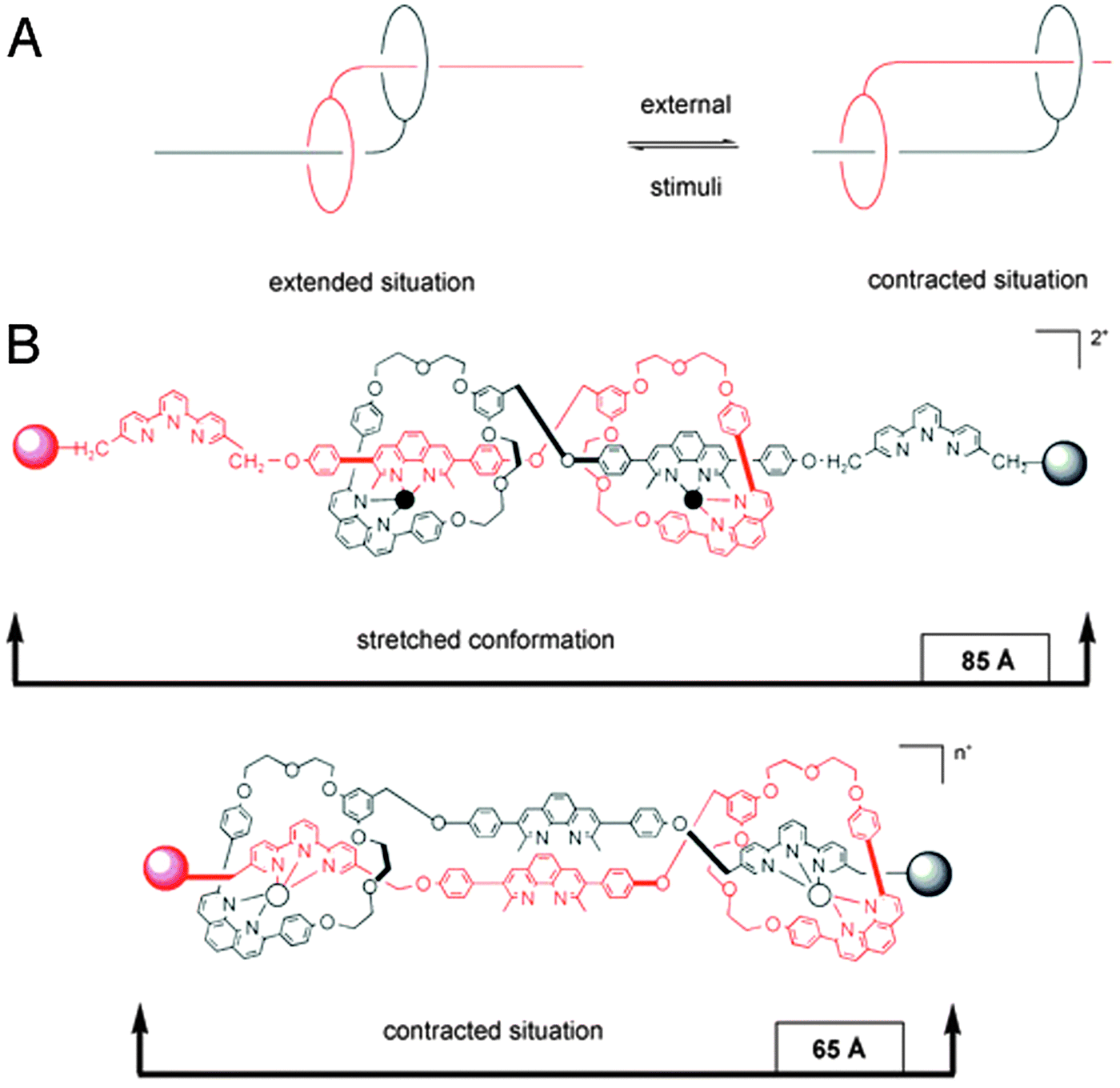 | ||
| Fig. 1 Early muscle mimics. (A) Schematic representation of the contraction and extension possible with [2]daisy chain rotaxanes. The filaments glide along one another. (B) The two states of the muscle mimic reported by the group of Sauvage. Extension and contraction are mediated by offering either 4-coordinate or 5-coordinate metal centers. Reprinted with permission (RSC).5 | ||
This [2]daisy chain was the first unimolecular linear array capable of stretching and contracting on demand. Model estimations showed that the length of the compound changed from 83 Å to 65 Å upon contraction, which in a happy coincidence roughly corresponds to the relative amount of contraction that natural muscles undergo (27%), as reported by the authors.4 The key reaction of the complex synthetic procedure that yields this molecular muscle was the double-threading step. It leveraged the coordinating capacity of copper(I) by allowing it to gather its desired ligands around it. Addition of a second fragment containing the terpyridine ligand and a stopper yielded the final muscle.
In addition to the demetalation/remetalation described above, Jimenez-Molero et al. envisioned a more elegant triggering signal to set the molecule in motion. They employed an electrochemical signal that could convert Cu(I) to Cu(II). Their intention was to have the five-coordinate Cu(II) fulfil the same role as the Zn(II) that was used in the remetalation strategy, that is, to have the change in coordination thermodynamics drive the molecular motion. Unfortunately, while Cu(II) was readily formed, the kinetic stability of the four-coordinate Cu(II) complex proved to be prohibiting to five-coordination, and thus to contraction.5
The reversible nature of most redox processes would have made such an electrochemical strategy especially appealing. Therefore it is a credit to the groups of Ho and Stoddart that they could achieve just that: a rotaxane muscle that contracts and expands in response to electrochemical oxidation or reduction.6 Not following the daisy chain approach, they settled on a [3]rotaxane constituted of a single palindromic axle carrying two rings (see Fig. 2A). These rings, based on cyclobis(paraquat-p-phenylene) (CBPQT4+), could be selectively positioned on different stations in the axle through previously established recognition chemistry: they have an initial preference for occupying the tetrathiafulvalene (TTF) stations positioned at the outsides of the axle. CBPQT4+ rings can also encircle naphthalene (NP) stations towards the centre of the axle, but this is less energetically favourable. However, when chemical oxidation of the TTF stations makes them dicationic, this repels the CBPQT4+ macrocycles and drives them towards the now favourable NP stations. A subsequent reduction of the TTF2+ stations back to their neutral TTF form allows the rings to diffuse back to their initially favoured positions. Thus, a [3]rotaxane was achieved where two rings could actively approach one another, driven by electrostatic repulsion, and where the initial state could be regenerated after passive diffusion (see Fig. 2A).
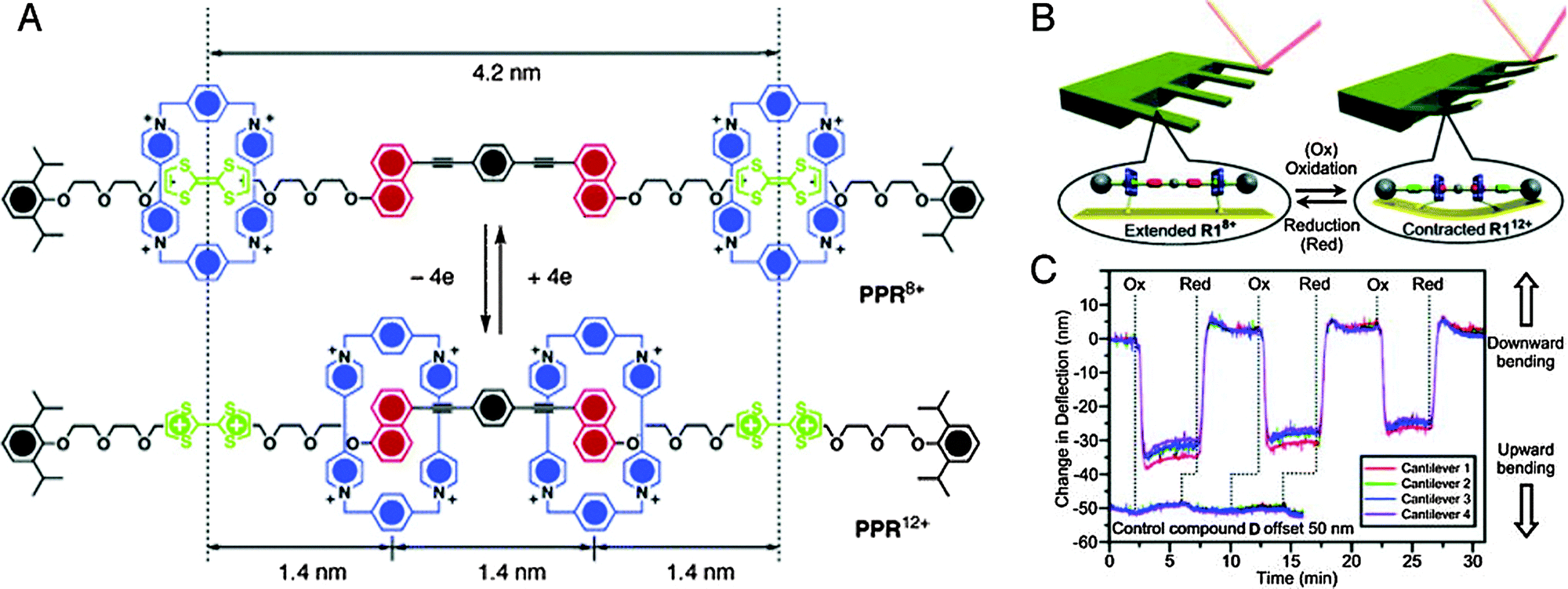 | ||
| Fig. 2 Muscle mimics that bend cantilevers. (A) Molecular structures of the palindromic [3]rotaxane in its extended (top) and contracted (bottom) state. The cyclobis(paraquat-p-phenylene) (CBPQT4+) macrocycle is blue, the tetrathiafulvalene (TTF) stations are green and the naphthalene (NP) stations are yellow. (B) By attaching the macrocycles to the surface of an array of cantilever beams, the cantilevers could be bent. (C) Repeated bending of the cantilever. Reprinted with permission.6,7 Copyright 2004, American Institute of Physics. Copyright 2005, American Chemical Society. | ||
In itself, the above did not yet mimic the action of a muscle. This comparison did become valid when the CBPQT4+-macrocycles were equipped with a disulphide tether to allow their immobilisation on a gold-coated surface.6 Thus, as shown in Fig. 2B, the linear [3]rotaxanes were assembled on a gold-coated silicon cantilever array sporting four parallel thin cantilevers. This array was placed in a fluid cell, and the spatial position of each cantilever was monitored. Upon introduction of aqueous Fe(ClO4)3 (oxidant), the cantilever beams were found to bend upwards. Follow-up addition of ascorbic acid as a reductant brought the cantilevers back to their original positions. This behaviour was observed for up to 25 cycles (Fig. 2C),7 with a slight attenuation that was ascribed to chemical and/or physical degradation of the applied monolayer of rotaxane molecules.
An interesting discussion of the actual forces involved in the bending of the cantilever is given in a follow-up full paper.7 There, the mean compression per anchored molecule was estimated to be a mere 0.001 pm, even though the inter-ring distances are altered by an estimated minimum of 1.2 nm after switching (with a maximum of 2.8 nm – the switching distance depends on whether the axle is fully stretched when TTF is neutral). Altogether, this illustrates how many molecules working together can assert a macroscopic effect, even when they are randomly aligned on an unordered surface.
Additional methods for triggering ‘muscle’ contraction have been reported in more recent publications. The group of Huang, for example, have reported a [2]daisy chain that is based on two interlocked alkane-appended copillar[5]arenes.8 When dissolved in chloroform, the muscle would be in its contracted state, controlled by dispersion forces between the alkane axles that drive the two rings away from each other. In a polar solvent such as DMSO, on the other hand, these forces were overcome by solvent–rotaxane interactions, and the rings would slide towards one another, extending the muscle. For this solvent-driven system, the occupation of stretched or contracted states was merely statistical, as opposed to the clear-cut switching in the systems described above. This probably stems from the lack of well-defined stations on the threaded axles. However, this lack of station-dependence concurrently offered an elegant upside: by varying the percentage of polar solvent, the muscle could be extended or contracted in an almost continuous fashion, just like natural muscle filaments can.8
The last system we will discuss here once again uses a different mode of switching, namely by changes in pH. Romuald et al. recently described what practically amounts to a [1]daisy chain.9 This complex interlocked system, shown in Fig. 3A, was obtained by conjoining the tails of a precursor [2]daisy chain. The resulting unimolecular system could be contracted and stretched, just like a linear [2]daisy chain, but in this case, the result was the widening or tightening of the macrocycle. In vertebrates, circular muscles such as sphincters are essential, which makes this unique example of a [1]daisy chain particularly interesting. Its molecular structure contains two dibenzo-24-crown-8 (DB24C8) rings that surround a single, interlinking thread. The thread contains two stations for each of the rings (four stations in total). At low pH, the two rings are located around the ammonium stations (green balls, see Fig. 3B) and the circular muscle lies in its relaxed conformation. After deprotonation of the ammonium stations, the two DB24C8 cycles slide towards the triazolium stations (pink balls in Fig. 3B) to dock there, which forces the molecule to adopt a contracted conformation. Reprotonation relaxed the circular muscle again. Such pH-switchable behaviour has been extensively covered by the group of Coutrot9 (see references therein) and it offers a complementary alternative to other methods of switching the state of molecular muscles.
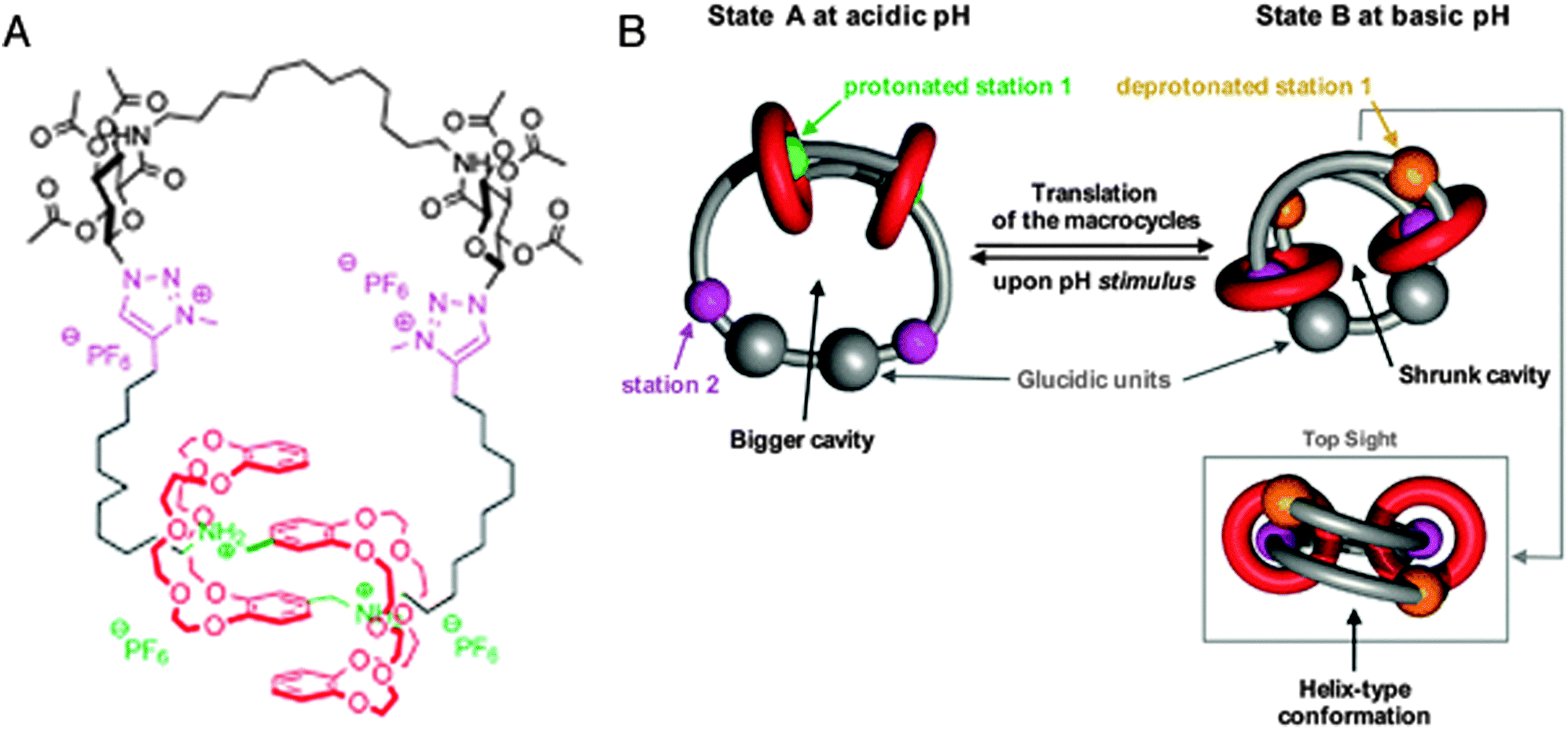 | ||
| Fig. 3 A circular muscle mimic. (A) Molecular structure with colour codes. The dibenzo-24-crown-8 (DB24C8) macrocycles are red, the ammonium stations are green, and the methyltriazole stations are purple. (B) Graphic representation of the contraction and relaxation as driven by pH. Reprinted with permission (RSC).9 | ||
2.2 Experiments with atomic force microscopy
The cantilevers that were described above as macroscopic objects that could be bent by linear rotaxane muscles (Fig. 2B) can also be used to do the reverse: by precisely moving a cantilever attached to a rotaxane, the state of the rotaxane can be changed. In cases like this, the cantilever is part of an atomic force microscope (AFM). In AFM, minute cantilevers that bear a tiny tip at their extremity are slid over a surface. The degree of bending of the cantilever is monitored using a photodiode, which detects the light of a laser beam that is continuously aimed at the AFM tip. Any change in the tip's orientation results in a different deflection of the beam. Surface features that the AFM tip encounters will cause the tip to bend through inter-atomic forces, and this is detected in the same way. As a result, an AFM can be used to map the ‘force profile’ of a surface, which generally translates quite well into a height map, allowing AFM to even detect single molecules that lie on a sufficiently flat surface. The interaction between AFM and rotaxanes, however, does not focus on the microscopy, but on the force: an AFM is ideally suited to measure forces on the smallest imaginable scale.In a collaboration between the groups of Leigh and Duwez, a surface-mounted rotaxane with two ‘stations’ in its axle was developed.10 This [2]rotaxane, shown in Fig. 4, consisted of a benzylic amide macrocycle mechanically locked onto a thread bearing both fumaramide and succinic amide-ester sites. Each of these could bind the macrocycle through up to four hydrogen bonds, but the macrocycle had a preference for the fumaramide station. The axle was stoppered by a diphenylethyl ester moiety on one end and a disulphide that would graft to a gold substrate on the other, and the macrocycle was equipped with a long poly(ethylene glycol) chain which could aspecifically adsorb onto an AFM tip. This adsorption allowed the direct manipulation of the macrocycle by moving the AFM tip: it proved possible to move the macrocycle from its preferred station up to the lower-affinity succinic one, and to sense its attempts to diffuse back (see Fig. 4).
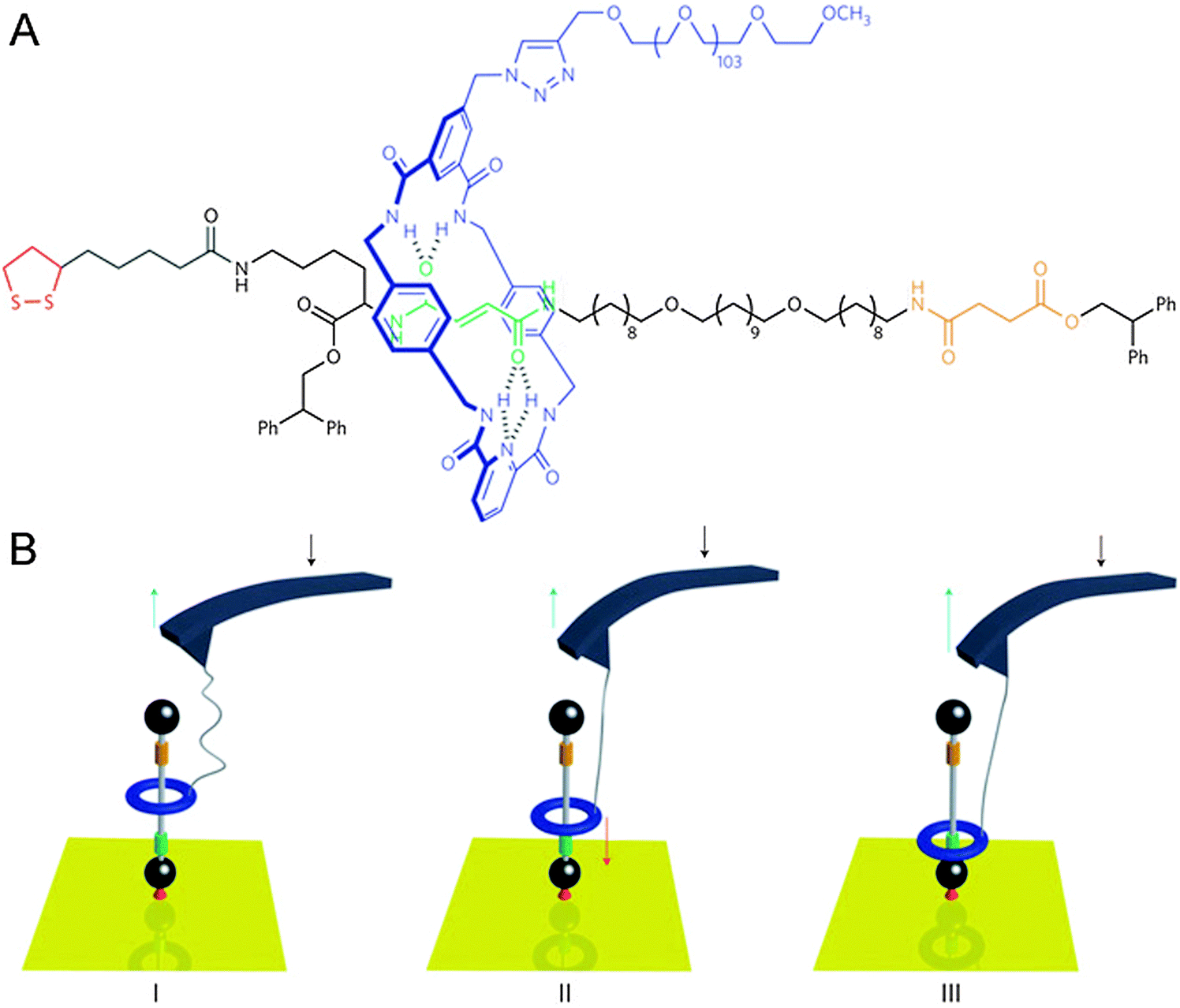 | ||
| Fig. 4 Schematic representation of an AFM cantilever interacting with a surface-bound rotaxane. (A) Molecular structure with colour codes that match the cartoon. (B) (I) Progressive release of the tension in the PEG tether after the benzylic amide ring (blue) had been pulled up to the less favourable succinamide station (yellow). (II) The force (red arrow) increases as a result of the ring shuttling back to its preferred fumaramide station (green). (III) The ring has rebound to the fumaramide site. Reprinted with permission from Macmillan Publishers Ltd: Nat. Nanotechnol.,10 copyright 2011. | ||
If left to freely equilibrate, any macrocycle in this system that was positioned on the succinic station would diffuse back towards the preferred fumaramidic station. In a way, such a macrocycle would always move in the same direction, subsequently generating a directional force. And indeed, the authors demonstrated that such biased Brownian motion of the molecular ring does exactly that. By sensing the force exerted on the AFM tip by the macrocycle, an estimation was made of the forces involved in this biased sliding. By exerting a counterforce with the AFM tip, they revealed that the macrocycle travels back to its preferred station even against an external load of up to 30 pN. This result showed that individual rotaxanes can generate directional forces of a magnitude comparable to those generated by naturally occurring molecular machines such as real muscles, which typically generate forces in between 5 and 60 pN, as discussed in the original report.10
An interesting application of combining AFM and rotaxanes was reported three years earlier by the group of Lindsay.11 They described a nanomechanical molecular “tape reader” rotaxane that could be used to probe the secondary structure of DNA. Their setup was also based on a surface-grafted rotaxane (see Fig. 5A). To construct it, a linear axle containing an adamantane station was built up on a surface. Then, β-cyclodextrin spontaneously docked onto the adamantane station, after which a stopper was attached to the axle, creating the rotaxane. Instead of a conventional stopper, a specifically designed thread of DNA was used. In its stretched conformation, this DNA could be threaded by the β-cyclodextrin, yet the DNA still acted as a stopper: its secondary structure assumed hairpin conformations (see Fig. 5B). These large structural features were too sterically bulky for the macrocycle to thread over. The purpose of this setup was to gain insight into the forces needed to unfold these hairpins by means of forcing them to thread through a small pore, i.e. through the opening of the β-cyclodextrin.
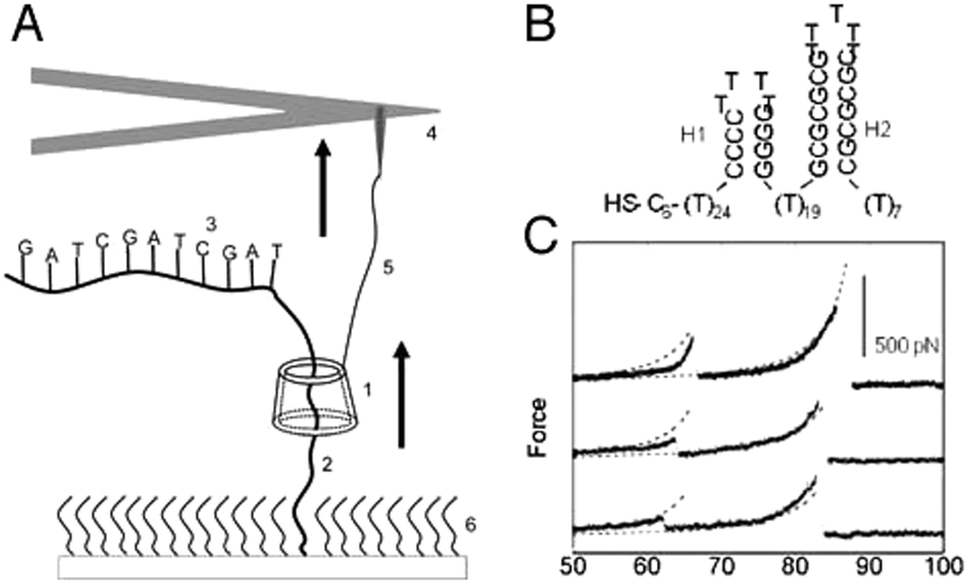 | ||
| Fig. 5 An AFM-rotaxane for the reading of DNA secondary structure. (A) The β-cyclodextrin (CD) ring (1) is pulled up over a PEG spacer (2) and onto a DNA oligomer (3) attached to the PEG. An AFM cantilever (not to scale) (4) is linked to the CD with a functionalised PEG linker (5). (B) Example of hairpin motifs that were probed. (C) Example of resulting force curves after measuring the hairpin shown in (B). Reprinted with permission (Wiley).11 | ||
To perform their experiment, the β-cyclodextrin was equipped with a protected thiol group that could be unveiled just prior to the experiment. This allowed the direct conjugation of the macrocycle to an AFM tip featuring covalently linked PEG terminated with vinylsulfone groups. The resulting covalent linkage allowed the exertion of forces up to 400 pN, which would not have been possible with the non-specific adsorption method used in the system described above. The resisting forces registered by the AFM tip after the β-cyclodextrin was set in motion were closely monitored, and it was found that a difference between various sizes of hairpins could be detected based on the resulting force curves (see Fig. 5C). It is impressive that this complex setup could indeed provide information about the secondary structure of such an important molecule as DNA. Regrettably, oligomers that contained alternating blocks of purines and pyrimidines exhibited no composition-dependent signal above the noise level. Therefore, this method does not yet appear to be suitable for actual sequence determination of DNA.11
2.3. Polyrotaxane materials
Having just described a rotaxane system where a cyclodextrin acted as the macrocycle, it is now time to describe a special class of rotaxanes: polyrotaxane materials. As their name implies, they consist of long axles threaded with multiple macrocycles, and multiple CDs threaded on PEG is one of the most thoroughly studied polyrotaxane materials to date.12 Due to their low cytotoxicity and controllable size, CD-based polyrotaxanes have allowed the formation of interesting materials. For example, by using a variety of scattering techniques, Mayumi and Ito have shown that molecular dynamics in this polyrotaxane are much slower than in free PEG.13 By admixing a bicyclic cross-linker, presumably two connected cyclodextrins, the polyrotaxanes formed gels with appealing material properties: after uniaxial strain, no spatial inhomogeneities were formed, causing the gel to revert to its original shape. This elasticity probably finds its root in the large degree of molecular freedom the PEG chains have, as they are cross-linked by dynamic sets of rings that are free to slide or move to alleviate local strain.13There are a variety of potential applications for CD-PEG polyrotaxanes in biotechnology. For example, by using drug-appended CDs threaded on a PEG with bio-degradable stoppers, the mechanically linked drugs were released when these stoppers were degraded inside cells, allowing multivalent delivery or controlled release, depending on the specifics of the polyrotaxane.12
Gene delivery is another possibility, this time employing cationic CDs. For example, the group of Thompson has shown that a four-arm PEG loaded with multiple cationic CDs can be used as a DNA transfection vector, to effect gene silencing.14 In all these cases, the rotaxane architecture is essential to the functionality of the reported materials, mostly through the material properties imparted by a rotaxane's internal freedom of movement. We would like to refer to the cited review for readers who want to learn more about polyrotaxanes.12
The final class of polyrotaxane materials we discuss here does not yet have a function by itself, apart from being quite powerful at achieving the organisation of rotaxanes. Various metal–organic frameworks exist where the organic linkers that connect the metal centres are at the same time axles that carry a macrocycle, as exemplified by the report from the groups of Schurko and Loeb,15 with more examples cited therein. Effectively, the metal centres are linked by rotaxanes to form a framework of up to millimetre size, resulting in highly ordered arrays of myriad macrocycles (Fig. 6A). This high degree of order allows various detection techniques to characterise the dynamics of the components. For example, solid state NMR was used to verify the dynamic movement of the rings inside the otherwise rigid framework. As predicted by the authors, “the ability to arrange mobile and functional molecular components in a highly dense and predictable array is a crucial step towards the generation of solid-state devices with multiple functions and properties,”15 making such polyrotaxane frameworks promising precursors to actually functional ones.
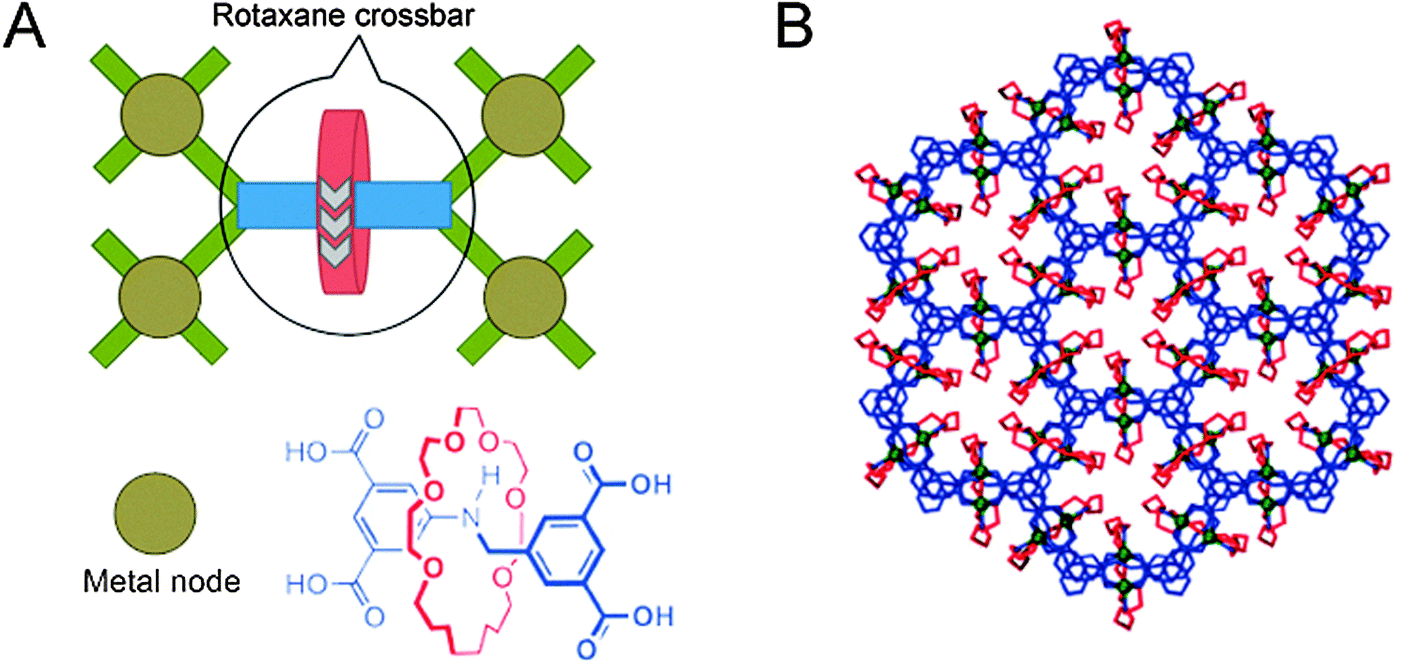 | ||
| Fig. 6 Schematic representation of a polyrotaxane metal–organic framework. (A) The [2]rotaxane, of which the structure is shown, acts as a tetravalent crossbar, coordinating four metal nodes (Cu). (B) Several linked polyhedra of the framework, viewed down the c-axis. Molecules are colour-coded as in the structure shown in (A). Adapted with permission from Macmillan Publishers Ltd: Nat. Chem.,15 copyright 2012. | ||
2.4 Other molecular machines
This final part of the section on rotaxanes and forces discusses a variety of molecular machines or motors,6 which in many cases are related to the artificial muscles described above. One of these systems is the molecular elevator developed by the groups of Stoddart and Credi.16 Their reported device consisted of a platform-like component that was mounted on a three-legged rig (Fig. 7A). Reminiscent of switchable linear muscles at first, this molecular machine is far more complex due to its multivalent nature. This elevator employed DB24C8 rings that were initially mounted on ammonia stations, not on bipyridinium (BIPY2+) stations that were also available in the axles. The DB24C8 rings were fused via an aromatic core to form a trimeric unit. Their axles, too, were trimeric in nature, being linked via another aromatic core. In an acidic environment, the ammonia stations were preferred because they allowed for the formation of hydrogen bonds with the rings. On addition of base, the ammonia station was deprotonated, and the DB24C8 rings moved to the BIPY2+ stations further away from the core. Exhibiting a clear-cut on-off reversible behaviour, reacidification would drive the platform back ‘up’ to the ammonia stations. Its microscopic reversibility makes this intricate rotaxane an exponent of current molecular machines.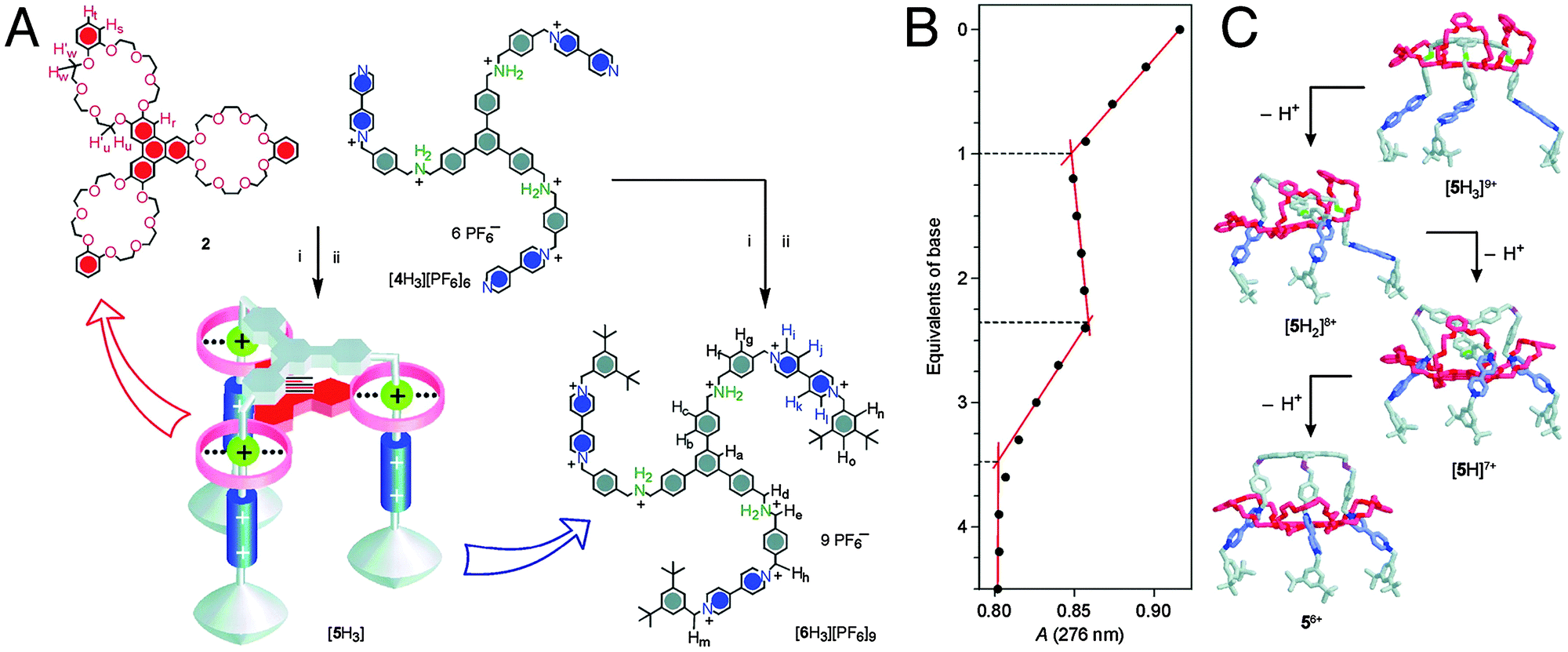 | ||
Fig. 7 A molecular elevator. (A) The trifurcated guest [4H3][PF6]6 and the tritopic host 2 can form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct. Changes in pH drive 2 from the green ammonium stations to the blue bipyridinium stations or back. (B) Changes in the absorbance at 276 nm during base titration revealed that each ‘leg’ of the elevator moved independently. (C) A model for the stepwise downward motion of the platform on successive deprotonation of its three ammonium stations. Adapted from J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science 2004, 303, 1845–1849, reprinted with permission from AAAS. :1 adduct. Changes in pH drive 2 from the green ammonium stations to the blue bipyridinium stations or back. (B) Changes in the absorbance at 276 nm during base titration revealed that each ‘leg’ of the elevator moved independently. (C) A model for the stepwise downward motion of the platform on successive deprotonation of its three ammonium stations. Adapted from J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science 2004, 303, 1845–1849, reprinted with permission from AAAS. | ||
From thermodynamic considerations, it could be established that the energy available for the relevant upwards (acidification) and downwards (base addition) movements amounted to approximately >4 and 21 kcal mol−1, respectively.16 Because the distance travelled by the platform upon each elevator trip was about 0.7 nm, the elevator thus potentially generated a force of up to 200 pN on its way down, which is more than one order of magnitude larger than the forces generated by natural actuators.
This elevator would not be comfortable for passengers, though: titration experiments shown in Fig. 7B revealed that the multivalent platform descends one ring at a time, as opposed to in one concerted slide (Fig. 7C). Multivalency in recognition, as exhibited by this elevator, is a well-established concept, but its past investigation has often been restricted to the acquisition of thermodynamic parameters, not taking the timing of events into account. This elevator provided the opportunity to study how three equivalent recognition sites responded to being simultaneously addressed within a single molecularly constrained environment.16 The results presented by Badjic et al. were a clear indicator of the one-at-a-time mechanism of multivalent processes. As described by the authors: “the molecular elevator is more reminiscent of a legged animal than it is of the passenger elevator.”
The elevator described above had a well-defined directionality of motion because the ‘pillars’ of the ‘elevator shaft’ were all attached to the central core in the same direction. This motion was always highly discrete, because of the finite length of the axles. Theoretically, infinite motion would be obtainable if one end of an axle were to be connected to its other end. This semantically leads us to the realm of catenanes, mechanically interlocked macrocyclic molecules. In this connection, the unidirectional rotary motor described by Hernández, Kay and Leigh deserves mentioning here.17 It contains two macrocycles which are appreciably different: one is clearly the ‘track,’ which would be a rotaxane's axle, and the other is the ‘shuttle,’ or the ring. The authors achieved movement of the shuttle around the track by harnessing biased Brownian motion, combined with the erection or removal of kinetic barriers.
In effect, the track contained a fumarate station and a succinimate station, similar to the system used in Fig. 4 (see ref. 17 for the exact molecular structures). The fumarate station had a higher affinity for the benzylic amide shuttle ring as long as it was in its e-configuration. Photoisomerisation to the z-configuration lowered this station's affinity by such an amount that the succinimate station on the other side of the track became the shuttle's preferred docking site. However, this station had two serine-derived moieties flanking it, each carrying bulky protecting groups on their hydroxyl groups. These two protecting groups were not identical, but both of them were too large for the shuttle to slide over. Using orthogonal deprotection/reprotection chemistry, one of the kinetic barriers could be temporarily removed, opening the path for the shuttle to slide to its preferred succinimate station. Addition of piperidine allowed the z-fumarate station to revert to its energetically favourable e-configuration, and after another round of deprotection/reprotection reactions, the shuttle would slide back to this station. By choosing which protecting group was removed or reattached at which moment, the authors could determine which direction the shuttle would slide towards, achieving directional rotation.
In this motor, the fuel spent to achieve locomotion was purely derived from Brownian motion. The energy that the system required from external sources was spent solely on achieving directionality and switching the motion's bias. The motor we describe next takes a green approach to this: it is solar-powered.18 Based on a bis-p-phenylene-34-crown-10 ring locked on an axle containing a bipyridinium and a dimethyl-bipyridinium station, this rotaxane was stoppered by a conventional stopper on the one side, but by a photosensitising Ru-complex on the other. Intuitively, the ring preferentially docked on the less bulky of the two abovementioned stations. To operate this motor, the photosensitiser would reduce the doubly charged bipyridinium station to a singly charged one. This strongly reduced its affinity for the ring, causing the latter to slide to the now more favourable dimethyl-bipyridinium site. Ingeniously, single electron reduction of this dimethyl-station never occurred due to poorer spectral overlap with the photosensitiser. Finally, as soon as the reduced station would decay back to its original state, the rotaxane's macrocycle diffused back to it, regenerating the original state of this elegant system. The authors calculated that the quantum yield of this sunlight-powered motor was 2%, but they also pointed out that the fuel was free.18
The final system described in this section combines aspects of many of the approaches discussed above. Berná et al. reported an approach to carry a droplet of liquid uphill using nothing but the rotaxanes that covered the incline's surface and a light source.19 Their system was based on a now familiar rotaxane: a benzylamidic macrocycle locked on an axle containing both a succinic and a fumaric station. The fumaric station had the highest affinity for the ring as long as it was in its e-configuration. Irradiation with UV-light (254 nm) converted the station to its z-isomer, lowering the station's ability to form energetically favourable hydrogen bonds with the macrocycle, causing the latter to slide towards the more flexible succinamide station. This station, though, was tetrafluorinated. Therefore, irradiation would indirectly lead to the rotaxane effectively becoming less polarophobic, because after irradiation, the macrocycle would slide over the previously solvent-exposed fluorine atoms and shield them from contact with other molecules. These rotaxanes were then physisorbed to a surface, creating a surface with a light-responsive degree of fluorination.
The mechanism of drop transport was based on this degree of fluorination: many solvents are known to be sensitive to surface-bound halogens and adjust their surface tension accordingly. As one side of a drop of diiodomethane and an adjacent section of the unwetted surface was irradiated (see Fig. 8), that part of the surface eventually came to expose less fluorine. The drop responded by decreasing its contact angle at that side, and it started wetting the less polariphobic surface. To keep up with its extending front, the back of the droplet was continuously forced to decrease its contact angle, up to a point where it was eventually forced to slide towards the front of the droplet to be able to revert to more favourable angles. This shift resulted in a net transport of the droplet. The effect was so pronounced that droplets could even be goaded up a 12° inclined surface covered with these photoresponsive rotaxanes, against the pull of gravity. For this review, this system introduced the concept of sheltering a specific moiety from interactions with its environment by engirdling it with a macrocyclic protective ring. The next chapter will explore this concept in more depth.
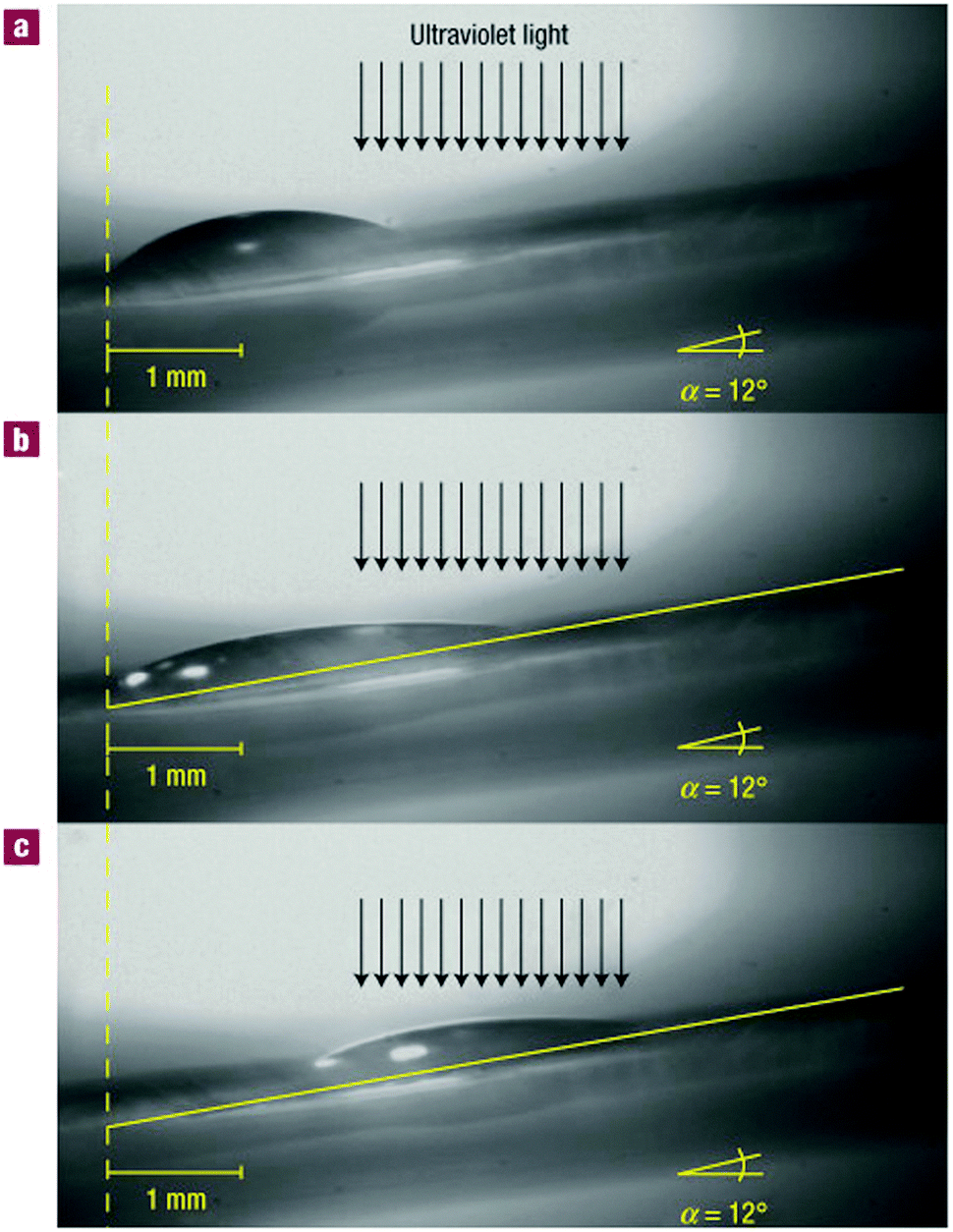 | ||
| Fig. 8 Macroscopic transport by surface-coated rotaxanes. Lateral photographs of a diiodomethane drop on a rotaxane-covered surface with a 12° incline. For clarity, a yellow line can indicate the surface of the substrate. (A) Before irradiation, the entire surface exposes fluoride. (B) After 160 s of irradiation (just before transport). The drop starts to wet the area where fluoride is covered by the macrocycle, which has shuttled in response to the irradiation. (C) After 245 s of irradiation (just after transport). Reprinted with permission from Macmillan Publishers Ltd: Nat. Mater.,19 copyright 2005. | ||
3. Regulated access or location
If you park a bicycle somewhere, it is common practice to use a chain lock for safety. This immobilises the rigid rods of the bike frame by attaching them to an external scaffold in an effort to prevent undesirable interactions of third parties with the bike. If no external scaffolding is present, sometimes multiple bicycles are locked together by a single lock to reduce their mobility. In effect, this common mechanically interlocked architecture of a ring around a stoppered bar regulates access to the bar: whoever controls the lock controls the bicycle. This simple philosophy has found its way into the molecular world as well, where we find demonstrations of rotaxanes with the macrocycle protecting the axle, or with the macrocycle keeping multiple axles in place. This section deals with systems where rotaxane architectures are used to control the location or accessibility of molecules.3.1 Protective rings
The green fluorescent protein (GFP) is a widely used fluorescent marker, and its success is partially due to its relative stability: a protective β-barrel encases the active fluorophore, shielding it from molecular interactions that may cause its quenching or bleaching. Compared to the encased fluorophore of GFP, squaraines may be even more desirable markers for they are near-IR dyes, enabling several biomedical applications. Unfortunately, the ‘naked’ dye (Fig. 9) is inherently susceptible to nucleophilic attack, which leads to loss of the chromophore. Another drawback is its tendency to aggregate in water, which quenches fluorescence. Both of these problems were solved by the group of Smith,20 who encapsulated the cyclobutene core of the squaraine dye inside an amide-containing macrocycle, forming a rotaxane. They found a greatly enhanced stability, which was due to the surrounding macrocycle that sat perfectly over both faces of the electrophilic moiety and blocked nucleophilic attack. That same moiety templated the assembly of the macrocycle, which was formed in one pot from precursor acid chlorides and diamines.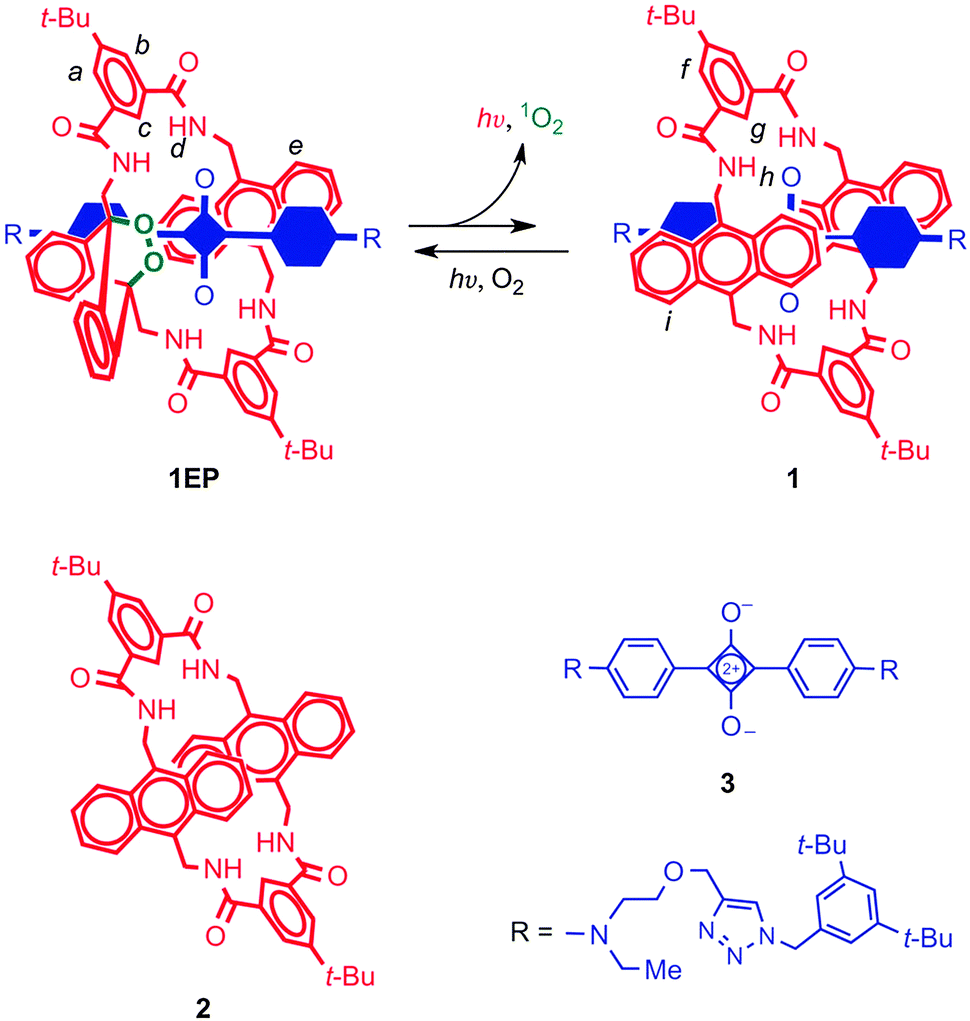 | ||
| Fig. 9 A squaraine dye (3, blue) is protected by a functional macrocycle (2, red) through a rotaxane architecture. The macrocycle can form an endoperoxide (1EP) which can spontaneously emit light at physiologically relevant temperatures. The encapsulated blue component in rotaxane 1 and 1EP is the squaraine 3. See text for more details. Reprinted with permission from Macmillan Publishers Ltd: Nat. Chem.,22 copyright 2010. | ||
In later developments, rotaxane formation was made more efficient by threading a preformed macrocycle over a squaraine axle that only carried one stopper, followed by subsequent conjugation of the second stopper.21 Also, mere sterically bulky stoppers were replaced by bacteria-targeting units that comprised two zinc coordinated 2,2′-dipicolylamine groups, allowing use of the rotaxane as a targeted dye.
Increasing the complexity of the system even further, the macrocycle itself had also been upgraded to a version carrying an anthracene diamine instead of a phenyl diamine moiety (Fig. 9).22 It is known that anthracenes are able to form endoperoxides, which emit visible light when they revert to their original state. Baumes et al. thus created a squaraine rotaxane featuring an endoperoxide macrocycle, which could be stored indefinitely at −20 °C, but which would slowly revert to the root anthracene-like state at 37 °C, emitting near-IR light in the process. The resulting complex still had the same useful near-IR fluorescence of the rotaxane dyes described above. Thus, the properties of fluorescence and heat-activated chemiluminescence were combined in a single rotaxane system. Such self-illuminating systems are particularly attractive because no irradiation is necessary, and there is no background emission. Prior chemiluminescence strategies involved short-lived molecular species that had to be generated in situ, making this a sizeable improvement. Further modifications of this interesting system's macrocycle will be discussed in Section 3.3. For an in-depth overview of fluorescent rotaxanes with similar or with many different characteristics, the reader is referred to the review by Ma and Tian.23
Other molecules that warrant protection are those that have a biological function that should only act in a highly localised fashion: a protected drug would not only be more stable, it would also be inactive as long as it was protected. For example, opioid growth factor (OGF) is a pentapeptide (YGGFM) that has a certain anti-tumour activity, but also influences pain perception. It would be beneficial if drugs such as this could be made available only in targeted tissues, as opposed to full systemic distribution. The groups of Leigh, Papot and Aucagne collaborated to develop a rotaxane with an OGF-based axle.24 Their synthesis started with a protected YGG that carried an N-terminal galactose stopper attached via a self-immolative linker. Benzylic amide macrocycles can be assembled around peptides with C-terminal glycines, and this was indeed done here (Fig. 10 gives a schematic representation using a different axle). Subsequently, the C-terminus was extended by the FM dimer, a bulky sequence that could function as a natural stopper. Upon treatment of this rotaxane with β-galactosidase, the galactose was cleaved from the complex, enabling the subsequent disassembly of the self-immolative linker. Thus, the enzymatic cleavage effectively removed one of the two stoppers, allowing the macrocycle to slide off of the peptide drug, making it biologically available.24 Such a strategy is promising, because antibody-mediated delivery of β-galactosidase is an established prodrug strategy, now granting the same specificity to this rotaxane prodrug. A later expansion on this work by Viterisi and Fernandes et al. employed a PEG-derivatised macrocycle to greatly increase the solubility of rotaxanes involving this macrocycle in aqueous media.25 Rendering the macrocycle more water-soluble simultaneously causes the threaded peptide axle to effectively become more soluble by association. This can be advantageous during the administration of pharmaceuticals such as these.
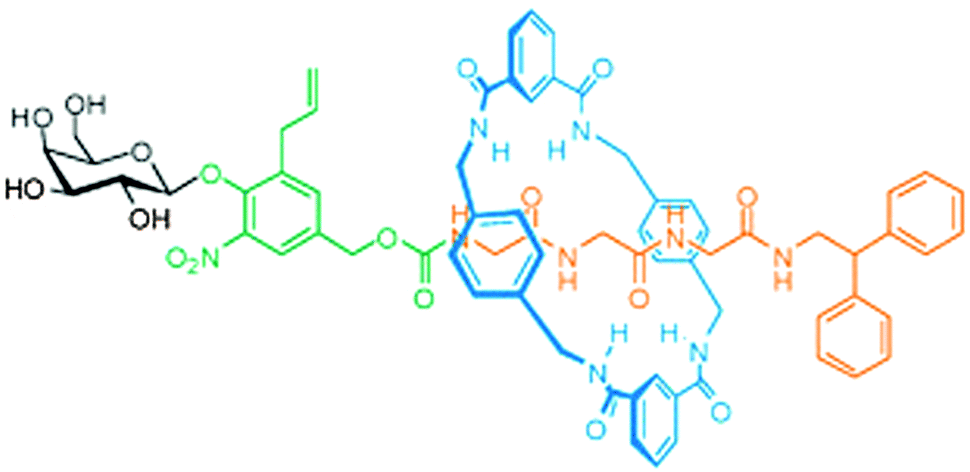 | ||
| Fig. 10 A rotaxane where the macrocycle (blue) shields a peptide (orange). The enzyme β-galactosidase can cleave the carbohydrate (black) to trigger the release of the peptide via a self-immolative linker (green). Reprinted with permission (RSC).25 | ||
3.2 Valves
Bicycles offer more analogies with rotaxanes than just locking them up with a chain: their pneumatic tires have compressed air in them, and this air is separated from the atmosphere by a valve. Such a valve may be nothing more than a tube with a little bolt protruding in the middle. A sufficiently large nut that is twisted tight onto this bolt seals the tube. Untwisting the nut reopens the valve. We have seen in the previous section how rotaxane macrocycles can form effective barriers on a molecular scale, and the section on molecular muscles has provided various examples of macrocycles that can be shuttled around an appropriately designed axle. These capabilities have been put to good use in the emulation of macroscopic valves by molecular rotaxane systems.The first example of this type was reported by the group of Stoddart and Zink.26 They made use of a CBPQT4+ macrocycle and an axle featuring a TTF and an NP station, shown in Fig. 11A and similar to the ring and axles shown in Fig. 2A. The axle had two different stoppers, of which one had a functional group that allowed its conjugation to the surface of mesoporous silica particles, in a setup similar to that shown in Fig. 11B. Redox control over the position of the macrocycle thus caused it to be either close to the surface of the particle (valve ‘closed’) or further away from it (valve ‘opened’). It was demonstrated that in the ‘open’ state, a fluorescent iridium complex could diffuse into the pores of the particle, and that a later ‘closing’ of the valves would trap the dye. The closed state functioned as desired: no leakage of trapped dye was observed by fluorescence spectroscopy monitoring as long as the valve remained closed. The addition of ascorbic acid reduced the axle's distant TTF2+ station and allowed the macrocycle to slide away from the surface-proximal NP station, opening the valves. An immediate and fast increase in Ir-fluorescence was observed, indicating that trapped molecules were once again free to diffuse into the solution. This nanovalve was based on an established molecular shuttle system, ensuring its reversibility and thus its reusability.26
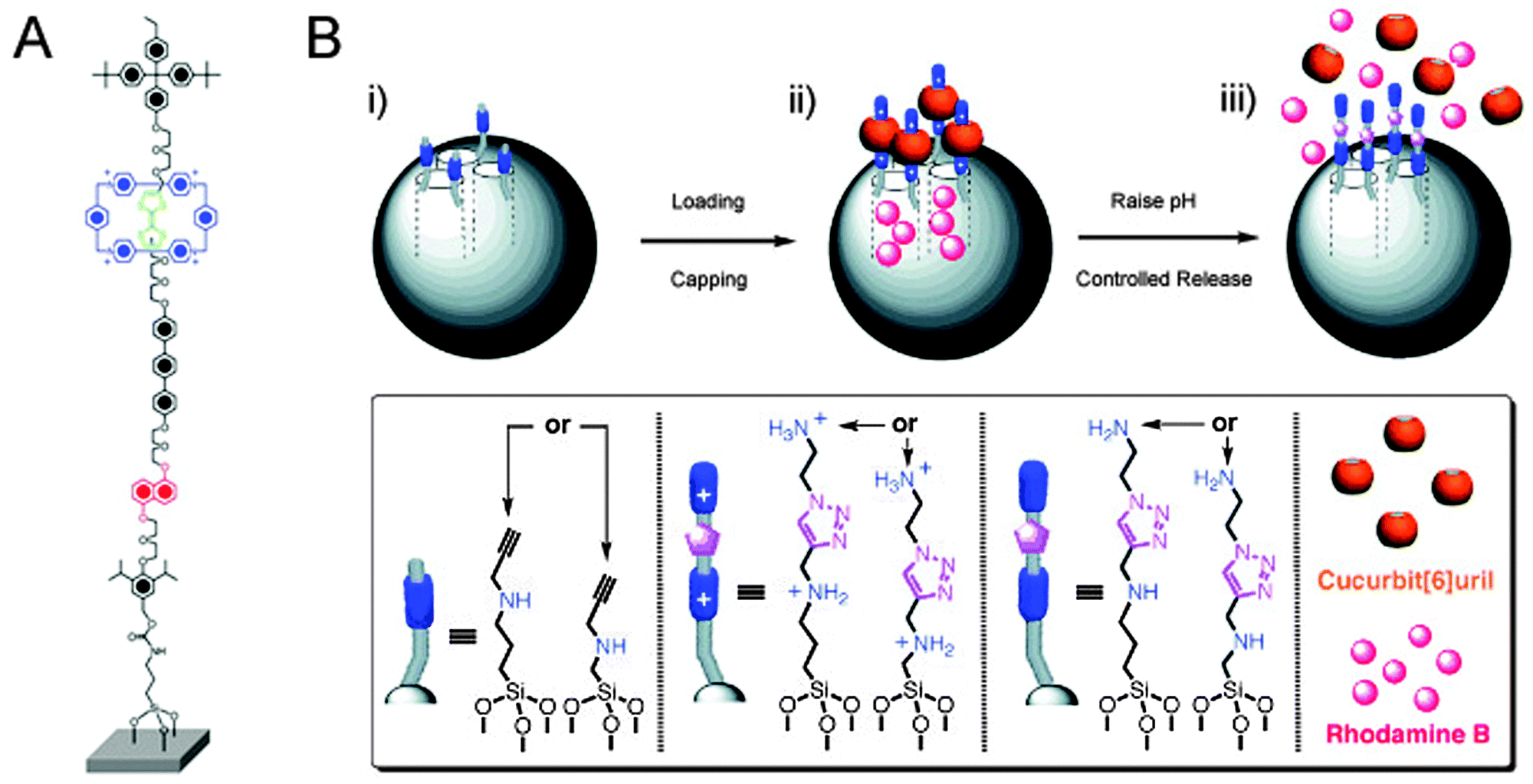 | ||
| Fig. 11 Rotaxanes as nanovalves. (A) Molecular structure of a redox-responsive nanovalve. From ref. 26. Reprinted with permission from AAAS. (B) Graphical representation of pH-switchable supramolecular nanovalves. (i) Alkyne-functionalised mesoporous silica nanoparticles (grey spheres) are loaded with rhodamine B and capped with CB[6], followed by washing away the excess of substrates, yielding (ii). In (iii), RhB molecules are released by switching off the ion–dipole interactions between the CB[6] rings and the bisammonium axles upon raising the pH value. Adapted with permission (Wiley).27 | ||
A further development of this concept switched away from redox-responsive rotaxanes, opting to employ pH-sensitive pseudo-rotaxanes instead (Fig. 11B).27 After loading mesoporous silica particles with rhodamine B as a model cargo, surface-grafted amino-acetylenes were used in a cucurbit[6]uril-catalysed alkyne-azide cycloaddition to extend the short stalks with a second amine under acidic conditions. This simultaneously trapped the cucurbituril macrocycles because they could not slide over the protonated amines, but they encircled the triazoles upon their formation. Thus, closed valves were created in a self-threading fashion. After washing away of excess reagents, the closed silica particles were assayed and were found to be non-leaking containers for the cargo when short axles were employed. On raising the pH to 10, the amines in the axle were deprotonated, and because there was no stopper on the far end of the axle, the cucurbituril could then freely slide off of its stalk. This opened the valved, and indeed released the cargo into the medium, establishing a nanovalve system that responded to changes in pH. The group of Kim reported an earlier version of this system,28 using a similar strategy and achieving the same goal; their system involved poly(ethylene imine) stalks, and multiple cyclodextrin macrocycles were threaded to close the valves.
An additional form of control was added to this system by Angelos and Yang et al., who used a similar setup.29 As an additional feature, the nanopores in their silica particles had their surfaces covered with covalently linked azobenzene moieties. These light-responsive molecules can reversibly and repeatedly switch between trans- and cis-conformations when irradiated by specific wavelengths of light. When these so-called nano-impellers were irradiated with 488 nm light, they would undergo a wagging motion, increasing the mobility inside the pores. It was found that merely opening the valve did not lead to the release of cargo molecules, but neither did merely ‘activating’ the nano-impellers. Triggering of both release mechanisms, i.e. changing both the pH and the light irradiation, was required for cargo release, causing these controlled release particles to exhibit AND-logic responses.29
Later research by Liu and Du showed that the pH-based trigger for nanovalve opening could be either accelerated or even replaced completely by competition with alternate guest molecules for the cucurbituril.30 Using a cucurbit[7]uril, the addition of varying doses of a C16-alkylated ammonium bromide or a 1,6-hexanediamine shifted the supramolecular equilibrium of cucurbituril inclusion, allowing the opening of the valves even at near-neutral pH. (This competition for inclusion in the macrocycle is possible in these systems because they do not involve true rotaxanes, but pseudo-rotaxanes lacking a terminal stopper.) Combined, the competitor concentration allowed the nanovalves to be opened at any pH value between neutral pH and the same pH 10 that was needed to open the valve without competitors present.30
3.3 Assemblers
A key step in the synthesis of many different rotaxanes is the moment where the different components assemble. It is either when the preformed macrocycle first threads over its preferred station before all stopper groups are put in place or when the components that will soon form a ring template themselves around a pre-formed axle. By their very nature, rotaxanes are assembled small organic molecules. This power of assembly has been exploited for various purposes. It has been used to enforce assembly of larger molecules, and it has found sensory applications, all of which are covered in this section. A single caveat: some of these systems are not true rotaxanes, but pseudo-rotaxanes. This is because many of the systems described in this section require a dynamic character, which as we will see is one of the advantages that pseudo-rotaxanes can have over actual rotaxanes.Block copolymers are an important class of polymers, because their asymmetry gives them unique properties. Unfortunately, obtaining block copolymers is not always as trivial as obtaining the separate, unlinked blocks. The group of Fustin has devised a strategy that uses a rotaxane architecture to link separate blocks together in a modular fashion.31 Their strategy starts off with a macrocycle carrying a 4-chloro-2,6-pyridinedicarboxamide motif. Through paladium-templating, this macrocycle could thread a singly stoppered axle featuring another pyridine moiety. The non-stoppered end of this axle terminated in an alkyne, so that any azide-terminated polymer block could be conjugated after threading using Cu(I)-catalysed alkyne-azide cycloaddition (CuAAC). For their proof of concept, they used azido-poly(methyl acrylate) terminated with the same stopper group present on the initially templating axle, thus converting the pseudo-rotaxane to an actual rotaxane. Then, a substitution on the macrocycle's halogen by sodium azide enabled a second round of CuAAC conjugation to attach a second block, acetylene-terminated PEG in this case. The resulting block-copolymer's identity (see Fig. 12A) was confirmed by MALDI-TOF and size exclusion chromatography. While the authors only achieved roundabout conjugation of an azido-polymer to an alkyne-polymer, they presented a novel concept for block-copolymer formation, and their block copolymers are sure to have interesting but as of yet unexplored inter-block mobility.31
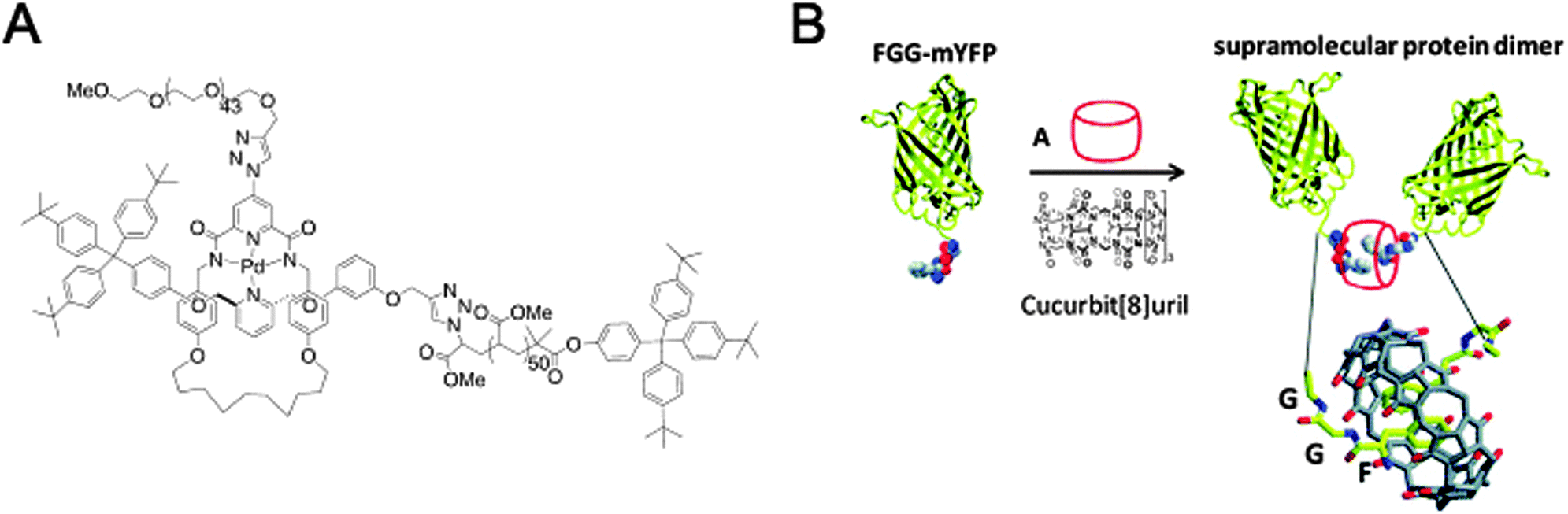 | ||
| Fig. 12 Macromolecular assembly through threading of macrocycles. (A) A diblock copolymer where the two blocks are linked by a mechanical bond as part of a [2]rotaxane. Reprinted with permission (Wiley).31 (B) Macrocycle-mediated dimerisation of two PheGlyGly-tagged proteins through the formation of a [3]pseudorotaxane. Reprinted with permission (RSC).32 | ||
Another system where a macrocycle brings together two macromolecules is based on a pseudo[3]rotaxane and was presented by the group of Brunsveld.32 Here, the ability of a cucurbit[8]uril macrocycle to host more than one guest inside its spacious cavity was exploited. Having identified the Phe-Gly-Gly (FGG) motif as a possible guest for such double occupancy, a yellow fluorescent protein (YFP) with an N-terminal FGG-tag was created. Indeed, addition of the cucurbituril to a solution of monomeric FGG-YFP induced dimerisation (Fig. 12B). Many interactions between cucurbiturils and (modified) proteins have been described, in some cases even with nanomolar binding affinities (the recognition of N-terminal 4-aminomethyl-phenylalanine by cucurbit[7]uril).33 Regardless of their appeal, though, they do fall just outside the scope of this review since neither true rotaxanes nor practical applications are involved. For more information, we encourage the reader to peruse the cited references and references given therein.32,33
The previous subject already touched on recognition as enabled by macrocycles, and on macrocycles hosting multiple guests, making this a suitable moment to return to the squaraine rotaxanes described in Section 3.1. As shown in Fig. 13A, macrocycles exist that will only thread squaraine in the presence of sodium ions.34 Complexation enhances the fluorescence of the dye, giving the entire system sodium-sensing abilities. Interestingly, potassium ions will displace the dye (along with the sodium ions), giving this pseudo-rotaxane an on–off type sensitivity as well if it is used in its preassembled state. The cited review gives more examples of this type of behaviour.21
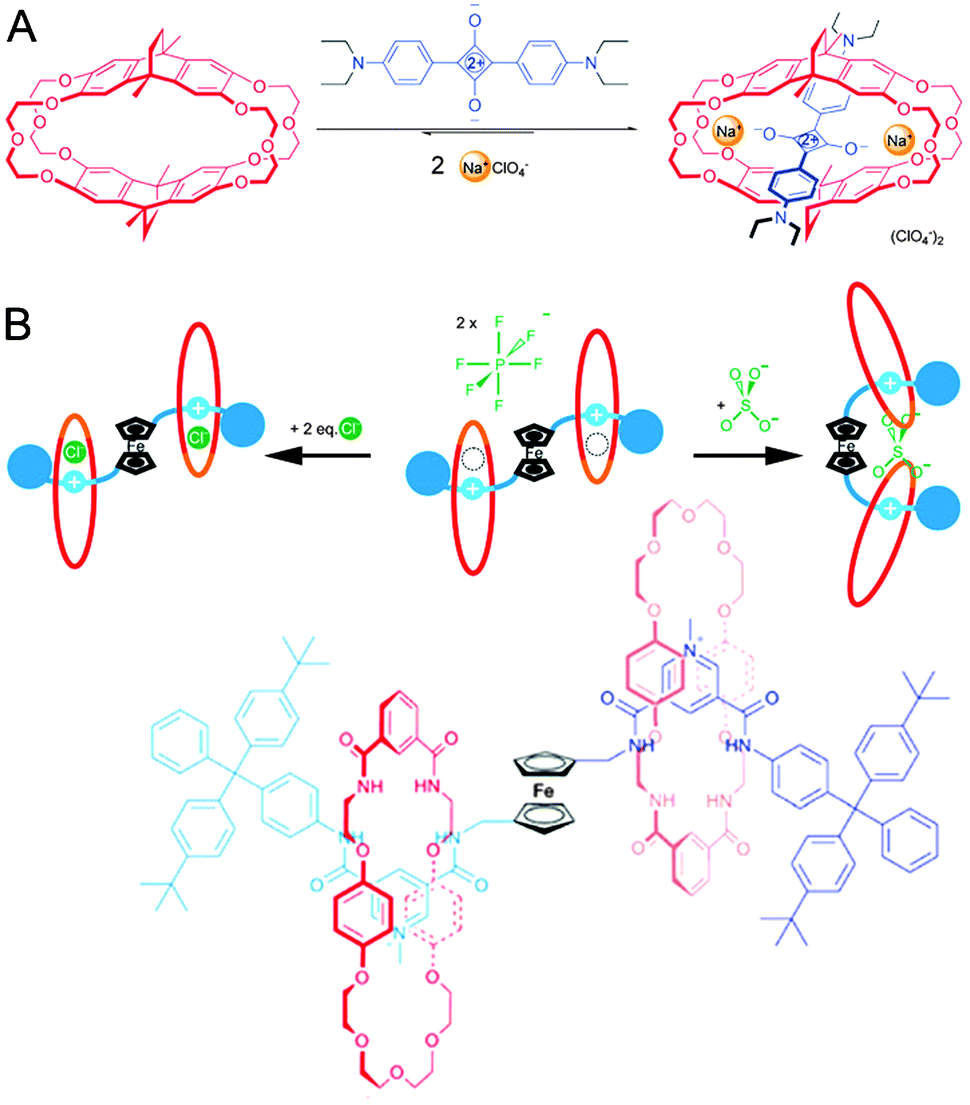 | ||
| Fig. 13 Sensors with a (pseudo)rotaxane architecture. (A) Crown ether cyclophane macrocycle (red) encapsulates squaraine (blue) only in the presence of Na+ (yellow). Adapted with permission (Wiley).34 (B) Schematic representation and structure of a [3]rotaxane that can sense either chloride or sulphate. The ferrocene ‘hinge’ (black) provides the required flexibility to the axle (blue). The macrocycles (red) are inherently mobile, allowing their repositioning. Adapted with permission (RSC).35 | ||
The ability to bind multiple guests is not limited to pseudo-rotaxanes: fully fledged rotaxanes can have similar capacities, as demonstrated in a report from the laboratory of Beer.35 The authors describe a [3]rotaxane that features a multifunctional axle with two pyridinium-type stations. In between the stations is a ferrocene moiety, and docked on the stations are two macrocycles featuring a crown-ether section and an isophthalamide anion recognition element. The pyridinium stations can join forces with the amides in the macrocycle to bind a single chloride ion inside each ring (see Fig. 13B). Facilitated by the rotational flexibility of the ferrocene moiety, the two rings can also approach each other to bind a sulphate dianion, giving this [3]rotaxane two separate modes of sensing. Now the versatility of the ferrocene-hinge becomes apparent: ferrocene is a very responsive group in voltammetry experiments, and the presence of any or none of the compatible anions (chloride or sulphate) could be determined by measuring a voltammogram of the rotaxane in solution. The rotaxane-based binding pocket for chloride and also the freedom of movement of the macrocycle around its axle to accommodate sulphate binding are both essential to the functionality of this interesting hinged sensor, illustrating the unique potential of rotaxanes.
4. Information
The ability to discretely and reversibly switch between states is the principle on which many current microelectronic devices operate. Logic operations require gates that can give clear outputs of either one state or another and that can switch this output based on dynamically varying inputs. The storage of data requires a material that can similarly be set to 0 or 1 and which then maintains this state for as long as possible to enable a readout of the original setting at a later point in time. As we have seen before in this review and as will be covered in the next section, rotaxanes are outstanding candidates for this behaviour.4.1 Molecular electronics
Molecular switch tunnel junctions (MSTJs) are a class of devices where a monolayer of switchable rotaxanes (Fig. 14A) is embedded in between two conducting electrodes (Fig. 14B).36 Several reports exist where this switchable monolayer was based on a [2]rotaxane featuring the by now well-known CBPQT4+ macrocycle on a two-station axle containing a TTF or TTF-like station and an NP station (Fig. 14A shows the TTF-like rotaxane). In its ground state (the ground state co-conformation, or GSCC), this bistable rotaxane has its ring positioned predominantly around the TTF station. Electrochemical oxidation of this station to the TTF2+ state drives the ring to the NP station, causing the rotaxane to adopt its metastable state co-conformation (MSCC). The electric conductivities of these two states, the GSCC and the MSCC, are not identical. This difference can be detected using the same electrochemistry that can switch the system, only at much lower voltages: by ‘interrogating’ the system with a small test voltage, the resulting current over the MSTJ can be measured. A rotaxane with a high conducting state (hypothesised to be the MSCC)36 will have a low resistance and thus produce a high current. Conversely, a rotaxane in its GSCC will not be as conductive, resulting in a lower current (Fig. 14C). By repeating these experiments with just the unthreaded rotaxane's axle, no switchable behaviour was discovered. This virtually proved that the spatial position of the CBPQT4+ ring, and thus the rotaxane architecture, plays a key role in the MSTJ's behaviour.36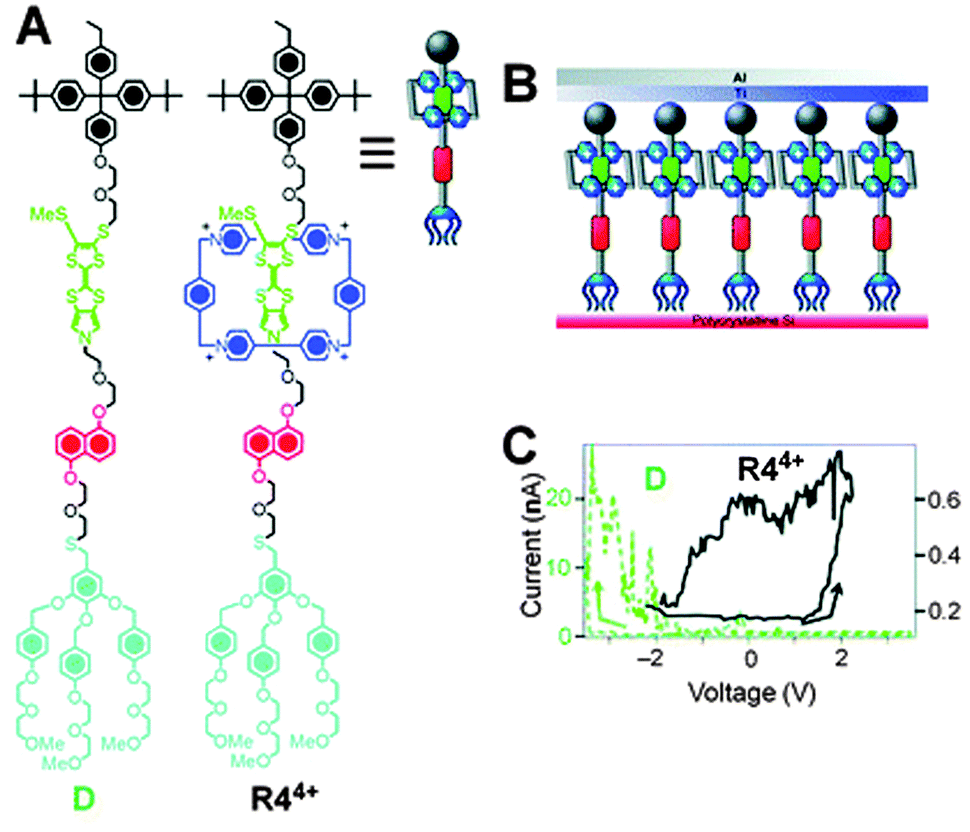 | ||
| Fig. 14 Molecular switch tunnel junctions (MSTJs). (A) Structural formulae of rotaxane R44+ and its axle D (for ‘dumbbell’) along with a graphical representation of R44+. (B) Schematic representation of the MSTJ devices, where a monolayer of rotaxanes is sandwiched by two electrodes. (C) Current–voltage characteristics expressed as remnant molecular signatures of devices (top) incorporating monolayers of R44+ (black trace). A control with the corresponding dumbbell component D (green trace) shows that the rotaxane architecture is essential. Adapted with permission (RSC).40 Data originally reported in ref. 36. | ||
An MSTJ is effectively a gate that can be opened or closed – it can respond to an applied voltage with conductivity or with resistance. This enables their application in logic gates, a fact that was recognised by the group of Heath when they published a pioneering proof of principle using a non-reversible rotaxane switch.37 By linking MSTJs in specific linear arrays, different types of logic gates were obtained, including an AND gate and an OR gate. Expanding the linear arrays to two-dimensional ones, an XOR gate was obtained using the reversible system described above.36 The creation of such molecular logic gates was hypothesised by the authors to have an excellent scalability, because the electronic properties of the solid state devices were very similar to those of the dissolved rotaxanes in dilute solution, implying that the operation of these devices remains similar when fewer molecules populate a junction.37
The precise molecular structure of these devices has a large influence on their operation. For example, by replacing the TTF station with a quasi-TTF station that has a lower affinity for the CBPQT4+ macrocycle, the equilibrium between the GSCC and the MSCC is shifted.38 A more than 1000-fold difference in the relaxation rate was detected when the ring was allowed to slide back to its GSCC, which has many, but mainly negative implications for controlling the power efficiencies of eventual actual devices that employ these molecular switches. Interestingly, in the aforementioned report it was shown that the readout of the electronic device could also be based on the rotaxane's colour, which is green when it is in its GSCC and red when it is in its MSCC.38
The two-dimensional arrays mentioned above were so-called crossbar systems, which are systems where multiple inputs are connected to multiple outputs via a matrix of individual switches. One of the most impressive achievements for rotaxane electronics is a report by Green and Choi and coauthors.39 They describe a crossbar system that was made up of 400 16 nm broad silicon wires overlapping 400 similar titanium wires. Each junction contained rotaxanes to form an MSTJ, resulting in a 160 kilobit ‘chip’. The entire circuit had dimensions of 13 × 13 μm, which is so small that no readout system was readily available, causing the researchers to use test electrodes that contacted two to four nanowires at a time. When testing the memory, the researchers performed a massive write of “1” to all bits (they switched the rotaxane from its GSCC to its MSCC) by applying a +1.5 V pulse. A +0.2 V bias was then used to read each bit. Subsequently, a −2.0 V pulse was applied to reset all bits to “0”, and the bits were read once more. It was found that about 50% of the bits responded to this treatment, and that again 50% of those switched beyond a self-imposed threshold value. Bits could be switched up to ten times, and the volatility of the memory (which is a measure of how long data remain stored, so in this system it is the lifetime of the MSCC) was found to be in line with other switchable rotaxanes (medium decay 75 minutes). This was a clear indication that the molecular switching mechanism was the actual basis for the memory. Despite the growing pains described above, this report has set a powerful example of what can be achieved with modern nanotechnology.39 For an in-depth review of the current state of the art concerning similar molecular electronics, we recommend the cited recent review by Coskun and coauthors.40
Cavallini and coauthors presented an alternative [2]rotaxane-based method for data storage, which technically speaking does not count as molecular electronics because no voltages or currents were involved.41 Their setup relied on an AFM and its interaction with a dropcast film of rotaxanes for the writing and the readout of the memory. These rotaxanes did not feature multiple stations (see Fig. 15). Instead, the system relied on interconversion between different rotational isomers of the rotaxane. Molecular modelling of the solid state rotaxane had revealed that two nearly degenerate surfaces existed, between which it was possible to switch with a relatively low activation energy (200.4 kJ mol−1). This energy could be supplied by an AFM tip tracking over the surface with a set point force of slightly more than 2 nN. Due to the peculiarities of nucleation, crystallisation and ripening, a series of dots appeared in response to the perturbation, with a dot size that was related to film thickness. When the AFM tip was tracked over the surface with a set point force lower than 2 nN, no such perturbation of the surface occurred, yet existing dots could be effectively detected. Once more the critical role of the rotaxane architecture was established through experiments where either the macrocycle or the axle was dropcast, and ‘ploughing’ of the AFM tip in the surface was also experimentally ruled out. The authors speculate that data densities of up to 15.5 Gbit per cm2 should be possible with this system,41 which are in the same range as the 100 Gbit cm−2 that was obtained with the MSTJs described above.39 While the preparation of the memory was much less technically demanding than for the MSTJs, readout by AFM is not as practical as integration in a microelectronic circuit.
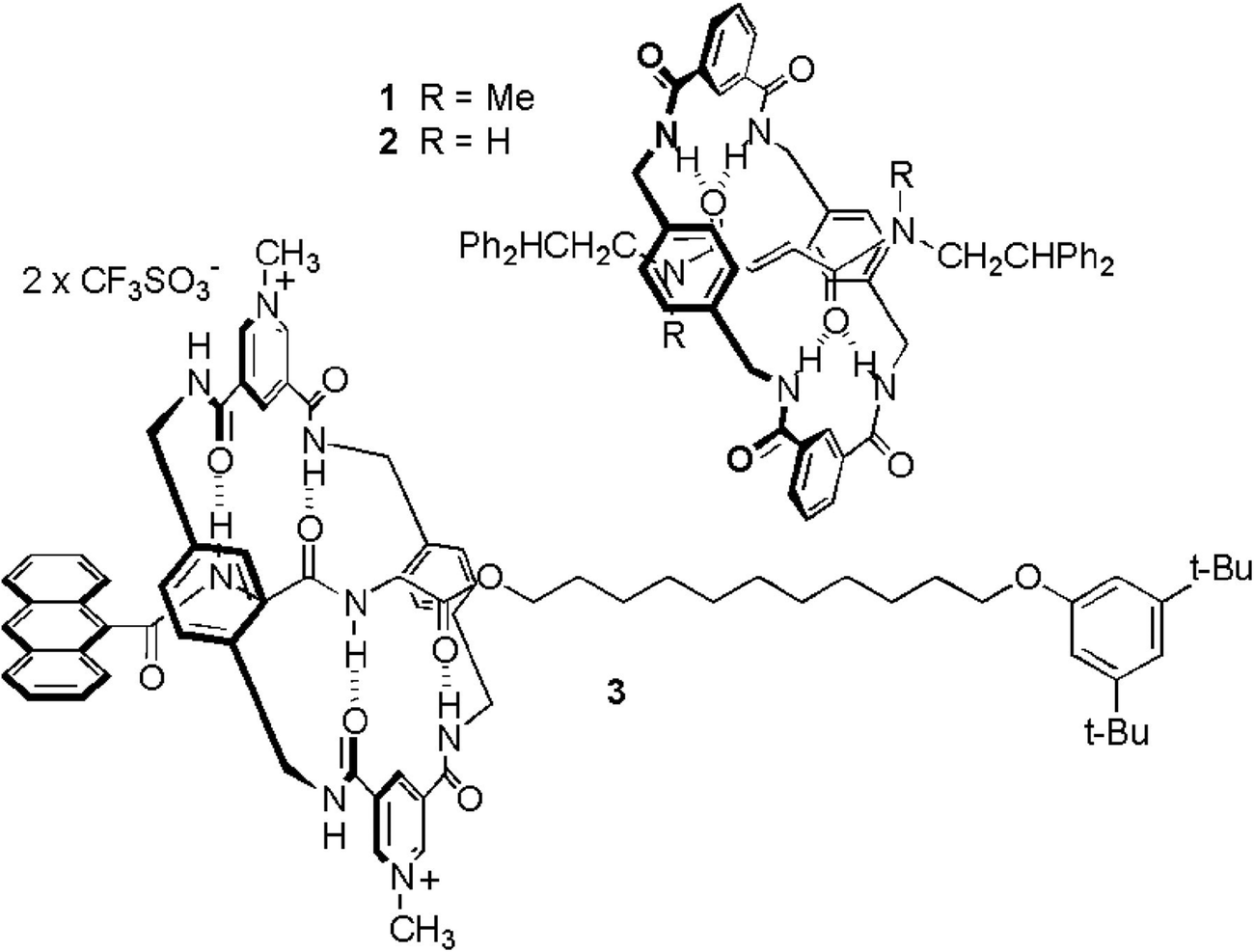 | ||
| Fig. 15 Rotaxanes used for the creation of surface patterns for data storage requiring AFM read/write operations. A slight force exerted by an AFM tip on a rotaxane-coated surface causes the rings to slide, macroscopically leading to a bulge on the surface where it was tapped. Gentler AFM-probing can detect the presence of such bulges, allowing binary patterns to be written and read. From ref. 41. Reprinted with permission from AAAS. | ||
4.2 Ratchets
A more esoteric concept of information is the physical principle of knowing the exact location of a particle. However, this is not as distant from everyday life as one might perhaps imagine: the molecular memory chip described above, for example, derives its storage capacity from the ability to maintain and report the location of a particle, with the particle being the macrocycle of a rotaxane. It is digitisation that allows such Boolean knowledge to be converted into more traditional analogue forms. Thus, information regarding the location of a particle can be of fundamental interest.In equilibrated systems, there is no way of knowing where an individual particular particle might be. For an ideal gas that fills a balloon one knows that all gas particles are inside the balloon, but their individual exact locations can only be described in probabilities. A situation involving less possible states is a [2]rotaxane where the axle features two docking stations with comparable affinities for the resident macrocycle. At a given moment in time, there is no telling on which station an individual cycle will be docked, but one can measure average distributions in terms of what percentage of macrocycles is where. Because the stations have similar affinities, there will be no clear predominance of one station's occupancy over the other.
The group of Leigh has presented a molecular information ratchet that was inspired by these ponderations.42 Ratchets are simple yet ingenious mechanical devices where a cogwheel is continuously engaged by a pawl or a tooth, allowing it to rotate in one direction only. The rotaxane ratchet featured a dibenzo-24-crown-8-based macrocycle mechanically locked onto an axle that featured two ammonium stations (see Fig. 16A). An α-methyl stilbene unit divided the axle into two ‘compartments’: with the stilbene in its e-isomeric form, the macrocycle could shuttle between the two stations based on Brownian motion. The z-isomer, on the other hand, was sufficiently bulky to become a de facto stopper group which the macrocycle could not cross, trapping the ring in its current compartment. The stilbene ‘gate’ could thus effectively be ‘opened’ (e) or ‘closed’ (z).
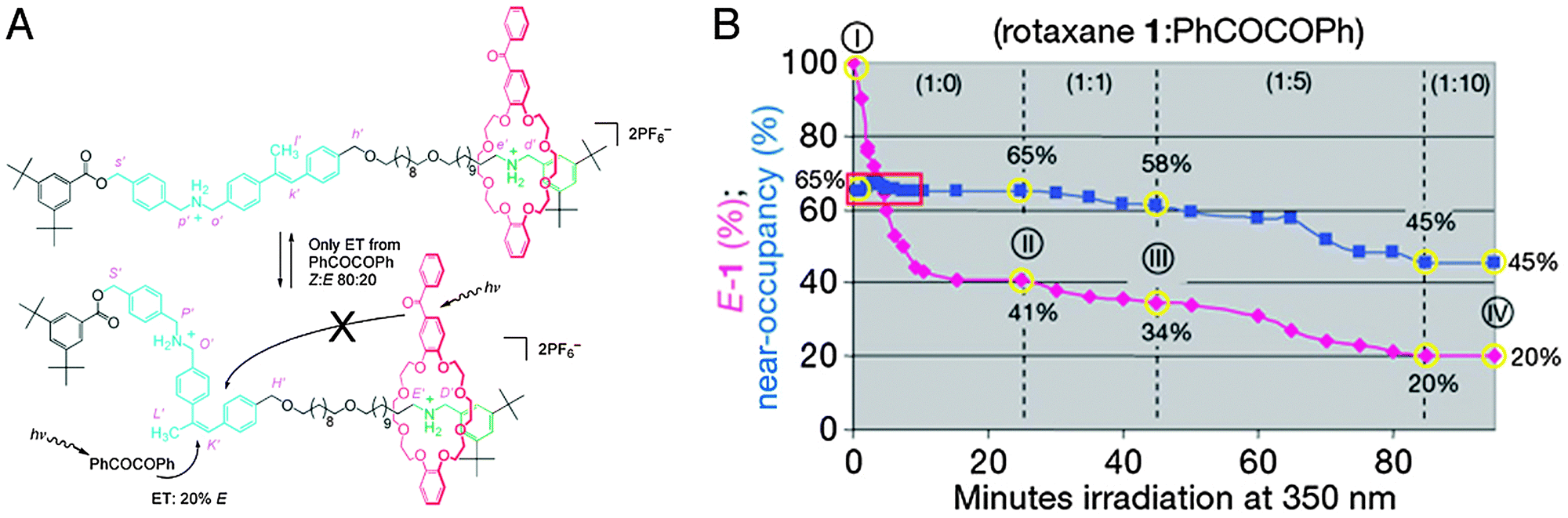 | ||
| Fig. 16 A molecular information ratchet, which in this case is a system where a population of rotaxanes has its macrocycle sliding away from their preferred stations, contrary to the thermodynamic equilibrium. (A) When the macrocycle (red) is on the ‘far’ binding site of the rotaxane (green) as depicted, it cannot open the stilbene gate. When it is on the ‘near’ station (blue, together with the stilbene gate), it can. The gate is kept predominantly ‘closed’ by dissolved PhCOCOPh. Thus, if the macrocycle is on the near gate, it can facilitate its own translocation to the far station, but not back again, moving the system away from equilibrium. (B) Change in the E:Z ratio (the amount of the ‘open’ gate, purple diamonds) and the near:far ratio (near-occupancy (%), blue squares) that occurs during irradiation of the ratchet: (I) pristine rotaxane; (II) after 25 min, no added PhCOCOPh; (III) after a further 20 min with 1 equiv. PhCOCOPh; and (IV) after a further 40 min with 5 equiv. PhCOCOPh plus a further 15 min with 10 equiv. PhCOCOPh. Reprinted with permission from Macmillan Publishers Ltd: Nature,42 copyright 2007. | ||
Stilbene moieties can be switched between their ‘open’ or ‘closed’ states through photon absorption. Direct absorption is possible, but a more efficient process is to employ photosensitisers. Two different sensitisers were employed: benzil was molecularly dissolved, and caused a Z:E ratio of 82:18 for the stilbene gate. Benzophenone was incorporated in the macrocycle and caused a 55:45 Z:E ratio. When irradiating at 350 nm, virtually all photoisomerisation took place through the photosensitisers.
The stilbene ‘gate’ was strategically placed asymmetrically in the axle so that it was much closer to one station (“near”) than to the other (“far”). Because energy transfer efficiency is strongly correlated to distance, the photosensitiser in the macrocycle could only be active when it was on the near station, and it would virtually not be active when the ring was on the far station. Because the sensitiser in the ring had an ‘opening’ effect on the gate, rings on the near station had a high chance of opening the gate and enabling their translocation to the far station. Rings on the far station, though, could no longer open the gate because they could no longer engage in phototransfer. The molecularly dissolved sensitiser predominantly ‘closed’ the gate, and its activity was independent of the position of the ring. Thus, a system was created where rings would automatically slide towards the far compartment without the affinities of the stations being influenced: their movement was ratcheted. In the equilibrium without freely dissolved benzil, 65% of the macrocycles occupied the near gate. Upon addition of one equivalent (compared to the amount of rotaxane) of the ‘closing’ benzil, this occupancy dropped to 58%. By moving up to 5 equivalents of benzil, only 45% of the macrocycles remained on the near station when a new equilibrium was reached. This clearly demonstrated that the equilibrium of the system could be perturbed without altering the system.42
A later report from the same group demonstrated a similar principle, this time using chemical energy to unbalance the equilibrium.43 A single hydroxyl group was placed in the middle of a symmetrical axle featuring two fumaramide stations. This hydroxyl ‘gate’ could be irreversibly closed by esterification with benzoic acid, which required a 4-dimethylaminopyridine (DMAP) type catalyst. It is not chiral as long as it is open, because both gates share equal time with the macrocycle, which is free to slide. Using DMAP, which is small and achiral, no ratcheting effect was observed: after closing the gate, the macrocycles had a 50:50 distribution over the two stations as evidenced by 1H-NMR (which was enabled by the tactical deuteration of one of the two stations) and chiral HPLC showed a 50:50 ratio of (r)/(s). However, switching to DMAP-derivatives featuring chiral and sterically bulky substituents resulted in 67:33 distributions, both for chirality and for station occupancy. Apparently, the bulky and chiral catalyst could more rapidly close a gate when the macrocycle was in a complementary position. This was corroborated by the fact that using the mirroring enantiomer led to an equally mirroring 33:67 distribution.
An intriguing expansion of this chemically driven ratchet was achieved when one of the stations was replaced by a succinamide station, which had a lower affinity for the macrocycle.43 The equilibrium for the ‘open’ state was 75:25 in favour of the fumaramide station, and using DMAP to close the gate resulted in a closely matching 74:26 distribution. By using the catalyst that favours the enantiomer where the ring is on the fumaramide station, this distribution could be driven up to 80:20. More compellingly, it could be driven away from its thermodynamic optimum to 63:37 by using the opposing catalyst. Thus, more than 15% of the macrocycles that were originally positioned on their preferred fumaramide sites were transported enthalpically uphill to the less favoured station by the chemically driven information ratchet.
That such ratcheting between two compartments is the basis for controlling longer distance movement was made clear when a three-compartment ratchet with an identical mechanism was reported.44 It featured three identical fumaramide stations and by using achiral DMAP as a catalyst, a 39:18:43 distribution over the three stations was achieved. These numbers indicate that the ring favoured end compartments, because if all stations were truly equal, a 25:50:25 distribution would be found using achiral DMAP. By using either of the two chiral catalysts, either a <1:21:79 or a 75:25:<1 distribution was obtained. Surprisingly, by using the racemic mixture of the chiral catalyst (a 50:50 mixture) the preference for end compartments was apparently inverted because a 10:77:13 distribution was found. This effect probably resulted from the steric bulk of the racemic mixture, as that is what differentiates it from achiral DMAP. The effect of an achiral yet bulky catalyst would be an interesting next step to study in this research.
5. Catalysis
Owing to their structural diversity and high stability, rotaxanes have been used for various purposes as discussed in the previous sections. However, the number of examples in which rotaxanes are applied as catalysts for chemical reactions is as of yet limited in spite of the initial great expectations. In the next section, the application of rotaxanes as catalysts will be discussed in two main parts: conventional and processive catalysis.5.1 Conventional catalysis
Pseudorotaxanes that are formed by crown ethers and ammonium salts have been used in transition-metal-catalysed asymmetric hydrogenation reactions by both Nishibayashi and Fan, simultaneously. Nishibayashi and coworkers designed a chiral ligand based on a pseudorotaxane skeleton by applying a crown ether moiety bearing an optically active 1,1′-binaphthalen-2,2′-ylphosphite group and a secondary ammonium salt functionalised with a diphenylphosphino group on a biphenyl ring.45 These two components were used in the enantioselective hydrogenation of methyl (Z)-α-acetamidocinnamates in the presence of [Rh(cod)2]PF6 resulting in high conversions (>99%) and high enantiomeric excess (e.e.) values (>90%). Fan and coworkers used a similar approach in their rhodium-catalysed asymmetric hydrogenation of α-dehydroamino acid esters,46 giving quantitative conversions and 73% e.e. after 3 h. Both hydrogenation reactions show that pseudorotaxane structures can be successfully applied as catalysts in enantioselective reactions; however the exact reaction mechanisms of the hydrogenations in the presence of the rotaxane catalyst were not unraveled.Harada and coworkers recently reported on the use of cyclodextrins (CDs) as an artificial molecular clamp that can initiate polymerisation of polyesters without cocatalysts or solvents.47 CD dimers formed by pairs of α- and β-CDs linked by a terephthalamide (TPA) spacer were used in the ring-opening polymerisation of δ-valerolactone (δ-VL). Studies revealed that the β-CD moiety acted as the active site of the dimer, converting the monomer into a polymer while the α-CD part served as a clamp that held the growing polymer chain (Fig. 17). The length of the linker between α- and β-CD units determined the polymerisation activity: if the linker length was too short, monomer recognition by the CD dimer was suppressed. However, overly long linkers caused the growing chain to no longer be held by the dimer.
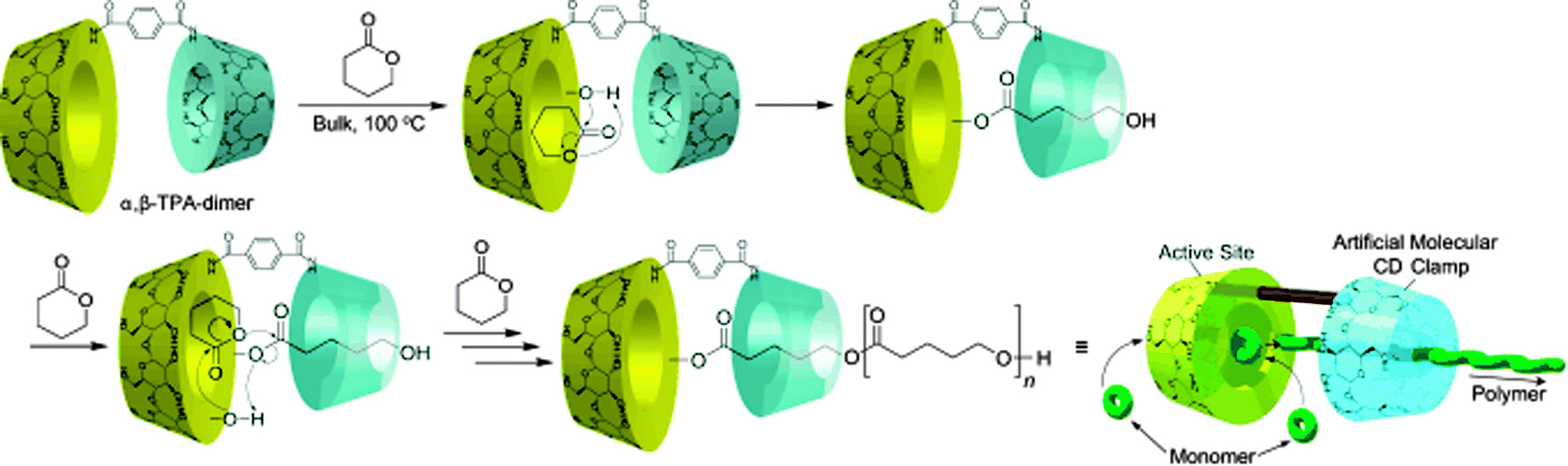 | ||
| Fig. 17 Proposed mechanism for the polymerization of δ-VL initiated by the α,β-TPA-dimer. Reprinted with permission (Wiley).47 | ||
A switchable organocatalyst based on a rotaxane structure has been developed by Leigh and coworkers,48 which functions through a similar protonation/deprotonation principle as some of the artificial muscles or valves described above. The catalytic site could be revealed by deprotonation of the rotaxane or vice versa. The application of this organocatalyst in the Michael addition of an aliphatic thiol to trans-cinnamaldehyde resulted in reasonably high yields (up to 83%). The rotaxane was used either in its active form or it was activated in situ by sliding the macrocycle. This introduced a further advantage of this catalytic rotaxane system: the rate of reaction could be effectively controlled through the addition of acid or base in situ, leading to any desired percentage of revealed, active catalyst at any time.
5.2 Processive catalysis
Processivity is a widely observed phenomenon in complex enzyme architectures in nature that facilitate the replication and degradation of biopolymers such as DNA and RNA.49 The principle of processivity ensures that the enzyme binds to the substrate and performs several catalytic rounds in a sequential manner before it dissociates from its substrate. The polymerases and λ-exonucleases that are responsible for DNA replication and degradation, respectively, embody the principles of processivity. Interestingly, these complex systems bear a rotaxane-like structure in which a long biomacromolecule is threaded through a protein macrocycle. In order to mimic these complex architectures found in nature by using synthetic rotaxanes, the challenge is the introduction of processivity to these small artificial molecular machines.Very recently Leigh and coworkers reported on the use of rotaxanes in the sequence-specific synthesis of peptides, which was a striking example in the field of processive rotaxane catalysts.50 Their rotaxane system consisted of a macrocycle, carrying a reactive arm and a thread functionalised with a predetermined sequence of building blocks, which determined the sequence of the peptide of interest (Fig. 18A). The catalytic part, which was located on the macrocycle, was a tethered thiol group. The thread contained terminal blocking groups that prevented the sliding off of the macrocycle before the peptide synthesis could be completed. It also contained three amino acid building blocks, which were attached to the thread by weak phenolic ester linkages (Fig. 18A). The synthesis of the peptide was initiated by the acid-catalysed deprotection of Boc and trityl groups (Fig. 18B) and reached completion in 36 h. This rotaxane-based artificial molecular machine is the first example of performing a reaction in a sequential processive fashion, which is a simple analogue of the synthesis of proteins on a ribosome.
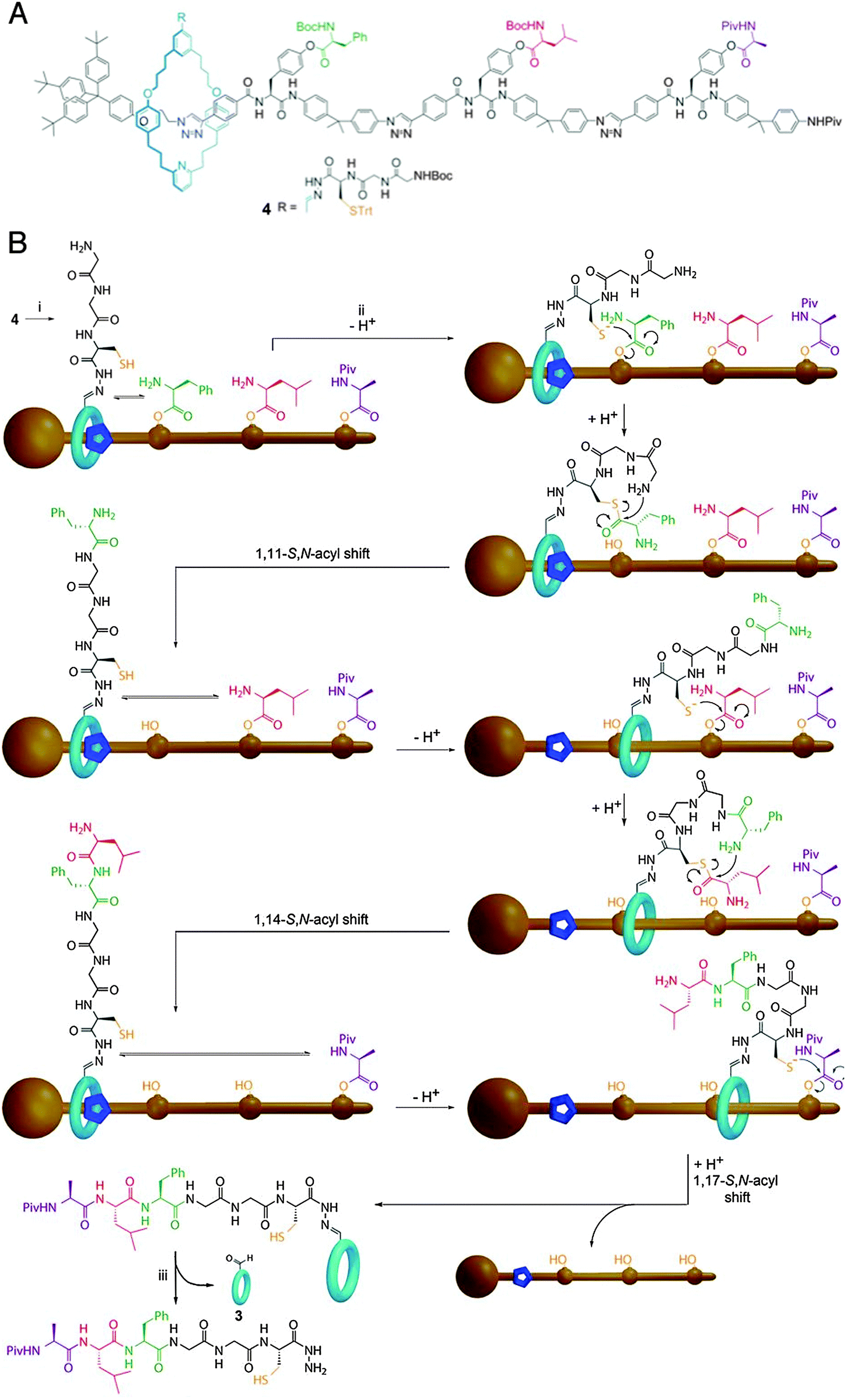 | ||
| Fig. 18 A sequentially operating rotaxane catalyst. (A) Structure of the small artificial rotaxane based molecular machine used in the sequential peptide synthesis. (B) Proposed mechanism for the peptide synthesis. See text for details. From ref. 50. Reprinted with permission from AAAS. | ||
Nolte, Rowan and coworkers have extensively studied processive catalysis in a system in which a porphyrin is covalently connected to a glycoluril moiety, resulting in a clip-like host molecule.51 Porphyrin clip molecules can be used as metal-free (H2-1) species or they can bind to metals such as manganese(III) or zinc(II) to form coordination complexes, referred to as Mn-1 or Zn-1. Furthermore, a polymer can perfectly thread through the cavity of the porphyrin clips giving rise to a (pseudo)rotaxane structure (Fig. 19A).51,52Mn-1 has been previously employed to epoxidise low molecular weight alkenes.51 By using bulky ligands to block the outer part of the catalyst, or by adding additional tails that could shield the outside,53 the epoxidation reaction could be rendered impossible on the outside, ensuring exclusive catalysis inside the cavity of the macrocycle. In the case of high molecular weight substrates, reactions within the cavity of Mn-1 showed high chemo- and stereoselectivity. For instance, the epoxidation of polybutadiene (MW ≈ 300 kDa) to form polybutadieneepoxide in the presence of 4-tert-butylpyridine as the bulky ligand and PhIO as an oxidant resulted in high trans/cis epoxide ratios (80% trans, 20% cis).51 Surprisingly, performing the epoxidation reaction under identical conditions by using a Mn-porphyrin without a cavity resulted in the opposite stereoselectivity (80% cis, 20% trans). Presumably, the sterically demanding cavity causes the differences in the stereoselectivity of the reaction. The pseudorotaxane catalyst described above serves as a simple synthetic mimic of the complex processive enzyme systems in nature, although the mechanism of the catalytic process, i.e. whether it follows a random or a sequential pathway, was not known.
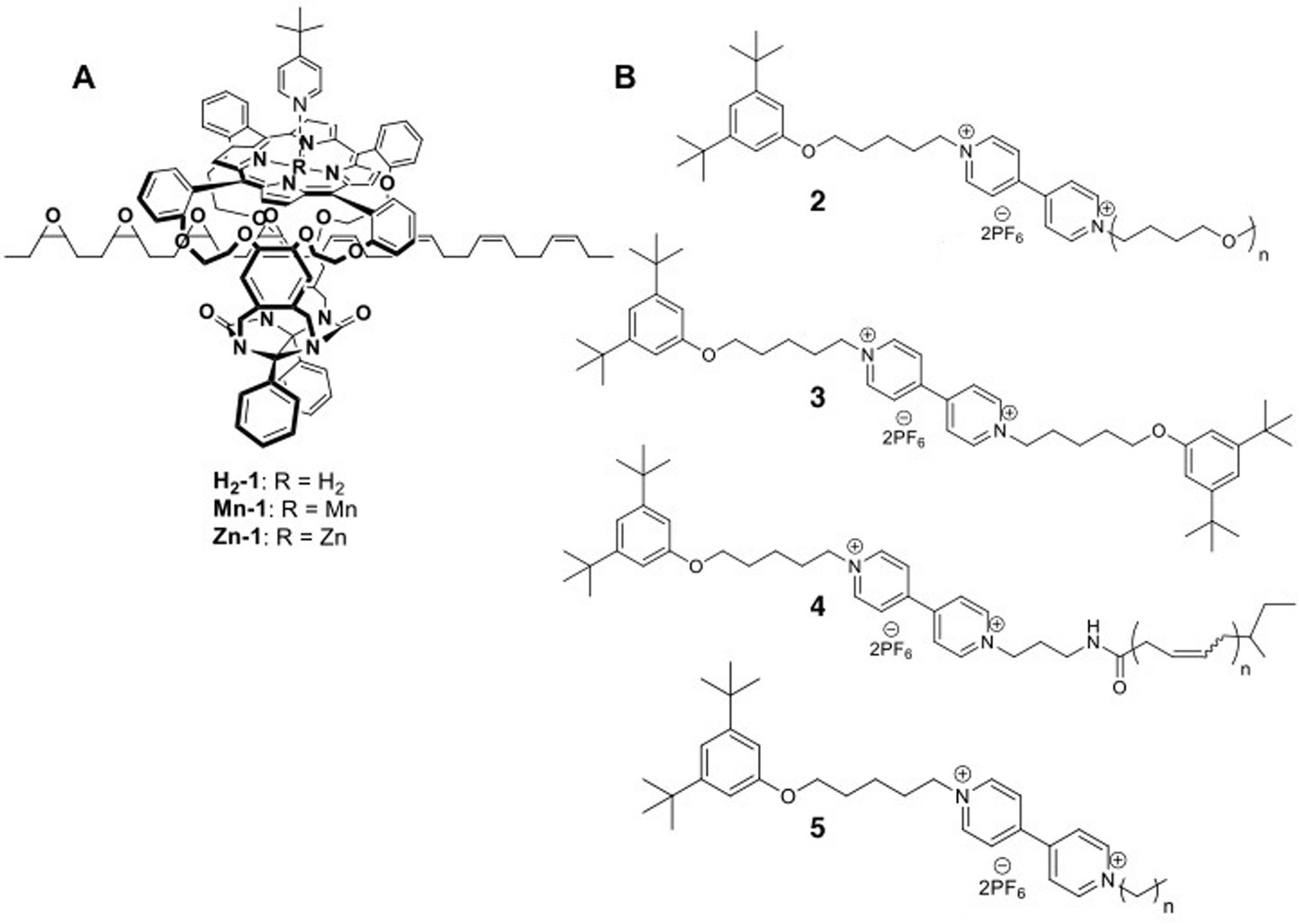 | ||
| Fig. 19 Porphyrin-capped clip-like macrocycles and their (polymeric) guests. (A) Structure of the pseudorotaxane catalyst with three compartments: manganese(III) incorporated porphyrin clip, pyridine ligand blocking the outside of the porphyrin clip and the polybutadiene which is epoxidized within the cavity. (B) General structures of polymers and diblocked viologen used in (de)threading experiments. | ||
An interesting question is now how the porphyrin catalyst finds the end of the polymer chain and threads on it. To elucidate this, threading studies were carried out with porphyrin clip H2-1 and a series of polymers. A polytetrahydrofuran chain (2) was equipped with an N,N′-dialkyl-4,4′-bipyridinium (viologen) unit, which is a ‘trap’ (a station with a high binding affinity) for the porphyrin, and with a bulky phenyl-based group that acted as a stopper (Fig. 19B).52 Fluorescence was found to be a sensitive tool to investigate the kinetics of the threading process: H2-1 is a fluorescent molecule and its fluorescence is fully quenched once it docks onto the viologen moiety.
Studies showed that the fluorescence quenching only occurs when the viologen bound to the porphyrin inside the cavity (Fig. 20). The rate constants for threading and dethreading (kon and koff) and the corresponding free energy values (ΔG≠on and ΔG≠off) were determined by monitoring the fluorescence emission intensity as a function of time. Studies revealed that the threading was a second-order process while dethreading followed first-order kinetics. It was also found that sufficient energy and the right orientation and conformation of the polymer chains are needed for the threading to fully occur. Furthermore, temperature-dependent measurements revealed that the entropic values of activation were strongly negative for all polymers and that the absolute value of the entropy of activation was found to increase with the length of the polymer chain, which is in line with an earlier proposed nucleation mechanism for threading. By measuring the rate of the threading (i.e. the rate of fluorescence quenching as a result of binding) as a function of the length of the polymers, the speed of the movement could be roughly estimated. This monitoring showed that the movement of H2-1 over the polymer chain is presumably a random sliding process, i.e. the porphyrin clip moves from one side to the other in a random fashion and sometimes may even leave the chain only to later bind again.52
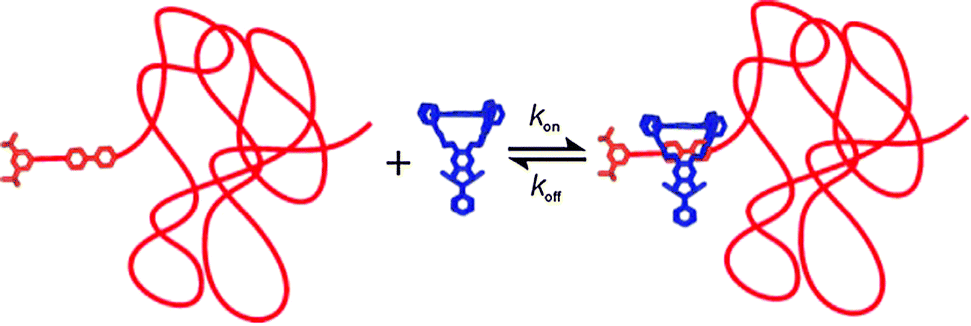 | ||
| Fig. 20 Schematic representation of the (de)threading equilibrium; the porphyrin clip reaches the viologen trap after moving along the polymer chain. kon describes the rate constant for threading, whereas koff is the rate constant for dethreading. Copyright (2006) National Academy of Sciences, USA.52 | ||
The mechanism of threading was further investigated with H2-1, Zn-1, viologen-functionalised polytetrahydrofuran (2), and polyalkanes (5) of different chain lengths (Fig. 21A).54 The trend in the decrease in kon was different for short chains (number of atoms in the chain between 5 and 8), oligomers (9–22) and polymeric chains (90–440). The rate decreased by a factor of two going from 5 to 6 atoms while it remained almost constant between 9 and 22 atoms. As for the polymeric chains (90–440), the rate decreased as a function of chain length but with a different trend than that for short chains.
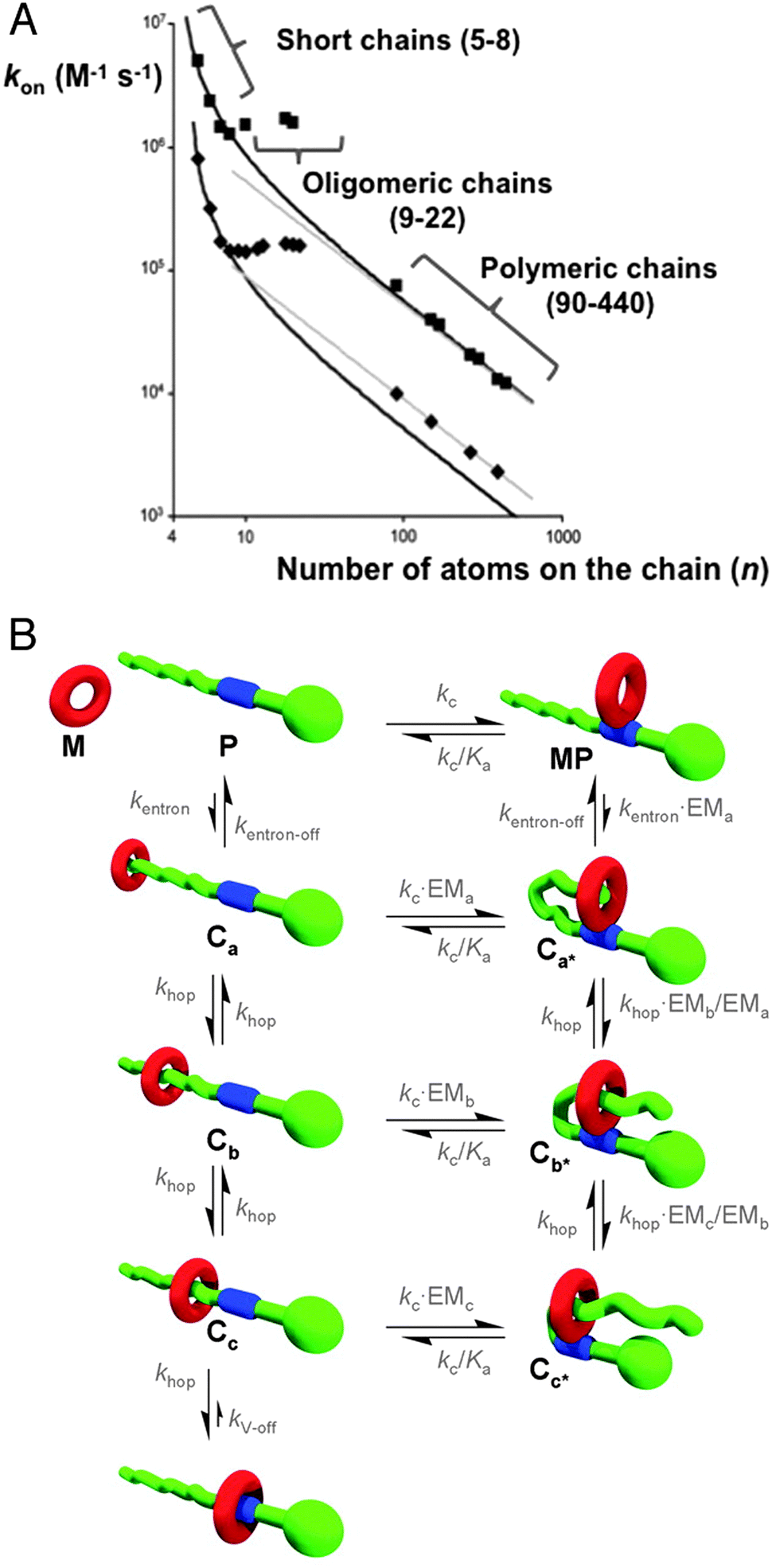 | ||
| Fig. 21 Threading mechanism. (A) Log–log plot of rate constants (square: H2-1, diamond: Zn-1) as a function of the number of atoms on the chain with the corresponding fits (gray line from fits obtained from the consecutive-hopping model; black line from fits obtained from the entron effect). (B) Proposed mechanism of threading via intra- and intermolecular pathways. From ref. 54. Reprinted with permission from AAAS. | ||
It was concluded that the threading mechanism of the polymeric chains could be described by a consecutive-hopping mechanism.54 In this model, initial binding is a second-order process and is described by a rate constant ‘kinitial’ which is independent of the chain length; thus entanglements do not play a role in the initial binding process in the present case. The macrocycle travels the whole chain, ultimately reaching the viologen trap where fluorescence is then quenched. The macrocycle can subsequently dissociate and return to sliding along the chain (kv-off). In the consecutive-hopping mechanism the speed of movement (khop) and the dynamics of translocation are not specified. Computed curves led to the grey line in Fig. 21A which depicts a good fit for the polymeric chains. The theoretical model describes the basics of the threading as well as the temperature dependency of the process; however it was not sufficient to describe the nonlinear trend in the rates of threading among the short chains and oligomers. To explain the different trend in the rates, the consecutive-hopping mechanism was combined with the so called ‘entron effect’ which suggests that a minimum number of atoms (entrons) in the chain are needed to fill the cavity of the macrocycle and that this is followed by the movement of the macrocycle through a hopping mechanism. According to this theory, 5 atoms are needed to initially fill the cavity. The predicted curve for rate constants as a function of number of atoms in the chain derived by using the new equation was found to be in excellent agreement with the experimental findings especially for H2-1 (black line in Fig. 21A).54 The second region (8–22), where the rate remains constant, was described by an intramolecular threading mechanism. This mechanism held that threading is initiated by the interactions between the viologen and the macrocycle outside of the cage, leading to accelerated rates (i.e. lowering the transition state energy and energetically forcing the process downhill toward the viologen trap) by means of transition state stabilisation. For the chains bearing 8 and more atoms (8–22), the viologen trap binds to the macrocycle from outside and the open end of the chain subsequently loops inside the cavity like a dog biting its own tail. Interestingly, in this case there is a driving force for the macrocycle to move along the polymer chain, namely the fact that the size of the folded ring, which is formed between the macrocycle and the polymer chain, becomes smaller when the threading process proceeds. The formation of the viologen–macrocycle complex from the outside facilitates the process like a ring closure reaction; when the loop becomes too small at the last 8 atoms (Cc* in Fig. 21B), the viologen dissociates from the macrocycle and the last 8 atoms enter the cavity in a normal fashion. This implies that the insertion of the last 8 atoms of the polymer is rate-limiting, which is in agreement with the experimental data showing that the observed rates for chains with number of atoms between 8 and 22 are almost the same.
This artificial system of threading a macromolecule through a hole is reminiscent of natural translocation systems in which initial interactions between a biopolymer protein and a receptor in the vicinity of the pore play an important role. Another natural system is that where clamp proteins encircle DNA, adopting a rotaxane-like configuration. Enzymes that associate with such a clamp automatically also associate with the DNA and can slide over it through the mobility of the clamp. These clamp–enzyme interactions enhance the processive behaviour of many DNA polymerases, such as that of the T4 bacteriophage.55 Clerx and van Dongen et al. have recently reported a mutated T4 clamp protein that was conjugated to an artificial catalyst: a manganese tripyridyl porphyrin that selectively oxidises the backbone of DNA after or before any AAA sequence. The resulting bio-hybrid catalyst could thus slide over DNA to nick it after specific sequences only, resembling an artificial endonuclease.55
The T4 clamp protein is trimeric and by itself it does not form a perfect circle because one of the subunit interfaces remains ‘open’ in solution. It is through this open interface that the clamp protein loads onto DNA. Once loaded, the C-terminus of the T4 DNA polymerase docks into this open interface, simultaneously closing up the ring and binding the polymerase to its template. The authors employed a peptide analogue of this C-terminus in an attempt to also complete the circle for their catalytic clamp. They found that the peptide indeed closed the open interface, and that this closed circle could no longer load onto DNA (see Fig. 22). Thus, the peptide allowed control over the processivity of the catalytic clamp: without it, the clamp could load onto DNA to perform processive catalysis, but no complex-formation was possible in its presence. It is unfortunate that the peptide apparently could not lock the clamp protein onto the DNA after complexation had already occurred. As an additional feature, a dedicated clamp-loading enzyme was used. In nature, this clamp-loader bootstraps the loading of the clamp onto specific clamp-loading sites. By pairing both proteins with a DNA template containing appropriately designed clamp-loading sites, the sliding direction of the catalytic clamp could be controlled. Thus, this report demonstrates that naturally occurring rotaxane-like processivity factors can be used to confer processivity to artificial catalysts.55
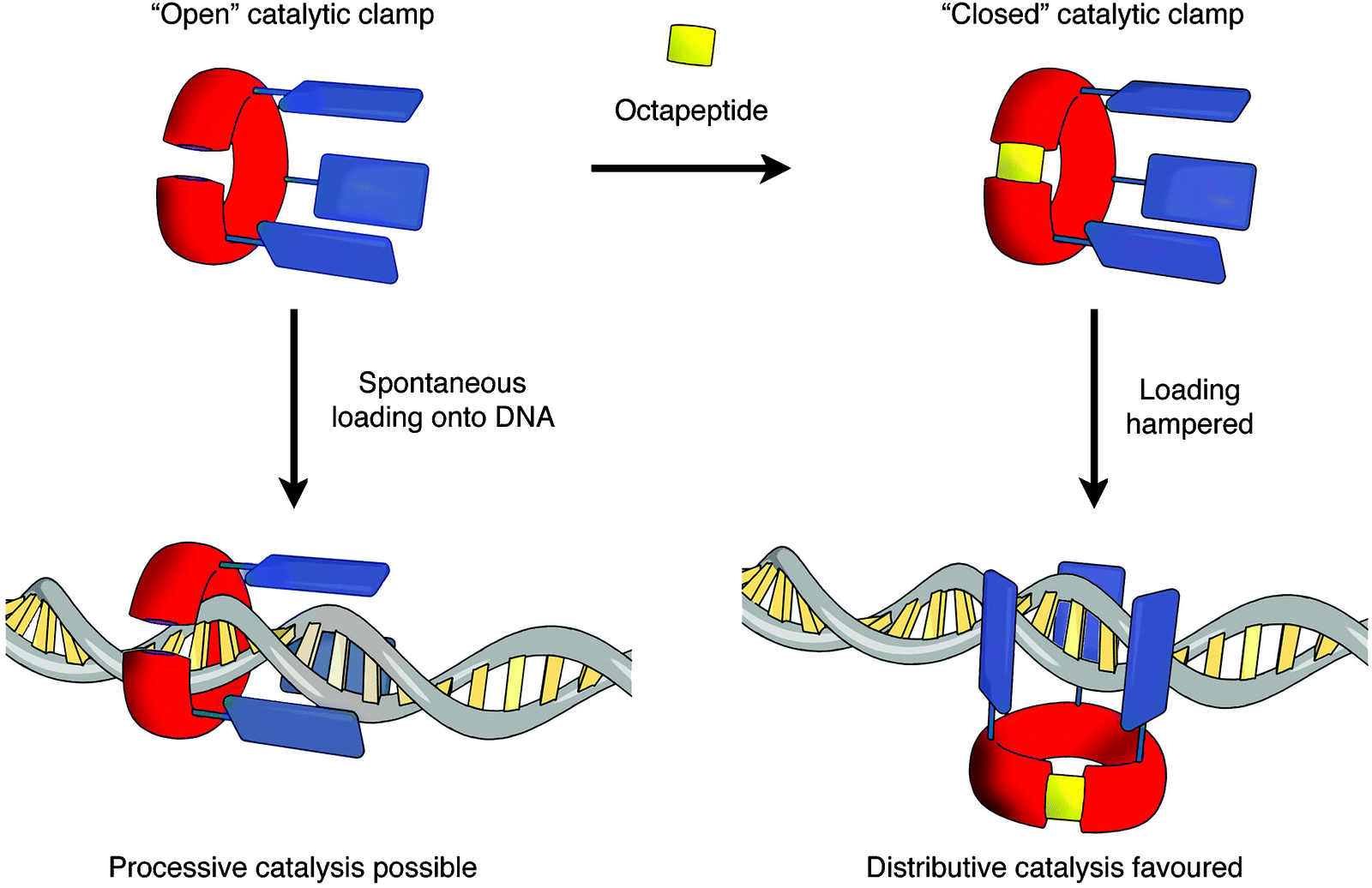 | ||
| Fig. 22 A bio-hybrid catalyst based on a clamp protein that naturally slides over DNA. Each subunit of the trimeric protein (red) carries a porphyrin catalyst (blue) that can catalyse sequence-specific DNA oxidation. The protein has an open interface through which it can load onto DNA, forming a rotaxane-like complex where the artificial enzyme can freely slide to perform processive catalysis. In the presence of a peptide that closes this open interface (yellow), loading is hindered and no rotaxane-like complex can be formed.55 | ||
The detailed understanding of the threading and association mechanisms in rotaxane systems as presented in the studies above is important for future applications of rotaxanes, e.g. for novel artificial machines and catalysts where threading is important.
6. Conclusion and outlook
Slowly but surely, mechanically interlocked architectures are starting to fulfil their promise as addressable components in all kinds of nanosystems that benefit from controlled motion between two or more defined states. On the waves of the nanotechnology revolution and the concomitant developments in new characterisation methods, such as scanning probe and fluorescence microscopy, unique insights into the working mechanisms of interlocked molecules are being obtained, even on the single molecule level. There is little doubt that this deeper understanding will lead to improvements in their molecular design and functionality. In addition, the gradual merging of the physical, chemical, and biological disciplines in the past decade has opened the way to a wealth of new opportunities to implement interlocked systems into new, hybrid materials and devices. It is here that the most exciting developments might be expected: by employing metallic or semiconductor scaffolds on which millions of interlocked molecules can make their movements cooperatively to create responsive materials on a macroscopic scale, and by taking nature as a blueprint to mimic processive enzymes, constructing a new generation of efficient catalysts.Acknowledgements
The authors have received funding from the Ministry of Education, Culture and Science (Gravity program 024.001.035.) J.A.A.W.E. thanks the Council for the Chemical Sciences of the Netherlands Organization for Scientific Research (CW-NWO) for a Vidi grant (700.58.423) and the European Research Council for an ERC Starting Grant (NANOCAT – 259064). R.J.M.N., S.F.M.v.D, and S.C acknowledge support from the European Research Council (ERC Advanced Grant, ALPROS-290886). R.J.M.N. was furthermore supported by The Royal Netherlands Academy of Science (Academy of Science Professorship).References
- V. Balzani, M. Gomez-Lopez and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405–414 CrossRef CAS.
- C. O. Dietrich-Buchecker and J. P. Sauvage, Chem. Rev., 1987, 87, 795–810 CrossRef CAS.
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC.
- M. C. Jiménez, C. Dietrich-Buchecker and J. P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284–3287 CrossRef.
- M. C. Jimenez-Molero, C. Dietrich-Buchecker and J.-P. Sauvage, Chem. Commun., 2003, 1613–1616 RSC.
- T. J. Huang, B. Brough, C.-M. Ho, Y. Liu, A. H. Flood, P. A. Bonvallet, H.-R. Tseng, J. F. Stoddart, M. Baller and S. Magonov, Appl. Phys. Lett., 2004, 85, 5391–5393 CrossRef CAS.
- Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H. R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough and M. Baller, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS PubMed.
- Z. Zhang, C. Han, G. Yu and F. Huang, Chem. Sci., 2012, 3, 3026–3031 RSC.
- C. Romuald, A. Ardá, C. Clavel, J. Jiménez-Barbero and F. Coutrot, Chem. Sci., 2012, 3, 1851–1857 RSC.
- P. Lussis, T. Svaldo-Lanero, A. Bertocco, C.-A. Fustin, D. A. Leigh and A.-S. Duwez, Nat. Nanotechnol., 2011, 6, 553–557 CrossRef CAS PubMed.
- B. A. Ashcroft, Q. Spadola, S. Qamar, P. Zhang, G. Kada, R. Bension and S. Lindsay, Small, 2008, 4, 1468–1475 CrossRef CAS PubMed.
- J. J. Li, F. Zhao and J. Li, Appl. Microbiol. Biotechnol., 2011, 90, 427–443 CrossRef CAS PubMed.
- K. Mayumi and K. Ito, Polymer, 2010, 51, 959–967 CrossRef CAS PubMed.
- A. Kulkarni, K. DeFrees, R. A. Schuldt, A. Vlahu, R. VerHeul, S.-H. Hyun, W. Deng and D. H. Thompson, Integr. Biol., 2012, 5, 115–121 RSC.
- V. N. Vukotic, K. J. Harris, K. Zhu, R. W. Schurko and S. J. Loeb, Nat. Chem., 2012, 4, 456–460 CrossRef CAS PubMed.
- J. D. Badjic, C. M. Ronconi, J. F. Stoddart, V. Balzani, S. Silvi and A. Credi, J. Am. Chem. Soc., 2006, 128, 1489–1499 CrossRef CAS PubMed.
- J. V. Hernández, E. R. Kay and D. A. Leigh, Science, 2004, 306, 1532–1537 CrossRef PubMed.
- V. Balzani, M. Clemente-León, A. Credi, B. Ferrer, M. Venturi, A. H. Flood and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1178–1183 CrossRef CAS PubMed.
- J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704–710 CrossRef PubMed.
- E. Arunkumar, N. Fu and B. D. Smith, Chem.–Eur. J., 2006, 12, 4684–4690 CrossRef CAS PubMed.
- J. J. Gassensmith, J. M. Baumes and B. D. Smith, Chem. Commun., 2009, 6329–6338 RSC.
- J. M. Baumes, J. J. Gassensmith, J. Giblin, J. J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025–1030 CrossRef CAS PubMed.
- X. Ma and H. Tian, Chem. Soc. Rev., 2010, 39, 70–80 RSC.
- A. Fernandes, A. Viterisi, F. Coutrot, S. Potok, D. A. Leigh, V. Aucagne and S. Papot, Angew. Chem., Int. Ed., 2009, 48, 6443–6447 CrossRef CAS PubMed.
- A. Fernandes, A. Viterisi, V. Aucagne, D. A. Leigh and S. Papot, Chem. Commun., 2012, 48, 2083–2085 RSC.
- T. D. Nguyen, H. R. Tseng, P. C. Celestre, A. H. Flood, Y. Liu, J. F. Stoddart and J. I. Zink, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10029–10034 CrossRef CAS PubMed.
- S. Angelos, Y.-W. Yang, K. Patel, J. Stoddart and J. Zink, Angew. Chem., Int. Ed., 2008, 47, 2222–2226 CrossRef CAS PubMed.
- C. Park, K. Oh, S. C. Lee and C. Kim, Angew. Chem., Int. Ed., 2007, 46, 1455–1457 CrossRef CAS PubMed.
- S. Angelos, Y. W. Yang, N. M. Khashab, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11344–11346 CrossRef CAS PubMed.
- J. Liu and X. Du, J. Mater. Chem., 2010, 20, 3642–3649 RSC.
- G. De Bo, J. De Winter, P. Gerbaux and C.-A. Fustin, Angew. Chem., Int. Ed., 2011, 50, 9093–9096 CrossRef CAS PubMed.
- D. T. Dang, J. Schill and L. Brunsveld, Chem. Sci., 2012, 3, 2679–2684 RSC.
- L. A. Logsdon, C. L. Schardon, V. Ramalingam, S. K. Kwee and A. R. Urbach, J. Am. Chem. Soc., 2011, 133, 17087–17092 CrossRef CAS PubMed.
- S. Y. Hsueh, C. C. Lai, Y. H. Liu, S. M. Peng and S. H. Chiu, Angew. Chem., Int. Ed., 2007, 46, 2013–2017 CrossRef CAS PubMed.
- N. H. Evans, C. J. Serpell and P. D. Beer, Chem. Commun., 2011, 47, 8775–8777 RSC.
- Y. Luo, C. P. Collier, J. O. Jeppesen, K. A. Nielsen, E. DeIonno, G. Ho, J. Perkins, H. R. Tseng, T. Yamamoto and J. F. Stoddart, ChemPhysChem, 2002, 3, 519–525 CrossRef CAS.
- P. Collier, E. W. Wong, M. Belohradsky, F. M. Raymo, J. F. Stoddart, P. J. Kuekes, R. S. Williams and J. R. Heath, Science, 1999, 285, 391–394 CrossRef.
- D. W. Steuerman, H. R. Tseng, A. J. Peters, A. H. Flood, J. O. Jeppesen, K. A. Nielsen, J. F. Stoddart and J. R. Heath, Angew. Chem., Int. Ed., 2004, 43, 6486–6491 CrossRef CAS PubMed.
- J. E. Green, J. W. Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. DeIonno, Y. Luo, B. A. Sheriff, K. Xu, Y. S. Shin, H. R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS PubMed.
- A. Coskun, J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botros and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 4827–4859 RSC.
- M. Cavallini, F. Biscarini, S. León, F. Zerbetto, G. Bottari and D. A. Leigh, Science, 2003, 299, 531 CrossRef CAS PubMed.
- V. Serreli, C. F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523–527 CrossRef CAS PubMed.
- M. Alvarez-Pérez, S. M. Goldup, D. A. Leigh and A. M. Slawin, J. Am. Chem. Soc., 2008, 130, 1836–1838 CrossRef PubMed.
- A. Carlone, S. M. Goldup, N. Lebrasseur, D. A. Leigh and A. Wilson, J. Am. Chem. Soc., 2012, 134, 8321–8323 CrossRef CAS PubMed.
- G. Hattori, T. Hori, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2007, 129, 12930–12931 CrossRef CAS PubMed.
- Y. Li, Y. Feng, Y.-M. He, F. Chen, J. Pan and Q.-H. Fan, Tetrahedron Lett., 2008, 49, 2878–2881 CrossRef CAS PubMed.
- Y. Takashima, M. Osaki, Y. Ishimaru, H. Yamaguchi and A. Harada, Angew. Chem., Int. Ed., 2011, 50, 7524–7528 CrossRef CAS PubMed.
- V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS PubMed.
- W. A. Breyer and B. W. Matthews, Protein Sci., 2001, 10, 1699–1711 CrossRef CAS PubMed.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Nature, 2003, 424, 915–918 CrossRef CAS PubMed.
- R. G. E. Coumans, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19647–19651 CrossRef CAS PubMed.
- C. Monnereau, P. H. Ramos, A. B. C. Deutman, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, J. Am. Chem. Soc., 2010, 132, 1529–1531 CrossRef CAS PubMed.
- A. B. C. Deutman, C. Monnereau, J. A. A. W. Elemans, G. Ercolani, R. J. M. Nolte and A. E. Rowan, Science, 2008, 322, 1668–1671 CrossRef CAS PubMed.
- S. F. M. van Dongen, J. Clerx, K. Nørgaard, T. G. Bloemberg, J. J. L. M. Cornelissen, M. A. Trakselis, S. W. Nelson, S. J. Benkovic, A. E. Rowan and R. J. M. Nolte, Nat. Chem., 2013 DOI:10.1038/nchem.1752.
| This journal is © The Royal Society of Chemistry 2014 |