Intramolecular C–C agostic complexes: C–C sigma interactions by another name
Michel
Etienne
*ab and
Andrew S.
Weller
*c
aCNRS, LCC (Laboratoire de Chimie de Coordination), 205, route de Narbonne, BP 44099, F-31077 Toulouse Cedex 4, France. E-mail: michel.etienne@lcc-toulouse.fr
bUniversité de Toulouse, UPS, INPT, LCC, F-31077 Toulouse Cedex 4, France
cDepartment of Chemistry, Chemistry Research Laboratories, University of Oxford, Oxford, OX1 3TA, UK. E-mail: andrew.weller@chem.ox.ac.uk
First published on 10th September 2013
Abstract
In this comprehensive review the developments in the synthesis, characterization and reactivity of complexes (s-, d- and f-block) in which a C–C single bond interacts with a metal centre are discussed: so called C–C⋯M agostic complexes. Such species are of significant interest with regard to structure and bonding, the activation of C–C single bonds and, thus, catalytic methods of C–C bond formation (or activation). Examples of C–C agostic complexes of early and later transition metals, actinides and group 1 metals are discussed, along with C–C agostic interactions in metallacyclobutanes. Examples of Si–Si⋯M, B–C⋯M and B–B⋯M agostic interactions are also presented. Throughout, the structural, spectroscopic and computational markers that indicate the likely presence of a C–C⋯M agostic interaction in a complex are highlighted.
 Michel Etienne | Michel Etienne is a Professor at the Université Paul Sabatier in Toulouse and group leader at the Laboratoire de Chimie de Coordination of CNRS. Research interests in the group concentrate on structural and mechanistic organometallic chemistry of early transition metal complexes directed at the activation of strong and inert bonds like CH or CC bonds. More recently the synthesis of highly fluorinated ligands has led to electrophilic copper and silver complexes capable of catalytically functionalizing alkanes and most prominently methane. Fruitful collaborations with groups involved in computational chemistry and catalysis make an important contribution to the research. |
 Andrew S. Weller | Andrew Weller holds a personal chair in Inorganic Chemistry at the University of Oxford and a tutorial Fellowship at Magdalen College. Research in the Weller group is based upon synthetic organometallic chemistry, and in particular the generation and stabilisation of transition metal complexes with a low coordination number or which are “operationally unsaturated”. Through this he is particularly interested in topics related to catalysis, C–H, C–C and B–H sigma complexes and subsequent activation and the self-assembly of metal fragments to form novel high-hydride content clusters. His research themes broadly encompass organometallic, inorganic chemistry and catalysis. |
1 Introduction
1.1. Sigma and agostic complexes and scope of review
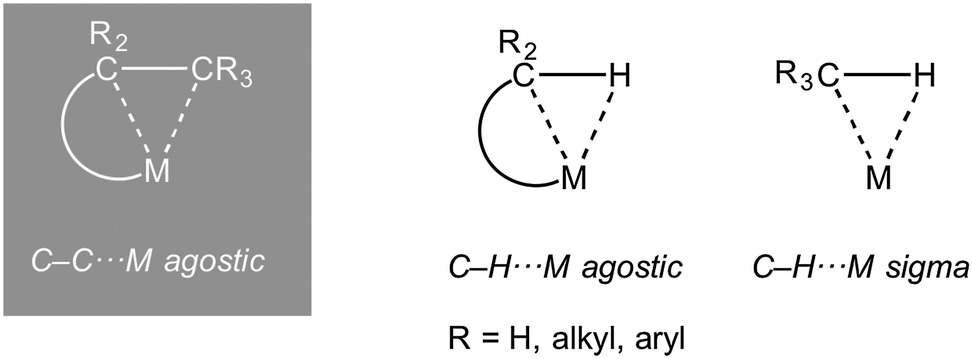
The interaction of a saturated, single, bond with a metal centre has now become an established bonding motif in organometallic chemistry and catalysis. Such complexes are characterized by the donation of an electron pair from a σ-bond from the ligand to the metal centre. They are perhaps best exemplified by the first well characterised examples to be reported, the metal dihydrogen complexes (Ln)M(η2-H2), which are also intermediates on the pathway of overall oxidative addition of H2 to a metal centre.1 Likewise, the role of C–H⋯M (agostic bonds) bonds in processes that involve overall C–H activation at a metal centre is now well established.2–5 Similarly C–C⋯M interactions have been proposed as intermediates or transition states in C–C cleavage reactions mediated by transition metals.6 Although, thermodynamically, C–C activation can be favoured over C–H activation when relative C–C, M–C, C–H and M–H bonds strengths are considered,7 kinetically C–C activation is disfavoured due to the relative inaccessibility of the directional C–C bond compared with C–H.8 For this reason, C–C activation by transition metal complexes generally involves substrates that are either intrinsically strained [e.g. unstrained C–C bonds E(C–C) ∼440 kJ mol−1, cf. biphenylenes, E(C–C) 274 kJ mol−1 or cyclopropanes, E(C–C) 255 kJ mol−1] or the C–C bond in question is held in close proximity to the metal centre.6,9–11The intramolecular 3-centre 2-electron (3c-2e) C–H⋯M interaction (Scheme 1) was originally defined as an agostic interaction by Brookhart and Green as: “…agostic will be used to refer specifically to situations in which a hydrogen atom is covalently bonded simultaneously to both a carbon atom and a transition metal atom.”2 Although this definition has subsequently been refined to include other (e.g. main group) metals,3 under rigorous use it does not include other intramolecular 3c-2e E–X⋯M interactions, such a C–C⋯M, or intermolecular interactions such as sigma-alkane (X = H, E = C),12–15 sigma borane (X = H, E = B)16,17 or sigma-silane complexes (X = H, E = Si);17,18 although these interactions can be grouped together under generic electron counting classification schemes.19 In this review that concentrates on the interaction of saturated C–C bonds with metal centres, to avoid the cumbersome, but ultimately rigorous, use of “sigma-interaction” for all C–C⋯M interactions when the community now recognises the concept of “agostic” interactions in the broadest sense to encompass all intramolecular X–E⋯M interactions, we chose to use the term “C–C agostic” when discussing intramolecular C–C⋯M, and related, interactions. Others have recently discussed the usage of the term agostic in a broader sense than originally defined.20–22
 | ||
| Scheme 1 | ||
As far as we are aware all the examples of complexes where C–C⋯M interactions have been characterised are intramolecular (i.e. the C–C bond under scrutiny might be considered to be a part of a chelating ligand). However intermolecular interactions, that is to say “true” C–C sigma-complexes, have been proposed on the basis of computational and kinetic studies in C–C activation processes. For example Zeise's dimer, [PtCl2(η2-C2H4)]2, is proposed to react with cyclopropane via a σ-C–C⋯M intermediate,23 σ-cyclopropane complexes have been suggested as intermediates in olefin cyclopropanation reactions,24 and calculations on the interaction of the C–C bond in ethane with Pt in the model complex [PtMe(PH3)2(η2-C2H6)]+ suggest a σ intermediate.25 Calculations on C–C cleavage in acetonitrile complexes of Rh suggest an intermediate with a Rh⋯C–C interaction;26 while Bergman27 and Jones28 have presented mechanistic data, derived from kinetic studies, that show that such intermediates are important in the rearrangement of rhodium cyclopropyl hydrido complexes to metallacyclobutanes.
In this review we discuss the developments in the synthesis, characterization and reactivity of complexes (s-, d- and f-block) in which a C–C single bond interacts with a metal centre. Such species are of significant interest with regard to structure and bonding, the activation of C–C single bonds and, thus, catalytic methods of C–C bond formation (or activation).6,9,11 We do not cover C–C activation processes in detail, unless they are directly connected to an observable C–C agostic intermediate.
2 Main group, early transition metals and actinides
2.1. Structure and bonding
Two early literature examples serve to highlight the main characteristics of this class of complex. In 1996,29 Schleyer et al. reported the solid-state structure of a hexameric alkoxy lithium compound [LiOC(Me)(c-C3H5)2]61 in which one of the α-C–C bond of a cyclopropyl group was coordinated to lithium. Short Li⋯C contacts [2.615(3), 2.644(3) Å] associated with an elongated C–C bond [1.519(3) Å, ca. 0.02 Å longer with respect to uncoordinated C–C bonds] characterized what was named “a “lithium-bonded” cyclopropyl edge” (Fig. 1a). The nature of the interaction between the Li cation and cyclopropane was addressed using DFT computational techniques. These indicated that the most stable structure was of C2v symmetry with Li+ coordinated to an elongated C–C bond (Fig. 1b). A Natural Population Analysis (NPA) indicated the nature of the interaction was essentially electrostatic.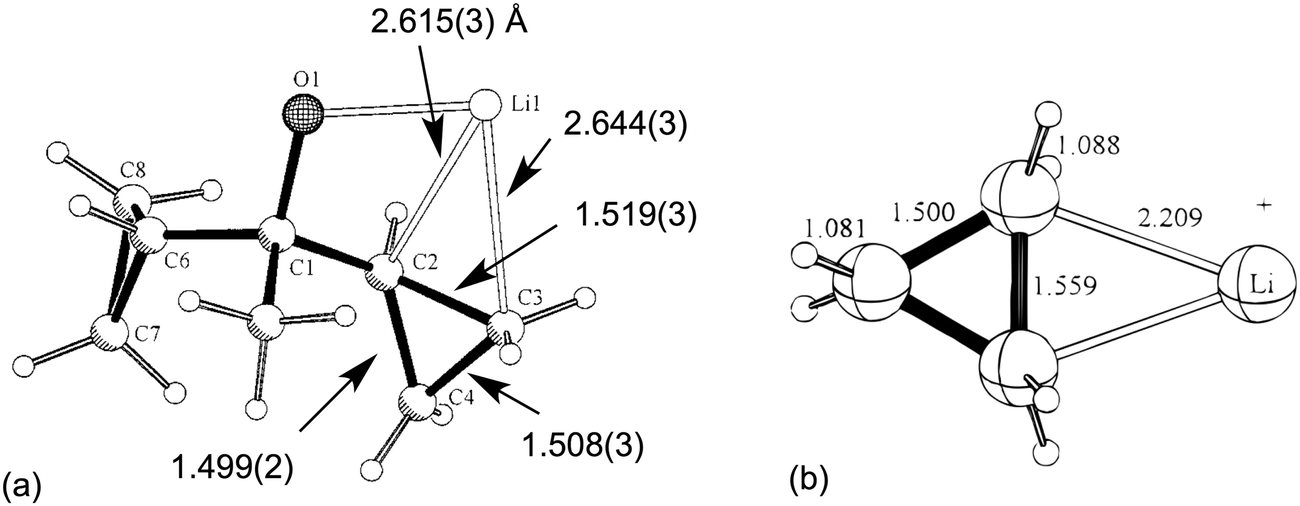 | ||
| Fig. 1 (a) Asymmetric unit in the X-ray crystal structure of [LiOC(Me)(c-C3H5)2]61 highlighting the Li⋯C–C close contacts. (b) The DFT optimized model Li+(c-C3H6). Key structural parameters are included. Adapted with permission from ref. 29. Copyright (1996) American Chemical Society. | ||
In 1998 Gleiter, Ernst et al. described a formally 14-electron titanium(II) complex resulting from the coupling of the cyclooctadienyl ligand in CpTi(C8H11)(PEt3) with three PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CSiMe3 units, CpTi{C8H11(PhCCSiMe3)3} 2.30 The solid-state structure of 2 as determined by X-ray crystallography revealed a somewhat congested and constrained hydrocarbyl ligand and coordination sphere around the titanium atom (Fig. 2). As a consequence of this at least one saturated carbon atom was found to be in close proximity to the titanium center [Ti–C 2.293(7) Å], and one of the associated C–C bonds was also significantly elongated [C–C 1.596(8) Å] suggesting an agostic C–C interaction. By use of 13C-labelled alkyne it was possible to determine the magnitude of 1JCC for several C–C bonds in 2, and a low 1JCC of 17.9 Hz was measured for the C–C bond that is proposed to engage in the agostic C–C⋯Ti interaction. Other JCC coupling constants in the hydrocarbyl ligand were also lower than expected. As a general guideline, 1JCC in unstrained alkanes is ca. 35 Hz (e.g. 34.5 Hz for ethane) and decreases in cycloalkanes as ring strain increases, e.g. 28.4 and 29.1 Hz in methylcyclobutane; 12.4 Hz in cyclopropane.31 Due to the topology of the hydrocarbyl ligand, no C–H bonds in 2 are oriented towards titanium, meaning that any C–H⋯Ti agostic interactions are likely to be relatively weak, at best. The nature of the interaction between the hydrocarbyl ligand and titanium was further probed by an NPA/NBO (natural bond orbital) analysis, which revealed a significant interaction between the long C–C bond (and its associated C–H bond) orbital and an empty orbital on titanium. Other saturated C–C bonds within the complex interacted more weakly with the titanium centre. This bonding analysis has been questioned on the basis of an in-depth QTAIM analysis of the topology of the electron density.32 No bond paths were identified between the saturated carbons close to titanium and the titanium itself using this methodology. No charge concentration directed to Ti from these close contact carbons was noted, and the electron density, Laplacian of the electron density and energy density for the hydrocarbyl ligand could be interpreted without hint of an interaction with Ti. Related titanium and zirconium complexes, which also result from oligomerization of alkynes such as PhCCSiMe3, show similar metric parameters to 2,33–35 although no additional support for the presence of C–C⋯M interactions was presented from either solution NMR spectroscopy or computational investigations. Another well characterized titanium complex related to 2, CpTi{C8H11(PhCCSiMe3)2}(PMe3) 3 (Fig. 2), also exhibited low JCC values for some C–C bonds, most prominently those adjacent to a coordinated C
CSiMe3 units, CpTi{C8H11(PhCCSiMe3)3} 2.30 The solid-state structure of 2 as determined by X-ray crystallography revealed a somewhat congested and constrained hydrocarbyl ligand and coordination sphere around the titanium atom (Fig. 2). As a consequence of this at least one saturated carbon atom was found to be in close proximity to the titanium center [Ti–C 2.293(7) Å], and one of the associated C–C bonds was also significantly elongated [C–C 1.596(8) Å] suggesting an agostic C–C interaction. By use of 13C-labelled alkyne it was possible to determine the magnitude of 1JCC for several C–C bonds in 2, and a low 1JCC of 17.9 Hz was measured for the C–C bond that is proposed to engage in the agostic C–C⋯Ti interaction. Other JCC coupling constants in the hydrocarbyl ligand were also lower than expected. As a general guideline, 1JCC in unstrained alkanes is ca. 35 Hz (e.g. 34.5 Hz for ethane) and decreases in cycloalkanes as ring strain increases, e.g. 28.4 and 29.1 Hz in methylcyclobutane; 12.4 Hz in cyclopropane.31 Due to the topology of the hydrocarbyl ligand, no C–H bonds in 2 are oriented towards titanium, meaning that any C–H⋯Ti agostic interactions are likely to be relatively weak, at best. The nature of the interaction between the hydrocarbyl ligand and titanium was further probed by an NPA/NBO (natural bond orbital) analysis, which revealed a significant interaction between the long C–C bond (and its associated C–H bond) orbital and an empty orbital on titanium. Other saturated C–C bonds within the complex interacted more weakly with the titanium centre. This bonding analysis has been questioned on the basis of an in-depth QTAIM analysis of the topology of the electron density.32 No bond paths were identified between the saturated carbons close to titanium and the titanium itself using this methodology. No charge concentration directed to Ti from these close contact carbons was noted, and the electron density, Laplacian of the electron density and energy density for the hydrocarbyl ligand could be interpreted without hint of an interaction with Ti. Related titanium and zirconium complexes, which also result from oligomerization of alkynes such as PhCCSiMe3, show similar metric parameters to 2,33–35 although no additional support for the presence of C–C⋯M interactions was presented from either solution NMR spectroscopy or computational investigations. Another well characterized titanium complex related to 2, CpTi{C8H11(PhCCSiMe3)2}(PMe3) 3 (Fig. 2), also exhibited low JCC values for some C–C bonds, most prominently those adjacent to a coordinated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (an allylic like fragment).36 In addition to the allylic unit, an elongated C–C bond is connected to the tertiary carbon attached to the α-carbon that is σ-bonded to titanium. This bridge-head β-carbon is in proximity with the titanium [Ti–C 2.539(3) Å] and the authors comment that these structural data point to a developing metallacyclobutane structure, much like those involved as intermediates in alkene metathesis. Such species will be described in more detail in Section 4.
C bond (an allylic like fragment).36 In addition to the allylic unit, an elongated C–C bond is connected to the tertiary carbon attached to the α-carbon that is σ-bonded to titanium. This bridge-head β-carbon is in proximity with the titanium [Ti–C 2.539(3) Å] and the authors comment that these structural data point to a developing metallacyclobutane structure, much like those involved as intermediates in alkene metathesis. Such species will be described in more detail in Section 4.
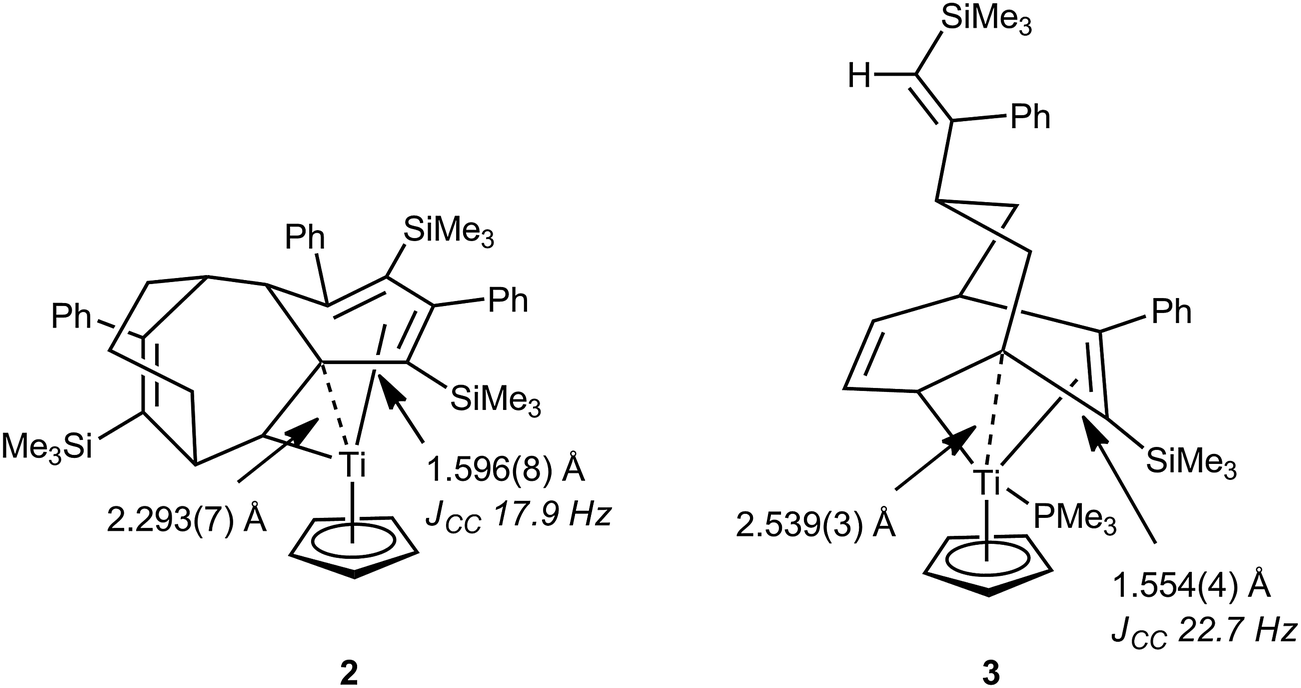 | ||
| Fig. 2 Diagrammed X-ray molecular structures of C–C agostic complexes 2 and 3 resulting from alkyne oligomerization at titanium centres, showing some key structural parameters. | ||
In both Li and Ti cases, whatever the intimate nature of the interactions between the metal and the hydrocarbon fragment, it is striking that these electrophilic metals appear to interact with C–C rather than C–H bonds. In the titanium cases there are very few C–H bonds present in the vicinity of the metal and, due to the topology of the ligands, they are not spatially well located to contribute to the bonding. Consequently the saturated C–C bonds come into close proximity to a titanium centre that has little choice but to interact with them. In the lithium case 1, it is a heavily strained and hence relatively destabilized C–C bond of a cyclopropyl group that is the preferred partner despite the presence of several C–H bonds. These two examples demonstrate that C–C⋯M agostic structures are likely to be observed in situations of steric confinement or electronic activation.6 This is the case for virtually all C–C agostic structures known to date, as we will show. Additionally, and depending on the need of the unsaturated metal centre, the surrounding C–H bonds may also contribute to the interaction with the metal, for example in a η3-C–C–H type interaction with the metal centre.26,37
The crucial impact of a distortion or constraint to induce a C–C rather than a C–H agostic structure is exemplified by a series of cyclopropyl complexes of niobium. Complexes of the type TpMe2NbClR(η2-alkyne), 4 (Fig. 3), exhibit a wealth of agostic interactions that could be defined in both the solid state and in solution, and additionally by computational studies.38 When R is a linear primary n-alkyl group α-C–H⋯M agostic structures are observed exclusively, whereas secondary straight-chain sec-alkyl sec-CnH2n+1 groups lead to equilibrium mixtures of alkyl rotamers exhibiting α- or β-C–H agostic structures, the β-C–H agostic structures being lower in energy.39 When a cyclic secondary c-alkyl group is considered, the preferences for either α- or β-C–H agostic structures are altered but an equilibrium that depends on the ring size between the moderately strained C4 cyclobutyl to the unstrained C6 cyclohexyl is still observed.40,41 In this system the agostic site is uniquely defined as being in the immediate vicinity of the Cl–Nb–Cα plane, and one alkyl substituent is located in the wedge between two pyrazole rings for steric reasons. However for a C3 ring, i.e. for a cyclopropyl group in TpMe2NbX(c-C3H5)(η2-MeCCMe) (X = Cl, 4a; Ph, 4b), the agostic site is not occupied by a C–H bond but rather by a Cα–Cβ bond.40,42,43 With X = Cl, this Cα–Cβ bond is a significantly longer than the other one [Cα–Cβ 1.539(4), 1.490(4) Å], and the cyclopropyl group is distorted towards the niobium [Nb–Cα–Cβ 109.7(2)°, 131.4(2)°]. With X = Ph, an X-ray diffraction experiment was unable to differentiate between the two CαCβ bonds, while for X = Me, 4c, the solid-state structure was disordered.43 Definitive assignment of C–C⋯M agostic structures comes from JCC measurements (natural abundance INADEQUATE NMR sequence) and DFT structural optimizations in concert with the computation of JCC.44Fig. 3 shows the example of TpMe2NbCl(c-C3H5)(η2-MeCCMe), 4a, and summarizes the experimental and computational results that probe the C–C⋯Nb interaction for this complex. It was found that the measured JCC for one of the C–C bonds of the cyclopropyl group was reduced to such an extent that it was less than the inherent line-width of the INADEQUATE experiment (ca. 3 Hz), and this was confirmed by computation to be the C–C bond involved in the C–C⋯Nb agostic bonding. Taking into account the difficulties inherent in the measurement of JCC it is gratifying that computation yields not only reliable trends in the variation of JCC but also interpretable absolute values.
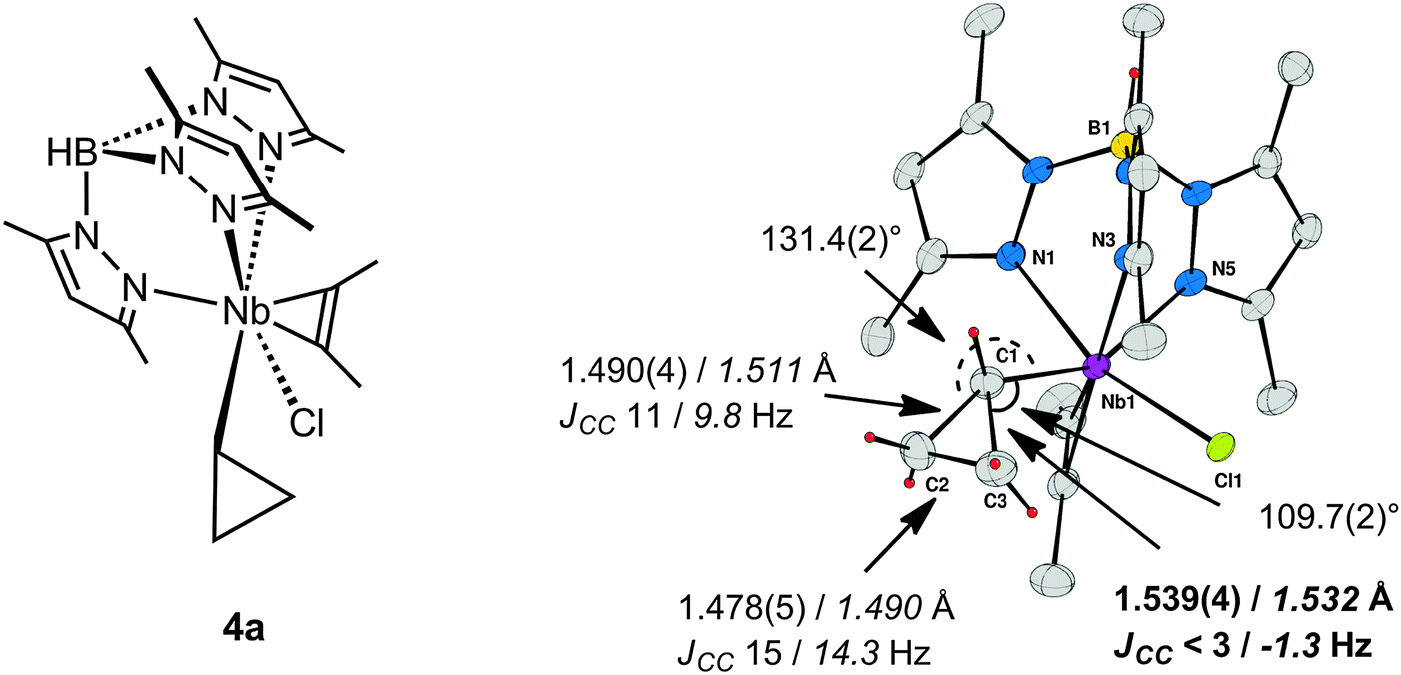 | ||
| Fig. 3 TpMe2NbCl(c-C3H5)(MeCCMe) 4a and its X-ray molecular structure with selected structural parameters, JCC (solution INADEQUATE), and DFT computed parameters in italics. | ||
The combination of two types of C–C⋯M agostic structures is observed in the heterobimetallic lithium yttrium complex Cp*2Y(μ-c-C3H5)2Li(thf) 5.45 In solution, the complex undergoes a fluxional process, and thus shows time-averaged C2v symmetry. However the complex is clearly non-symmetric in the solid-state (Fig. 4a), with one cyclopropyl group being tilted toward the yttrium (group B) and the other towards the lithium (group A). The X-ray crystal structure is accurately reproduced by DFT optimization, and both experiment and computation indicate an elongation of the C–C bonds oriented towards Li or Y (Fig. 4b).
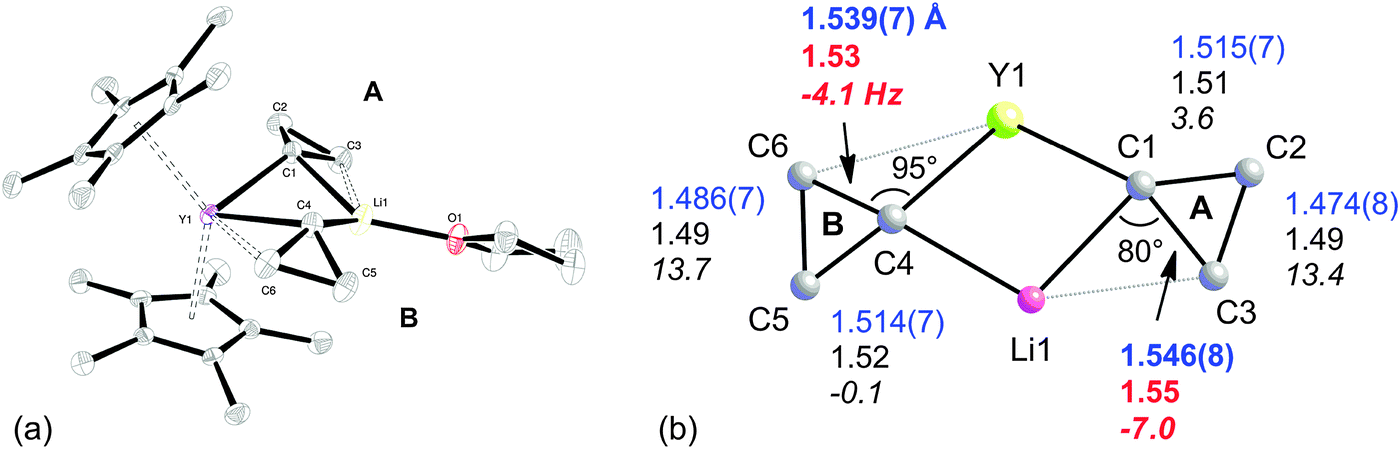 | ||
| Fig. 4 (a) X-ray molecular structure of Cp*2Y(μ-c-C3H5)2Li(thf), 5. (b) Experimental (blue), computed (black/red) structural parameters and JCC (italics) of the core of Cp*2Y(μ-c-C3H5)2Li(thf), 5. Adapted from ref. 45. | ||
This fluxional behaviour of 5 prevents the informative measurement of JCC for the cyclopropyl groups. However, for those C–C bonds that are oriented towards the metals, the computed coupling constants indicate a considerable lowering of the JCC for these bonds on benchmarking with cyclopropane. For cyclopropyl group A (Fig. 4b), the situation is reminiscent of that found by Schleyer et al. for the lithium compound [LiOC(Me)(c-C3H5)2]6, 1.29 Indeed revisiting the computed model complex Li+(c-C3H6) confirmed that the C–C bond interacting with Li+ had a low JCC (−0.27 Hz), similar to both 4a and 5.
An NPA/NBO analysis further reveals that the geometrical distortions of the cyclopropyl groups in complex 5 have different origins (Fig. 5). It is found that the C–C agostic distortion of cyclopropyl group A towards lithium has a marked electrostatic contribution, which is perhaps not surprising given the closed shell nature of Li+: Li+ bears a 0.911 au charge whereas C3, the closest β-carbon, bears a higher negative charge than distal C2 [−0.537 vs. −0.463 au, respectively]. This is also reminiscent of the interaction present in 1, as discussed above. For the cyclopropyl group B, the two β-carbons C5 and C6 bear the same negative charge and there is a second-order perturbative stabilization between the σ-CαCβ and a d orbital on yttrium, suggesting a significant covalent character for this interaction. The NBO analysis reveals that the α-C–C interaction is partnered by a weaker β-C–H interaction giving an η3-C–C–H agostic structure. The bonding scheme in 5, together with the observation of α-C–H, β-C–H and α-C–C agostic rotamers in the Nb systems discussed above, suggests there is a continuum of situations ranging from purely C–H or C–C agostic structures depending on the requirements of these electrophilic metals i.e. their nature and coordination sphere. Indeed, early computational work on the model cation [TiCl2(C(CH2CH3)CH2)]+ revealed such interactions, in as much as there is a β-C–C⋯Ti agostic interaction accompanied by two γ-C–H agostic interactions.46
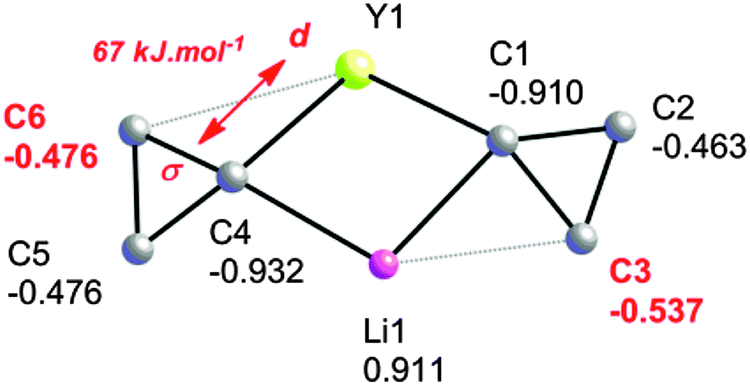 | ||
| Fig. 5 NPA/NBO analysis of the bonding in 5. | ||
The importance of higher energy, strained, C–C bonds in promoting C–C agostic interactions is exemplified in the deltate uranium complex formed from CO reduction by Cp*(COT#)U(THF) [COT# = 1,4-bis(triisopropylsilyl)cyclooctatetraenediyl] to give [Cp*(COT#)U]2(μ-κ1,κ2-c-C3O3) 6.47 The molecular structure as determined by X-ray crystallography (Fig. 6) reveals an elongation of a C–C bond [1.436(7) Å] that is in close contact with a formally U(IV) centre [U–C = 2.654(4), 2.670(4) Å]. Evidence for a 3c-2e C–C⋯U agostic interaction comes from a DFT computational study that both reproduced the distortion of the {C3O3}2− fragment and provided a fragment analysis for the bonding. This latter analysis shows in-plane bonding interaction of the C–C lobe of the HOMO − 2 of the organic fragment with an empty f orbital on the uranium. The HOMO − 2 is also C–O antibonding and U–O bonding (Fig. 7).
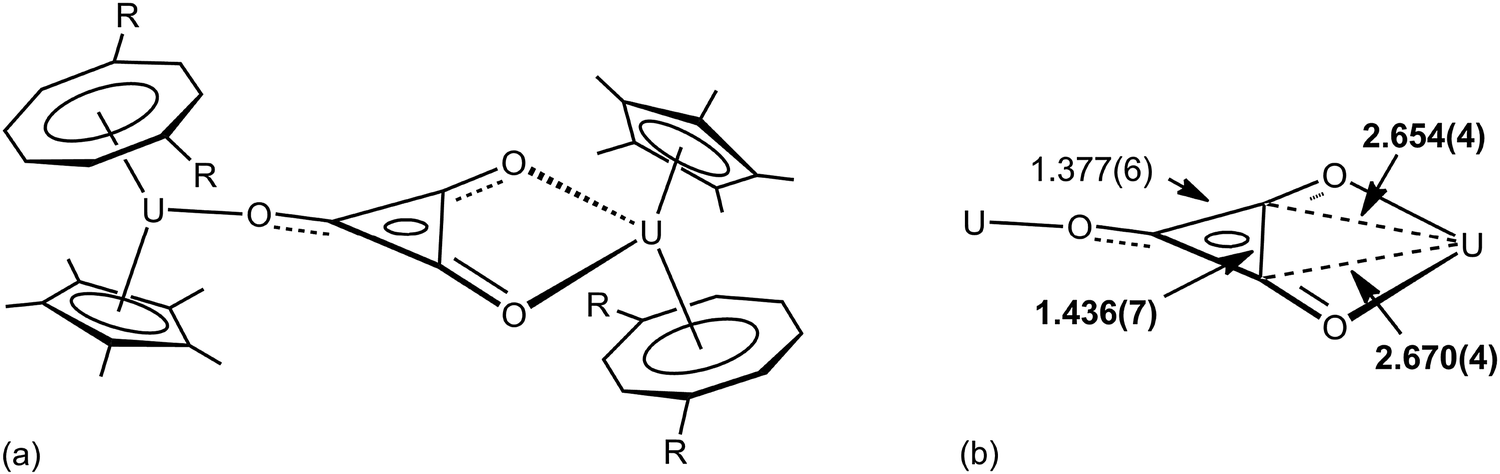 | ||
| Fig. 6 (a) Structure of [Cp*(COT#)U]2(μ-κ1,κ2-c-C3O3) 6, R = Si-i-Pr3 (b) selected structural metrics (Å). | ||
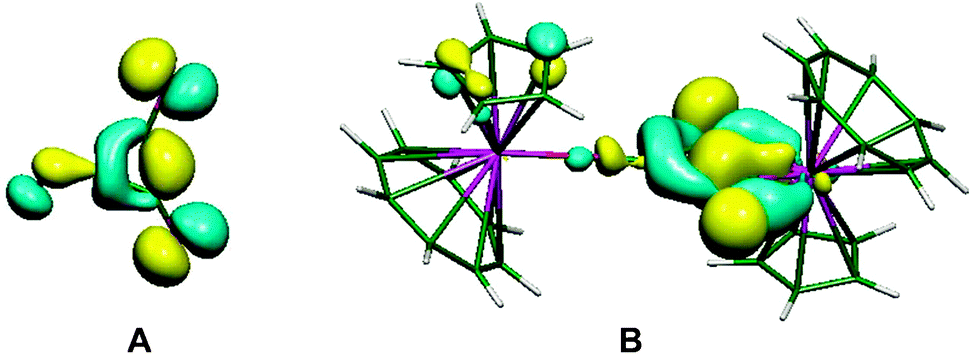 | ||
| Fig. 7 (A) Fragment orbital of the C3O3 core of 6, (B) molecular orbital describing the C–C agostic interaction. From ref. 47. Reprinted with permission from AAAS. | ||
Before closing this part, there is at least one example where “non-classical” bonding could be described C–C agostic.48 The alkyne complexes (C5Me5−nHn)2Ti(η2-Me3SiCCSiMe3) (n = 3, 4, 5) react with R2Si(CCSiMe3)2 (R = Me, Ph) to give 1-titana-4-sila-3,5-bis(trimethylsilyl)cyclohexa-2,5-dienes 7 (Fig. 8). The short Ti–Cα bonds [1.981(4)–1.998(3) Å depending on the substituents] and short Cα–Cα contacts [1.821(4)–1.933(2) Å] suggest an unusual bonding situation. According to a molecular orbital description, the HOMO is a 3c-2e bond delocalized on Ti and both Cα that the authors coin a “Δ bond”. This is somewhat reminiscent of complex 6.
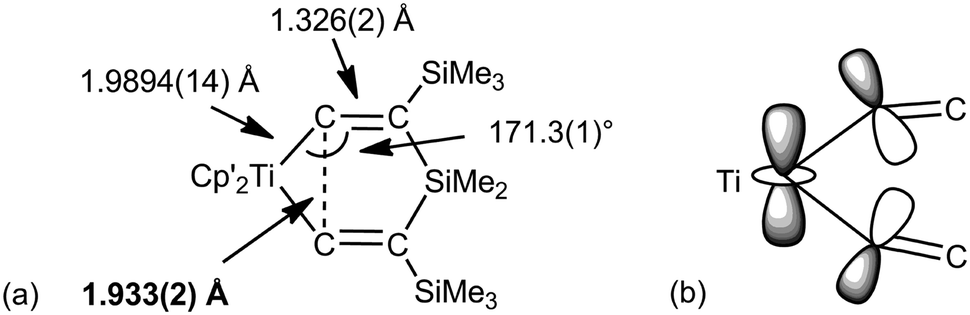 | ||
| Fig. 8 (a) Structure of (C5Me4H)2Ti{CCSiMe3}2SiMe2 (Cp′ = C5Me4H) 7. (b) Pictorial view of the HOMO highlighting the 3c-2e interaction. | ||
2.2. Reactivity of C–C agostic complexes of the main group, early transition metals and actinides
Whereas the occurrence of C–C agostic interactions have been postulated and investigated computationally during several types of mechanistic discussions, including those on C–C bond cleavage, exceedingly few reports on the reactivity of the complexes described in the previous section have appeared. One might anticipate that the C–C agostic structure would describe an arrested C–C activation along a reaction coordinate leading to C–C bond cleavage, i.e. an activated C–C bond. Although this picture is likely true for late transition metal complexes and metallacyclobutanes (see later), the inaccessibility of higher oxidation states for earlier transition, main group and f-block metals precludes reactivity via direct C–C bond cleavage (an overall oxidative addition). On the contrary all the structural and theoretical data currently available for these, formally, d0 species suggest that the C–C agostic structures represent the final, thermodynamic, product. Indeed only the TpMe2NbX(c-C3H5)(η2-MeCCMe) complexes have been shown to undergo further reactivity, but activate C–H bonds rather than C–C.TpMe2NbMe(c-C3H5)(η2-MeCCMe), 4c (Fig. 9), reacts with benzene to yield first methane and the cyclopropyl phenyl complex TpMe2NbPh(c-C3H5)(η2-MeCCMe) 4b, which subsequently, and more slowly, loses cyclopropane to form the diphenyl complex TpMe2NbPh2(η2-MeCCMe) 8.43 An experimental and computational mechanistic study indicated that both reactions occur by rate determining intramolecular β-H abstractions yielding highly reactive unsaturated η2-cyclopropene TpMe2Nb(η2-c-C3H4)(η2-MeCCMe) A and η2-benzyne TpMe2Nb(η2-c-C6H4)(η2-MeCCMe) B intermediates for the first and second reactions, respectively (Fig. 9).49 These unsaturated intermediates then activate the C–H bonds of benzene via a 1,3-addition to give the products. Whereas the reaction of unsaturated benzyne derivatives is known, that with η2-cyclopropene or η2-alkene is much less common as early transition metal complexes tend to activate hydrocarbon C–H bonds via α-H abstraction/1,2-addition pathways. It has not been possible to demonstrate the link between the existence of the C–C agostic interaction and the reactivity of the cyclopropyl complexes. Instead, the strength of the Nb(η2-cyclopropene) interaction was suggested to be the key to the observed reactivity.49
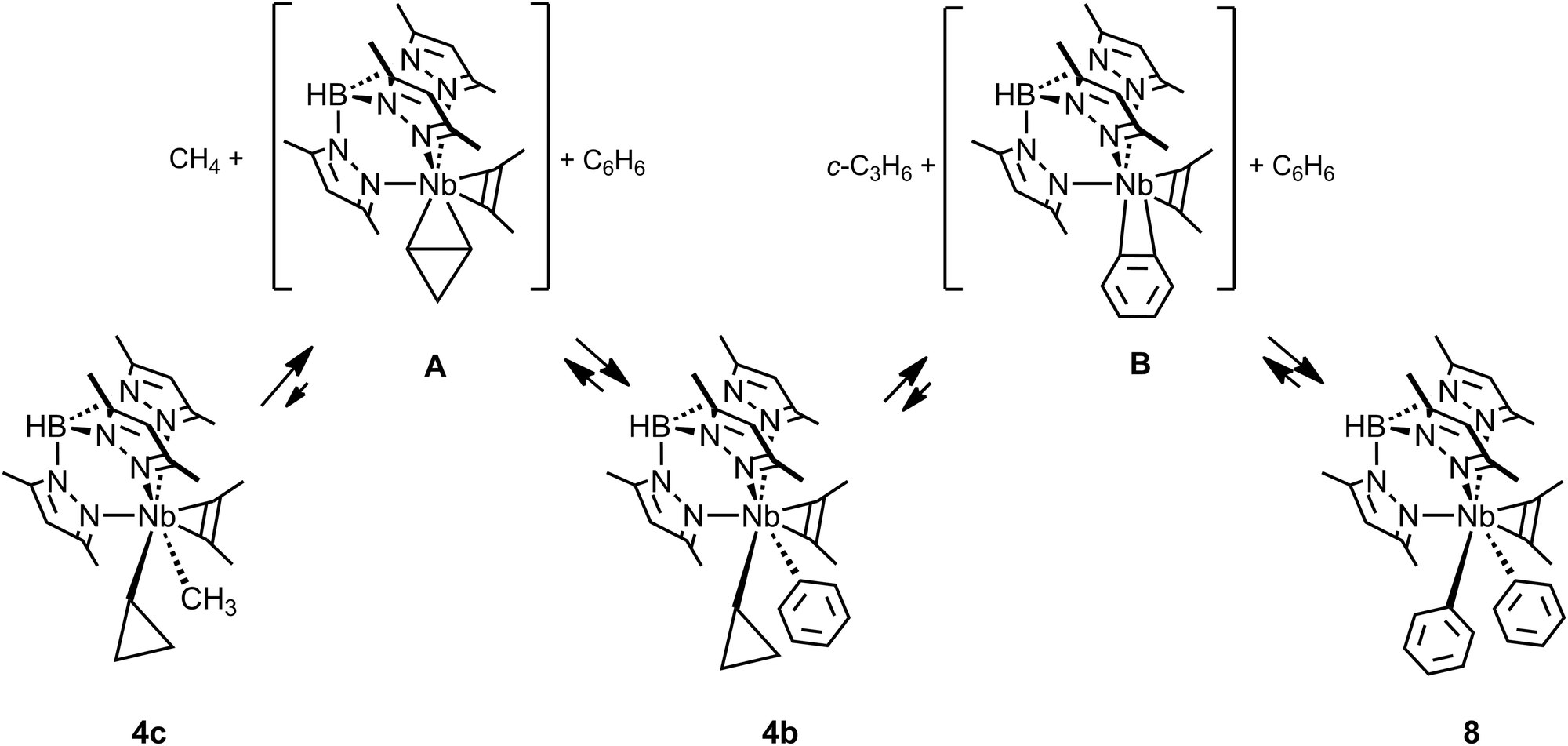 | ||
| Fig. 9 Benzene C–H bond activation by TpMe2NbMe(c-C3H5)(η2-MeCCMe), 4c. | ||
The competition between benzylic and aromatic C–H bond activation in several methylbenzenes (mesitylene, xylenes) by the unsaturated η2-cyclopropene TpMe2Nb(η2-c-C3H4)(MeCCMe) intermediate A have been studied.50 Aromatic C–H bond activation is preferred provided there is no ortho methyl group otherwise benzylic C–H bond activation occurs. Accordingly a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of aryl TpMe2Nb(c-C3H5)(3,5-C6H3Me2)(MeCCMe) and benzylic TpMe2Nb(CH2-3-C6H4Me)(c-C3H5)(MeCCMe) complexes is observed upon reaction of TpMe2NbMe(c-C3H5)(MeCCMe) with 1,3-C6H4Me2. Evidence for steric effects in this selectivity came from computational studies.
:1 mixture of aryl TpMe2Nb(c-C3H5)(3,5-C6H3Me2)(MeCCMe) and benzylic TpMe2Nb(CH2-3-C6H4Me)(c-C3H5)(MeCCMe) complexes is observed upon reaction of TpMe2NbMe(c-C3H5)(MeCCMe) with 1,3-C6H4Me2. Evidence for steric effects in this selectivity came from computational studies.
3 Late-transition metals
3.1. C–C sigma interactions in Binor-S complexes
For the later transition metals perhaps the best represented contiguous series of complexes that contain C–C agostic interactions are those of group 9 in which a saturated organic fragment (Binor-S) and a Rh(III) centre are combined (Binor-S = 1,2,4,5,6,8-dimetheno-s-indacene): [Rh(Binor-S)(PR3)][BArF4], 9a–eFig. 10 (ArF = 3,5-(CF3)2C6H3). In these complexes the close approach of a strained C–C bond in a cyclopropyl fragment on Binor-S with a {Rh(PR3)}+ fragment is forced by geometric constraint. A number of complexes of Rh have been prepared using a variety of phosphines, although these phosphines all occupy a rather narrow cone-angle range, for example PiBu3 (cone angle 143°) and PtBu3 (182°) do not support the formation of such complexes, whereas PiPr3 (160°) and PCy3 (170°) do.51–53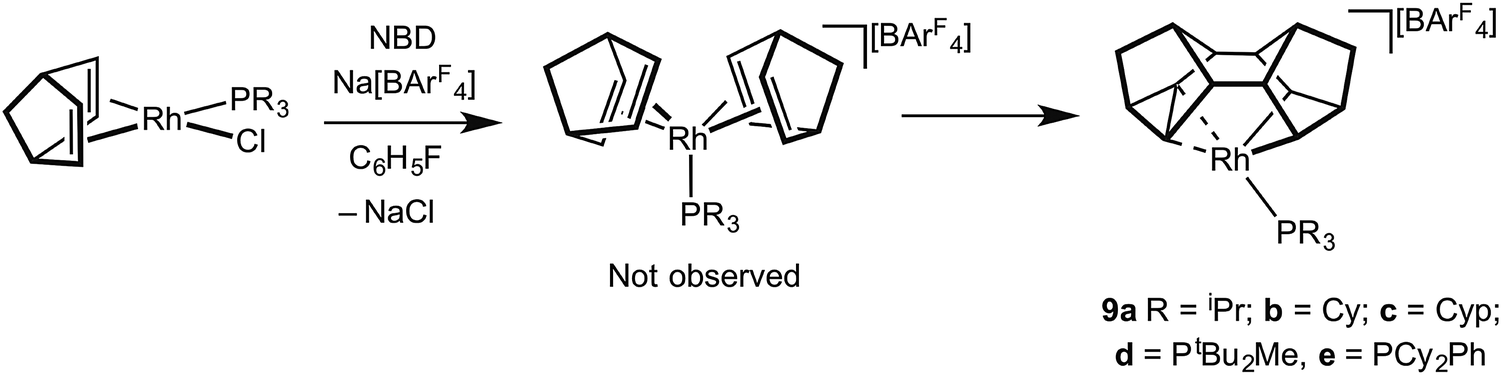 | ||
| Fig. 10 Synthesis of Rh-complexes containing the Binor-S fragment. | ||
Fig. 11 shows the solid-state structure, key structural metrics and selected NMR data for the iPr complex, 9a. The structure of the cation shows a Binor-S fragment that has formally undergone a C–C bond activation of one cyclopropyl group in Binor-S to form a rhodametallacyclobutane (Rh/C11/C16/C15). There is a weak agostic C–H⋯Rh interaction [Rh–C2 2.901(3) Å] and, crucially, a close approach of the C–C single bond of the cyclopropane unit in the Binor-S fragment [Rh–C: 2.352(3) Å, 2.369(3) Å]. This C–C bond is lengthened from that found in a close analogue of free Binor-S [1.604(4) Å versus 1.497(6) Å] indicative of an interaction with the Rh centre. Complexes 9b–e also show very similar motifs.52,53 Formally these cationic metal centres can be formulated as being 16-electron Rh(III) d6, with four of these valence electrons coming from C–H⋯Rh and C–C⋯Rh agostic interactions. In solution, at low temperature, the 13C{1H} NMR spectrum of 9a reveals a coupling between 103Rh and the carbon atoms involved in the agostic interaction [J(RhC) = 9 Hz] which is considerably smaller than that associated with the Rh–C 2c–2e bonds in the metallacyclobutane [J(RhC) = 22 Hz], indicative of a weak but significant interaction between the Rh-centre and the C–C bond. Due to the symmetry (Cs) present in the molecule JCC for the C–C agostic bond could not be measured.
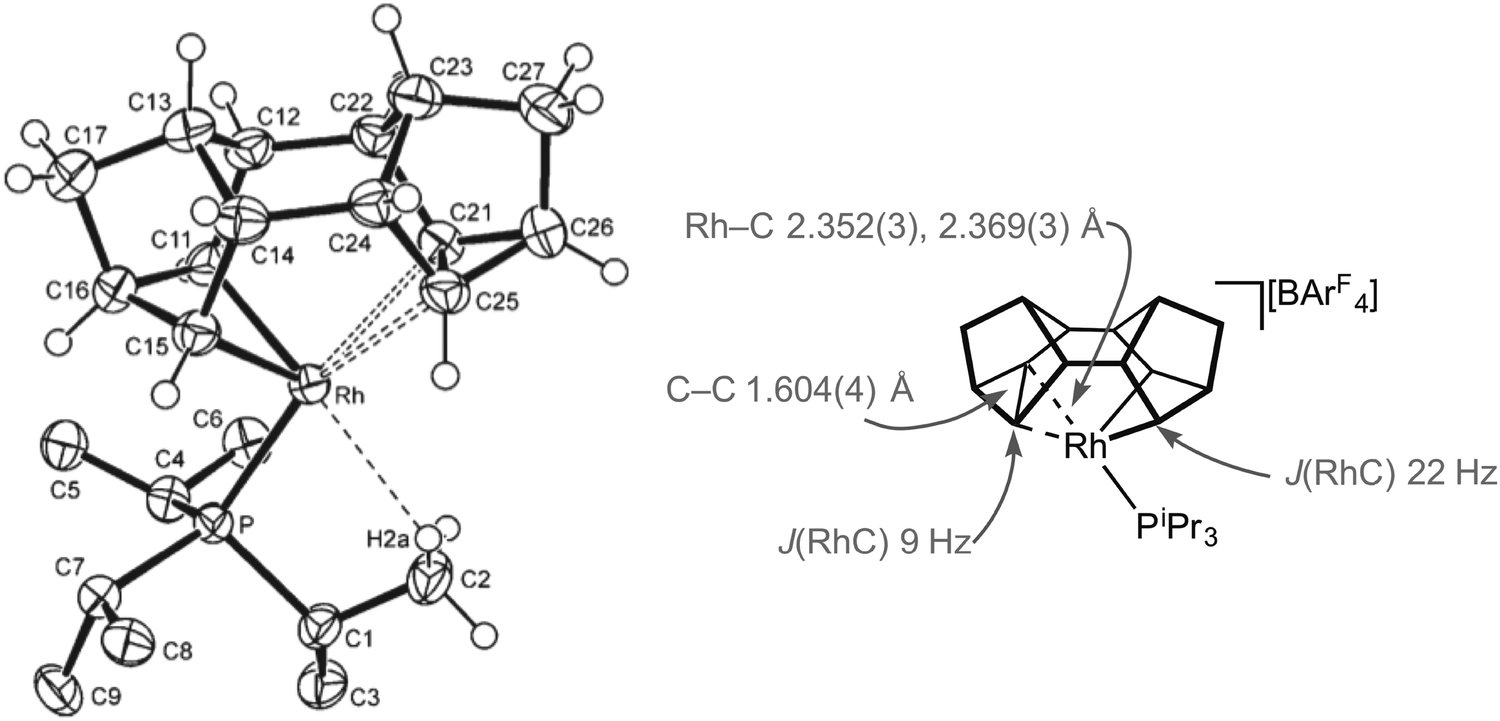 | ||
| Fig. 11 Structure of complex 9a, alongside selected structural and NMR data. | ||
DFT calculations, combined with a computational topological analysis of the electron density in 9a using Bader's Atoms in Molecules approach, demonstrates that there is a weak covalent interaction between a C–C bond of the cyclopropane and the Rh(III) centre by the presence of both bond and ring critical points between salient atoms.51 A more detailed combined experimental and computational charge density study on the [HCB11Me11]− salt of 9b54 confirms this, and also demonstrates a weakening of the C–C bond in the σ-interaction and a distortion of the charge concentration associated with this bond towards the Rh. A curved bond path from the C–C bond to the metal centre is also characteristic of the contribution of a covalent, delocalised, 3c–2e bonding interaction (Fig. 12). Overall, four key markers, assembled from solution (NMR), structural metrics (X-ray) and electron density analysis (experiment and computation), point to these complexes being genuine agostic C–C complex of Rh: (i) a close approach of the C–C bond to Rh; (ii) a lengthening of this C–C bond; (iii) coupling being observed between the carbon atoms in the C–C unit and the metal; (iv) experimentally and computationally determined charge density that shows both bond and ring critical points, a weakening of the C–C bond involved in the interaction and bond ellipticity profiles that are suggestive of an electron density shift from the C–C bond to Rh.
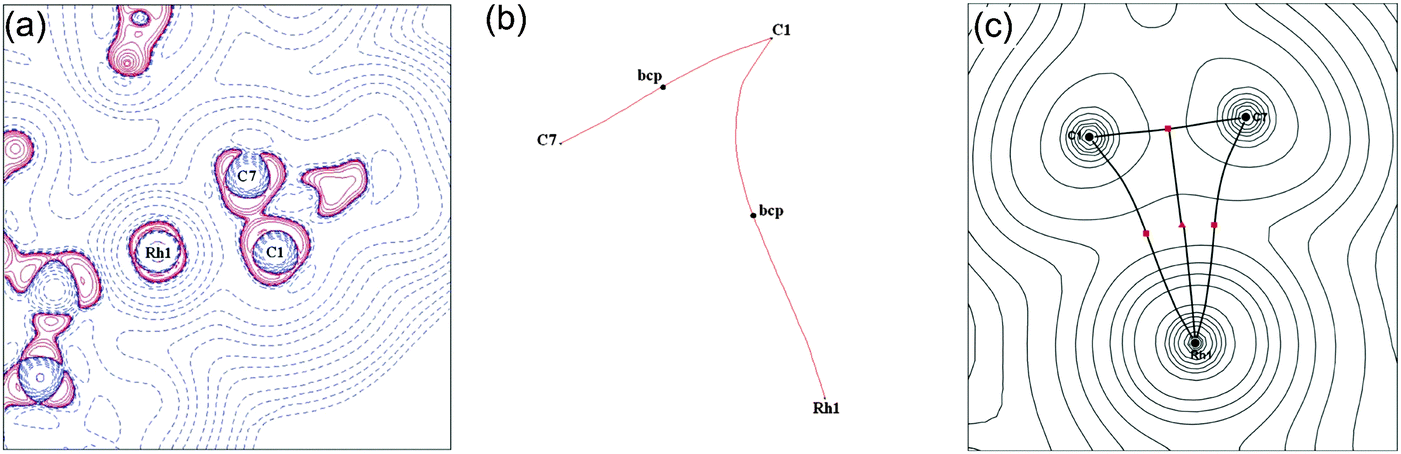 | ||
| Fig. 12 (a) Laplacian of the electron density drawn in the plane of the Rh and the C–C sigma interaction (C1/C7) for 9b; (b) bond paths between Rh and C1/C7; (c) calculated electron density, critical points and bond paths. Reprinted from ref. 54 with permission from the Royal Society of Chemistry. | ||
The mechanism by which these complexes are formed has been investigated using DFT methods using PMe3 models of the Rh and Ir (vide infra) complexes.55 Although the overall process is a simple 4+4 cyclodimerisation of the two NBD ligands, the calculated pathway (Fig. 13) involves oxidative C–C bond formation to give D, followed by two rearrangements, viaE, to give the final product F. The rate determining step calculated for Rh (C to D) is considerably lower in energy than that calculated for Ir (C to D and E to F are almost isoenergetic and ∼25 kJ mol−1 higher in energy than for M = Rh) consistent with the experimental observations that C is not observed when M = Rh, but can be isolated for M = Ir (vide infra).51,56 The higher barrier for the Ir-congener presumably reflects the stronger Ir–C bonds that need to be manipulated in this transformation. For M = Ir the equivalent complex to C transforms onto the C–C agostic complex (F) on gentle heating.
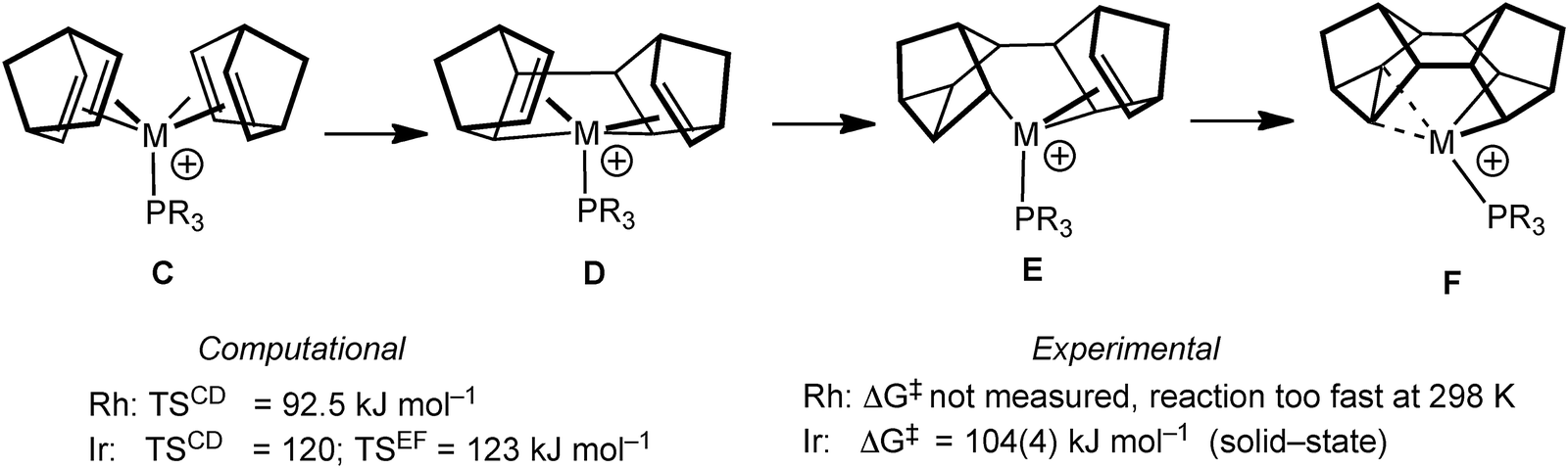 | ||
| Fig. 13 Computationally-derived mechanism for the formation of Binor-S. | ||
As pointed out in the introduction, complexes that show C–C agostic interactions are of direct relevance to C–C activation processes, that formally arise from oxidative cleavage of the C–C bond. Complexes 9 undergo rapid, and reversible, C–C activation in solution, which can be frozen out at low temperature. This process makes the two sides of the Binor-S fragment equivalent at room temperature, and calculations (on a PH3 model) suggest that this proceeds through a Rh(V) tetraalkyl intermediate, rather than a Rh(I) complex that contains two C–C⋯Rh agostic interactions (Fig. 14). The activation barrier has been determined experimentally for this process, ΔG‡(298 K) = 44.8 ± 4 kJ mol−1 ΔS‡ = −18.2 ± 4 JK−1 mol−1, which correlates reasonably with that determined from the computational model.52 This barrier is lower than other activation energies experimentally determined for C–C activation,57,58 consistent with the strained C–C bond in the cyclopropyl unit.
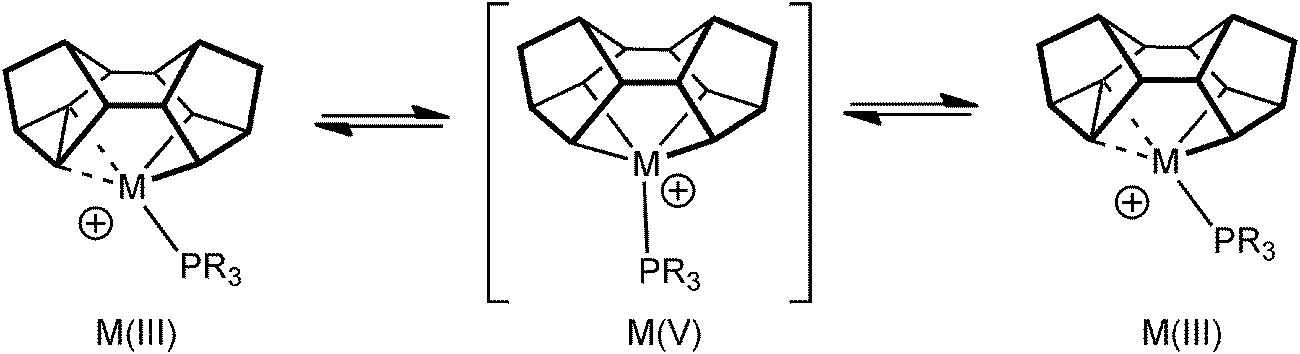 | ||
| Fig. 14 The fluxional process occurring in [M(Binor-S)(PR3)]+ fragments. | ||
It is well established in the organometallic chemistry of sigma complexes that for heavier members of a triad the products of oxidative cleavage are more accessible.59 For example [Rh(η5-C5Me5)(PMe3)H(H2)][BArF4] is formulated as a Rh(III) hydride–dihydrogen complex,60 while [Ir(η5-C5Me5)(PMe3)H3][BF4] is an Ir(V) trihydride.61 Having established the synthetic routes and coordination chemistry of the Rh(III)–Binor S complexes it was thus clearly of interest to study the Ir-congeners as such a complex might show increased C–C activation of the agostic bond. In contrast to the Rh-chemistry, the bis-NBD complex 10 that is a precursor to the Binor-S complexes can be isolated for M = Ir, as predicted by computational methods (Fig. 13 and 15).55,56 Although 10 does proceed slowly to form the corresponding Binor-S complex 11, that shows a C–C⋯Ir agostic interaction, both 10 and 11 decompose at a similar rate in solution, meaning that isolating pure material is difficult. Instead, by simply performing the reaction in the solid-state complex 11 can be produced pure, allowing for its characterisation by both solution techniques and X-ray crystallography. The molecular structure of 11 as determined at 100 K is very similar to that determined for 9a (Rh), with the major difference being that the C–C agostic bond distance is significantly longer for 11 than in 9a [1.704(5) Å and 1.610(2) Å respectively]. Unexpectedly, complex 11 showed a progressive lengthening of this C–C bond when the solid-state structures were determined at increasingly warmer temperatures, so that at 250 K it is 1.833(13) Å. The equivalent distance in 9a is essentially unchanged over this temperature range. This was interpreted as being due to dynamic disorder in the solid-state for 11 that establishes a temperature dependent equilibrium between agostic C–C⋯Ir [Ir(III)] and bis alkyl [Ir(V)] complexes (Fig. 16). Calculations show that the barrier to C–C activation to form a M(V) complex is much lower for Ir than for Rh, consistent with that only the Rh(III) complex is observed in the solid-state. Consistent with this, in solution 11 is fluxional even at low (200 K) temperature while for 9a the higher energy of the Rh(V) intermediate means that this process can be arrested at 200 K.
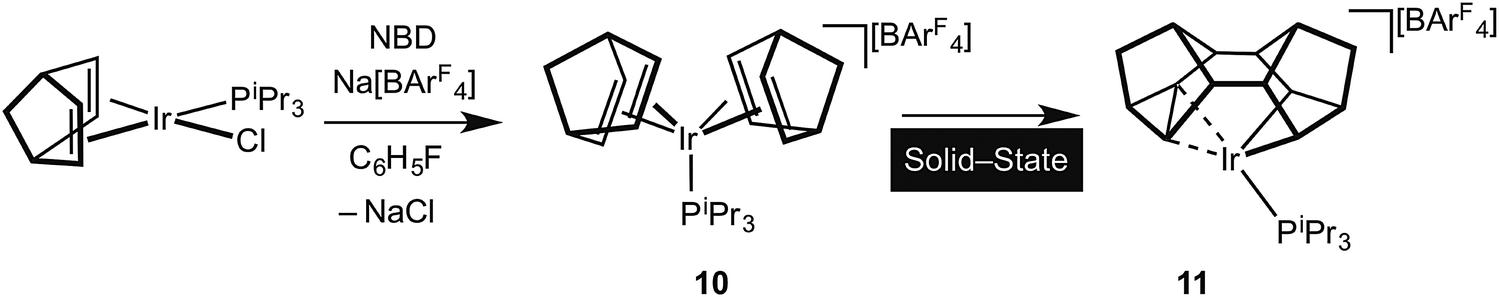 | ||
| Fig. 15 Synthesis of 11 in the solid-state. | ||
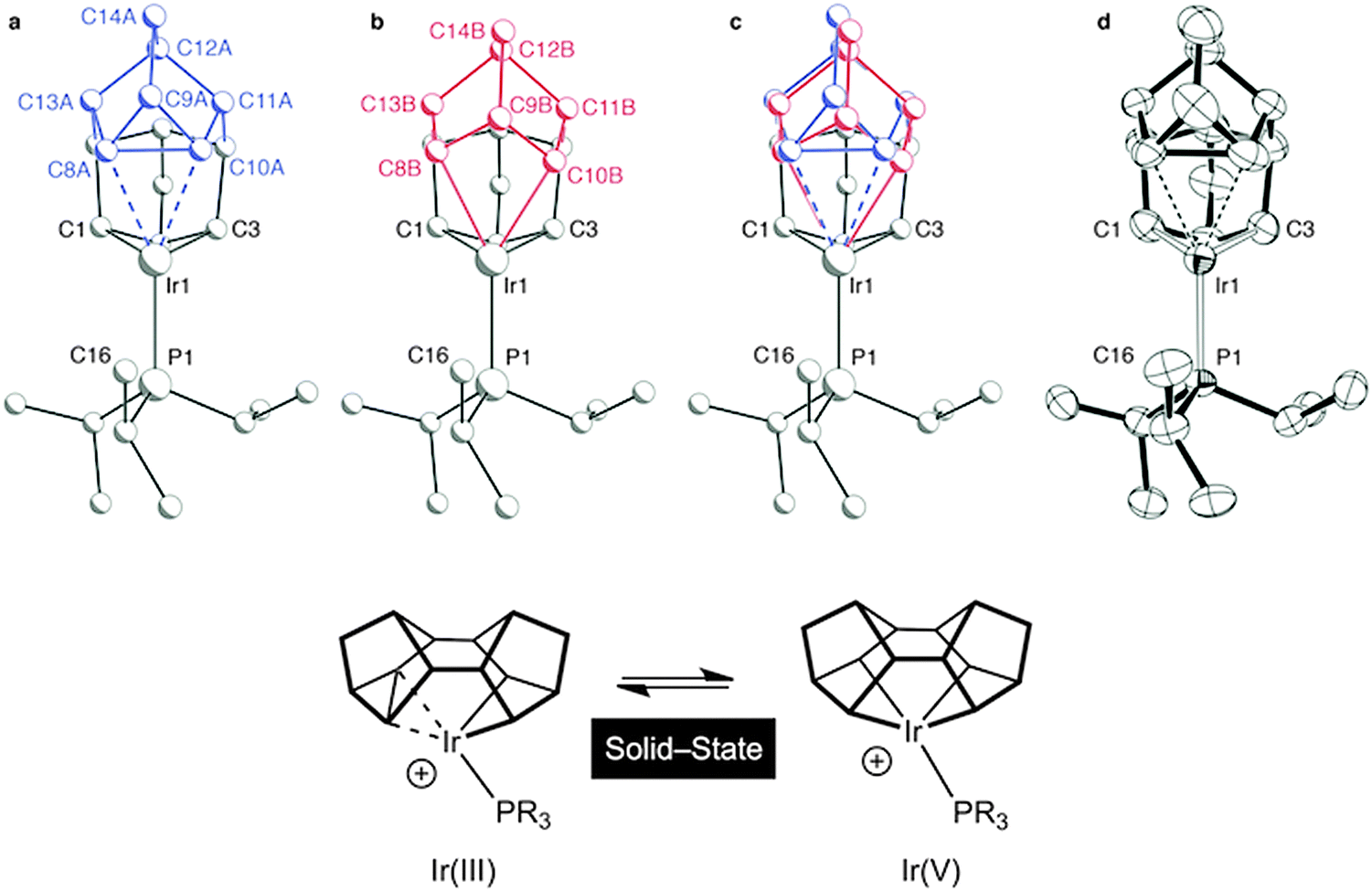 | ||
| Fig. 16 Disorder models for the molecular structure of 11 in the solid-state: ball-and-stick representations of the individual disordered components (a) and (b), the combined model (c), and an ORTEP plot (thermal ellipsoids drawn at the 50% probability level) without disorder modeling (d). Modified from ref. 56. | ||
The Rh–Binor complexes, 9, are also useful, and readily available, synthons for the delivery of a {Rh(PR3)}+ fragment, as addition of an exogenous Lewis bases results in reductive elimination of the Binor-S fragment. Although these C–C agostic complexes undergo a fluxional process that invoke a Rh(V) intermediate they react as if a latent source of Rh(I). This suggests the fluxional process and onward reactivity do not proceed through a common intermediate, and calculations support this.55 Examples of the use of these complexes as synthons for Rh(I) {Rh(PR3)}+ include (Fig. 17): addition of cyclooctadiene/benzene to afford 12;51 amine–borane addition leads to the novel Rh2 dimer 13 in which B–H activation has occurred;62 addition of tris-isopropyl phosphine gives a 14-electron complex, 14, that has an agostic C–H interaction, that itself undergoes C–H activation (dehydrogenation);63 pincer ligands add to form Rh(I) complexes such as 15 that are themselves synthons for the generation of dihydrogen complexes;64 a rare example of a Rh(I) tricarbonyl species 16 form on the addition of CO;51 and H2 adds to form Rh(III) dihydride species, 17, which is trapped by fluorobenzene solvent.51
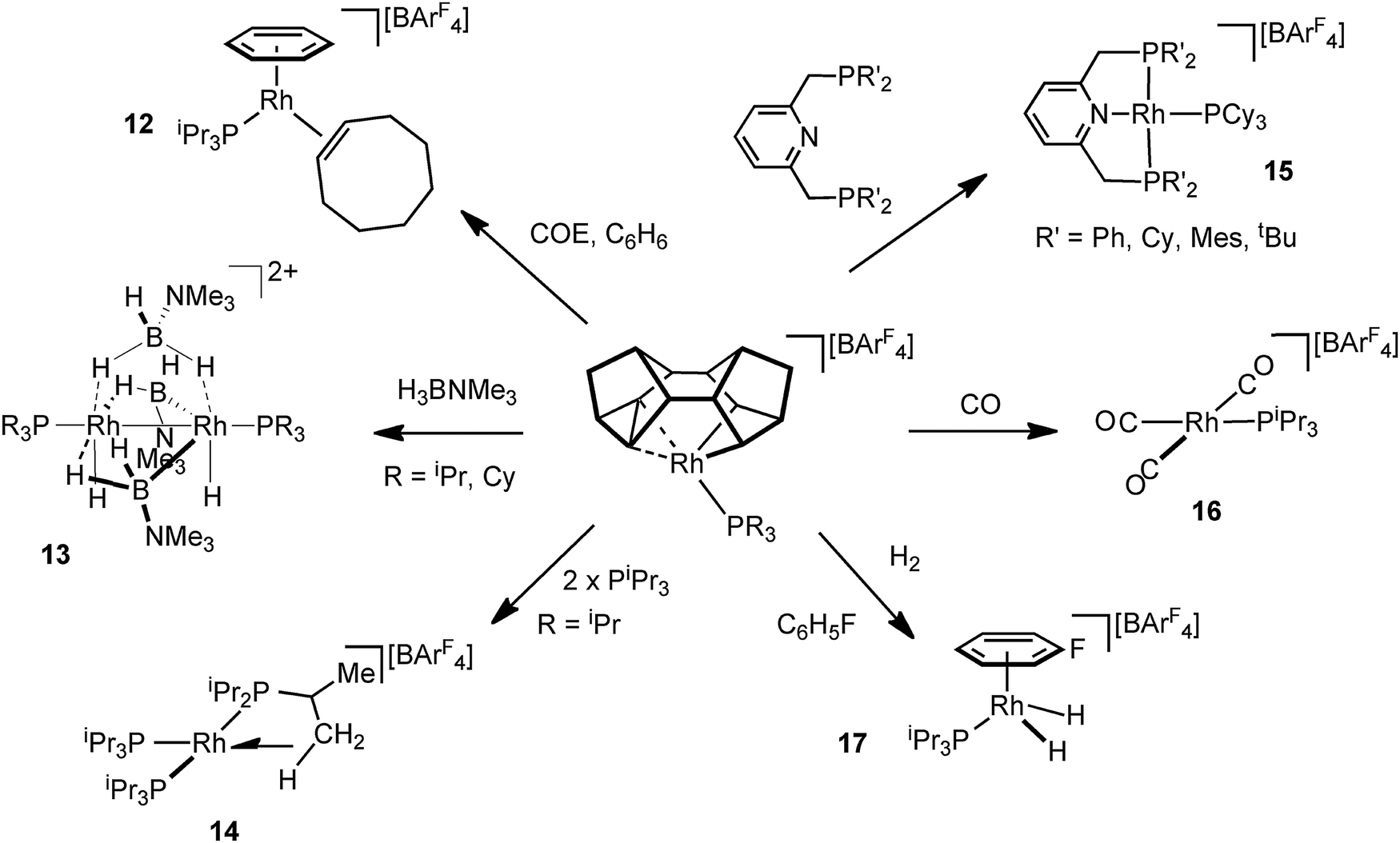 | ||
| Fig. 17 Using [Rh(Binor-S)(PR3)]+ as a latent source of {Rh(PR3)}+. | ||
3.2. Pincer ligand complexes that support C–C sigma interactions
Early reports by Milstein and co-workers on C–C activation in pincer complexes showed that complexes in which a pincer ligand forces a methyl group to be in close proximity to a Rh(I) centre can promote C–C bond cleavage;6,65–67 while there is also good evidence for the formation of agostic complexes that might act as intermediates on the pathway to C–C activation (Fig. 18).6,67 For example complex 18 comes from addition of CO to the corresponding Rh(I)–methyl complex, resulting in a 1,2-methyl shift to the arene, the microscopic reverse of C–C activation. The structure of the ethyl analogue shows that there is an approach of the CH2 carbon to the Rh-centre [2.817 Å] which is closer than the sum of the Van der Waals radii, hinting at a C–C⋯Rh interaction. However, this is still a long distance, especially when compared to other complexes of Rh with C–C interactions (e.g.9), and caution should always be exercised in defining the presence or absence of an agostic interaction on the basis of the separation between atoms alone.68 NMR data does, however, suggest a contribution from an agostic C–C⋯M interaction. The 13C{1H} NMR spectrum of 18 shows a small J(RhC) coupling constant [2.3 Hz] for the ipso-carbon suggestive of a weak interaction with the metal. These data, alongside others, suggest that 18 is best represented as a mixture of arenium (18) and arene/agostic-C–C (18′) resonance forms.65 Likewise complexes 19 and 20 have been characterised by NMR spectroscopy, although 103Rh–C coupling constants for the agostic interactions were presumably too small to be observed.69,70 Interestingly, very recent experimental and computational studies on related C–C activation in rhodium complexes of the thiophosphoryl-based pincer ligand, 1-CH3-2,6-(SPiPr2)2C6H3 show that an intermediate with a η3-C–C–H interaction is on the pathway to C–C activation.37 The Pt(II) complex 21 complex has been spectroscopically characterised, which is formed on reaction of MeOTf with the corresponding Pt(II)–CH3 pincer complex.71 In particular complex 21 is characterised as having an agostic C–C⋯Pt interaction by a 13C{1H} NMR spectrum in which a larger than expected 195Pt–C coupling constant [982 Hz] for the Pt–CH3 group is observed, that is indicative of a weakly bound trans ligand; while weak 195Pt coupling is observed to both the ipso carbon [14 Hz] and the aryl methyl [56 Hz] groups, consistent with an interaction between the Pt centre and the C–C unit.
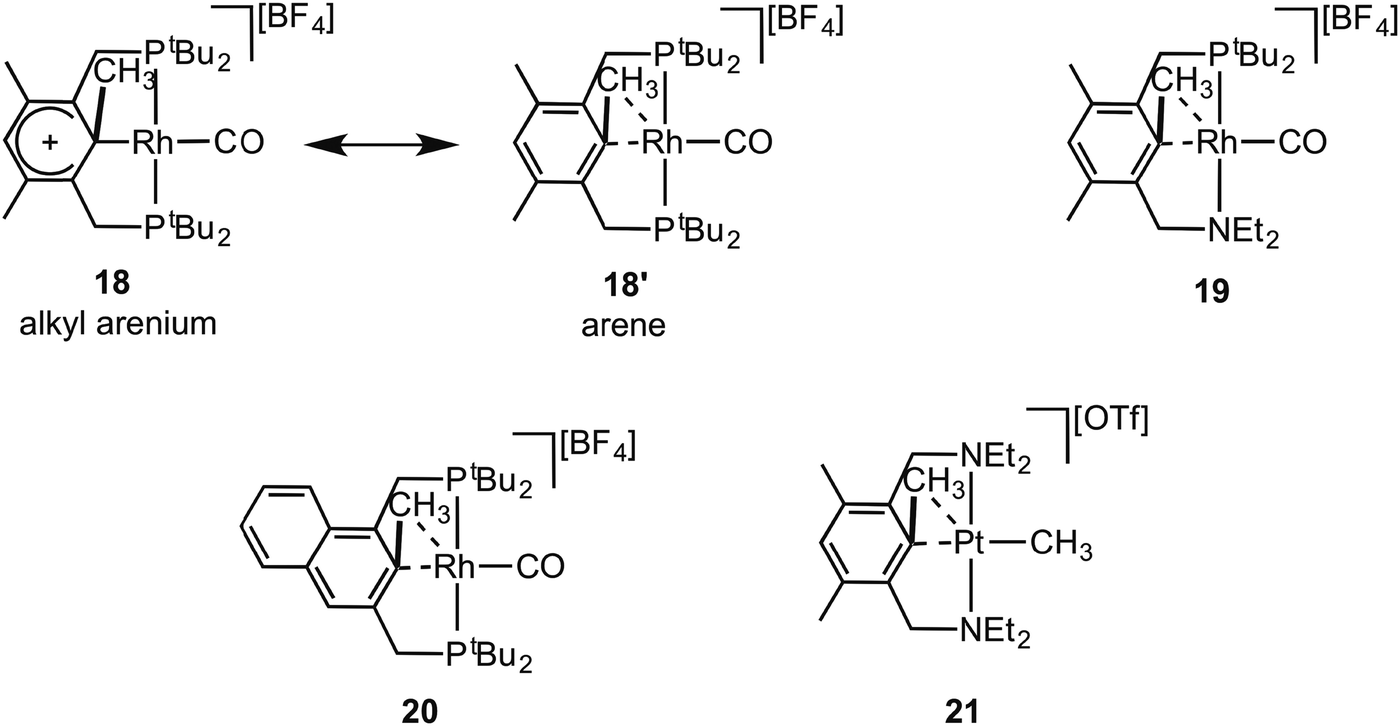 | ||
| Fig. 18 Examples of C–C agostic complexes with pincer ligands. | ||
3.3. Complexes with E–E or E–X interactions other than C–C
Metal systems that show close approaches of saturated single bonds are not limited to those of C–C⋯M. Complexes with Si–Si⋯M interactions with Pt and Pd centres have been reported. The first of these reported to show a Si–Si sigma (agostic) bond with a metal centre was that of Tanaka, 22, which has a tri-palladium complex in which there are six silicon atoms in close approach to the central palladium atom (Fig. 19).72 Although originally described as a Pt(VI) complex with 6 × 2c 2e Pt–SiR3 interactions,73 the alternative representation that is now accepted to be closer to the actual bonding situation is as a central Pd(II) square planar centre, in which there are two agostic Si–Si interactions with the metal centre. DFT calculations that show a significant Si–Si bond, a more square planar [i.e. Pd(II)] than octahedral [i.e. Pd(IV)] geometry, diamagnetism [calculations indicate that Pt(VI) might be expected to be paramagnetic] all point to this new interpretation as being correct.74–76 Interestingly this work also suggests the reinterpretation of a earlier-reported dinuclear Ni–silyl complex that might also contain Si–Si⋯Ni agostic interactions.77 The three-coordinate Pt complex, Pt[NHC(Dip)2](SiMe2Ph)223 (NHC = N-heterocyclic carbyne; Dip = 2,6-diisopropylphenyl), that shows a trigonal planar Y-shaped geometry with an acute Si–Pt–Si angle [80.9(1)°] and a short Si–Si distance [2.980(5) Å], was originally described as a Pt(II) complex in which there was a significant amount of sigma bonding between the Si-atoms. This structure was thus described as being a “snapshot” on the reaction pathway of reductive elimination of R3Si–SiR3 from a Pt(II) center.78 A more recent detailed computational analysis (including 195Pt chemical shift calculations) broadly supports this interpretation, but places the complex as sitting further along the reductive elimination pathway, being better described as a Pt(0) σ-disilane complex in which there is significant back donation from the Pt(0) into the σ* orbitals of the R3Si–SiR3 unit.79 Perhaps, then, this is best described as a rare example of a true sigma complex, rather than agostic. Si–Si⋯Cu agostic interactions have been reported, in which a trans-spanning ligand, Ph2P(C6H4)Me2Si–SiMe2(C6H4)PPh2, interacts with a Cu(I) metal centre, 24. The bonding, as analysed by DFT methods, is dominated by a Si–Si σ-donation to Cu.80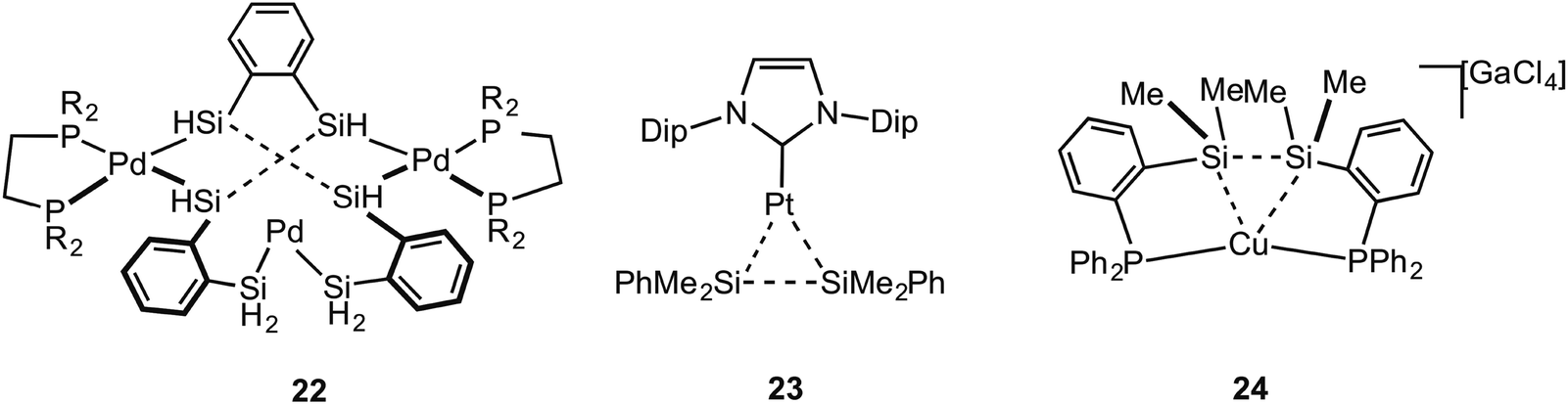 | ||
| Fig. 19 Examples of Si–Si⋯M agostic interactions. | ||
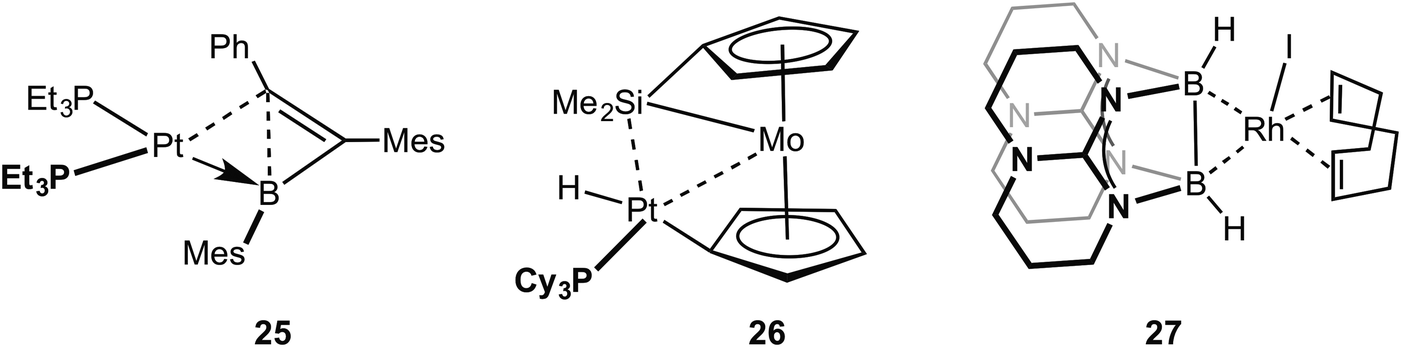
Braunschweig and co-workers have reported that B–C single bonds can also partake in agostic interactions with Pt metal centres, i.e. complex 25 in which an unsupported borirene ligand interacts with the metal centre through a B–C single bond (Fig. 20).81 Similar to 23, calculations, NMR evidence, electrochemistry and X-ray crystallography suggest 25 is best represented as a Pt(0) metal centre in which donation from a B–C σ bond is supported by Pt to B dative bonding, leading to significant activation (although not cleavage) of the B–C bond. A sigma interaction between a Mo–Si single bond and a Pt(II) centre in a disilamolybdenocenophane complex with the {Pt(PCy2)} fragment, complex 26, has been suggested on the basis of a structural and computational analysis (e.g. square planar Pt centre, elongated Mo–Si distance, Wiberg bond indices).82 This structure is suggested to lie on the pathway to oxidative cleavage of the Mo–Si bond to form a Pt(IV) centre. Himmel and co-workers have recently reported on late transition metal complexes (Rh and Cu) of doubly base-stabilised diborane(4) [HBhpp]2 (hpp = 1,3,4,6,7,8-hexahydro-2H-pyrimido-[1,2-α]pyrimidinate) that have dominant unsupported B–B⋯M sigma interactions, e.g. RhI(COD)(HBhpp)227. These complexes have been characterised on the basis of DFT [topological analysis of electron density, AIM], X-ray crystallography [lengthened B–B distances], Infra Red [B–H stretching frequencies that are very similar to free ligand], NMR spectroscopy [especially δ(BH) that shows only a small chemical shift change on coordination]. Interestingly, for complexes of earlier transition elements (e.g. Cr) the dominant interaction is from B–H⋯M agostic interactions.83 Related B–B⋯Fe agostic interactions in a {Fe(CO)2} complex with a catenated FeB4 unit has recently been reported.84 The bonding in the five membered FeB4 “inorganometallic”85 species is also reminiscent of the bonding in organometallic metallacyclobutanes, discussed next.
 | ||
| Fig. 20 Examples of E–X⋯M agostic interactions. | ||
Complexes in which there is a close approach of a Si–C bond to a metal centre are widespread, especially rare-earth and early transition metals, and are not covered in this review.46,86–88
4 Metallacyclobutanes
Very early on after the Chauvin mechanism for alkene metathesis was established, distinctive spectroscopic (1H and 13C NMR) and geometrical features for early transition metallacyclobutanes were noted, as summarized by Feldman and Schrock,89 and later extended to ruthenium-based intermediates.90 Full recognition that this data could be interpreted as α,β-CCC or, maybe more appropriately, α,β,α′-CCC agostic interactions has come more recently (Fig. 21).91 Just like C–H⋯M agostic interactions facilitate the insertion of an alkene into a M–C bond during alkene polymerization in some systems,3 unsaturated metallacyclobutane complexes that are intermediates in alkene metathesis (A, Fig. 22) often exhibit α,β,α′-CCC agostic structures, or at least such structures are energetically within reach. Electronically saturated metallacyclobutanes do not contain C–C agostic interactions, and do not catalyze alkene metathesis.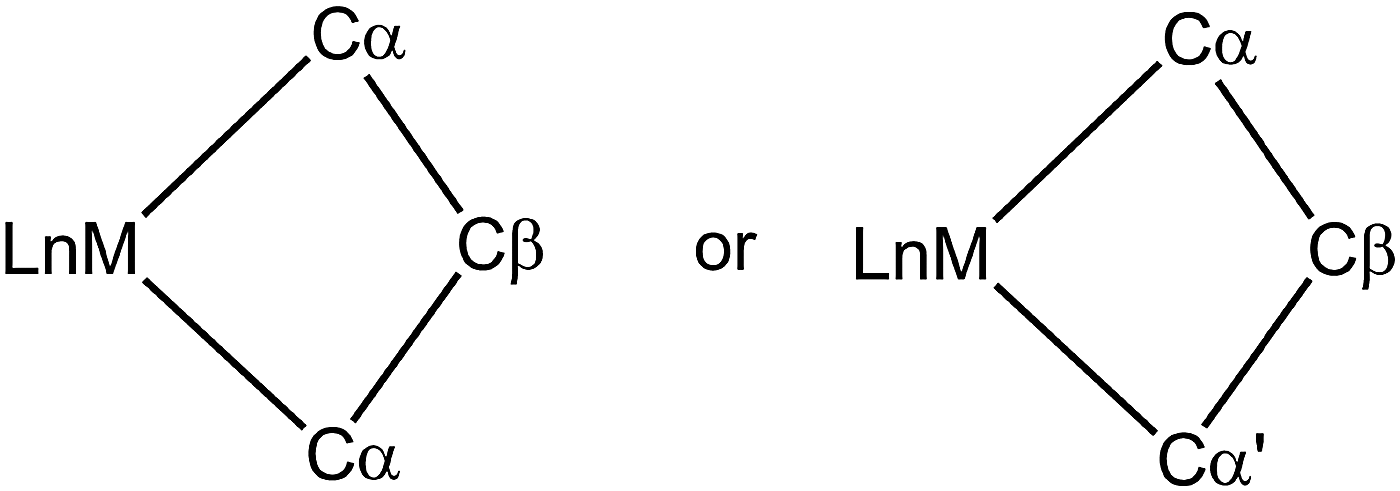 | ||
| Fig. 21 Nomenclature for the α,β-CCC or α,β,α′-CCC agostic structure in metallacyclobutanes. | ||
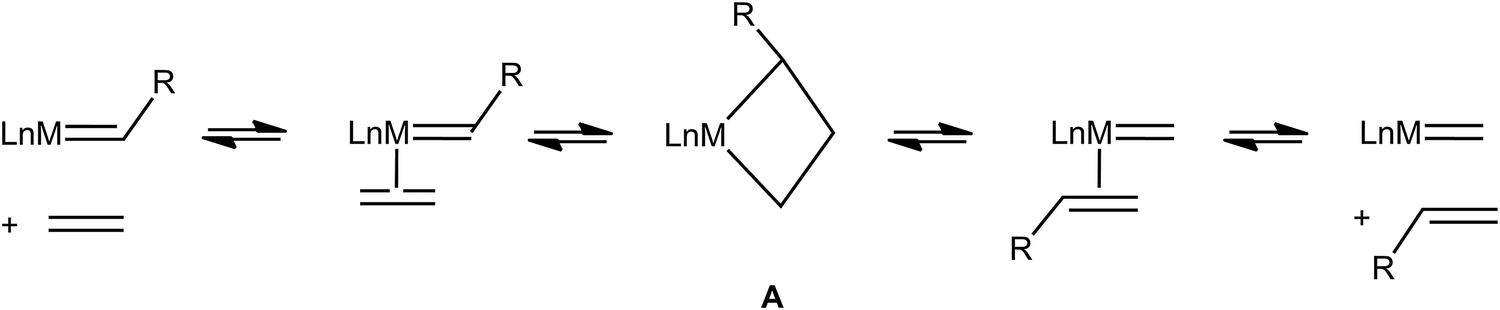 | ||
| Fig. 22 A general scheme for alkene metathesis showing the involvement of metallacyclobutanes A as intermediates. | ||
The NMR data for group 4 metallacyclobutane complexes in the metallocene family Cp′2M(CHRCHR′CHR″) (Cp′ = Cp, C5H5Me, Cp*; M = Ti, Zr, Hf; R, R′, R″ = H or hydrocarbyl) show distinctive high field shifts of Hβ and Cβ relative to those for Hα and Cα. Although there are some variations due to different substitutions at carbon, the resonance for Hβ occurs around δ 0 when that for Hα is observed in the range δ 1.8–2.5. In the 13C NMR spectra, Cβ is shielded by ca. 10 ppm [reaching δ −12 in Cp2Ti(CH2CH2CH2)] whereas Cα typically resonates above δ 65. This difference in chemical shifts between Cα and Cβ is commonly higher than Δδ 50 ppm and can be up to 80 ppm. 1JCH do not vary significantly, being in the range 125–137 Hz. More recently, 1JCαCβ have been measured for representative complexes. Cp2Ti(CH2CMe2CH2) 28, Cp*2Ti[CH2CH(i-C3H7)CH2] 29 and Cp*2Zr[CH2CH(i-C3H7)CH2] exhibit 1JCαCβ of 22.0, 24.2 and 20.7 Hz respectively.36 Along with geometrical data, this parameter has been used to assign C–C⋯M agostic interactions in comparison with related group 6 metallacyclobutanes that show higher, and normal magnitudes of JCC.92 Experimental electron density studies also support the C–C agostic interpretation.92 Free cyclobutanes have 1JCC values of ca. 27–30 Hz,31 with lower values being observed for highly strained molecules or when an electropositive substituent is present like in dimethylsilacyclobutane where 1JCC is 24.6 Hz.93 Therefore caution must be exercised when interpreting the JCC data. Structural data, and high resolution electron density measurements in particular, together with computational analyses point to the presence of an α,β,α′-CCC agostic interaction (vide infra). In contrast 18-electron group 6 metallocyclobutanes are not expected to have C–C (or C–H) agostic interactions. 13C NMR data for Cp2Mo[CH2CH(CH2-i-C3H7)CH2] (shielded Cα, δ −7.7, 1JCH 133 Hz; deshielded Cβ, δ 50.4, 1JCH 124 Hz) and Cp2W[CH2CH(CH2C3H5)CH2] (Cα, δ −23.7, 1JCH 130 Hz; Cβ, δ 52.7, 1JCH 125 Hz) with normal 1JCαCβ of 32.1 and 31.2 Hz, respectively, clearly indicate the absence of agostic interaction. This is confirmed by unremarkable metrical parameters in the solid-state.36
The NMR data for group 5 and 6 metallacyclobutanes that are not metallocenes are more diverse due to the existence of geometrical isomers for five coordinate species that can be trigonal bipyramid (TBP) or square planar (SP).89 In both series, the SP complexes show Hβ and Hα resonances at relatively close chemical shifts to one another, with Hβ observed at slightly higher field than Hα for tantalum complexes [viz. the tris(alkoxo)tantalum complex Ta[CH2CH(t-Bu)CHPh](O-2,6-C6H3-i-Pr2)3, δ 2.17–3.10 for Hα; δ 1.57 for Hβ],94 and Hβ downfield of Hα for tungsten complexes based on the W(NAr)(OR)2 motif [viz. W(CH2CH2CH2)(N-2,6-C6H3-i-Pr2)[OCMe2(CF3)]230-SP, δ 2.29 for Hα; δ 4.30 for Hβ; W[CH2CH(t-Bu)CH2](N-2,6-C6H3-i-Pr2)[OCMe2(CF3)]2, δ 1.12–2.36 for Hα; δ 2.62 for Hβ].95 In the 13C NMR spectra, Cα is observed downfield of Cβ with Cα at ca. δ 80 and Cβ ca. δ 40 for the tantalum complexes (Δδ 40 ppm), whereas for tungsten complexes Δδ is typically 20 ppm with Cα observed at ca. δ 45 and Cβ at δ 25. For both metals, 1JCH values are not significantly different for Cα and Cβ being measured in the range 125–135 Hz. 1JCαCβ have been obtained for five metallacyclobutanes based on W(NAr)(OR)2.96,97 They are found to lie in a narrow range of 28–33 Hz, e.g. for 30-SP and W[CH(t-Bu)CH2CH2](N-2,6-C6H3-i-Pr2)(O-t-Bu)2 respectively, a normal range for cyclobutane derivatives.31 For the TBP complexes, the NMR data are strikingly different.89 Hβ and Cβ are significantly shielded as compared to Hα and Cα. Typical chemical shifts ranges for tantalum complexes are δ 0 and δ 4 for Hβ and Hα respectively, and δ 0 and δ 100 for Cβ and Cα respectively. 1JCH appear in a normal range 125–135 Hz. For tungsten complexes these ranges extend to δ −1 and δ 5 for Hβ and Hα respectively, and 13C NMR chemical shifts remain similar to the tantalum case. 1JCH values appear slightly higher in the tungsten complexes (ca. 150 Hz). Remarkably, addition of 13C2-ethylene, *C2H4, to the neopentylidene complex W(CH-t-Bu)(N-2,6-C6H3-i-Pr2)[OCMe2(CF3)]2 yielded an interconverting mixture of SP and TBP isomers of W(*CH2*CH2*CH2)(N-2,6-C6H3-i-Pr2)[OCMe2(CF3)]2, 30-SP and 30-TBP, resulting from alkene metathesis.96,97 The TBP isomer exhibited resonances at δ 98.8 and 3.6 for Cα and Cβ, respectively, with a conspicuously low 1JCαCβ of 13 Hz attributable to an α,β,α′-CCC agostic interaction. These observations are summarized in Fig. 23.
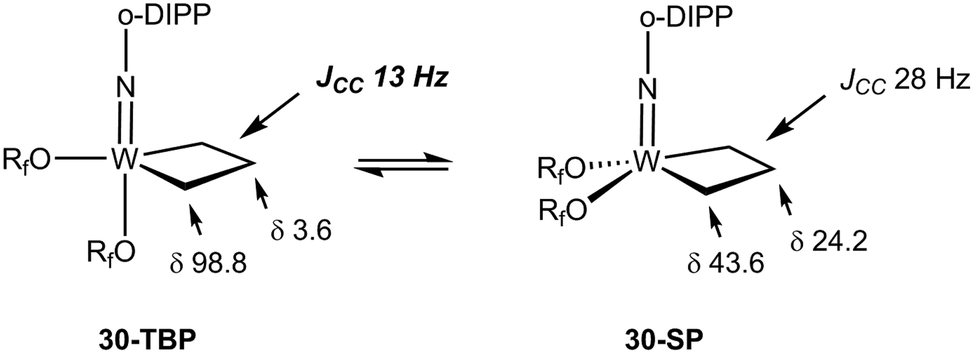 | ||
| Fig. 23 Summary of 13C NMR data for tungstacyclobutanes (Rf = CMe2(CF3), o-DIPP = 2,6-C6H3-i-Pr2) emphasizing the low 1JCC for the TBP isomer. | ||
Similar observations have been made on Mo(*CH2*CH2*CH2)(N-2,6-C6H3-i-Pr2)[OCMe2(CF3)]2.98 A TBP structure is deduced from the low temperature NMR data (1H NMR), Hβ (δ −0.26, −1.13) downfield of Hα (ca. δ 5.0); 13C NMR, Cβ (δ −2.28) upfield of Cα (δ 104.1). There is a low 1JCαCβ of 11 Hz that can be ascribed to an α,β,α′-CCC agostic interaction. The complex Mo[*CH2*CH2CH(t-Bu)](N-2,6-C6H3-i-Pr2)[O-2,6-C6H3-i-Pr2]2 exists as a mixture of SP (95%) and TBP (5%) at low temperature. The SP isomer exhibits 1JCαCβ of 49 and 55 Hz while the TBP isomer shows a low 1JCαCβ of 22 Hz.98 Similar observations are available for chiral diolatomolybdenum complexes.99,100
Overall, the NMR parameters for these early transition metallacyclobutanes are directly connected to structural data. Early on,89 it was recognized that some unsaturated metallacycles were kite-shape four-membered rings with short M–Cα bonds, long Cα–Cβ bonds, wide Cα–Cβ–Cα′ angles and short M⋯Cβ contacts, viz. the TBP isomers of the tantala,89 tungstacyclobutanes95–97,101 and the group 4 metallocenes,36,102,103 and related complexes.104 They are indeed associated with low 1JCαCβ values and are C–C agostic. Those metallacycles having more regular, less distorted metrical parameters, viz. the SP isomers of the tantala, molybda and tungstacyclobutanes or 18-electron metallocenes of molybdenum or tungsten,36 have normal 1JCαCβ and are not CC agostic. A comparison of selected early transition metallacycles structures is provided in Fig. 24 for metallocenes and in Fig. 25 for other metallacyclobutanes.
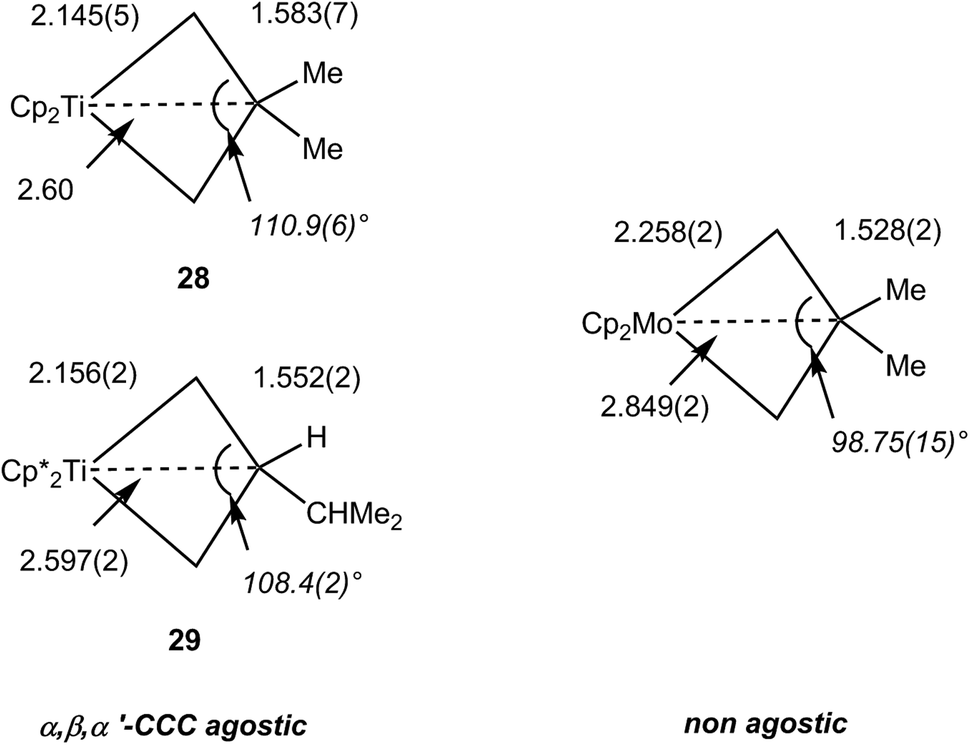 | ||
| Fig. 24 Typical metrical data for metallacyclobutanes in metallocene complexes (distances in Å). | ||
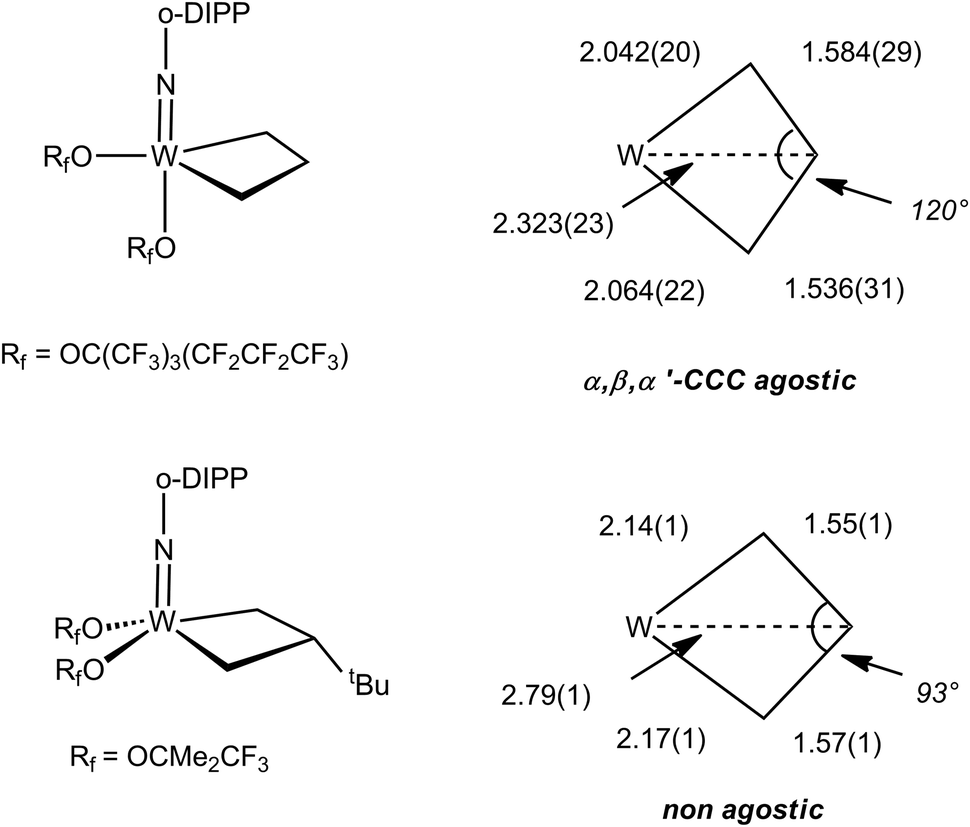 | ||
| Fig. 25 Typical metrical data for tungstacyclobutanes (distances in Å). | ||
A topological analysis of the experimental electron density using the AIM approach has been carried out on (η5-C5H4Me)2Ti(CH2CMe2CH2) 28-Me, a C5H4Me analogue of 28 (Fig. 26).92 Both elongated Cα–Cβ bonds [1.5723(3); 1.5772(3) Å] show lower electron densities and less negative Laplacian at their bond critical points than other single C–C bonds. The ellipticity along the Cα–Cβ bond paths is strongly asymmetric at the bond critical point and it shows maxima on the Cα side of the bonds. At maximum ellipticity, the electron density is distorted towards the titanium (black arrow in Fig. 26c). DFT calculations at the B3LYP and 6-311++G** basis set for Ti reproduced the experimental data. No distortion of the ellipticity profile is computed for a related non-agostic molybdacyclobutane complex.
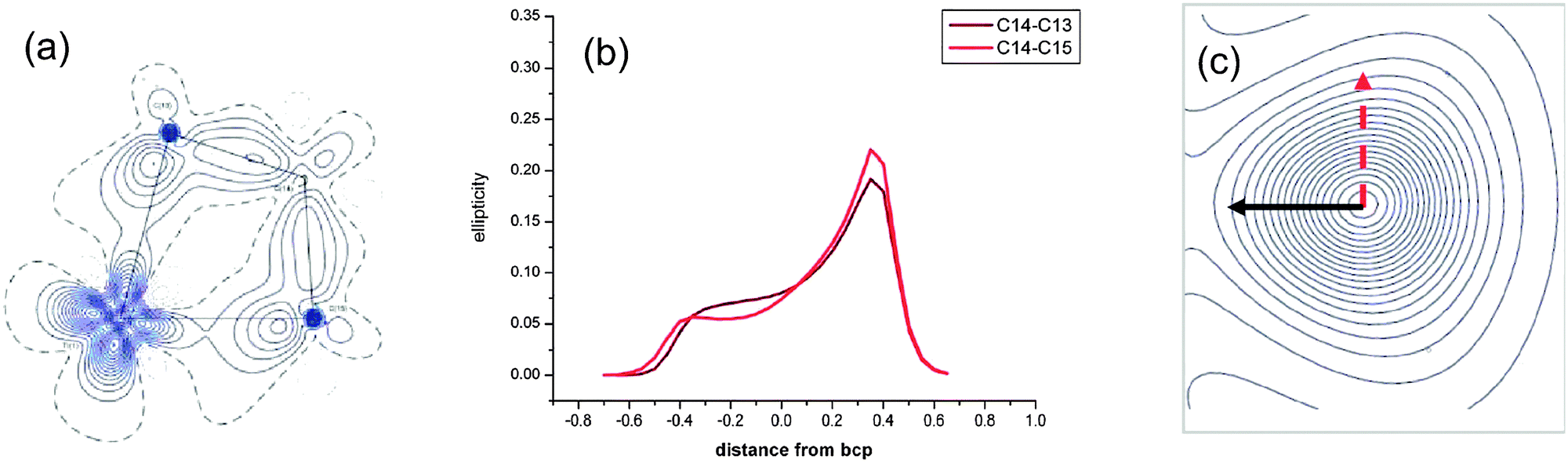 | ||
| Fig. 26 (a) Experimental density in the TiCαCβCα′ plane, (b) ellipticity profile along CαCβ bond paths and (c) contour map of the electron density at maximum ellipticity along a CαCβ bond path of (η5-C5H4Me)2Ti(CH2CMe2CH2) 28-Me. Adapted with permission from ref. 92. Copyright (2009) American Chemical Society. | ||
Similar NMR data were obtained for ruthenacyclobutanes that are intermediates in alkene metathesis by Grubbs type catalysts. Treatment of [(IH2Mes)Cl2RuCHPCy3][B(C6F5)4] with two 2 equivalents of 13C-labeled ethylene, *C2H4, at −50 °C yielded [*CH2CHPCy3][B(C6F5)4] and the 14-electron ruthenacyclobutane complex (IH2Mes)Cl2Ru(*CH2*CH2*CH2) 31. The NMR data for 31 are summarized in Fig. 27: in the 1H NMR spectrum Hα (δ 6.62) appears downfield of Hβ (δ −2.65) whereas in the 13C NMR spectrum, Cα (δ 94.0) is deshielded with respect to Cβ (δ 2.30) with a large chemical shift difference. JCHα and JCHβ of 165 and 155 Hz, respectively, are high and a low 1JCαCβ of 15.0 Hz supports the presence of an α,β,α′-CCC agostic interaction.90,105
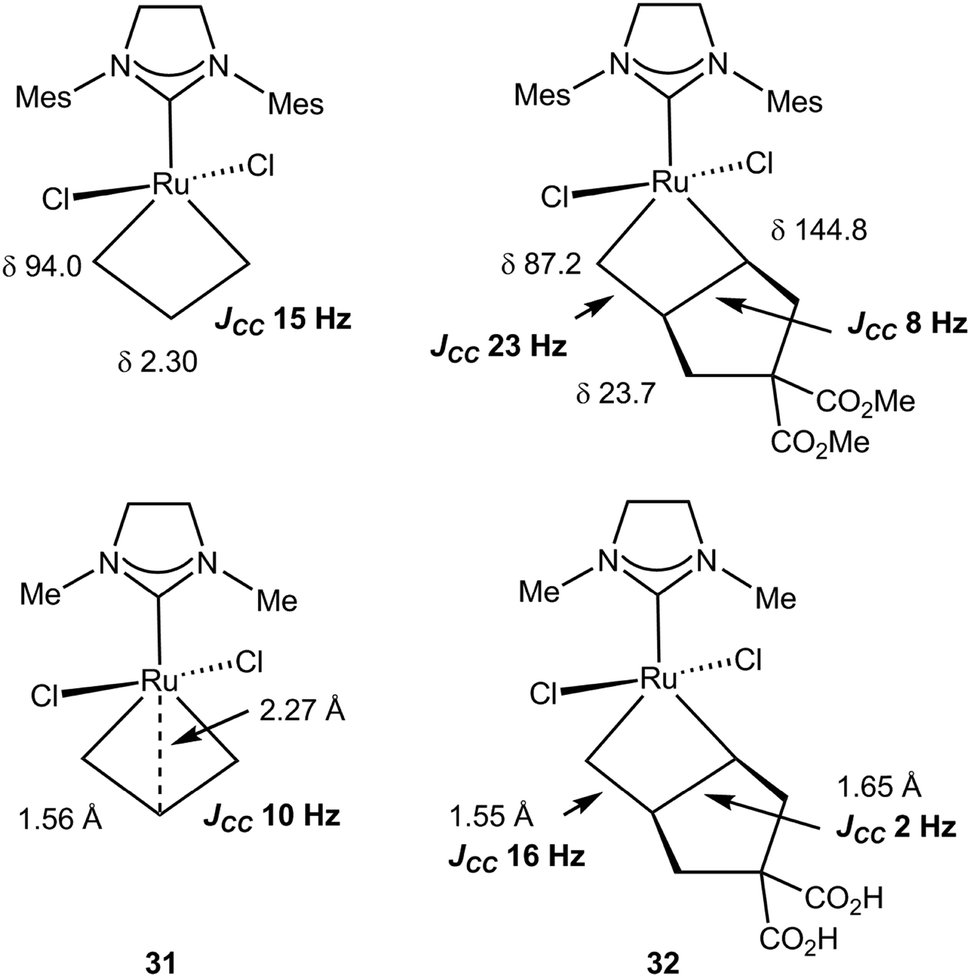 | ||
| Fig. 27 13C NMR and structural data for ruthenacyclobutanes 31 and 32: experiment (top) and computation (below). | ||
Similar chemistry using dimethylcyclopent-3-en-1,1-dicarboxylate in the presence of ethylene produced an unsymmetrical ruthenacyclobutane 32 (Fig. 27).106 Analysis of the spectroscopic data indicates the more substituted Cα–Cβ bond would be the most weakened. Indeed evidence is provided that a secondary alkylidene complex with an η2-vinyl end group is formed, not an alkene methylidene complex, when ring opening occurs, i.e. ROM would be easier than RCM. The structures of 31 and 32 were optimized using DFT and the NMR parameters computed (BP86/QZP).107 They emphasize the characteristic features of a kite-shape metallacycle with a close Ru⋯Cβ contact as expected from spectroscopic data. Reasonable absolute values and very good trends (differences) are observed for the chemical shifts and the JCC. Correlation of long CC bonds with reduced JCC is noted. This also correlates with the ease of ring opening of the unsymmetrical ruthenacyclobutane 32. Computation confirms the preferential formation of the product arising from ROM rather than RCM.
Remarkably a unified description of the bonding scheme in all the agostic metallacyclobutanes has been reached with the use of DFT calculations.49,50,91,107 Most calculations build upon the study of the electronic structure of models of Grubbs' first and second generation ruthenacyclobutanes. Details vary with the structural models, the functionals and basis sets used in the DFT calculations but suffice to say here that overall geometries exhibit the characteristic short M–Cα bonds, long Cα–Cβ bonds and markedly obtuse Cα–Cβ–Cα′ angles resulting in short M⋯Cβ distances. When X-ray structures are known, most significantly for agostic metallacycles of Cp′2Ti and TBP-W(NR)(OR′)2 but also for non agostic counterparts or isomers, i.e. metallacycles of SP-W(NR)(OR′)2, the structural parameters are reproduced reasonably well. Two key molecular orbitals (MO) involved to describe the α,β,α′-CCC agostic interaction are depicted in Fig. 28 where L can be PH3 or PMe3 (as models for PCy3) or IMe (as a model for IMes). A first MO (a) involves the interaction of a dz2 orbital on Ru with an in-phase combination of p-type orbitals on the propane-1,3-diyl. This MO accounts for a direct interaction between M and Cβ. Another MO (b) involves the interaction of a dxz-type orbital on M with an antisymmetric σ-Cα–Cβ–Cα orbital i.e. a formally four-center-2e interaction (4c,3e). Qualitatively similar metrical data (viz. a short M–Cβ distance, a large Cα–Cβ–Cα′ angle) and fully comparable interactions (apart from a π component) have been computed for the highly electron-deficient titanacyclobutadiene CpTi(CHCHCH) as well as in the tungstacyclobutadiene WCl3(CHCHCH),108 a realistic model of WCl3[C(tBu)CMeCMe],109 a potential intermediate in alkyne metathesis.
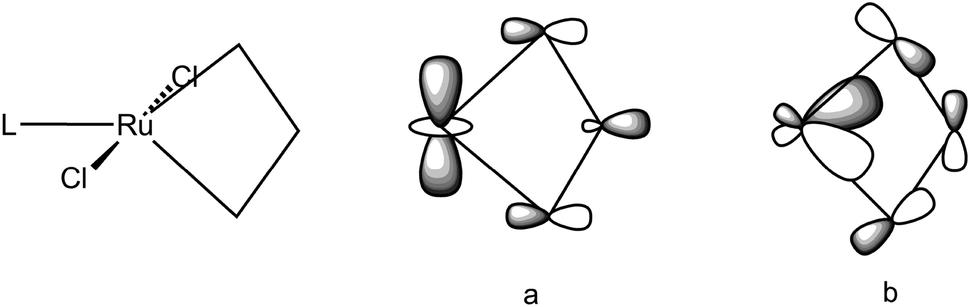 | ||
| Fig. 28 Main orbital interactions responsible for the agostic distortion in metallacyclobutanes. | ||
These interactions are responsible of the short Ru⋯Cβ distance and the long Cα–Cβ bonds in 31 and 32. Although no BCP was found,110 bond orders between Ru and Cβ are in the range 0.20–0.32 depending on method and model emphasizing the direct Ru⋯Cβ interaction when those for the interaction between Cα–Cβ are smaller (0.09). Similar interpretations are valid for other metallacycles.110,111 As seen experimentally for the titanium complex 28-Me (see above),92 computed electron density at the M–Cα BCP is high when that between Cα–Cβ is low. Ellipticity at Cα–Cβ BCP is accordingly large with a distortion towards M. A decreased covalent character for Cα–Cβ is seen from a modestly negative Laplacian at BCPs. By comparing agostic and non-agostic counterparts, upper limits for the strength of the agostic interaction in the metallacycles was estimated to be as high as 154 kJ mol−1 for Grubb's systems (M = Ru), 120 kJ mol−1 for Schrock's systems (M = W) and down to 57 kJ mol−1 for systems with M = Ti.110
α,β,α′-CCC agostic interactions stabilize metallacycles that are involved in alkene metathesis. In the case of ruthenium, such complexes have been observed at low temperatures. Exchange of α- and β-carbons occurs without loss of ethylene via a proposed ethylene methylidene complex (ΔG = 51 kJ mol−1 at −50 °C).90,105,112 A much slower exchange with free ethylene is also observed as an associative process where ring opening is not the rate-limiting step. As mentioned above, the preferential formation of the product arising from ROM rather than RCM was observed and computation indicated this occurred at the most activated Cα–Cβ bond.106,107 For tungsten, it has been proposed that an alkylidene alkene intermediate would be more accessible from the agostic TBP isomer.97 Loss of an olefin would not be possible from a non-agostic SP isomer. TBP isomers are favored by electron-withdrawing alkoxides whereas steric effects lead to ring distortion. Both effects would be important for easy exchange with free olefin and for minimizing energy differences between different 5-coordinate structures thereby leading to active catalysts.
5 Conclusions
Although they might be considered as exotic species, we hope that this review demonstrates that complexes in which a C–C saturated bond is in close approach to a metal centre, forming a C–C agostic interaction, should now be considered as an established chemical bonding motif, complementing ubiquitous C–H agostic interactions, and other sigma interactions. The successful identification of such C–C⋯M interactions should ideally be based on a number of complementary structural, spectroscopic and computational markers; and although not every complex is amenable to analysis by all of these approaches, the following might serve as a guide to establishing their presence or absence:(i) A close approach of the C–C bond to the metal;
(ii) A lengthening of this C–C bond as determined by X-ray crystallography or computation;
(iii) A reduced JCC coupling constant for the bond in question;
(iv) Additional coupling being observed between the carbon atoms in the C–C unit and the metal, where appropriate;
(v) Computational evidence for an interaction between the C–C bond and the metal. In particular, experimentally and computationally determined charge density that shows both bond and ring critical points, a weakening of the C–C bond involved in the interaction and bond ellipticity profiles that are suggestive of an electron density shift from the C–C bond to the metal.
As the area develops, through the efforts and ingenuity of new synthetic and catalytic approaches, it will be interesting to see if C–C agostic interactions can be harnessed in the same manner that metal dihydrogen, C–H agostic, silane and borane complexes have been used to promote catalytic bond activations.56
Acknowledgements
M.E. acknowledges fruitful discussions with Prof. John E. McGrady (Inorganic Chemistry Laboratories, University of Oxford) and Prof. Feliu Maseras (ICIQ, Tarragona and Universitat Autonoma de Barcelona) and financial support from the ANR. A. S. W. acknowledges then EPSRC and the Royal Society for funding, and stimulating discussions on this topic with Prof. J. C. Green (Oxford).References
- G. J. Kubas, Metal Dihydrogen and σ-bond complexes, Kluwer, New York, 2001 Search PubMed.
- M. Brookhart and M. L. H. Green, J. Organomet. Chem., 1983, 250, 395 CrossRef CAS.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908 CrossRef CAS PubMed.
- M. Albrecht, Chem. Rev., 2010, 110, 576 CrossRef CAS PubMed.
- I. Omae, J. Organomet. Chem., 2011, 696, 1128 CrossRef CAS PubMed.
- (a) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef; (b) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed.
- J. A. M. Simoes and J. L. Beauchamp, Chem. Rev., 1990, 90, 629 CrossRef CAS.
- P. E. M. Siegbahn and M. R. A. Blomberg, J. Am. Chem. Soc., 1992, 114, 10548 CrossRef CAS.
- C. H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC.
- C. Perthuisot, B. L. Edelbach, D. L. Zubris, N. Simhai, C. N. Iverson, C. Müller, T. Satoh and W. D. Jones, J. Mol. Catal., 2002, 189, 157 CrossRef CAS.
- K. Ruhland, Eur. J. Org. Chem., 2012, 2683 CrossRef CAS.
- W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553 CrossRef CAS PubMed.
- J. A. Calladine, S. B. Duckett, M. W. George, S. L. Matthews, R. N. Perutz, O. Torres and K. Q. Vuong, J. Am. Chem. Soc., 2011, 133, 2303 CrossRef CAS PubMed.
- R. D. Young, A. F. Hill, W. Hillier and G. E. Ball, J. Am. Chem. Soc., 2011, 133, 13806 CrossRef CAS PubMed.
- S. D. Pike, A. L. Thompson, A. G. Algarra, D. C. Apperley, S. A. Macgregor and A. S. Weller, Science, 2012, 337, 1648 CrossRef CAS PubMed.
- J. F. Hartwig, C. N. Muhoro, X. He, O. Eisenstein, R. Bosque and F. Maseras, J. Am. Chem. Soc., 1996, 118, 10936 CrossRef CAS.
- G. Alcaraz and S. Sabo-Etienne, Coord. Chem. Rev., 2008, 252, 2395 CrossRef CAS PubMed.
- S. Lachaize and S. Sabo-Etienne, Eur. J. Inorg. Chem., 2006, 2115 CrossRef CAS.
- J. C. Green, M. L. H. Green and G. Parkin, Chem. Commun., 2012, 48, 11481 RSC.
- W. Scherer and G. S. McGrady, Angew. Chem., Int. Ed., 2004, 43, 1782 CrossRef CAS PubMed.
- W. Scherer, V. Herz and C. Hauf, Struct. Bonding, 2012, 146, 159 CrossRef CAS.
- E. Clot and O. Eisenstein, Struct. Bonding, 2004, 113, 1 CrossRef CAS.
- (a) R. J. Puddephatt, Coord. Chem. Rev., 1980, 33, 149 CrossRef CAS; (b) A. Abo-Amer and R. J. Puddephatt, Inorg. Chem. Commun., 2011, 14, 111 CrossRef CAS PubMed.
- C. B. Rodríguez-García, A. Oliva, R. M. Ortuño and V. Branchadell, J. Am. Chem. Soc., 2001, 123, 6157 CrossRef PubMed.
- G. S. Hill and R. J. Puddephatt, Organometallics, 1998, 17, 1478 CrossRef CAS.
- M. E. Evans, T. Li and W. D. Jones, J. Am. Chem. Soc., 2010, 132, 16278 CrossRef CAS PubMed.
- R. A. Periana and R. G. Bergman, J. Am. Chem. Soc., 1986, 108, 7346 CrossRef CAS.
- D. D. Wick, T. O. Northcutt, R. J. Lachicotte and W. D. Jones, Organometallics, 1998, 17, 4484 CrossRef CAS.
- B. Goldfuss, P. v. R. Schleyer and F. Hampel, J. Am. Chem. Soc., 1996, 118, 12183 CrossRef CAS.
- R. Tomaszewski, I. Hyla-Kryspin, C. L. Mayne, A. M. Arif, R. Gleiter and R. D. Ernst, J. Am. Chem. Soc., 1998, 120, 2959 CrossRef CAS.
- L. Krivdin and G. Kalabin, Prog. Nucl. Magn. Reson. Spectrosc., 1989, 21, 293 CrossRef CAS.
- R. F. W. Bader and C. F. Matta, Inorg. Chem., 2001, 40, 5603 CrossRef CAS PubMed.
- B. G. Harvey, A. M. Arif and R. D. Ernst, J. Organomet. Chem., 2006, 691, 5211 CrossRef CAS PubMed.
- B. G. Harvey, A. M. Arif and R. D. Ernst, J. Mol. Struct., 2008, 890, 107 CrossRef CAS PubMed.
- A. M. Wilson, A. L. Rheingold, T. E. Waldman, M. Klein, F. G. West and R. D. Ernst, J. Organomet. Chem., 2009, 694, 1112 CrossRef CAS PubMed.
- B. G. Harvey, C. L. Mayne, A. M. Arif and R. D. Ernst, J. Am. Chem. Soc., 2005, 127, 16426 CrossRef CAS PubMed.
- M. Montag, I. Efremenko, Y. Diskin-Posner, Y. Ben-David, J. M. L. Martin and D. Milstein, Organometallics, 2012, 31, 505 CrossRef CAS.
- M. Etienne, J. E. McGrady and F. Maseras, Coord. Chem. Rev., 2009, 253, 635 CrossRef CAS PubMed.
- (a) M. Etienne, R. Mathieu and B. Donnadieu, J. Am. Chem. Soc., 1997, 119, 3218 CrossRef CAS; (b) J. Jaffart, M. Etienne, F. Maseras, J. E. McGrady and O. Eisenstein, J. Am. Chem. Soc., 2001, 123, 6000 CrossRef CAS PubMed.
- J. Jaffart, M. L. Cole, M. Etienne, M. Reinhold, J. E. McGrady and F. Maseras, Dalton Trans., 2003, 4057 RSC.
- M. Besora, F. Maseras, J. E. McGrady, P. Oulié, D. H. Dinh, C. Duhayon and M. Etienne, Dalton Trans., 2006, 2362 RSC.
- J. Jaffart, M. Etienne, M. Reinhold, J. E. McGrady and F. Maseras, Chem. Commun., 2003, 876 RSC.
- P. Oulié, C. Boulho, L. Vendier, Y. Coppel and M. Etienne, J. Am. Chem. Soc., 2006, 128, 15962 CrossRef PubMed.
- C. Boulho, T. Keys, Y. Coppel, L. Vendier, M. Etienne, A. Locati, F. Bessac, F. Maseras, D. A. Pantazis and J. E. McGrady, Organometallics, 2009, 28, 940 CrossRef CAS.
- Y. Escudie, C. Dinoi, O. Allen, L. Vendier and M. Etienne, Angew. Chem., Int. Ed., 2012, 51, 2461 CrossRef CAS PubMed.
- N. Koga and K. Morokuma, J. Am. Chem. Soc., 1988, 110, 108 CrossRef CAS.
- O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, Science, 2006, 311, 829 CrossRef CAS PubMed.
- M. Horacek, P. Stepnicka, R. Gyepes, I. Cisarova, J. Kubista, L. Lukesova, P. Meunier and K. Mach, Organometallics, 2005, 24, 6094 CrossRef CAS.
- C. Boulho, P. Oulié, L. Vendier, M. Etienne, V. Pimienta, A. Locati, F. Bessac, F. Maseras, D. A. Pantazis and J. E. McGrady, J. Am. Chem. Soc., 2010, 132, 14239 CrossRef CAS PubMed.
- C. Boulho, L. Vendier, M. Etienne, A. Locati, F. Maseras and J. E. McGrady, Organometallics, 2011, 30, 3999–4007 CrossRef CAS.
- S. K. Brayshaw, J. C. Green, G. Kociok-Köhn, E. L. Sceats and A. S. Weller, Angew. Chem., Int. Ed., 2006, 45, 452 CrossRef CAS PubMed.
- S. K. Brayshaw, E. L. Sceats, J. C. Green and A. S. Weller, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6921 CrossRef CAS PubMed.
- A. B. Chaplin and A. S. Weller, J. Organomet. Chem., 2013, 730, 90 CrossRef CAS PubMed.
- H. A. Sparkes, T. Kramer, S. K. Brayshaw, J. C. Green, A. S. Weller and J. A. K. Howard, Dalton Trans., 2011, 40, 10708 RSC.
- Y. Wu, L. Jin, Y. Xue, I. M. Lee and C. K. Kim, J. Comput. Chem., 2010, 31, 2248 CrossRef CAS PubMed.
- A. B. Chaplin, J. C. Green and A. S. Weller, J. Am. Chem. Soc., 2011, 133, 13162 CrossRef CAS PubMed.
- M. Gandelman, A. Vigalok, L. Konstantinovski and D. Milstein, J. Am. Chem. Soc., 2000, 122, 9848 CrossRef CAS.
- N. M. Brunkan, D. M. Brestensky and W. D. Jones, J. Am. Chem. Soc., 2004, 126, 3627 CrossRef CAS PubMed.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578 CrossRef CAS PubMed.
- F. L. Taw, H. Mellows, P. S. White, F. J. Hollander, R. G. Bergman, M. Brookhart and D. M. Heinekey, J. Am. Chem. Soc., 2002, 124, 5100 CrossRef CAS PubMed.
- T. M. Gilbert and R. G. Bergman, J. Am. Chem. Soc., 1985, 107, 3502 CrossRef CAS.
- A. B. Chaplin and A. S. Weller, Angew. Chem., Int. Ed., 2010, 49, 581 CrossRef CAS PubMed.
- A. B. Chaplin, A. I. Poblador-Bahamonde, H. A. Sparkes, J. A. K. Howard, S. A. Macgregor and A. S. Weller, Chem. Commun., 2009, 244 RSC.
- A. B. Chaplin and A. S. Weller, Organometallics, 2011, 30, 4466 CrossRef CAS.
- A. Vigalok, B. Rybtchinski, L. J. W. Shimon, Y. Ben-David and D. Milstein, Organometallics, 1999, 18, 895 CrossRef CAS.
- D. Morales-Morales and C. G. M. Jensen, The Chemistry of Pincer Compounds, Elsevier Science, 2011 Search PubMed.
- A. Vigalok and D. Milstein, Acc. Chem. Res., 2001, 34, 798 CrossRef CAS PubMed.
- F. Maseras and R. H. Crabtree, Inorg. Chim. Acta, 2004, 357, 345 CrossRef CAS.
- M. Gandelman, L. J. W. Shimon and D. Milstein, Chem.–Eur. J., 2003, 9, 4295 CrossRef CAS PubMed.
- C. M. Frech and D. Milstein, J. Am. Chem. Soc., 2006, 128, 12434 CrossRef CAS PubMed.
- B. L. Madison, S. B. Thyme, S. Keene and B. S. Williams, J. Am. Chem. Soc., 2007, 129, 9538 CrossRef CAS PubMed.
- W. Chen, S. Shimada and M. Tanaka, Science, 2002, 295, 308 CrossRef CAS PubMed.
- In their original report Tanaka, Shimada and coworkers acknowledge that the alternative representation of 22 as having a Pd(II) centre with a two sigma Si–Si interactions is possible.
- G. Aullón, A. Lledós and S. Alvarez, Angew. Chem., Int. Ed., 2002, 114, 2036 CrossRef.
- E. C. Sherer, C. R. Kinsinger, B. L. Kormos, J. D. Thompson and C. J. Cramer, Angew. Chem., Int. Ed., 2002, 41, 1953 CrossRef CAS.
- G. I. Nikonov, Angew. Chem., Int. Ed., 2003, 42, 1335 CrossRef CAS PubMed.
- S. Shimada, M. L. N. Rao, T. Hayashi and M. Tanaka, Angew. Chem., Int. Ed., 2001, 40, 213 CrossRef CAS.
- G. Berthon-Gelloz, B. de Bruin, B. Tinant and I. E. Markó, Angew. Chem., Int. Ed., 2009, 48, 3161 CrossRef CAS PubMed.
- N. Takagi and S. Sakaki, J. Am. Chem. Soc., 2012, 134, 11749 CrossRef CAS PubMed.
- P. Gualco, A. Amgoune, K. Miqueu, S. Ladeira and D. Bourissou, J. Am. Chem. Soc., 2011, 133, 4257 CrossRef CAS PubMed.
- H. Braunschweig, P. Brenner, R. D. Dewhurst, I. Krummenacher, B. Pfaffinger and A. Vargas, Nat. Commun., 2012, 3, 872 CrossRef PubMed.
- H. Braunschweig, P. Brenner, M. Gross and K. Radacki, J. Am. Chem. Soc., 2010, 132, 11343 CrossRef CAS PubMed.
- A. Wagner, E. Kaifer and H.-J. Himmel, Chem.–Eur. J., 2013, 19, 7395 CrossRef CAS PubMed.
- H. Braunschweig, Q. Ye, A. Vargas, R. D. Dewhurst, K. Radacki and A. Damme, Nat. Chem., 2012, 4, 563 CrossRef CAS PubMed.
- Inorganometallic Chemistry, ed. T. P. Fehlner, Springer, New York, 1992 Search PubMed.
- G. Nikonov, J. Organomet. Chem., 2001, 635, 24 CrossRef CAS.
- H. Fan, B. C. Fullmer, M. Pink and K. G. Caulton, Angew. Chem., Int. Ed., 2008, 47, 9112 CrossRef CAS PubMed.
- C. S. Tredget, E. Clot and P. Mountford, Organometallics, 2008, 27, 3458 CrossRef CAS and references therein.
- J. Feldman and R. R. Schrock, Prog. Inorg. Chem., 1991, 39, 1 CrossRef.
- P. E. Romero and W. E. Piers, J. Am. Chem. Soc., 2005, 127, 5032 CrossRef CAS PubMed.
- C. H. Suresh and N. Koga, Organometallics, 2004, 23, 76 CrossRef CAS.
- S. Scheins, M. Messerschmidt, M. Gembicky, M. Pitak, A. Volkov, P. Coppens, B. G. Harvey, G. C. Turpin, A. M. Arif and R. D. Ernst, J. Am. Chem. Soc., 2009, 131, 6154 CrossRef CAS PubMed.
- B. Wrackmeyer and W. Biffar, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1979, 34, 1270 Search PubMed.
- K. Wallace, A. Liu, J. Dewan and R. Schrock, J. Am. Chem. Soc., 1988, 110, 4964 CrossRef CAS.
- R. R. Schrock, R. T. DePue, J. Feldman, C. J. Schaverien, J. C. Dewan and A. H. Liu, J. Am. Chem. Soc., 1988, 110, 1423 CrossRef CAS.
- J. Feldman, W. M. Davis and R. R. Schrock, Organometallics, 1989, 8, 2266 CrossRef CAS.
- J. Feldman, W. M. Davis, J. K. Thomas and R. R. Schrock, Organometallics, 1990, 9, 2535 CrossRef CAS.
- R. R. Schrock, J. S. Murdzek, G. C. Bazan, J. Robbins, M. DiMare and M. O'Regan, J. Am. Chem. Soc., 1990, 112, 3875 CrossRef CAS.
- W. C. P. Tsang, R. R. Schrock and A. H. Hoveyda, Organometallics, 2001, 20, 5658 CrossRef CAS.
- W. C. P. Tsang, J. Y. Jamieson, S. L. Aeilts, K. C. Hultzsch, R. R. Schrock and A. H. Hoveyda, Organometallics, 2004, 23, 1997 CrossRef CAS.
- C. Schaverien, J. Dewan and R. Schrock, J. Am. Chem. Soc., 1986, 108, 2771 CrossRef CAS.
- J. B. Lee, G. J. Gajda, W. P. Schaefer, T. R. Howard, T. Ikariya, D. A. Straus and R. H. Grubbs, J. Am. Chem. Soc., 1981, 103, 7358 CrossRef CAS.
- B. G. Harvey, A. M. Arif and R. D. Ernst, J. Chem. Crystallogr., 2011, 41, 1391 CrossRef CAS PubMed.
- K. Mashima, M. Kaidzu, Y. Nakayama and A. Nakamura, Organometallics, 1997, 16, 1345 CrossRef CAS.
- P. E. Romero and W. E. Piers, J. Am. Chem. Soc., 2007, 129, 1698 CrossRef CAS PubMed.
- E. F. van der Eide, P. E. Romero and W. E. Piers, J. Am. Chem. Soc., 2008, 130, 4485 CrossRef CAS PubMed.
- C. N. Rowley, E. F. van der Eide, W. E. Piers and T. K. Woo, Organometallics, 2008, 27, 6043 CrossRef CAS.
- C. H. Suresh and N. Koga, Organometallics, 2006, 25, 1924 CrossRef CAS.
- S. F. Pedersen, R. R. Schrock, M. R. Churchill and H. J. Wasserman, J. Am. Chem. Soc., 1982, 104, 6808 CrossRef CAS.
- C. H. Suresh, J. Organomet. Chem., 2006, 691, 5366 CrossRef CAS PubMed.
- C. H. Suresh and M. H. Baik, Dalton Trans., 2005, 2982 RSC.
- A. G. Wenzel and R. H. Grubbs, J. Am. Chem. Soc., 2006, 128, 16048 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |