Merging the best of both worlds: hybrid lipid-enveloped matrix nanocomposites in drug delivery
Koen
Raemdonck
a,
Kevin
Braeckmans
ab,
Jo
Demeester
a and
Stefaan C.
De Smedt
*a
aGhent Research Group on Nanomedicines, Laboratory of General Biochemistry and Physical Pharmacy, Faculty of Pharmaceutical Sciences, Ghent University, Harelbekestraat 72, B-9000 Ghent, Belgium. E-mail: stefaan.desmedt@ugent.be; Fax: +32 9 264 81 89; Tel: +32 9 264 80 76
bCenter for Nano- and Biophotonics, Ghent University, Belgium
First published on 8th October 2013
Abstract
The advent of nanotechnology has revolutionized drug delivery in terms of improving drug efficacy and safety. Both polymer-based and lipid-based drug-loaded nanocarriers have demonstrated clinical benefit to date. However, to address the multifaceted drug delivery challenges ahead and further expand the spectrum of therapeutic applications, hybrid lipid–polymer nanocomposites have been designed to merge the beneficial features of both polymeric drug delivery systems and liposomes in a single nanocarrier. This review focuses on different classes of nanohybrids characterized by a drug-loaded polymeric matrix core enclosed in a lipid shell. Various nanoengineering approaches to obtain lipid–polymer nanocomposites with a core–shell nanoarchitecture will be discussed as well as their predominant applications in drug delivery.
 Koen Raemdonck | Koen Raemdonck obtained his MS degree in Pharmaceutical Sciences at Ghent University in 2004. In the same year he became a doctoral fellow of the IWT-Vlaanderen. He initiated his PhD research in the laboratory of General Biochemistry and Physical Pharmacy under the supervision of Prof. Stefaan De Smedt. His doctoral research evaluated the application of nanogels for the controlled delivery of nucleic acids. Since 2010, he has been a postdoctoral fellow of the Research Foundation-Flanders (FWO-Vlaanderen). He received the biennial National Prize of the Belgian Society of Pharmaceutical Sciences in 2011. His current research is focused on bio-inspired delivery approaches for small RNA therapeutics. |
 Kevin Braeckmans | Kevin Braeckmans obtained an MS in Physics at Ghent University (Belgium) in 1999, and received his PhD from Ghent University (Belgium) on advanced optical microscopy methods, for which he received first prize (FBBF) in 2005. He stayed in the group of Prof. Braüchle at the Ludwig-Maximilians Universität München, developing algorithms for single particle tracking. Later, he returned to Ghent University as a FWO postdoctoral researcher. In 2008 he became a professor in the Faculty of Pharmaceutical Sciences of Ghent University where he leads the Biophotonic Imaging Group, focusing on biophotonic methods for studying the interaction of nanoparticles with biological barriers and improving nanomedicine mediated drug delivery. |
 Jo Demeester | Joseph Demeester graduated in Pharmaceutical Sciences at Ghent University in 1974, earned a PhD in Pharmaceutical Sciences in 1980 and became professor in 1989. He became laureate of the Belgian Royal Academy of Sciences in 1980 and First Laureate of the Travel grant of the Ministry of Education in 1981. He did postdoctoral research on light scattering and rheology at the Institute of Physical Chemistry of the University of Graz. Since 1994 he has been the Director of the International Centre for Standards of the International Pharmaceutical Federation (F.I.P.) and since 1998 he has been an expert in the Group on Biological Products of the European Pharmacopeia. In 2003, he became President of the F.I.P. Enzyme Commission. |
 Stefaan De Smedt | Stefaan De Smedt studied pharmacy at Ghent University (Belgium) and graduated in 1995. He joined the pharmaceutical development group of Janssen Research Foundation. In 1999 he became a professor at Ghent University where he chairs the Ghent Research Group on Nanomedicines. Since 2004 he has been serving as European Associate Editor of the Journal of Controlled Release. His research is at the interface between drug delivery, biophysics, material sciences and advanced optical imaging. He received the 2006 CRS Young Investigator Award and the 2010 APV Research Award for Outstanding Achievements in Pharmaceutical Sciences. He is the scientific founder of spin-off Memobead Technologies. |
1. Introduction
Nanotechnology has found widespread use in pharmaceutical and biomedical applications, including improved delivery of small molecule drugs, macromolecular therapeutics and/or imaging agents. To improve the overall therapeutic index of existing low molecular weight therapeutics, their encapsulation in nanosized carriers has demonstrated great benefit. Likewise, to attain the full therapeutic potential of novel biopharmaceuticals such as proteins, peptides and nucleic acids, formulating them in nanoparticles (NPs) is generally required.1–10 Rational design of drug-loaded nanocarriers, so-called nanomedicines, may aid in overcoming physicochemical limitations associated with free drugs, i.e. solubility and stability in aqueous media. NPs should additionally allow facile encapsulation of drug cargo with high efficiency and prevent premature drug release. Ultimately, NPs ideally encompass multiple functionalities devised to overcome the various extra- and intracellular barriers imposed by the human body. The latter implies avoiding fast renal clearance, protecting the active payload against (enzymatic) degradation, bypassing the body's immune defenses, improving pharmacokinetics toward the diseased tissue and, in case an intracellular target is envisioned, actively promoting intracellular drug delivery.1–3,8,11 If successful, nanomedicines may thus enhance the fraction of the administered dose that effectively reaches the intended target site and mitigate potential off-target toxicity. Moreover, in the light of personalized medicine, flexible design of targeted drug carriers that enable tailored controlled release of drugs or synergistic drug combinations with distinct physicochemical properties could provide a definite asset.12The dominant types of conventional nanomedicines to date, i.e. liposomes7,13 and polymeric NPs,2,8 generally have failed to combine all of the complex requirements outlined above. Liposomes can be defined as self-assembled vesicles comprising one or multiple concentric lipid bilayers that enclose an aqueous core. The versatility of liposomes is reflected in their ability to carry both hydrophilic and hydrophobic drugs in the aqueous lumen and lipid bilayer, respectively, their outstanding safety profile and ease of surface functionalization with hydrophilic polymers like poly(ethylene glycol) (PEG) and/or ligands to obtain long-circulating targeted nanomedicines. Decades of research has converged in the clinical application of liposomal formulations for a wide variety of drug molecules.7,13 In spite of the many favorable characteristics they exhibit, liposomes may experience low loading efficiencies, especially for hydrophobic drugs. In addition, depending on their composition, liposomes may suffer from poor stability in vivo, resulting in unwanted burst drug release.13–15 Polymeric NPs may provide a valuable alternative to overcome some of the above-mentioned limitations, as they generally demonstrate outstanding drug loading capacity for drugs and/or contrast agents with diverging physicochemical properties. Moreover, progress in polymer chemistry enables the design of NPs with ample control over their nanoarchitecture and biophysical properties which facilitates application in controlled and triggered drug release strategies. On the other hand, issues have been raised regarding the biocompatibility of certain polymeric materials and the potential heterogeneity and low density of chemical surface functionalization.2,8,16–18
To address the multifaceted drug delivery challenges outlined above, several research groups have turned to the design of hybrid lipid–polymer nanocomposites with the primary aim to combine the most valuable features of both polymeric drug delivery systems and liposomes in the design of lipid–polymer nanocomposites.14,19–22 Many classes of lipid-coated nanomaterials have been described in the literature. For example, lipopolyplexes can be defined as self-assembled ternary electrostatic complexes comprising a nanosized polyanion:polycation complex (polyplex) subsequently coated with (oppositely charged) lipids.23,24 The lipid bilayer/multilayer coat confers additional colloidal stability to the ensemble and serves to incorporate key functional moieties, as demonstrated in numerous publications by the groups of Leaf Huang25–29 and Hideyoshi Harashima.30–34 This type of hybrid nanocomposites has most often been employed to encapsulate and deliver nucleic acid therapeutics. A remarkable example, highlighting the drug delivery potential of these hybrid NPs for siRNA, has been put forward by Peer et al. They succeeded in designing multifunctional antibody-targeted lipopolyplexes to direct anti-inflammatory siRNA toward gut-associated leucocytes following systemic administration to tackle experimentally induced colitis in mice.23 Furthermore, in the emerging field of nanotheranostics, aiming to merge both therapeutic and imaging modalities in a single nanostructure, inorganic NPs like semiconductor nanocrystals (quantum dots) and magnetic NPs have been provided with a lipid coat to optimize their in vivo application in fluorescence or magnetic resonance imaging (MRI) respectively.35–37 Although of outstanding interest for drug delivery, the nanocomposites described above are beyond the scope of this review. Here, we will predominantly focus our attention on lipid-enveloped polymer matrix nanoparticles, typically defined by a core–shell architecture in which a (drug-loaded) matrix core is surrounded by a lipid shell. Polymeric matrix nanoparticles can be defined as colloidal 3D polymer networks with a size ranging from 10 nm to 1 μm, in which the drug can be physically complexed/dispersed/dissolved or chemically coupled to the polymer chains.3 In contrast to conventional polyplexes, in a matrix NP the encapsulated therapeutic compound typically does not structurally contribute to NP formation.38
The consequential advantages of combining lipids and polymers in a hybrid drug delivery platform are situated at many levels.14,19–22 Efficient drug encapsulation can be achieved both in the polymeric core and in the surrounding lipid envelope. Drug release can be controlled by polymer degradation but also modulated by the presence of the lipid coat that acts as a diffusional barrier. In this way, unwanted drug leakage from the nanocarrier can be prevented. Furthermore, the lipid layer may hinder the influx of water, tempering polymer hydrolysis and slowing down drug release. The lipid shell may impart many other valuable traits such as the ease of incorporating PEGylated lipids and targeting ligands, the obscuration of toxicity associated with the polymer core, the improvement of colloidal stability and enhancement of intracellular drug delivery. In addition, the polymer matrix core may also contribute to the structural integrity of the lipid coat.
Our major aim is to discuss various inspiring reports on lipid–polymer nanocomposites that successfully implemented this synergistic drug delivery approach, focusing on lipid-coated solid hydrophobic polyester NPs, mesoporous silica NPs (MSNPs) and hydrophilic hydrogel nanoparticles (nanogels). Different preparation methods to obtain lipid–polymer nanocomposites with a core–shell nanoarchitecture will be briefly described and novel nanoengineering approaches will be highlighted. Finally, we present an overview of drug delivery applications, mainly in the context of cancer therapy and vaccination.
2. General considerations on nanomedicinal drug delivery
Upon instillation of NPs in a biological medium or in contact with target cells, the size, shape and surface properties of the NP will largely define its factual behavior at the so-called nano–bio interface.39–41 With regard to the polymer–lipid core–shell NPs described here, this implies that the external lipid shell (and the modifications it harbors) will significantly determine the dynamic interactions with the myriad of biological components the hybrid NP will encounter. Following parenteral administration, in general unmodified NPs are rapidly removed from the blood circulation via the mononuclear phagocytic system (MPS), by which NPs will predominantly accumulate in the liver and spleen.40–42 The latter is facilitated in a passive manner via NP extravasation through the sinusoidal capillaries in these organs that are characterized by a fenestrated discontinuous endothelium and in an active fashion by the deposition of particular blood proteins that opsonize the NP surface and mediate recognition and clearance by hepatic/splenic phagocytes. Interaction with circulating proteins can additionally evoke NP aggregation, causing them to be trapped in the lung capillary bed.40 To reduce MPS clearance and maintain colloidal stability, it has become common practice to shield the NP surface with a hydrophilic stealth layer, most commonly composed of poly(ethylene glycol) (PEG), which is registered by the US Food and Drug Administration (FDA) as a GRAS polymer (“Generally Recognized As Safe”).2,43 In this way, the blood circulation time can be significantly extended, which is of particular interest to augment the passive extravasation of nanomedicines in solid tumors and sites of inflammation, as these are characterized by an increased vascular permeability. Besides the defective angiogenesis in cancerous tissues, accounting for the leaky vasculature, also the lymphatic drainage is (partially) impaired thus contributing to NP accumulation in the tumor interstitium. This so-called enhanced permeation and retention (EPR) effect may allow the infiltration of nanosized drug carriers up to ∼400 nm in diameter,1,44 although this is largely dependent on tumor type and microenvironmental factors (Fig. 1). Even within the same tumor, the endothelial permeability may be quite heterogeneous.2,40,45 In general, to experience optimal EPR, NPs should be larger than 10 nm to avoid glomerular filtration by the kidneys and should not exceed 200 nm in size.1,41 Nevertheless, it is known that within this size range the NPs are also able to penetrate into the liver tissue.10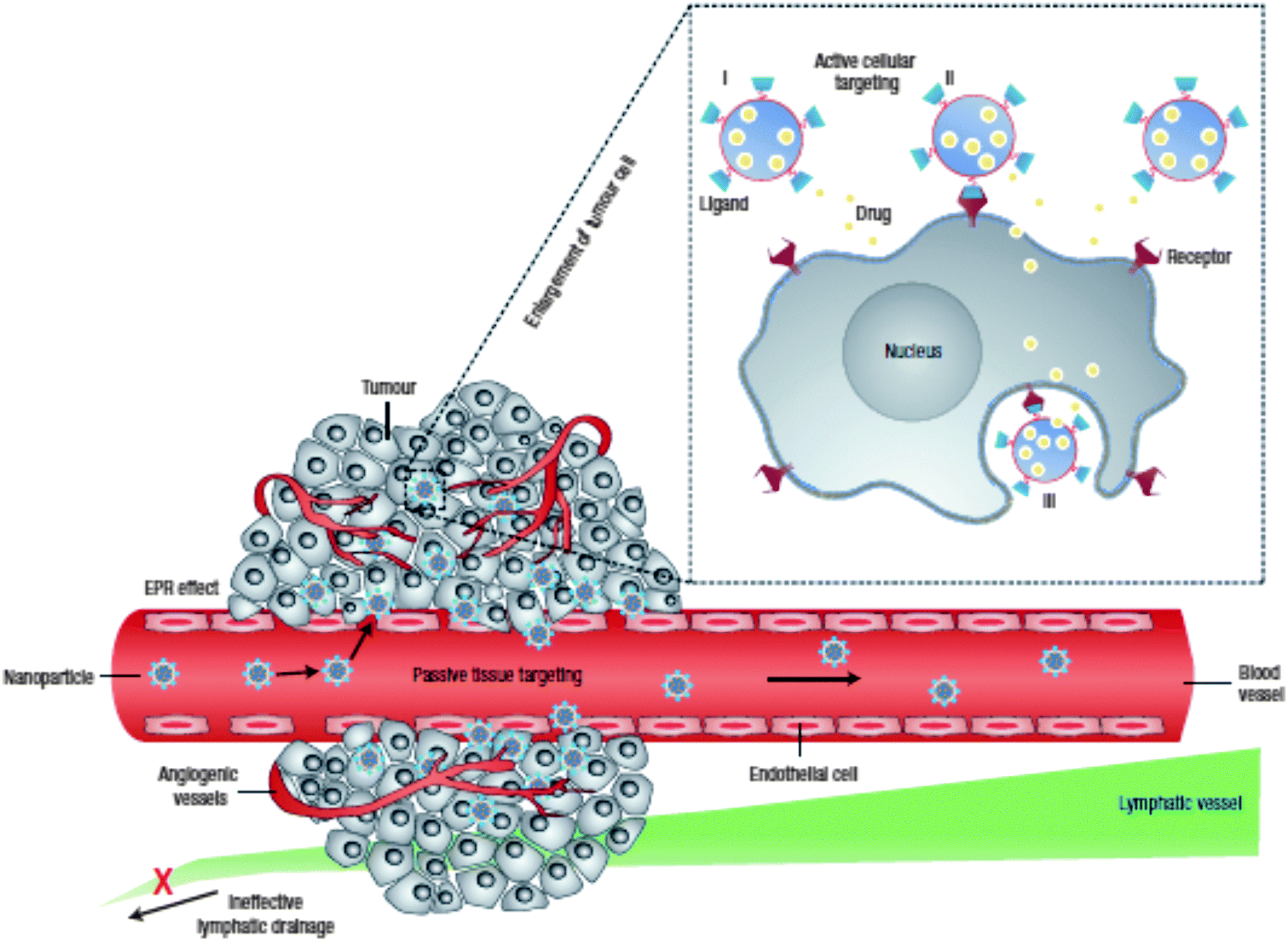 | ||
| Fig. 1 Combined passive and active tumor targeting. Enhanced tumor tissue accumulation of nanomedicines is achieved via the leaky vasculature and impaired lymphatic drainage, i.e. the enhanced permeation and retention effect (EPR). Decorating the nanomedicines with targeting ligands allows the specific binding of tumor cells followed by receptor-mediated internalization. Drug release can occur in the extracellular environment or directly in the cell after endocytic uptake. The latter delivery method leads to higher intracellular drug concentrations that can bypass efflux-based drug resistance. (Reprinted with permission from Macmillan Publishers Ltd: Nature Nanotechnology,1 Copyright 2007.) | ||
In the situation where an extracellular molecular target is envisioned, controlled release of the active compound in the target tissue interstitium may suffice for therapeutic benefit. However, many drugs require delivery across the cellular barriers. Hydrophobic small molecule anticancer drugs, when released from their nanocarrier in the tissue extracellular matrix, are able to passively diffuse across the plasma membrane of diseased cells to interact with their intracellular target. On the other hand, this uncontrolled arbitrary nature of drug delivery may hamper maintaining active tissue drug levels within an acceptable range and could promote the development of a multi-drug resistant (MDR) phenotype.1,46,47 Cancer cell drug resistance is dominated by the activity of MDR membrane transporter proteins that mediate efflux of a variety of chemotherapeutic drugs. Cancer cells that lack drug efflux mechanisms or only moderately express these transporters will preferentially be affected by the drug, eventually promoting selective survival of a drug resistant cell population. Altogether, these claims advocate targeted intracellular drug delivery via nanomedicines to increase intracellular drug concentrations and bypass efflux pump mediated drug resistance1,10 (Fig. 2).
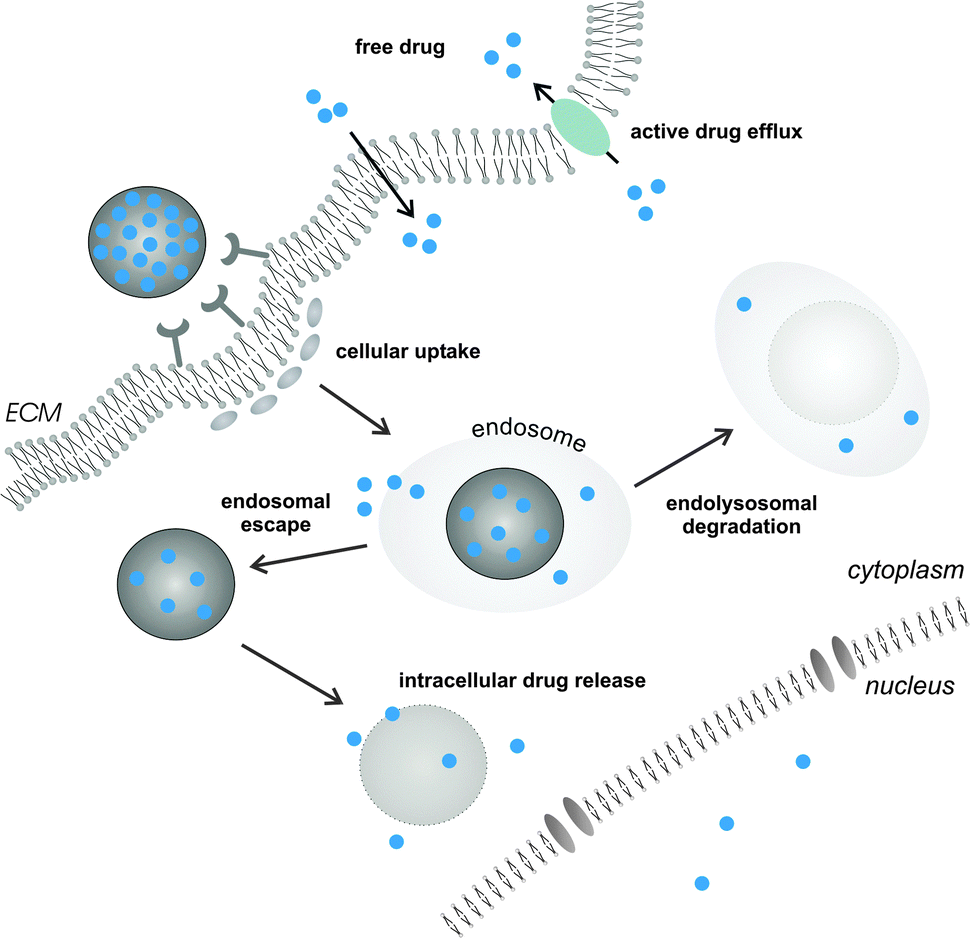 | ||
| Fig. 2 Overview of the predominant intracellular barriers encountered in nanomedicinal drug delivery. Following endocytic uptake, nanomedicines are confined within endosomal vesicles. Escape from the endosomes is required for drugs that need to be delivered in the cell cytoplasm or nucleoplasm and to avoid endolysosomal degradation (e.g. for nucleic acid therapeutics). Small lipophilic drugs can passively permeate across the plasma membrane or endosomal limiting membrane when released in the extracellular matrix (ECM) or endosomal lumen, respectively. Cancer cells can adopt a multidrug-resistant (MDR) phenotype by expressing membrane transporters (e.g. p-glycoprotein) that actively expel various chemotherapeutics, thus lowering the intracellular drug concentration. | ||
Likewise, the novel class of biopharmaceuticals mainly comprises large, hydrophilic and charged molecules (e.g. peptides and nucleic acids) that require the aid of an optimally designed nanocarrier to reach the intracellular site of action. It is well-established that nanomedicines as a rule are internalized via distinct endocytic entry portals as a function of cell type and physicochemical properties.5,6,48,49 For instance, NP size is an important regulator that influences cellular uptake and subsequent intracellular trafficking.48 Larger NPs (>500 nm) are preferentially engulfed via phagocytosis by specialized cell types of the immune system, i.e. macrophages, monocytes and dendritic cells.48,50 Of note, in (cancer) vaccination or anti-inflammatory treatments these cells may in fact be the desired targets for drug delivery when aiming to modulate local immune responses and incite humoral and/or cellular (anti-tumor) immunity.51–55 Smaller NPs, in the acceptable size range for in vivo biodistribution, can be internalized via a clathrin-mediated or clathrin-independent mechanism.48,50 On the one hand, cell uptake can be triggered by non-specific hydrophobic or electrostatic interactions with the target cell membrane. Alternatively, surface functionalization of NPs with bioligands, such as antibodies,53,56 peptides57,58 or aptamers,59–61 may allow for cell-specific recognition and internalization via receptor-mediated endocytosis. Following endocytic uptake, drug-loaded NPs tend to accumulate in endolysosomes, packed with acid hydrolases, or are prone to exocytosis in which the luminal content of multivesicular late endosomes is again expelled into the surrounding medium.62 To avoid the endolysosomal degradation pathway, many strategies to promote drug delivery into the cytosol have been reported in the literature. Small lipophilic drugs, able to passively permeate across the endosomal limiting membrane, can escape from the endosomal compartment unaided.2 For cytosolic delivery of (large) hydrophilic compounds, nanocarriers have to rely on their intrinsic pH buffering capacity, osmotic swelling and/or fusogenic activity to disrupt endosomal membrane integrity.6,63 A very recent report, aiming to unravel the intracellular processes that govern the efficiency of liposome-mediated small interfering RNA (siRNA) delivery, demonstrated that prolonging the intracellular retention of the NPs in late endosomes substantially improved RNA interference (RNAi) mediated gene silencing.62 This study suggests that strategies able to bypass standard endocytic recycling might prove beneficial toward cytosolic drug delivery, especially when endosomal escape is the main limiting factor. Altogether, it is of key importance to combine a detailed understanding of the underlying extra- and intracellular barriers together with innovations on the interface of materials chemistry, nanotechnology, pharmacy and medicine.48,64–66 Novel insight into the biological behavior of nanomedicines related to successful drug delivery should fuel the rational design of next generation nanomedicines in order to unleash the full benefits of nanomedicinal drug delivery. The favorable features associated with lipid–polymer matrix nanocomposites for drug delivery across the many barriers and advances in lipid–polymer nanoengineering might therefore act in concert to accelerate the lab-to-clinic transition of nanomedicinal products.
3. Lipid–polymer nanoengineering
Given the often complex composition of lipid–polymer nanocomposites and the broad range of techniques that are available for their production, a myriad of hybrid nanoformulations can be constructed. The properties of the bulk material and the applied synthesis approach will largely govern the physicochemical properties of the resulting NPs (e.g. size, stability, drug encapsulation and release) and hence also their biological performance as a drug delivery carrier. Broadly, the currently available synthesis techniques can be classified into two main categories, i.e. a two-step and a single-step approach.19 Both can be defined as bottom-up approaches, taking advantage of the inherent properties of the building blocks to obtain the desired supramolecular structure.67 Commonly, a two-step synthesis involves the formation of polymeric matrix NPs and liposomes via separate protocols after which they are merged into lipid–polymer core–shell nanohybrids in the final step. Importantly, a two-step synthesis also implies that the size and shape of the resulting nanohybrid will be largely governed by that of the polymeric template. Although multicomponent hybrid NPs may definitely expand the performance of traditional nanomedicines, one of the major limitations is inadequate control over the production process. For this reason, ample research effort has also gone toward the optimization of single-step protocols, mostly based on the controlled self-assembly of the various molecular components in a one-pot synthesis. Both preparation methods, if available, will be detailed below for lipid-coated polyester NPs, MSNPs and nanogels, prior to discussing potential drug delivery applications in Section 4.3.1. Lipid-coated polyester nanoparticles
Synthetic polymeric NPs, e.g. constructed from the polyester poly(lactic acid) (PLA) and its copolymer with poly(glycolic acid), i.e. poly(lactic-co-glycolic acid) (PLGA), have been studied extensively for drug delivery purposes over the last few decades.2,3,8,68–70 Their biodegradable and biocompatible features largely account for their widespread use in biomedical applications and justify the approval of PLGA micro- and nanoparticles for human use in drug delivery by the FDA and the European Medicine Agency (EMA).2,69 PLGA and PLA NPs slowly degrade via surface erosion in an aqueous environment due to hydrolysis of the ester linkages in the (co)polymer backbone, to produce the original monomers lactic acid and glycolic acid. The latter can be further metabolized in the human body, which explains the favorable toxicity profile of these polyesters.71 A multitude of therapeutic molecules have already been encapsulated in the polymer matrix, ranging from small hydrophobic chemotherapeutics to large hydrophilic macromolecules, such as proteins and nucleic acids. The kinetics of drug release from PLGA/PLA-based matrices is mainly governed by polymer matrix degradation and drug diffusion and can be controlled over periods ranging from hours to weeks.70 Conjugating drug molecules directly to the PLA/PLGA chains allows to prolong drug release as a function of ester hydrolysis without the risk of premature diffusive drug release. The latter might be of particular value for sub-100 nm particles that are endowed with a large surface-to-volume ratio and are typically associated with a high risk of substantial burst release of drug molecules adsorbed on the NP surface.72,73A variety of methodologies are available to fabricate polymeric NPs, commonly based on the self-assembly of preformed (co)polymers via an emulsion–solvent evaporation or nanoprecipitation technique (Fig. 3), in which the latter usually yields the smallest sized NPs.2,3,69,70 Both techniques typically lead to spherical matrix NPs. Of note, DeSimone's group reported on an alternative soft lithographic technique termed PRINT (Particle Replication in Non-wetting Templates) to engineer monodisperse submicron PLGA particles of distinct yet well-defined sizes and shapes (Fig. 4).74,75 The PRINT process employs perfluorinated polyether elastomeric molds for imprint lithography in which the shape of each individual particle is determined by the cavities present in the mold.76
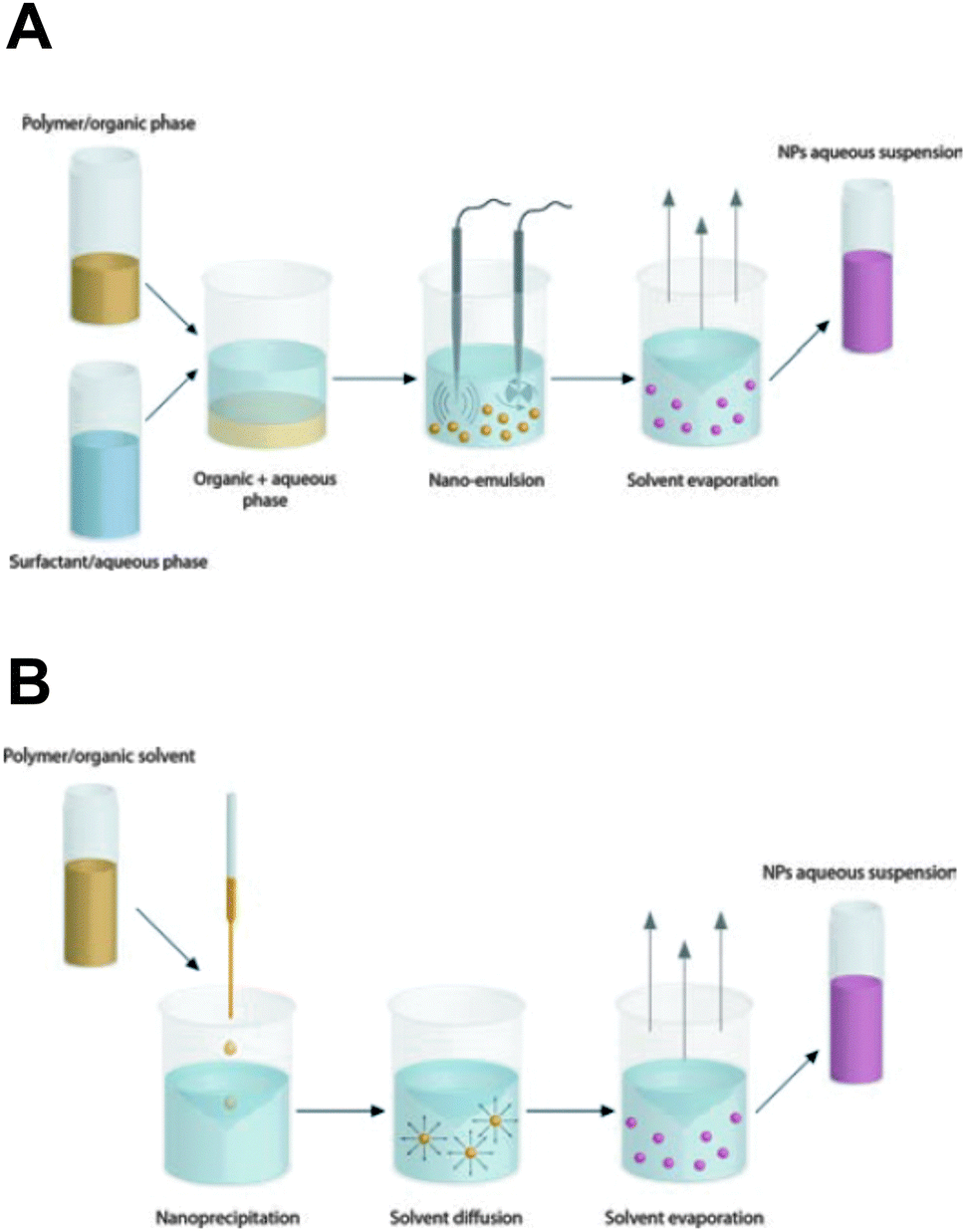 | ||
| Fig. 3 The most common techniques for the preparation of polymeric nanoparticles are (A) emulsion–solvent evaporation and (B) nanoprecipitation. The former method involves the emulsification of a non-water miscible organic phase (containing polymer and hydrophobic drugs) in an aqueous phase supplemented with appropriate surfactants. Subsequent removal of the organic solvent transforms the nanoscopic emulsion droplets into solid drug-loaded polymeric NPs. In contrast, the nanoprecipitation technique requires the dissolution of polymer and drug in a water-miscible organic solvent that is added dropwise into the water phase (the non-solvent). Nanoprecipitation occurs through rapid solvent diffusion and subsequent evaporation. (Reproduced with permission from ref. 3. Copyright 2012 The Royal Society of Chemistry.) | ||
 | ||
| Fig. 4 Particle replication in non-wetting templates (PRINT) allows fabrication of monodisperse PLGA nano- and microparticles of well-defined size and shape. Scale bars (A) 5 μm, (B) 4 μm, (C) 3 μm, (D) 10 μm, (E) 3 μm, and (F) 20 μm. (Reprinted with permission from ref. 74. Copyright 2011 American Chemical Society.) | ||
Multiple functionalities can be incorporated into polymeric NP design77 to assist in drug encapsulation, drug release, in vivo biodistribution, cellular targeting and intracellular trafficking, which has recently been comprehensively reviewed by others.2,3,8 One particular strategy to modulate the surface of polymeric NPs, and hence also influence their behavior at the nano–bio interface, is by depositing a stabilized (phospho)lipid layer onto the hydrophobic NP core. As for liposomal NPs, this lipid layer can be employed to anchor amphiphilic PEGylated lipids to the surface of the particles and/or conjugate targeting moieties. However, another important motivation to invest in the development of these more complex lipid–polymer hybrids lies within their anticipated improved in vivo stability, increased drug loading and added control over the drug release process.2,19–21 The most straightforward approach to obtain lipid–polymer NPs with a core–shell morphology is by mixing preformed polymeric NPs with preformed liposomes at the desired vesicle to nanoparticle ratio.78–81 Electrostatic interaction between the anionic PL(G)A surface and cationic liposomes is frequently applied as the driving force behind the lipid coating process.79,80 Unilamellar liposomal vesicles can easily be prepared via extrusion, sonication or high pressure homogenization.7 The same techniques are often also applied to the NP–liposome mixture to provide additional energy input to the mixing process and facilitate lipid reorganization and fusion onto the NP surface. Alternatively, drug loaded core NPs can also be used to directly hydrate a preformed lipid film, followed by an appropriate sizing method, to obtain lipid-coated polymer hybrids.73,82,83 Irrespective of the applied method, the mixing of polymeric NPs and lipids usually occurs at a temperature exceeding the gel-to-liquid phase transition temperature of the lipid.
The production of lipid–polymer hybrid NPs via two (or more) consecutive steps may lead to marked batch-to-batch variation, which could conceivably alter the drug delivery performance and thus hamper clinical translation. Many research groups therefore invested in the optimization of single-step formulation strategies, allowing a more feasible and reproducible synthesis of lipid–polymer hybrids with better control over the physicochemical characteristics of the final hybrid NP construct. The single-step approach implies the mixing of polymer and lipid solutions prior to nanoprecipitation or emulsion–solvent evaporation to self-assemble the lipid-coated polymer NPs. Herein the used phospholipids themselves act as stabilizers instead of the conventionally used poly(vinyl alcohol) (PVA) or polypropylene oxide–polyethylene oxide block copolymers.2,84–86 Depending on the preparation method used, solid NPs with lipid monolayer, bilayer or multilayer are produced. The group of Farokhzad optimized the single-step self-assembly of drug-loaded PLGA NPs encapsulated within a lecithin lipid monolayer containing interspersed PEGylated phospholipids via a modified nanoprecipitation method. To this end, an acetonitrile solution of PLGA and hydrophobic drug was mixed with an aqueous ethanolic (PEGylated) lipid dispersion, preheated above the lipid transition temperature. The mixture was subsequently stirred for 2 h to allow self-assembly of lipid-coated PLGA NPs via solvent-diffusion and evaporation of the organic solvent.60,87 The presence of a lipid monolayer resulted in reduced drug release rates as a function of lipid coverage, as demonstrated for the small hydrophobic anticancer drug docetaxel, likely due to the delayed drug diffusion across the lipid layer and the reduced water penetration inside the PLGA core slowing down ester hydrolysis.60,87 PEGylated lipid–polymer NPs can also be fabricated using PEGylated amphiphilic polyesters as precursor materials instead of applying PEGylated lipids. Cationic lipid–polymer hybrid NPs were thus produced via a single-step nanoprecipitation of cationic lipid with methoxy-poly(ethylene glycol)-block-poly(lactide) (mPEG–PLA). The resulting particles consist of a hydrophobic PLA core with a non-fouling PEG shell and a monolayer of cationic lipid at the core–shell interface.88 In contrast to the lipid-monolayer described above obtained via nanoprecipitation, Bershteyn et al. could clearly visualize lipid-bilayer coated particles via cryo-TEM imaging, obtained with their optimized emulsion–solvent evaporation protocol. The authors could show that the finally obtained surface structure of the hybrid NPs – distinguishing single bilayer shell, multilayer onion or flower-like lipid configurations – was highly dependent on the amount of lipids used and the lipid composition.89 However, a major impediment to the nanoprecipitation and emulsion–solvent evaporation technique is the poor encapsulation of hydrophilic therapeutics, e.g. pharmaceutical proteins and peptides, which can be resolved by virtue of a double (water-in-oil-in-water) emulsification process.2,3,69 Encapsulation of ovalbumin (OVA) as a model protein antigen in lipid-coated PLGA NPs obtained via a double-emulsion solvent evaporation self-assembly process was successfully demonstrated by Stephan and coworkers. In comparison with liposomes of equal composition, the lipid-coated PLGA NPs could encapsulate higher amounts of the OVA peptide and showed markedly delayed release kinetics.90
From a manufacturing perspective, the assembly of multiple molecular components with distinct physicochemical properties into complex nanocomposites could additionally obstruct transition from lab-scale to large-scale synthesis. To facilitate the scaling up of the fabrication process, various nanoengineering approaches have recently been explored. For instance, a single step sonication method was proposed in order to render the nanoprecipitation production process less laborious and time-consuming. By replacing the commonly applied heating, vortexing and solvent evaporation steps60 with a single bath sonication step, the required production time to obtain lipid-monolayer coated PLGA NPs via nanoprecipitation could be significantly reduced without affecting their physicochemical properties.91 Nevertheless, this approach is practicable only at the lab-scale and batch-to-batch variations in particle synthesis still cannot be excluded. In response to these constraints, the same authors described the production of lipid–polymer hybrids via a continuous flow-confined mixing protocol, using a multi-inlet vortex reactor (MIVR) (Fig. 5).92 The latter device has already been evaluated for the large-scale production of a multitude of NPs.84 A typical MIVR geometry is composed of two or four radially symmetric inlets converging into a cylindrical mixing chamber. Separate inlets organize a continuous flow of organic phase (containing the dissolved hydrophobic polymers) and antisolvent aqueous phase (containing the dispersed lipids) into the central mixing area, initiating instantaneous nanoprecipitation and self-assembly of homogeneous lipid-coated polymer NPs.92 Employing this efficient and rapid mixing method, Fang and coworkers achieved NP production rates >10 g h−1, again without compromising the physicochemical characteristics relative to equivalent hybrid NPs obtained via the previously mentioned lab-scale sonication method.92 Alternatively, microfluidic-based approaches have been gaining momentum in recent years for the well-controlled, reproducible and large-scale manufacturing of polymer NPs via self-assembly.93 Much of the incentive in this context originated from the engineering of microfluidic designs that enable rapid mixing of solvent and antisolvent to obtain small and monodisperse polymeric NPs.93–95 Microfluidic channels can be engineered to encompass internal micromixing structures that enable rapid mixing of focused liquid streams flowing through these microchannels. Valencia and coworkers applied the so-called Tesla micromixing structure to regulate the nanoprecipitation of lipid-coated PLGA NPs.96 In analogy with the MIVR mixing geometry described above, separate inlet streams hydrodynamically focus a flow of organic solvent and aqueous antisolvent through a microchannel with internal Tesla structure to induce rapid homogeneous mixing, resulting in the formation of (PEGylated) lipid–PLGA nanospheres with high reproducibility.96 Aiming to optimize the high-throughput synthesis of these lipid–polymer hybrids, a three-inlet three-dimensional (3D) microfluidic platform was designed that generates two symmetric microvortices at the inlet intersection, controlling rapid mixing of PLGA polymer and lipid (Fig. 6). Interestingly, this platform demonstrated a productivity of up to 3 g h−1 at an optimized flow rate and PLGA concentration, significantly outperforming production rates obtained with the Tesla mixing design.97 Altogether, the compelling technological advances described here appear very promising toward mass production of complex lipid–polymer nanocomposites with acceptable reproducibility and may stimulate to a great extent their clinical translation.
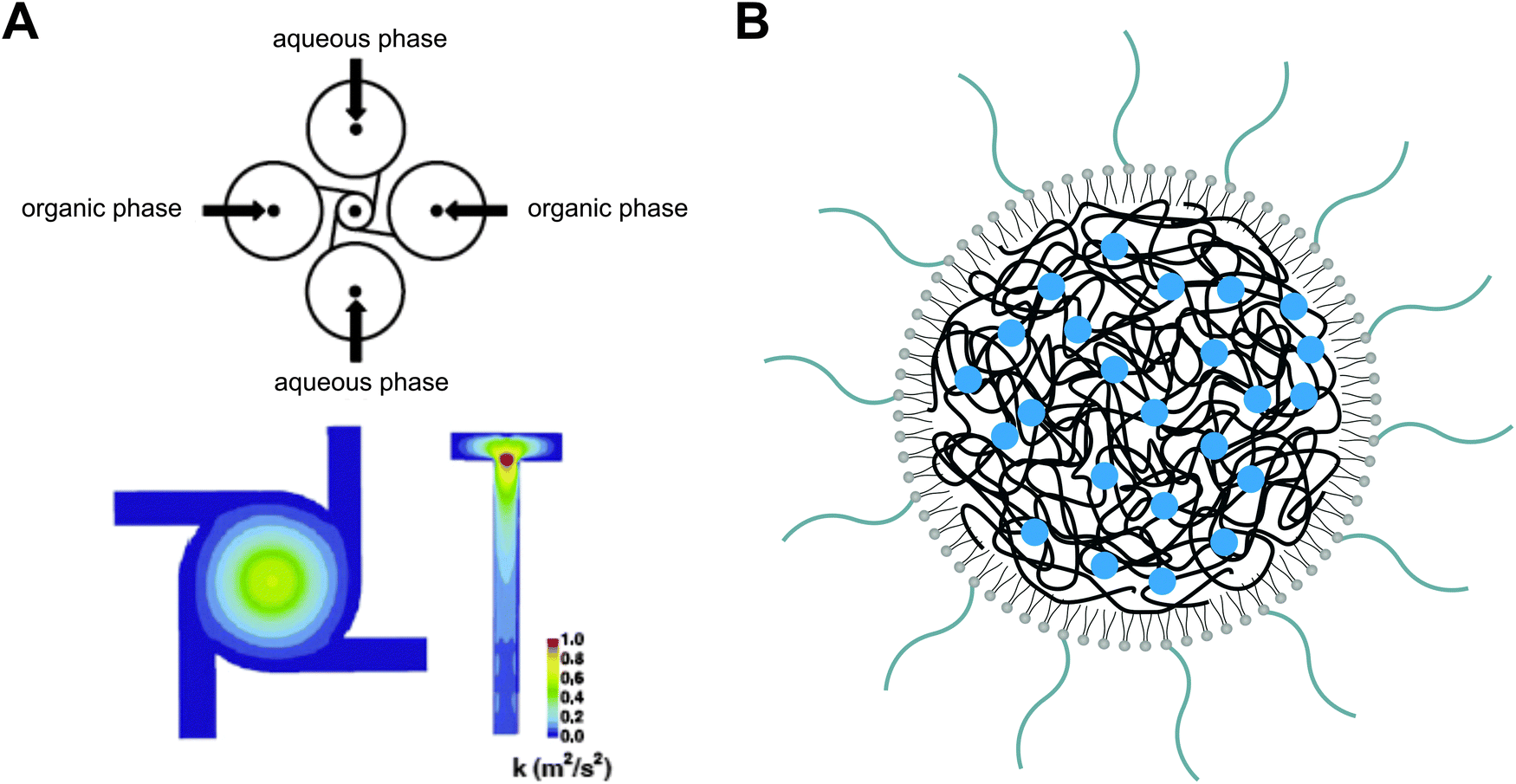 | ||
| Fig. 5 Graphical representation of a multi-inlet vortex reactor (MIVR) as a continuous mixing geometry for polymer nanoprecipitation. (A) In an MIVR geometry, the highest energy mixing of organic (solvent) and aqueous (anti-solvent) phase is observed at the outlet of the mixing chamber. Adapted from ref. 84 with permission from Elsevier, copyright 2011. (B) Schematic drawing of drug-loaded polymeric nanoparticles coated with a PEGylated lipid monolayer, as typically obtained via a nanoprecipitation protocol. Redrawn from ref. 92. | ||
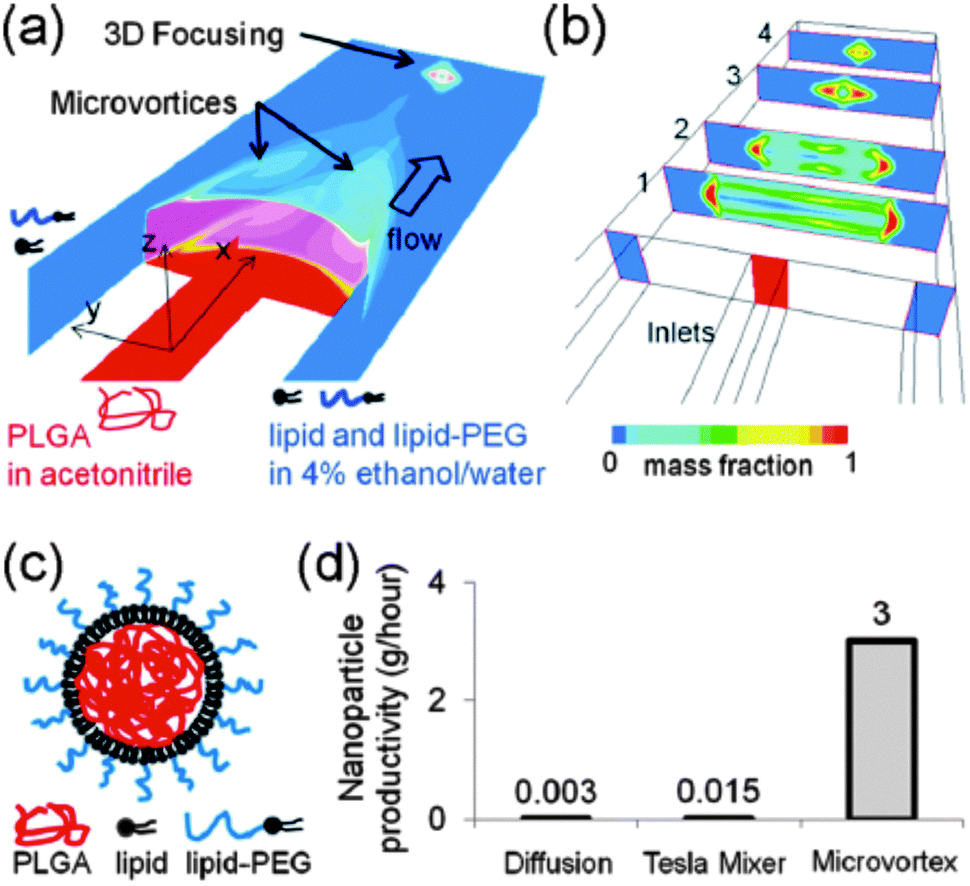 | ||
| Fig. 6 Microfluidic-based nanoprecipitation method allowing the fast and reproducible synthesis of PEGylated lipid-enveloped PLGA nanoparticles with low polydispersity. (a and b) Schematic representation of a three-inlet microfluidic platform yielding microvortices for rapid yet controlled mixing of PLGA polymer and lipid. (c) Structure of the obtained hybrid nanoparticles. (d) Microvortex mixing demonstrates substantially improved production rates when compared with other preparation methods. (Reprinted with permission from ref. 97. Copyright 2012 American Chemical Society.) | ||
3.2. Lipid-coated mesoporous silica nanoparticles
The use of inorganic NPs, e.g. constructed from silica, for drug delivery purposes has increased substantially during the last few decades.67,98 Following the successful deposition of (phospho)lipid bilayers onto planar silica substrates99 and μm-sized silica beads,21 many research groups initiated the decoration of silica nanoparticles with a so-called supported lipid bilayer (SLB).21,100–102 Mornet and co-workers described the successive steps in the adsorption of small unilamellar vesicles (SUVs) of varying composition onto preformed silica nanoparticles via cryotransmission electron microscopy (cryo-TEM). SUV adsorption, deformation and rupture at the lipid–particle interface precede the formation of bilayer patches that subsequently coalesce to form a continuous lipid bilayer shell around the anionic silica template. It was clearly visualized that the SLB closely followed the contours of the silica core. Electrostatic interaction facilitated this process as judged by the superior SLB formation using cationic or zwitterionic lipids opposed to lipid mixtures with a high net negative charge.100 In the latter report solid silica NPs were produced according to the well-known Stöber process. However, from a drug delivery perspective mesoporous silica nanoparticles (MSNPs) are clearly preferred.103 Generally, MSNPs are produced via a modified silica condensation protocol, in which supramolecular assemblies of (cationic) surfactants above their critical micelle concentration (CMC) (i.e. spherical micelles and liquid crystalline mesophases) are applied as soft templates during the sol–gel conversion. Following condensation of the silica precursor around the surfactant head groups and surfactant removal via calcination or solvent extraction, silica NPs are formed with a well-defined uniform internal porous structure.104 Excellent reviews are available describing the various preparation protocols of MSNPs in more detail.103,105–107 MSNPs are endowed with a tailorable pore size (1.5 to several tens of nanometers), a high specific surface area and a large internal pore volume which are advantageous for the encapsulation of drugs and imaging agents.106 A broad spectrum of drugs can be retained in the mesopores as a function of pore size and pore structure as well as direct physicochemical interaction between the guest molecules and the (modified) MSNP surface.103,105–107 Consistent with early reports on solid silica NPs, successful lipid modification of MSNPs has been demonstrated extensively.57,108–113 Sealing the MSNP pores by virtue of a SLB could provide added protection of the precious drug cargo against the harsh in vivo microenvironment and assist in preventing premature drug release in the extracellular matrix. The latter is illustrated by the fast release of a protein drug payload at physiological pH from bare MSNPs (reaching 100% release within 12 h) while the lipid-coated MSNPs did not release relevant amounts of the protein as measured over several days.113 The group of Jeffrey Brinker demonstrated the cargo loading and concomitant pore sealing of conventional negatively charged MSNPs and cationic MSNPs modified with 3-[2-(2-aminoethylamino)ethylamino]-propyltrimethoxysilane (AEPTMS) through fusion of oppositely charged liposomes on the MSNP surface, to obtain the so-called protocells. Again, electrostatic interaction appeared to be the predominant factor driving SLB formation.108,110 Unfortunately, cationic lipids are notorious for their potential in vitro and in vivo toxicity (see also Section 5)114–116 and charged nanomaterials are generally more prone to nonspecific interactions.6,39,41 To reduce nonspecific binding and cellular toxicity, both anionic and cationic MSNPs were incubated with liposomes consisting of zwitterionic lipids, cholesterol and PEGylated lipids (Fig. 7).57 In an alternative approach, Wang and coworkers modified the surface of preformed MSNPs with long hydrophobic tails through reaction with 13-(chlorodimethylsilylmethyl)-heptacosane. The hydrophobized MSNPs were subsequently capped with (PEGylated) phospholipids in an organic solvent mixture. Following solvent evaporation and rehydration in a salt-rich buffer, phospholipid-coated MSNPs are formed via self-assembly of the hydrophobic heptacosyldimethylsilyl groups grafted on the MSNP surface with the fatty acyl chains of the added phospholipids. The resulting PEGylated phospholipid-capped MSNPs displayed superior colloidal stability and resistance to nonspecific protein absorption as compared with unmodified MSNPs.109,117 Likewise, Koole et al. functionalized silica-coated QDs with octadecanol, prior to their dispersion in a chloroform–methanol mixture containing PEGylated lipids. Upon emulsification in an aqueous buffer and evaporation of the organic solvent via a heating step, a monolayer of PEGylated lipids was deposited on the surface of the hydrophobized silica NPs.35 Roggers and coworkers covalently modified MSNPs with a dipalmitoyl monolayer to facilitate phospholipid coating using a comparable solvent evaporation protocol.118 Organic solvents were also employed by Cauda et al., who incubated preformed drug loaded MSNPs in a 40% (v/v) ethanolic lipid solution in order to obtain a defect-free lipid bilayer coating. Upon dilution of the MSNP dispersion in water, decreasing the ethanol content to 5%, the dissolved phospholipids self-assembled into an SLB shell around the MSNP core. This procedure was successfully evaluated for both zwitterionic and cationic lipid mixtures.119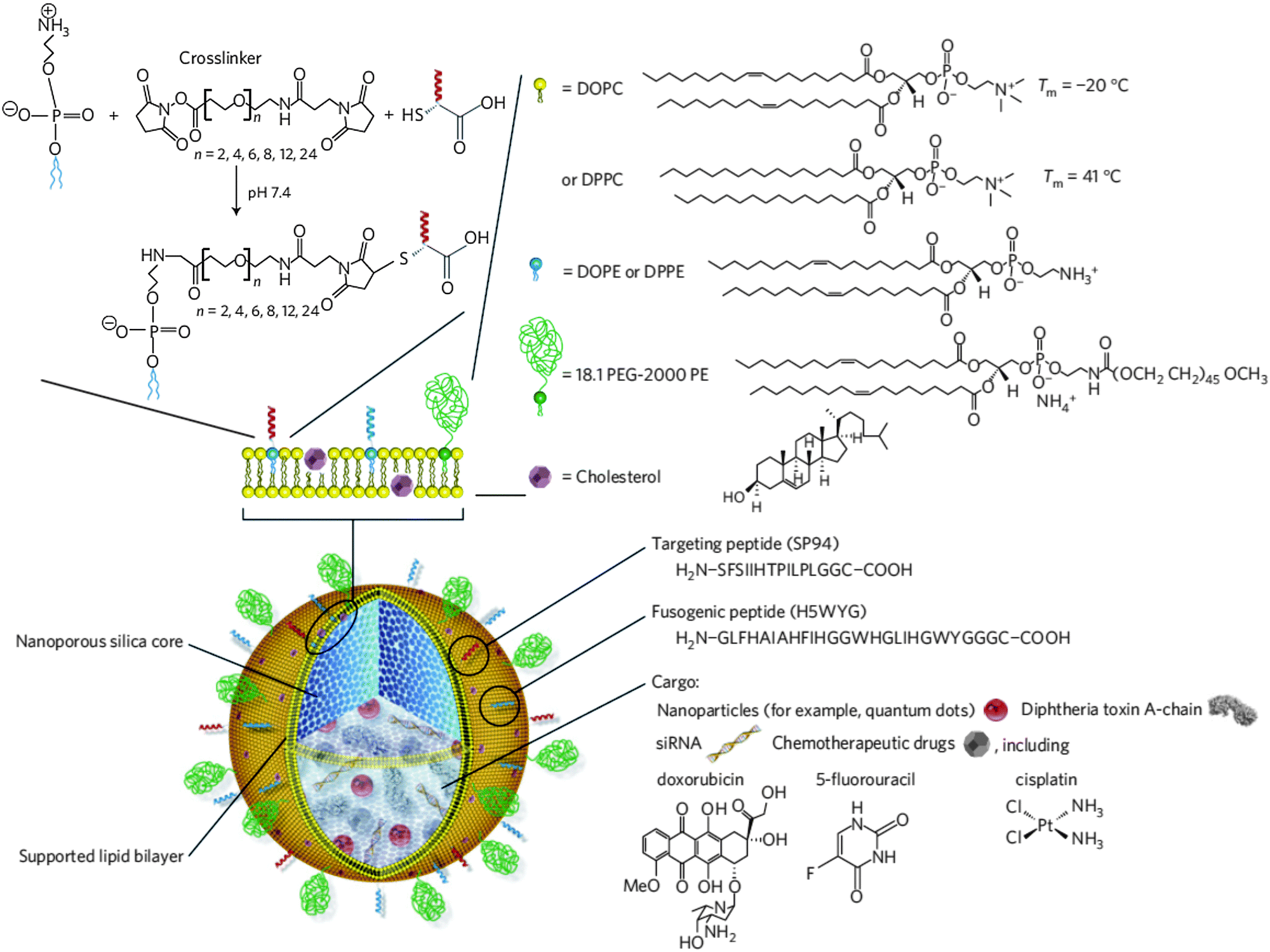 | ||
| Fig. 7 Schematic representation of lipid-enveloped mesoporous silica nanoparticles (termed protocells). The nanoporous silica core serves as a matrix type carrier for the encapsulation of a variety of therapeutic cargo and/or imaging agents. The silica core is surrounded by a supported lipid bilayer (SLB), mainly comprising zwitterionic lipids in a fluid (1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)) or more rigid gel-like phase (1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC)). The SLB is further supplemented with 30 wt% cholesterol, 5 wt% PEGylated lipids and 1–5 wt% 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) or 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) the primary amines of which are consumed for covalent anchoring of targeting and fusogenic peptides via a heterobifunctional PEG spacer. (Reprinted with permission from Macmillan Publishers Ltd: Nature Materials,57 copyright 2011.) | ||
3.3. Lipid-coated hydrogel nanoparticles
Hydrogels can be defined as 3D networks of hydrophilic polymers that are capable of absorbing large quantities of water or biological fluid, while maintaining their internal network structure. Hydrogel networks can be constructed by virtue of chemical and/or physical crosslinks between natural or synthetic polymers. These crosslinks are the driving force behind the structural stability of the hydrogel by preventing the dissolution of the hydrophilic polymers in the aqueous microenvironment.120,121 Since their introduction for biological use more than 50 years ago,122 at present hydrogels are favored for many biomedical and pharmaceutical purposes, including controlled drug delivery.120,123–125 Hydrogels may exist in many geometries such as macroscopic gels (e.g. scaffolds, cylinders), microscopic gels (microgels) and hydrogel nanoparticles (nanogels). While the literature has predominantly focused on macro- and microscopic hydrogels for extracellular drug delivery applications, during the last decade nanogels have gained momentum as drug delivery vehicles.126 Nanogels maintain equal network properties compared to their micro- and macroscopic counterparts, but their nanosized dimensions render them useful for intravenous administration (e.g. for tumor targeting) and intracellular drug delivery. In addition, their small size guarantees a rapid response to external stimuli, making them ideal candidates for triggered drug delivery.126 To date, nanogels have mainly been employed as carriers for low molecular weight chemotherapeutics, although in recent years the focus has shifted somewhat to macromolecular drug delivery, including proteins and nucleic acids. Many recent review papers comprehensively describe the plethora of available crosslinking methods, nanogel synthesis techniques and hydrogel drug release mechanisms, which will not be covered here.126–128 Instead we will focus on nanogel–lipid core–shell composites.Extensive work was done by several groups on the lipid coating of hydrogel microspheres for controlled drug release or as an artificial cell mimic.21,129–136 The adsorption of a lipid bilayer onto the microgel surface can be promoted by electrostatic interaction between charged microgels and oppositely charged liposomes.130,132 Alternatively, introduction of hydrophobic anchors at the microgel surface could drive the self-assembly of a (phospho)lipid bilayer in a subsequent step.129,134,135 However, only a limited number of publications can be found that extrapolate these findings to nanosized dimensions. Nevertheless, already more than two decades ago, a hybrid lipid–nanogel drug delivery system was introduced, originally constructed to mimic low density lipoproteins (LDL).137 The nanostructures, which were named SupraMolecular BioVectors (SMBV™), consisted of a crosslinked natural polysaccharide core layered by a (protein modified) phospholipid shell and were fabricated via a two-step synthesis. Preformed dextran nanogel cores were initially acylated by grafting fatty acids of distinct chain length onto their surface. Acylated cores and phospholipids were dispersed in ethanol and subsequently injected into an aqueous medium at elevated temperature, resulting in the deposition of a phospholipid monolayer around the fatty acid modified nanogel core.137 The SMBV concept was further extended to charged polysaccharide cores via the introduction of quaternary ammonium or succinate/phosphate groups and at a later stage the two-step synthesis was simplified by omitting the acylation step prior to phospholipid coating.54,138,139 Alternatively, dextran NPs have been coated with phospholipids via direct hydration of a dried lipid film followed by high-energy sonication and extrusion.140
The majority of the synthesis methods used to prepare nanogels afford limited control over nanogel size and/or size distribution and are often incompatible with labile macromolecular therapeutic compounds such as pharmaceutical proteins.128 Protocols for lipid coating of nanogels again mainly consist of multiple consecutive steps that need separate optimization which makes the preparation process laborious and difficult to control. Thus, single-step preparation protocols allowing concomitant loading of drugs with disparate physicochemical properties and robust control over nanoparticle dimensions are highly sought after. Of particular interest in this regard is the exploitation of liposomes as nanoscaled reactors for the selective hydrogel formation in their aqueous lumen. To this end, unilamellar liposomes are formed in the presence of a monomer solution (e.g. via a thin-film hydration method accompanied by freeze-thawing, sonication or extrusion), followed by a dilution-step and initiation of hydrogel crosslinking (Fig. 8). Dilution of the liposomal dispersion is of key importance to prevent macroscopic hydrogel formation and thus to ensure selective monomer crosslinking in the liposome interior. Using this method, Van Thienen et al. synthesized biocompatible PEG nanogels as well as biodegradable dextran nanogels within a zwitterionic liposomal template.141–143 Kazakov et al. reported on the selective formation of thermo- and pH-sensitive nanogels consisting of poly(N-isopropylacrylamide-co-1-vinylimidazole).144,145 On the other hand, Schillemans et al. described the use of a detergent-dilution method to construct the liposomal nanoreactor and relied on ascorbic acid to inhibit the free radical polymerization process in between the liposomal vesicles.146 In another recent report, Park et al. applied this liposomal template-assisted method in a protocol with sequential steps in which preformed lyophilized liposomes were hydrated with a monomer solution.147 To initiate hydrogel crosslinking, the latter reports made use of a water-soluble photoinitiator that was included in the monomer solution prior to liposome formation, enabling UV-triggered photopolymerization to create a chemically crosslinked hydrogel network. Alternatively, physically crosslinked alginate nanogels were also successfully produced. Hereto, liposomes encapsulating sodium alginate in their aqueous lumen were incubated in a solution of calcium chloride above the transition temperature of the lipid bilayer. Permeation of divalent Ca2+ ions across the lipid bilayer shell could then initiate ionic gelation of the negatively charged polysaccharide chains.148
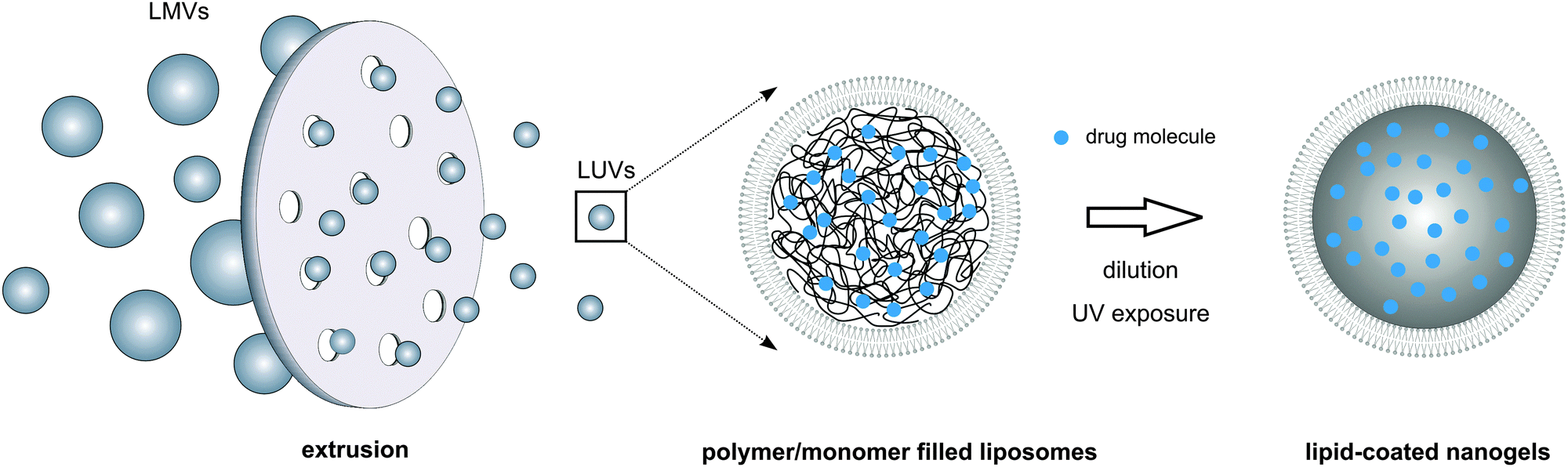 | ||
| Fig. 8 Liposomal-template assisted synthesis of hydrogel particles yielding lipid-coated nanogels. Hydration of a thin lipid film with a (meth)acrylate functionalized monomer–polymer solution leads to a dispersion of large multilamellar vesicles (LMVs) with a broad size distribution. Extrusion of the LMVs through a polycarbonate membrane with narrow pores is used to convert the obtained multilamellar vesicles into monodisperse large unilamellar vesicles or LUVs, carrying the (meth)acrylated monomer–polymer in their aqueous core. Following dilution, radical polymerization can be initiated, e.g. via UV illumination in the presence of a photoinitiator, resulting in the formation of lipid-coated nanogels. | ||
3.4. Post-synthesis surface modifications
As briefly outlined in the introduction and comprehensively reviewed by others,39,41 the surface characteristics of a drug-loaded NP will impinge on how they interact with their biological environment upon in vivo administration. Decorating the NP surface with a hydrophilic stealth layer (e.g. using PEG) may safeguard colloidal stability and extend blood circulation. As indicated above, for the construction of lipid-coated matrix NPs with a core–shell nanoarchitecture, this mostly implies the use of PEGylated lipids during NP synthesis. However, some protocols may require the post-synthesis shielding with a PEG outer layer. Su et al. reported on the fabrication of pH-responsive poly(β-amino ester) NPs enveloped in a cationic PEGylated lipid shell.149 To this end, both a double-emulsion solvent evaporation method and a solvent diffusion nanoprecipitation method were investigated. The authors found that adding PEGylated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-PEG) during the nanoprecipitation protocol markedly reduced NP yield. To resolve this issue, the DSPE-PEG was incorporated into the lipid bilayer via a post-insertion process.149 Incubation of preformed conventional liposomes with a dispersion of PEG-modified lipids above the critical micellar concentration (CMC) may indeed result in transfer of PEG-lipids from the micellar phase into lipid bilayers in a time- and temperature-dependent manner.150,151Although of great importance, the presence of a hydrophilic polymer layer generally precludes efficient endocytic uptake and processing by target cells due to reduced interaction with the cell plasma membrane.30,152 Inserting PEGylated lipids with distal end functional groups into the NP supported SLB allows further post-synthesis modification of the NP surface with bioactive moieties aiming to combine stealth properties with active cell targeting.2,3 For instance, thiol–maleimide surface chemistry has been widely employed in this context by virtue of PEGylated lipids carrying maleimide end-groups. In this way, lipid-coated PL(G)A NPs could be bestowed with cellular or tissue targeting capability by covalently coupling cysteine-terminated peptides,72 thiolated 2′-OMe RNA aptamers61 or disulfide reduced monoclonal antibody fragments.56 Likewise, Look et al. prepared stealth liposomes using amine-terminated DSPE-PEG in order to covalently link nondepleting CD4 targeting antibodies using sulfo-NHS/EDC carbodiimide-based coupling chemistry. These immunoliposomes were subsequently used for the preparation of so-called nanolipogels via the liposomal-template assisted synthesis method (see also Section 4.3). Post-synthesis carbodiimide-assisted modification was also applied by others to obtain lipid-coated PLGA NPs functionalized with the αvβ3 integrin-targeting Arg-Gly-Asp (RGD) peptide.58 Some lipids carrying less fragile targeting moieties, such as DSPE-PEG-folate, can also readily be used during hybrid NP synthesis in the presence of organic solvents.117,153–155
Functional moieties can also be covalently anchored directly to the SLB surface as demonstrated by Brinker's group for their silica based protocells (Fig. 7). They coupled bioactive (fusogenic or targeting) peptides, premodified for conjugation with a glycine–glycine spacer and a C-terminal cysteine, to the primary amines of phosphoethanolamine (PE) polar headgroups via an amino-to-sulfhydryl heterobifunctional succinimidyl-[(N-maleimido-propionamido)-tetracosa-ethyleneglycol] ester (SMPEG24) crosslinker.57,112,113 The outstanding cellular targeting efficacy achieved with these protocells clearly certifies that the presence of 5 wt% DSPE-PEG in the silica supported SLB, necessary to maintain colloidal stability, does not sterically interfere with ligand–receptor binding.57 Zheng et al. reported on the modification of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), in which the PE headgroup was reacted with a maleimidobenzoic acid N-hydroxysuccinimide ester (MB-NHS), with thiolated transferrin (Tf). The obtained Tf-DOPE micelles were subsequently employed for post-insertion into nanoprecipitated lipid-coated PLGA NPs.156
As detailed above, the majority of the NP production protocols encompass harsh preparation conditions including among others the use of organic solvents, the induction of shear stress via sonication, and heating, that might well be detrimental for labile drug molecules, e.g. nucleic acids and proteins. For this reason, some authors prefer to add the therapeutic compound post-synthesis. This strategy is frequently employed for the electrostatic complexation of anionic nucleic acids onto the cationic surface of lipid–polymer hybrids.82,88,149,157 In this way, not only the possible degradation of the therapeutic nucleic acid is avoided during synthesis, it may also remedy the low drug loading often observed for these large hydrophilic compounds, e.g. in unmodified PLGA NPs.69,158,159 On the other hand, it remains questionable to what extent this NP loading procedure can protect the nucleic acids against enzymatic degradation and preterm decomplexation in vivo. In contrast, Bershteyn et al. used a covalent coupling strategy to link thiolated ovalbumin (OVA) to the surface of lipid-enveloped PLGA NPs via the maleimide functional group of DSPE-PEG-mal. To this end, the ovalbumin antigen was premodified using a heterobifunctional crosslinker N-succinimidyl-S-acetyl-(thiotetraethyleneglycol), the succinimidyl moiety of which can react with OVA primary amines. The sulfhydryl group was deacetylated with hydroxylamine prior to thiol–maleimide coupling. Interestingly, in the final step, lipophilic danger molecules were also post-inserted in the lipid shell of antigen-conjugated hybrids.52
4. Drug delivery applications of lipid–polymer matrix nanocomposites
4.1. Lipid-coated polyester nanoparticles in drug delivery
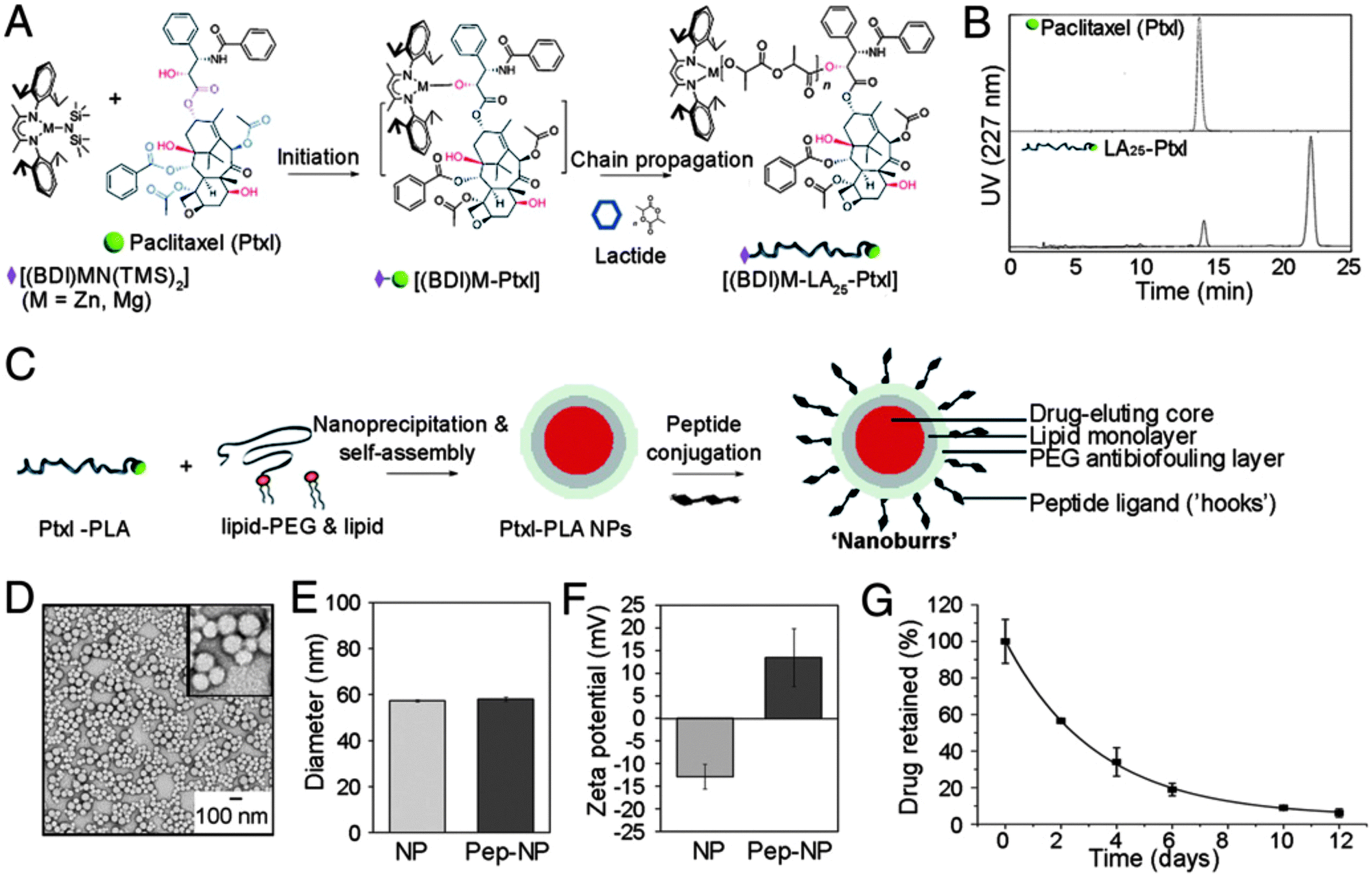 | ||
| Fig. 9 Overview of synthesis of lipid–PLGA core–shell ‘nanoburr’ particles. (A) Synthesis scheme of the paclitaxel (Ptxl)–poly(lactic acid) (PLA) conjugate. (B) RP-HPLC on the Ptxl and Ptxl–PLA conjugate. (C) Illustration of hybrid nanoparticle formation via a single-step nanoprecipitation method. The nanoparticles were modified post-synthesis with targeting peptides via maleimide–thiol chemistry. (D–F) Morphological and physicochemical characterization of the nanoburrs as a function of peptide conjugation. (G) In vitro release profile of Ptxl obtained with nanoburrs. (Reprinted with permission from ref. 72. Copyright 2010.) | ||
 | ||
| Fig. 10 Synthesis scheme of the hydrolysable paclitaxel–gemcitabine conjugate. (Reprinted with permission from ref. 162. Copyright © 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.) | ||
In another combinatorial strategy, Wang and coworkers translated the concept of chemoradiotherapy, i.e. the synchronized treatment with both chemotherapy and radiotherapy, to lipid–PLGA nanomedicines. The utilization of a lipid–polymer hybrid NP platform60 for chemoradiation, which they fittingly dubbed ChemoRad NPs, enabled the encapsulation of radio-isotopes without affecting the NP surface characteristics or interfering with drug encapsulation efficiency and drug release kinetics.59 The latter was accomplished by the integration of phospholipids, modified with the chelator diethylenetriaminepentaacetate (DTPA), in the PEGylated lipid monolayer that surrounds the docetaxel loaded PLGA core. By employing the A10 RNA aptamer as a targeting ligand, ChemoRad NPs demonstrated selective delivery of docetaxel and yttrium90 (90Y) to prostate-specific membrane antigen (PSMA) overexpressing prostate cancer cells and showed synergistic cell killing when compared with targeted monotherapy.59 Likewise, folate-targeted ChemoRad NPs were engineered by others for the concurrent delivery of paclitaxel and 90Y. Following their initial identification of folate-targeted and docetaxel-loaded hybrid NPs as effective radiosensitizers,154 the authors aimed to evaluate co-delivery of chemo- and radiotherapeutics in a mouse model of ovarian cancer peritoneal metastasis. Intraperitoneal injection of targeted ChemoRad NPs significantly outperformed their non-targeted counterparts and combinatorial chemoradiotherapy achieved the most effective therapeutic outcome.153 Folate-targeted, PEGylated and DTPA modified lipid–PLGA NPs were also proposed as targeted nanotheranostic agents, in which the DTPA chelator accommodates gadolinium (Gd3+) as an MRI-contrast agent.83
PLGA NPs have also been employed for the simultaneous loading of chemotherapeutics and thermo-optical agents.166 Within the latter group, the FDA-approved near-infrared (NIR) fluorescent dye indocyanin green (ICG) has been frequently applied in photodynamic and photothermal therapy.166,167 Upon absorption of NIR photons, ICG has been demonstrated to emit the excitation energy as heat, a trait that can be exploited to induce local hyperthermia.166 This localized heating effect is believed to synergize with standard chemotherapy by sensitizing cancer cells to its cytotoxic activity.168 Aiming to combine both chemotherapy and photothermal therapy in a single nanoparticle delivery system, Zheng and coworkers recently developed PLGA–lecithin–PEG NPs encapsulating a mixture of doxorubicin and ICG employing a single-step sonication method. They found that the combined chemophototherapy entailed a synergistic tumor cell apoptosis (Fig. 11) and tumor growth inhibition in a mouse MCF-7 xenograft model.169 In this report, the in vivo administration of the lipid-coated PLGA NPs was performed via intratumoral administration, thus bypassing much of the pre-existing barriers associated with systemic intravenous injection.
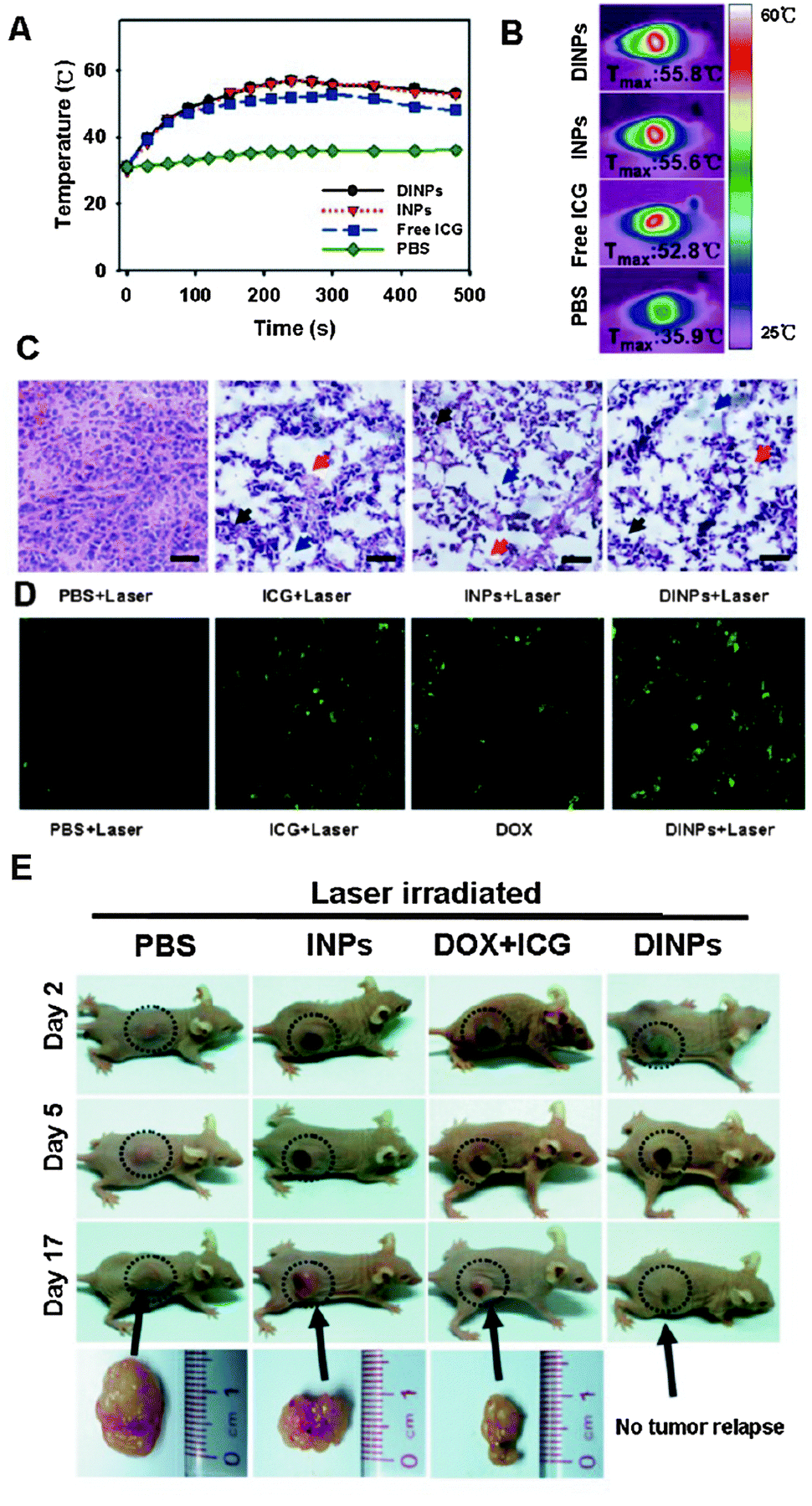 | ||
| Fig. 11 Intratumoral injection of lipid–PLGA nanohybrids loaded with doxorubicin and ICG (DINPs) in nude mice MCF-7 breast cancer xenografts. (A and B) Laser irradiation causes local hyperthermia due to ICG. (C) Thermally induced tissue damage and (D) tumor cell apoptosis (green staining, TUNEL assay) following laser irradiation in the presence of free ICG, ICG-loaded NPs (INPs) or DINPs. (E) Chemo-photothermal combination treatment with DINPs caused a marked anticancer effect, outperforming free drug and INP monotherapy. (Adapted with permission from ref. 169. Copyright 2013 American Chemical Society.) | ||
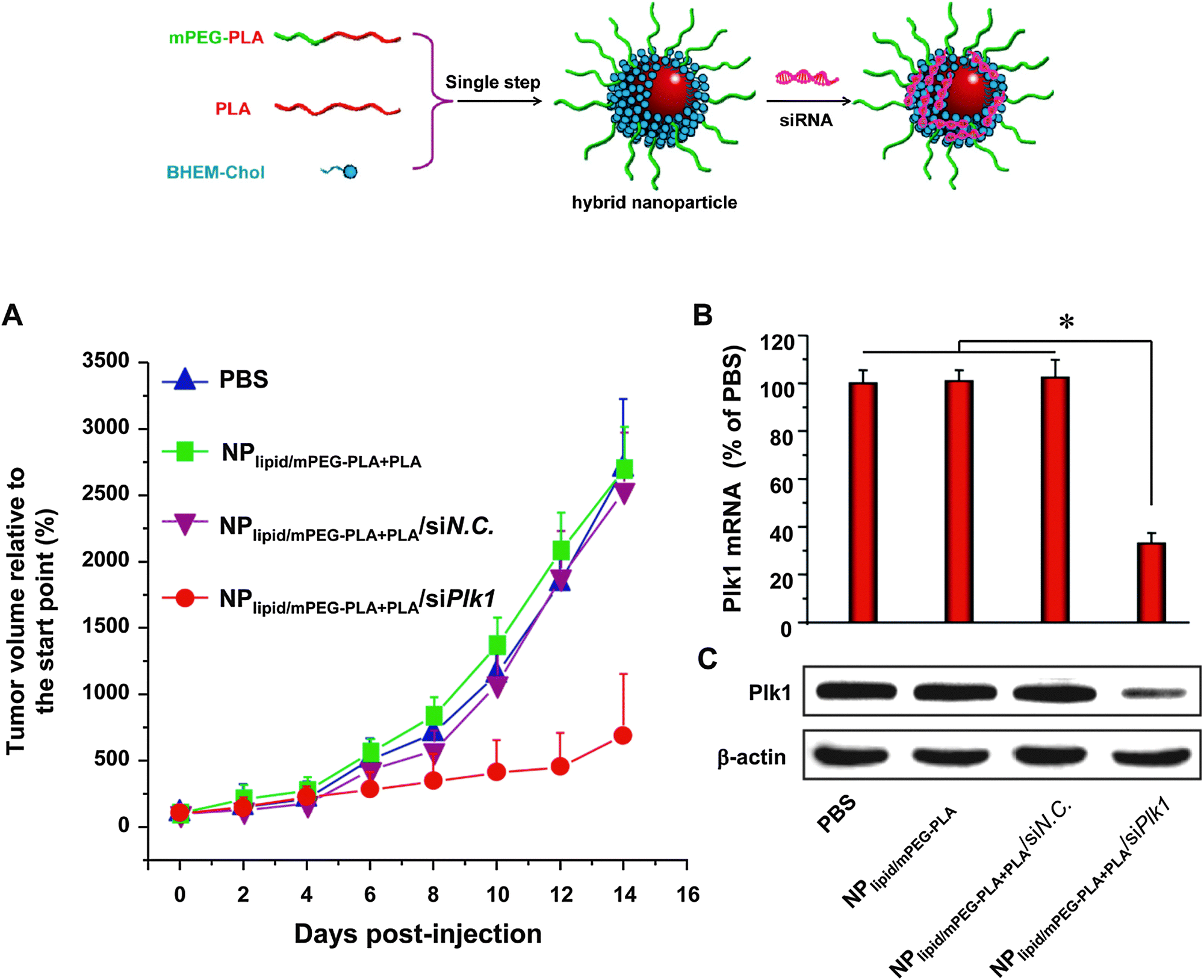 | ||
| Fig. 12 Formation of lipid–polymer hybrid nanoparticles via a single-step nanoprecipitation of poly(lactic acid) (PLA) and the amphiphilic PEG–PLA conjugate in the presence of the cationic lipid N,N-bis(2-hydroxyethyl)-N-methyl-N-(2-cholesteryloxycarbonyl aminoethyl) ammonium bromide (BHEM-Chol). The cationic lipid monolayer at the interface of the PLA hydrophobic core and the PEG hydrophilic shell allows surface complexation of siRNA targeting the polo-like kinase 1 (Plk1) oncogene. RNAi silencing of Plk1 in vivo following systemic administration significantly suppressed tumor growth. (Adapted with permission from ref. 88. Copyright 2012 American Chemical Society.) | ||
Aiming for topical siRNA delivery to treat inflammatory skin disorders, Desai et al. recently described co-delivery of anti-TNFα siRNA and capsaicin via formulation in PLGA NPs (encapsulating the hydrophobic capsaicin), enveloped within a lipid shell consisting of cationic amphiphiles carrying cyclic pyrrolidinium head groups. The latter enables efficient complexation of the negatively charged siRNA.157 Several strategies have been described in the literature to stimulate siRNA loading in PL(G)A matrices by precomplexation with cationic polymers.176–178 Interestingly, Yang and coworkers applied a proprietary cationic lipid to drive siRNA encapsulation in PLGA matrices during a modified double emulsion solvent evaporation method using PVA as the final stabilizer. In the course of the primary emulsification step, the cationic lipids tightly self-assemble at the water–oil interface, stabilizing the aqueous droplets in which the siRNA will be complexed to the cationic lipid headgroups. In this way, encapsulation efficiencies exceeding 90% are attained and the drug loading weight ratio almost reached 5%.159 Shi et al. extended this method to obtain differentially charged lipid–polymer–lipid nanostructures with a hollow core for siRNA encapsulation due to the addition of a mixture of DSPE-PEG and lecithin acting as stabilizers during the second emulsion step and solvent evaporation (Fig. 13). These hybrid lipid–polymer NPs were evaluated for siRNA delivery in vivo in a murine subcutaneous xenograft model and could evoke moderate luciferase silencing upon a single intratumoral injection.158
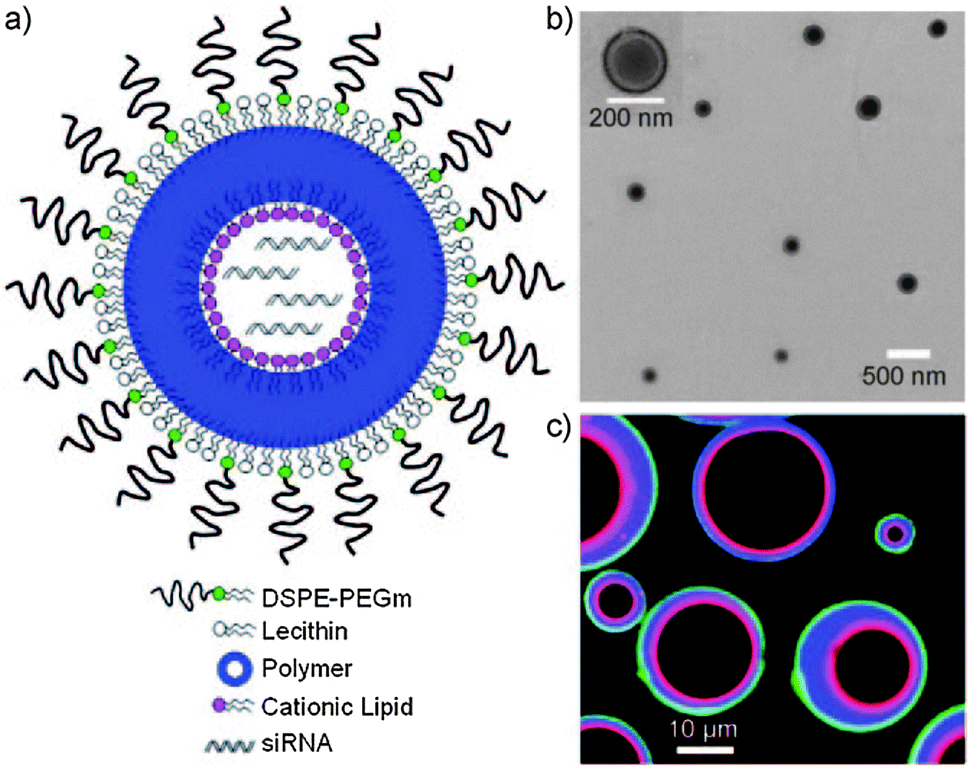 | ||
| Fig. 13 Lipid–PLGA–lipid hybrid nanoparticles with core–shell morphology designed for siRNA delivery. With the aid of positively charged lipids, a high encapsulation efficiency of the anionic siRNA is achieved in the nanoparticle hollow core. (a) Schematic representation and (b) transmission electron microscopy (TEM) image of the hybrid nanoparticle construct. (c) Confocal image of hybrid microparticles showing the outer PEG–lipid monolayer in green, the inner cationic lipid monolayer in red and the in-between PLGA layer in blue. (Reprinted with permission from ref. 158. Copyright © 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.) | ||
In recent years, it has been documented that besides NP size and surface chemistry, NP shape can also have a profound effect on biodistribution, particokinetics and cellular internalization and that nanospheres are not by definition superior over other NP designs.39,76,179,180 Recognizing the importance of nanogeometry in NP interactions at the nano–bio interface, monodisperse needle-shaped PLGA particles (with dimensions of 80 × 320 nm) were fabricated by virtue of the PRINT process introduced in Section 3.1. It has indeed been demonstrated that rod-like NPs with a high aspect ratio are more efficiently internalized by cells.179 The non-spherical PLGA NPs were utilized for siRNA encapsulation without the aid of cationic lipids or polymers. In this way, an siRNA encapsulation efficiency nearly reaching 50% was achieved. Nevertheless, to assist in cellular internalization, a cationic lipid coat was layered around the particles post-synthesis. Three different prostate cancer cell lines were successfully transfected with siRNA targeting the KIF11 (Eg5) gene which encodes a kinesin-like motor protein that is of essential importance during cell mitosis. Selective downregulation of this protein caused a marked decrease in cell viability in all three cell lines tested. Regrettably, no in vivo data are available yet for this drug delivery platform.75 However, it is conceivable that additional optimization, e.g. by bestowing the particles with targeting and/or stealth properties, will be indispensable for successful in vivo translation.
4.2. Lipid-coated mesoporous silica nanoparticles in drug delivery
The multifunctional character of lipid–polymer nanocomposites was impressively illustrated by Brinker's group that reported on drug-loaded MSNPs functioning as nanoscopic templates for the deposition of a phospholipid SLB.57 The resulting hybrid NPs were ultimately termed ‘protocells’, referring to their elementary resemblance of a cellular construct. The nanoporous silica core enabled encapsulation of high levels of various therapeutic (e.g. dsRNA, doxorubicin, diphtheria toxin A-chain) and diagnostic agents (e.g. quantum dots) with distinct physicochemical properties. Next, the drug-loaded core was sealed off with a single lipid bilayer mainly composed of zwitterionic and PEGylated lipids with the primary aim to protect the therapeutic cargo from premature release and reduce aspecific binding to nontarget cells. To facilitate specific cell targeting and intracellular drug delivery, the lipid shell was further functionalized with a targeting peptide (SP94, targeting human hepatocellular carcinoma (HCC)) and a pH-responsive peptide (stimulating disruption of both endosomal membrane and SLB at endolysosomal pH) respectively (Fig. 7). Importantly, by wrapping a fluid-phase SLB around the MSNP core, the lipid-anchored targeting peptides retain their lateral mobility within the bilayer. It was demonstrated that this particular feature allowed multivalent receptor-mediated binding of target cells with minimal surface densities of peptide ligand. Combining exceptional target cell specificity and drug loading capacity with proficient cytosolic delivery, a 106-fold improvement in cancer cell killing was achieved over conventional liposomes using protocells carrying a cocktail of doxorubicin, 5-fluorouracil and cisplatin.57 Unfortunately, to date, no proof-of-concept data on this targeted drug delivery platform in validated animal models are available yet and thus the in vivo targeted drug delivery performance of protocells still remains elusive.181 In fact, it has recently been disclosed that the deposition of a protein corona upon instillation of targeted nanomedicines in a biological medium may prominently alter their ultimate targeting efficacy,182,183 a finding that warrants further in vivo evaluation of targeted protocells, or any other targeted NP for that matter.As stated above, next to encapsulation of low molecular weight chemotherapeutics, MSNPs can also be tuned to support loading with high molecular weight biomacromolecules. To this end, MSNPs with a bimodal pore morphology were designed by using a mixture of two different types of surfactants as structure-directing agents. This particular templating strategy resulted in MSNPs containing large surface-accessible pores (∼10–30 nm) interconnected by smaller pores (∼5 nm).184 The larger pore diameter should allow the penetration and retention of more bulky therapeutic molecules such as proteins and nucleic acids in the MSNP matrix. Targeted protocells constructed with bimodal MSNP cores as described above were thus further pursued for the intracellular delivery of siRNA112 and the deglycosylated ricin toxin A-chain (RTA).113 In order to better accommodate the negatively charged siRNA and RTA in MSNPs, the latter were made cationic by incorporating the amine-containing silane AEPTMS, which significantly augments the MSNP loading capacity for both therapeutics. Moreover, AEPTMS modified MSNPs show faster dissolution kinetics under physiological conditions, promoting the release of encapsulated cargo.57,112,113 Ashley et al. encapsulated an equimolar mixture of distinct siRNAs in the protocell core, designed to silence different members of the cyclin protein superfamily.112 Cyclins are key regulatory proteins in various stages of the cell cycle that sustain proliferation of malignant cells through activation of cell-cycle dependent kinases.185 Targeted protocells could achieve maximal cyclin knockdown in the HCC cell line Hep3B with siRNA concentrations in the low pM range and thereby significantly outperform standard cationic liposomes. Importantly, the protocell-induced RNAi effect could evoke growth arrest and apoptosis in HCC cells at a particle:cell ratio ∼10, without affecting the viability of normal control hepatocytes lacking expression of the antigen that is recognized by the protocell-anchored targeting peptide.112 Likewise, Epler et al. demonstrated the selective induction of apoptosis in target HCC cells by cytosolic delivery of the protein toxin RTA.113 The catalytic ricin toxin A subunit inhibits protein synthesis by cleaving a specific N-glycosidic bond in 28S ribosomal rRNA, thereby irreversibly blocking mRNA translation.186 Protocells could achieve a half-maximal inhibition of protein synthesis at a RTA concentration ∼5 pM, demonstrating a 100-fold more potent inhibitory activity than RTA loaded control liposomes with an equal lipid composition as the protocell's SLB (Fig. 14).113
 | ||
| Fig. 14 Protocell-guided toxin delivery. (A) Mesoporous silica nanoparticles (MSNPs), modified with an amine-containing silane (AEPTMS) were loaded with the deglycosylated ricin toxin A chain (RTA), capped with a fluid-phase PEGylated phospholipid bilayer and modified with both a targeting (SP94) and an endosomolytic peptide, in line with reports by Ashley et al.57,112 (B) Transmission electron microscopy (TEM) image (scale bar = 50 nm) showing a MSNP core with bimodal porosity.184 The inset graph (scale bar = 200 nm) shows a scanning electron microscopy (SEM) image of a microscopic MSNP to visualize the surface-accessible nanopores. (C) Inhibition of protein synthesis induced by targeted RTA delivery in a hepatocellular carcinoma (HCC) cell line. (Reproduced with permission from ref. 113. Copyright © 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.) | ||
The protocells described above are particularly well designed toward intracellular drug delivery as they respond to the gradual acidification in the endosomal lumen following receptor-mediated endocytosis and subsequent endosomal maturation.63 Endosomal pH likely reduces electrostatic and dipolar interactions between the cargo loaded MSNP core and the PE/PC lipid polar headgroups, resulting in SLB destabilization. In addition, protonation of the lipid-anchored fusogenic peptide disrupts the endosomal membrane enabling cytosolic drug release.57,112,113 Besides variations in pH, the intracellular reductive environment can also be exploited to control drug delivery.63 To this end, thiolated MSNPs were decorated with a lipid bilayer, the inner leaflet of which was covalently attached to the MSNP surface via disulfide bonds. Triggered release of a fluorescent tracer molecule was demonstrated upon disulfide reduction and lipid bilayer shedding.118 This drug delivery concept however requires further intracellular confirmation with a model drug. Importantly, drug loaded NPs containing disulfide bonds will preferentially disassemble in the reductive cytoplasm rather than in endocytic compartments, again highlighting the importance of endosomal escape.6 Schloßbauer and coworkers recently reported on an alternative external light-triggered approach, i.e. photochemical internalization (PCI), to overcome the endosomal barrier.187 PCI is based on the selective accumulation of amphiphilic photosensitizers (PS) in the limiting bilayer of endocytic vesicles. Excitation of the PS compound with light of the appropriate wavelength is followed by its reaction with oxygen and the subsequent formation of reactive oxygen species (ROS), primarily singlet oxygen (1O2). This highly reactive intermediate can cause oxidative damage to cellular components, but this effect is mainly confined to the local production site of the singlet oxygen, owing to its short range of action and short lifetime. This localized effect will therefore selectively disrupt the endosomal membranes, releasing the endocytosed macromolecules or NPs into the cytosol.188,189 Since its discovery in 1999,190 PCI has been successfully applied to stimulate the cytosolic delivery of several types of macromolecules (peptides, proteins, nucleic acids) incorporated in non-viral carrier systems.191–193 However, one of the drawbacks of this technique on a cellular level is the inherent cytotoxicity associated with the localization of PS molecules in the plasma membrane and other organelles before illumination. To avoid this limitation, Schloßbauer et al. proposed to covalently couple the PS to the surface of drug loaded MSNPs prior to their encapsulation in a SLB. In this way, the PCI effect can be confined to endolysosomes that contain one or more lipid-coated MSNPs in their lumen following cellular internalization. The authors could show that upon illumination of the PS, protoporphyrin IX, the formed 1O2 sequentially disrupts both the SLB and the endosomal bilayer. It was demonstrated in a human hepatoma cell line that this two-step phototoxic effect could result in the successful cytosolic release of distinct membrane-impermeable drugs.187 On the other hand, Teng et al. recently reported on protoporphyrin IX loaded, folate-targeted and phospholipid-functionalized MSNPs for anti-cancer photodynamic therapy, exploiting the light-triggered ROS induction to selectively induce apoptosis in cancer cells.117
The application of an external magnetic field can also be employed as a physical stimulus to promote drug delivery.170 To this end, superparamagnetic iron oxide nanocrystals (SPIONs, ∼20 nm) have been encapsulated inside MSNPs (∼100 nm) before capping them with a zwitterionic DOPC lipid bilayer. Subjecting the particles to an alternating magnetic field causes local heating which affects SLB permeability and triggers drug release.194 Although the SPION–MSNP hybrids seem to be reasonably well tolerated by various human cell lines, the usefulness of these stimuli-responsive NPs toward intracellular drug delivery still requires experimental validation. Alternatively, SPIONs have been widely investigated as MRI contrast agents, possibly rendering such particles also valuable toward theranostic applications.195
4.3. Lipid-coated hydrogel nanoparticles in drug delivery
Pioneering research on the lipid coating of microscopic gel particles, e.g. performed by the groups of Needham and De Smedt, clearly illustrates the potential of combining stimuli-responsive hydrogel particles with a lipid bilayer coating for triggered drug delivery.130,132,133,196 Kiser and Needham designed anionic pH-responsive poly(methacrylic acid) microgels for doxorubicin loading and decorated these microgels with a phospholipid shell to control the drug delivery process. The microgels were loaded with doxorubicin in a swollen state at physiological pH, exploiting the anionic character of the gel to electrostatically bind the cationic doxorubicin. Decreasing the pH of the medium condensed the drug loaded microgels due to the protonation of the methacrylic acid groups. The condensed microgels were subsequently capped with a phospholipid shell that served as a diffusion barrier to avoid drug leakage and to stabilize the microgel in its condensed state upon re-immersion in a pH neutral buffer. Permeabilization of the SLB via electroporation or lipid destabilizing surfactants instigated pH-dependent swelling of the microgel core, disruption of the lipid bilayer and release of doxorubicin.132,133 In another example, De Geest et al. coated biodegradable anionic dextran microgels with an oppositely charged phospholipid layer and demonstrated that the increased swelling pressure, as a function of the degradation of the microgel core, could rupture the surrounding lipid membrane.130,196 In the same group, Van Thienen et al. documented the liposomal-template assisted synthesis of equivalent degradable dextran nanogels for protein delivery.142 In contrast to the earlier observations at a microscopic scale, it was demonstrated that the lipid coat of these nanosized core–shell particles stays layered around the nanogel core during its degradation. Likely the resulting build-up of internal osmotic pressure is insufficient to overcome the tensile strength of the lipid membrane.141 On the other hand, the swelling pressure of the degrading core could increase the drug permeability of the surrounding lipid layer, as it was shown that the release kinetics of model proteins could be tailored from days to weeks depending on the crosslink density of the gels and the presence of a lipid coat.142 The SupraMolecular BioVectors (SMBV™) designed by Biovector Therapeutics, which also contain a polysaccharide dextran–maltodextrin core, have been evaluated for delivery of various therapeutics.19 For example, antigen-loaded SMBVs have been put forward as nanoparticulate vaccines for intranasal administration, mimicking natural viral pathogens in terms of both size (60–80 nm) and antigen presentation. The SMBVs generated mucosal and serosal immunity in (pre-)clinical assessment as vaccination strategy against influenza A.54 Next to proteins, also antisense oligonucleotides have been encapsulated in SMBVs.197Lipid-enveloped dextran NPs were additionally investigated as a nanotheranostic platform by Erten and coworkers. The dextran nanogel matrix, loaded with doxorubicin, was constructed around an iron oxide core as an MRI contrast agent. The iron oxide–dextran nanogels were endowed with a PEGylated lipid coat to enhance in vivo biocompatibility. Furthermore, to maximize the physical integrity of the lipid coat, a fraction of acetylated DOPE lipids were incorporated to enable mild UV-triggered crosslinking of the SLB. The theranostic NPs were investigated in a murine tumor xenograft model where an enhanced MRI contrast, indicative of NP accumulation, could be observed in tumors through a dorsal skinfold window chamber.140,195
Very recently, the group of Tarek Fahmy impressively showed the implementation of nanosized lipid-coated polymeric gels (termed ‘nanolipogels’) for the immunotherapeutic treatment of metastatic melanoma (Fig. 15).147 The success of cancer immunotherapy largely depends on the immunosuppressive nature of the tumor microenvironment. Many tumors can develop tolerance via specific resistance mechanisms leading to immune evasion and failure of immunotherapeutic strategies. Novel immunotherapeutic approaches should therefore aim at improving tumor immunogenicity in order to attain an effective anti-tumor immune response.198,199 The accumulation of tolerogenic cytokines, such as transforming growth factor-β (TGF-β), in the tumor tissue interstitial space is one of the hallmarks that mitigate anti-tumor immunity.198,200 Therapeutic strategies aimed at blocking the function of these immunosuppressive factors should be able to reinforce T-cell mediated tumor cell killing.198 Park et al. therefore envisioned the combinatorial controlled delivery of a commercially available TGF-β receptor-I inhibitor together with IL-2, an immunostimulating cytokine that activates endogenous cytotoxic T cells. However, a major impediment to this co-delivery strategy is the co-encapsulation of high molecular weight, water-soluble IL-2 and the small hydrophobic TGF-β receptor-I antagonist in a single nanocarrier. To enable a concomitant sustained release of both molecules, the authors designed nanolipogels using PEGylated liposomes as a nanoreactor for the selective photopolymerization of methacrylated β-cyclodextrin and the terminally diacrylated poly(lactic acid-co-ethylene glycol-co-lactic acid) biodegradable macromer.147 Cyclodextrins have been frequently applied for the formulation of poorly water-soluble drugs by virtue of their ability to form molecular inclusion complexes with hydrophobic molecules.11 This particular feature is exploited here to stably encapsulate the TGF-β receptor-I inhibitor in the liposome interior together with the IL-2 cytokine that is sterically entrapped in the degradable polymer gel matrix. Controlled degradation of the latter enabled sustained delivery of both therapeutics. In a murine model of metastatic melanoma, the combination therapy entailed superior survival benefit over monotherapy with either of the two following systemic administration. It was demonstrated that the synergistic anti-cancer effect was the result of enhanced infiltration of both activated cytotoxic CD8+ T cells and natural killer (NK) cells.147
 | ||
| Fig. 15 Schematic overview of the synthesis protocol of drug-loaded nanolipogels. (a) Methacrylated β-cyclodextrins are employed for the solubilization of the hydrophobic TGF-β inhibitor SB505124. (b) Preformed and lyophilized PEGylated liposomes are loaded with the methacrylated cyclodextrin inclusion complex, a biodegradable diacrylated poly(lactic acid-co-ethylene glycol-co-lactic acid) polymer and the cytokine IL-2. The PEGylated liposomes next serve as nanosized templates for the selective UV-induced photopolymerization of the (meth)acrylated compounds, thus creating a hybrid core–shell nanolipogel particle. (Reprinted with permission from Macmillan Publishers Ltd: Nature Materials.147 Copyright 2012.) | ||
In a follow-up publication, the nanolipogel formulation was also evaluated for the sustained delivery of the immunosuppressant mycophenolic acid (MPA) for the treatment of the autoimmune disease systemic lupus erythematosus (SLE). Likewise, the co-polymerization of β-cyclodextrins in the biodegradable gel-like core enabled efficient loading of the hydrophobic MPA. Intraperitoneal administration of MPA-loaded nanolipogels significantly extended survival in a murine lupus model in contrast to free drug administration. Although the involvement of CD4+ T cells in SLE pathophysiology has been well established, the formulation of MPA in CD4-targeted nanolipogels did not give rise to a clinical benefit compared to their non-targeted counterparts. Biodistribution studies indicated that both nanoformulations are preferentially captured by macrophages and dendritic cells in lymphoid organs. Delivery of MPA to DCs resulted in decreased secretion of inflammatory cytokines, likely explaining the in vivo therapeutic outcome.53
4.4. Bio-inspired and bio-mimetic lipid-coated nanoparticles in drug delivery
Synthetic nanomedicines often fail to surmount the numerous biological barriers en route to their (intracellular) drug target. Poor efficacy in delivering therapeutic concentrations of a drug into the target tissue or cell and eminent (immune)toxicity frequently hamper the clinical translation of fundamental nanomedicine concepts.201 Fuelled by these limitations, currently a growing interest exists in the implementation of bio-inspired and bio-mimetic materials and drug delivery approaches.202,203 Naturally occurring micro- and nanoparticulate systems (e.g. lipoproteins,204,205 extracellular vesicles,206 mammalian cells90) are believed to possess specific features, including improved in vivo stability, biocompatibility and intrinsic cell–tissue targeting, from which drug delivery carriers could benefit.Recent progress in nanotechnologies and emerging knowledge on the properties of biological particulates should jointly pave the way toward successful hybrid bio-mimetic nanocomposites.203 In the context of core–shell lipid–polymer hybrid NPs, strategies are materialized to endow (synthetic) polymeric NPs with a phospholipid bilayer of natural origin. Red blood cells (RBCs) have already been extensively explored as drug delivery carriers owing to their biocompatibility and their extended in vivo circulation time (∼120 days).203,207 Inspired by these favorable characteristics, Hu et al. recently reported on the coating of polymeric PLGA NPs with erythrocyte membranes to adopt long-circulating stealth properties in the bloodstream.78 The latter is in part mediated by the cell surface expression of specific membrane proteins that function as ‘markers of self’, such as the CD47 glycoprotein that inhibits MPS clearance via binding of the signal regulatory protein-α (SIRP-α) on phagocytes.208,209 The RBC membrane-coated polymeric NPs were prepared via a two step process in which PLGA NPs are mechanically extruded together with preformed erythrocyte membrane-derived nanovesicles to induce fusion of the latter on the NP surface (Fig. 16). Following tail vein injection of this novel formulation, it was demonstrated that the circulation half-life of the RBC membrane-coated PLGA NPs was prolonged ∼2.5 fold over PLGA NPs functionalized with conventional PEGylated lipids.78 PEG-conjugated phospholipids, which are also mimics of a cell's glycocalyx, are still regarded as the gold standard for the engineering of lipid-based stealth nanomedicines. However, it has indeed become clear that a hydrophilic polymer brush coating cannot completely prevent the adsorption of serum proteins capable of inducing phagocytosis and accelerated blood clearance.208,210–212 Moreover, activation of complement has been described for PEG–phospholipid conjugates,213,214 indicating that a hydrophilic PEG shell does not guarantee immunological inertness and warranting further investigation of better and safer alternatives for NP surface engineering. Exploiting the long-circulating properties of the RBC bio-mimetic nanocomposites, the authors very recently could show their biomedical value in blood detoxification through specific absorption of membrane damaging pore-forming toxins into the erythrocyte shell.215
 | ||
| Fig. 16 Shielding of PLGA nanoparticles with erythrocyte-derived lipid membranes. Repeated extrusion steps applied to a physical mixture of preformed nanoparticles and empty red blood cell (RBC)-derived membrane vesicles result in RBC lipid-bilayer coating of the PLGA nanoparticle cores. (Reprinted with permission from ref. 78. Copyright 2011.) | ||
The decoration of drug loaded NPs with a bio-inspired lipid bilayer may also be advantageous toward drug delivery on the intracellular level. De Backer et al. recently reported on the coating of siRNA loaded biodegradable polysaccharide nanogels with a naturally derived pulmonary surfactant.216 The pulmonary surfactant, covering the entire alveolar surface of mammalian lungs, may potentially interact with nanomedicines upon inhalation therapy and deep lung deposition.217 It is therefore of key importance to carefully evaluate the effect that alveolar surfactants might have on the biological performance of drug-loaded NPs. To this end, the authors first constructed stable surfactant-covered siRNA nanogels. Although the coating with pulmonary surfactants substantially inhibited cellular internalization of the nanogels in lung epithelial cells and alveolar macrophages, their gene silencing potential in both cell types was maintained or even improved. These intriguing data suggest that pulmonary surfactants may enhance the fraction of internalized siRNA that is delivered into the cytosol, possibly mediated via a specific endocytic entry route or distinct intracellular trafficking following cellular uptake.216 Accordingly, the RNAi and gene transfer activity of nucleic acid loaded biodegradable NPs, fabricated from diamine modified poly(vinyl alcohol) grafted with PLGA, was significantly improved in the presence of a pulmonary surfactant.218,219 Here, the ternary nanocomposites were formulated using a single-step solvent displacement technique, using the lung surfactant as a surface altering component during the preparation procedure. The latter reports mainly ascribe the improved siRNA/DNA delivery to the surfactant-enhanced cellular internalization, as opposed to the observations by De Backer et al.
Biodegradable antigen-loaded PLGA micro- and nanoparticles have been widely investigated in vaccination regimens, mimicking the particulate nature of invading microorganisms to stimulate internalization by antigen presenting cells (APCs).69,220 Moreover, the controlled release of the encapsulated antigens enables tight control over the duration of antigen exposure in order to minimize tolerance and stimulate an effective and long-lasting immune response.69,221,222 Conversely, coupling the antigens to the surface of preformed particles shows better resemblance with the natural multivalent antigen presentation on the surface of microbial pathogens. Building further on this biomimetic antigen delivery concept, Bershteyn et al. designed phospholipid enveloped PLGA micro- and nanoparticles with the model antigen covalently linked to PEGylated lipids stably anchored in the phospholipid shell.52 In an attempt to enhance the immune response, the lipophilic danger signals monophosphoryl lipid A (MPLA) and α-galactosylceramide (α-GC) were additionally inserted in the lipid coating as adjuvants.223,224 Both lipid-coated PLGA micro- and nanoparticles evoked high titers of antigen-specific IgG using only nanogram antigen doses, which could be even further improved by the pathogen-mimetic presentation of the above-mentioned lipophilic adjuvants. Moreover, the reported particulate vaccination platform significantly outperformed a conventional vaccination strategy with soluble protein antigen combined with the particulate adjuvant alum or a mixture of MPLA/α-GC as molecular adjuvants.52 Similarly, as briefly mentioned in Section 4.3, SMBVs were also proposed as particulate antigens. Herein the charged polysaccharide core could serve for encapsulation of (intracellular) antigens while membrane-associated antigens could be integrated into the surrounding lipid bilayer, thus imitating the natural presentation of antigenic epitopes on the surface of microbial pathogens. In addition, the incorporation of nucleic acid-derived, protein-derived and lipophilic adjuvants has been suggested of which the latter are also assumed to be inserted in the lipid layer of the SMBV.54
5. Toxicological considerations on lipid–polymer matrix nanoparticles
It is becoming increasingly clear that careful NP design is key to reach the goal of both safe and effective nanomedicinal drug delivery. Although the major goal of drug encapsulation in targeted nanomedicines is to reduce off-target toxicity, the toxicity inferred by the nanocarrier itself cannot be ignored and also requires thorough assessment. The NP composition, physicochemical properties (size, shape, porosity and surface charge) as well as residual chemical or biological impurities may contribute to a toxic response. Nanotoxicity can be evoked via many distinct mechanisms including, but not limited to, the induction of reactive oxygen species (ROS), cellular membrane damage and immunotoxicity. Below, several important findings are highlighted in the context of lipid–polymer hybrid NPs. For a more detailed insight into the field of nanotoxicology the reader is referred to several excellent and comprehensive reviews available in the literature.225–229It is known that upon in vivo administration, NPs will interact with and bind various proteins, thereby creating the so-called protein corona on their surface.182 The identity of this corona is mainly influenced by the nature of the microenvironment (blood circulation, interstitial fluid, cytosol, etc.) and the physicochemical characteristics of NPs.230 Hydrophobic and charged NPs are especially prone to the deposition of a stable protein corona upon immersion in biological fluids.227,230 Importantly, the adsorption of proteins to a NP surface will influence its interaction with target cells and modulate NP internalization.182 The cellular uptake mechanism of NPs might be related to the observed toxic response since it determines the intracellular localization of the NP as well as the total (intra)cellular dose. As a consequence, factors that affect the extent and/or the mechanism of cellular internalization, e.g. the presence of a protein corona, likely also influence the level of nanotoxicity.225 Additionally, the proteins that interact with NPs might undergo conformational changes, thereby impairing the biological function or inducing NP recognition by cells of the immune system.40,225 Likewise, the induction of NP aggregation through interparticle protein bridging as well as NP opsonization contributes to NP scavenging by MPS phagocytes in vivo, possibly leading to immunotoxicity.40 It is conceivable that endowing the polymeric NPs with a neutral lipid coat will improve the overall tolerability of the formulation, as it obscures the hydrophobicity and charge of the underlying NP. Adding PEG–lipids to the lipid envelope is believed to further enhance the biocompatibility by mitigating unspecific interactions and decreasing phagocytic clearance.40 However, immunotoxicity still remains an issue given that all liposomes have the intrinsic potential of inducing complement activation, possibly leading to type I hypersensitivity reactions and fast MPS clearance, with negatively or positively charged lipid bilayers showing the highest reactivity.231 Further underscoring the importance of charge, Kedmi et al. documented significant toxicity in vivo in mice following intravenous injection of cationic lipids, as demonstrated by a marked hepatotoxic response and weight loss, in contrast to their neutral and negatively charged counterparts.232 As briefly mentioned in Section 4.4, even the insertion of PEGylated lipids can trigger complement activation.231,233 Moghimi et al. demonstrated that the negatively charged phosphodiester group within the PEG–phospholipid was responsible for this effect, since complement activation could be prevented by methylation of this anionic moiety or by application of neutral PEG–lipid conjugates.234,235 Furthermore, it has been demonstrated that the intravenous injection of a first dose of PEGylated liposomes could trigger the emergence of anti-PEG IgM, which initiates rapid clearance of a second liposome dose.212,236,237 This so-called accelerated blood clearance (ABC) phenomenon has recently been reviewed in detail.238 In addition, decorating the surface of (PEGylated) lipid–polymer nanohybrids with targeting ligands and/or therapeutics might further add to the concern of immune reactivity.181
Also at the cellular level, the size and the surface characteristics of NPs are the predominant factors that dictate interactions with cells or cellular components and hence drive toxicity.226 Many drug loaded nanoparticles carry a net positive surface charge to allow electrostatic interaction with the negatively charged cell membrane and to trigger adsorptive endocytosis. However, it has been demonstrated that the attachment of positively charged matrix NPs to the plasma membrane can lead to its concentration dependent deformation and rupture. The resulting influx of extracellular calcium may aggravate cellular toxicity by contributing to mitochondrial dysfunction and proapoptotic signaling.239 Induction of membrane damage at the level of erythrocytes can cause significant hemolysis, e.g. as demonstrated for silica NPs as a function of the surface density of available silanol (SiOH) groups, NP porosity and surface charge.240,241 MSNPs are believed to be less toxic than their solid non-porous counterparts, given that the controlled porosity reduces the contact area with biological membranes and lowers the surface density of silanol groups, which are believed to be largely responsible for the observed toxicity.105 Given the involvement of the surface characteristics of the NPs in the observed cellular toxicity, one can anticipate that masking the surface of the polymeric NPs by coating them with neutral or oppositely charged phospholipids could alleviate their cytotoxicity. Again, the use of cationic lipids for coating should be avoided given that transfer of cationic lipids to the plasma membrane can equally cause membrane destabilization, Ca2+ influx and oxidative stress.116 Moreover, it has been demonstrated that cationic lipids can evoke a pro-inflammatory response through activation of toll-like receptor 4 (TLR4).232 As highlighted earlier, many drugs require cytosolic delivery (e.g. nucleic acid therapeutics) and therefore depend on the efficiency with which their nanocarrier is able to mediate escape from the endosomal compartments following endocytic internalization. Endosomal escape is typically accompanied by rupture or permeabilization of the endosomal/lysosomal membrane, thereby releasing the lysosomal protease cathepsin B which is known to trigger inflammasome activation and apoptosis.242–244 The impact of cationic lipid-based nanomedicines on intracellular signaling pathways and cellular toxicity has recently been summarized in a comprehensive review by Lonez et al.245 Cytosolic accumulation of polycationic material can in turn interfere with protein function or induce impairment of cellular organelles.246
To avoid accumulation of polymeric matrix NPs in cells and tissues upon repeated administration, it is of equal importance that they are composed of biodegradable constituents. For instance, as mentioned in Section 3.1, PLGA and PLA NPs can be fully degraded via ester hydrolysis into the toxicologically acceptable products lactic and glycolic acid.71 MSNPs show faster dissolution kinetics compared to solid non-porous Stöber silica NPs owing to the higher total surface area, leading to nontoxic soluble silicic acid products.105 Hydrogel NPs can be rendered biodegradable by selecting (enzymatically or hydrolytically) degradable polymer building blocks or incorporating degradable crosslinks into the 3D polymeric network.120 Of note, the possible adverse biological interactions mediated by the emerging (polymeric) degradation products should also be taken into account.
As nanomedicines will interact with various cells and biomolecules upon in vivo administration, it is crucial that the toxicological impact of these interactions is carefully assessed. Summarizing the findings above, the potential toxicity of lipid-coated polymer NPs likely correlates well with what has been independently described for their lipid and polymer constituents. As a general rule, the use of charged (mainly polycationic) components is preferably avoided. Although the use of a (PEGylated) lipid coat can offer additional protection at the extracellular level and the level of the cell membrane, activation of complement and the resulting immunotoxicity cannot be completely ruled out. It is therefore advisable to meticulously document the nanotoxicological profile for every new NP design prior to clinical application, in order to identify potential side effects. To this end, novel standardized and high-throughput methodologies that allow a multiparametric assessment of (cellular) toxicity together with in vitro models that enable a more reliable prediction of in vivo toxicity could prove highly valuable.225,228,229,239
6. Conclusions and future outlook
A plethora of nanoparticles (NPs) with diverse nanoarchitectures has been witnessed to date with the aim to improve the efficacy and safety of encapsulated drugs. Biocompatible nanomedicines that enable a more selective delivery of therapeutics to diseased cells while mitigating exposure of healthy tissues are highly sought after. However, overcoming the countless barriers a drug-loaded NP encounters en route to its biological target remains a daunting task. Both polymeric NPs and liposomes have already demonstrated in vivo potential, but still suffer from inherent shortcomings. Capitalizing on recent advances in nanotechnology together with a more fundamental insight into the behavior of NPs upon in vivo administration should pave the way to the engineering of better and safer nanomedicines. Lipid-enveloped polymer matrix NPs mainly aim to merge the advantages of both materials in a single nanocarrier and at the same time tackle their limitations. These lipid–polymer nanohybrids provide a flexible platform affording ample control over their physical, chemical and biological attributes. This degree of flexibility is of the utmost importance to bypass the numerous extra- and intracellular biological hurdles and to provide a suitable drug delivery solution for the ever expanding collection of pharmaceuticals and adjuvants with greatly diverging physicochemical properties. In addition, the complex pathophysiology encountered in various diseases also demands a more complex integrative drug delivery approach. Lipid–polymer hybrid NPs have proven to be particularly suited toward targeted drug combination therapy in order to enhance therapeutic efficacy and reduce drug resistance. As polymeric NPs and liposomes have already been widely investigated for stimuli-responsive and image-guided drug delivery, these traits can also be implemented in the engineering of lipid–polymer hybrids to gain spatiotemporal control over the drug delivery process and further expand their use in theranostic applications.On the other hand, the currently witnessed transition toward more sophisticated multifunctional nanocarriers also entails novel challenges that may hamper clinical translation. From a manufacturing perspective, questions arise on the feasibility to scale-up the often labor-intensive multistep production process while meeting quality control standards and safeguarding cost-effectiveness. Most nanoformulations are designed for parenteral administration and thus require aseptic production, which could further add to the overall production costs.19 Therefore, it remains imperative to devote ample attention to clinical benefit by evaluating how these innovative drug delivery systems could outperform current state-of-the-art nanomedicines. An important step forward is the development of single-step and continuous (microfluidic) synthesis of lipid-coated matrix NPs, thereby reducing batch-to-batch variation while maintaining a sufficiently high production rate.92,96,97,247 However, this might not be as feasible for all types of lipid-coated nanomaterials and likely requires independent optimization. To ensure large-scale pharmaceutical production, the horizontal scaling-out of the production process through the use of multiple small-scale production units running in parallel could be favored over conventional scale-up of a single production unit.14
In the advent of nanotoxicology, it will also become increasingly important to make an in-depth assessment of the pharmacokinetic, pharmacodynamic and (immuno)toxicological profile. It is conceivable that multicomponent nanocarriers, constructed from materials with divergent properties, might pose higher risk for carrier-mediated off target toxicity. The emphasis should therefore be put as much as possible on biocompatible and biodegradable materials. The latter may also spur future investigation of bio-mimetic lipid–polymer nanocarriers as a biofunctional yet biocompatible alternative.
Altogether, as judged by the emerging research output on lipid–polymer nanocomposites, these hybrid nanocarriers evoked much enthusiasm among the drug delivery community. With further research investments on the level of manufacturing and pre-clinical assessment in appropriate animal models, we are confident that these multifaceted drug delivery systems could have broad clinical impact.
Acknowledgements
KR is a postdoctoral fellow of the Research Foundation-Flanders (FWO-Vlaanderen). Financial support by the Ghent University Special Research Fund and the FWO is acknowledged with gratitude.Notes and references
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- N. Kamaly, Z. Y. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- K. Raemdonck, R. E. Vandenbroucke, J. Demeester, N. N. Sanders and S. C. De Smedt, Drug Discovery Today, 2008, 13, 917–931 CrossRef CAS PubMed.
- K. Raemdonck, T. F. Martens, K. Braeckmans, J. Demeester and S. C. De Smedt, Adv. Drug Delivery Rev., 2013, 65, 1123–1147 CrossRef CAS PubMed.
- K. Remaut, N. N. Sanders, B. G. De Geest, K. Braeckmans, J. Demeester and S. C. De Smedt, Mater. Sci. Eng., R, 2007, 58, 117–161 CrossRef PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- E. Terreno, F. Uggeri and S. Aime, J. Controlled Release, 2012, 161, 328–337 CrossRef CAS PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- M. E. Davis and M. E. Brewster, Nat. Rev. Drug Discovery, 2004, 3, 1023–1035 CrossRef CAS PubMed.
- X. Q. Zhang, X. Xu, N. Bertrand, E. Pridgen, A. Swami and O. C. Farokhzad, Adv. Drug Delivery Rev., 2012, 64, 1363–1384 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- K. Hadinoto, A. Sundaresan and W. S. Cheow, Eur. J. Pharm. Biopharm., 2013 DOI:10.1016/j.ejpb.2013.07.002.
- N. Maurer, D. B. Fenske and P. R. Cullis, Expert Opin. Biol. Ther., 2001, 1, 923–947 CrossRef CAS PubMed.
- B. Naeye, H. Deschout, M. Roding, M. Rudemo, J. Delanghe, K. Devreese, J. Demeester, K. Braeckmans, S. C. De Smedt and K. Raemdonck, Biomaterials, 2011, 32, 9120–9127 CrossRef CAS PubMed.
- B. Naeye, H. Deschout, V. Caveliers, B. Descamps, K. Braeckmans, C. Vanhove, J. Demeester, T. Lahoutte, S. C. De Smedt and K. Raemdonck, Biomaterials, 2013, 34, 2350–2358 CrossRef CAS PubMed.
- M. M. van Schooneveld, E. Vucic, R. Koole, Y. Zhou, J. Stocks, D. P. Cormode, C. Y. Tang, R. E. Gordon, K. Nicolay, A. Meijerink, Z. A. Fayad and W. J. M. Mulder, Nano Lett., 2008, 8, 2517–2525 CrossRef CAS PubMed.
- B. Mandal, H. Bhattacharjee, N. Mittal, H. Sah, P. Balabathula, L. A. Thoma and G. C. Wood, Nanomedicine, 2013, 9, 474–491 CrossRef CAS PubMed.
- S. Tan, X. Li, Y. Guo and Z. Zhang, Nanoscale, 2013, 5, 860–872 RSC.
- A. L. Troutier and C. Ladaviere, Adv. Colloid Interface Sci., 2007, 133, 1–21 CrossRef CAS PubMed.
- L. Zhang and L. Zhang, Nano LIFE, 2010, 1, 163–173 CrossRef CAS.
- D. Peer, E. J. Park, Y. Morishita, C. V. Carman and M. Shimaoka, Science, 2008, 319, 627–630 CrossRef CAS PubMed.
- P. Vader, B. J. Crielaard, S. M. van Dommelen, R. van der Meel, G. Storm and R. M. Schiffelers, J. Controlled Release, 2012, 160, 211–216 CrossRef CAS PubMed.
- Y. C. Chen, S. R. Bathula, J. Li and L. Huang, J. Biol. Chem., 2010, 285, 22639–22650 CrossRef CAS PubMed.
- S. Chono, S. D. Li, C. C. Conwell and L. Huang, J. Controlled Release, 2008, 131, 64–69 CrossRef CAS PubMed.
- S. Li and L. Huang, Gene Ther., 1997, 4, 891–900 CAS.
- S. D. Li, Y. C. Chen, M. J. Hackett and L. Huang, Mol. Ther., 2008, 16, 163–169 CrossRef CAS PubMed.
- S. D. Li and L. Huang, Biochim. Biophys. Acta Biomembr., 2009, 1788, 2259–2266 CrossRef CAS PubMed.
- H. Hatakeyama, H. Akita and H. Harashima, Adv. Drug Delivery Rev., 2011, 63, 152–160 CrossRef CAS PubMed.
- H. Hatakeyama, H. Akita, E. Ito, Y. Hayashi, M. Oishi, Y. Nagasaki, R. Danev, K. Nagayama, N. Kaji, H. Kikuchi, Y. Baba and H. Harashima, Biomaterials, 2011, 32, 4306–4316 CrossRef CAS PubMed.
- I. A. Khalil, K. Kogure, S. Futaki, S. Hama, H. Akita, M. Ueno, H. Kishida, M. Kudoh, Y. Mishina, K. Kataoka, M. Yamada and H. Harashima, Gene Ther., 2007, 14, 682–689 CrossRef CAS PubMed.
- K. Kogure, H. Akita, Y. Yamada and H. Harashima, Adv. Drug Delivery Rev., 2008, 60, 559–571 CrossRef CAS PubMed.
- H. Akita, R. Ishiba, H. Hatakeyama, H. Tanaka, Y. Sato, K. Tange, M. Arai, K. Kubo and H. Harashima, Adv. Healthcare Mater., 2013, 2, 1120–1125 CAS.
- R. Koole, M. M. van Schooneveld, J. Hilhorst, K. Castermans, D. P. Cormode, G. J. Strijkers, C. D. Donega, D. Vanmaekelbergh, A. W. Griffioen, K. Nicolay, Z. A. Fayad, A. Meijerink and W. J. M. Mulder, Bioconjugate Chem., 2008, 19, 2471–2479 CrossRef CAS PubMed.
- W. J. Mulder, G. J. Strijkers, G. A. van Tilborg, D. P. Cormode, Z. A. Fayad and K. Nicolay, Acc. Chem. Res., 2009, 42, 904–914 CrossRef CAS PubMed.
- S. J. Soenen, M. Hodenius and M. De Cuyper, Nanomedicine, 2009, 4, 177–191 CrossRef CAS PubMed.
- B. Naeye, K. Raemdonck, K. Remaut, J. Demeester and S. C. De Smedt, Curr. Top. Med. Chem., 2012, 12, 89–96 CrossRef CAS.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and T. L. Andresen, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 481–503 CrossRef CAS PubMed.
- S. Mitragotri and J. Lahann, Adv. Mater., 2012, 24, 3717–3723 CrossRef CAS PubMed.
- J. W. Yoo, E. Chambers and S. Mitragotri, Curr. Pharm. Des., 2010, 16, 2298–2307 CrossRef CAS.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- F. Yuan, M. Dellian, D. Fukumura, M. Leunig, D. A. Berk, V. P. Torchilin and R. K. Jain, Cancer Res., 1995, 55, 3752–3756 CAS.
- S. K. Hobbs, W. L. Monsky, F. Yuan, W. G. Roberts, L. Griffith, V. P. Torchilin and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4607–4612 CrossRef CAS.
- M. M. Gottesman, Annu. Rev. Med., 2002, 53, 615–627 CrossRef CAS PubMed.
- M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 48–58 CrossRef CAS PubMed.
- D. Vercauteren, J. Rejman, T. F. Martens, J. Demeester, S. C. De Smedt and K. Braeckmans, J. Controlled Release, 2012, 161, 566–581 CrossRef CAS PubMed.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS PubMed.
- T. G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS PubMed.
- F. Leuschner, P. Dutta, R. Gorbatov, T. I. Novobrantseva, J. S. Donahoe, G. Courties, K. M. Lee, J. I. Kim, J. F. Markmann, B. Marinelli, P. Panizzi, W. W. Lee, Y. Iwamoto, S. Milstein, H. Epstein-Barash, W. Cantley, J. Wong, V. Cortez-Retamozo, A. Newton, K. Love, P. Libby, M. J. Pittet, F. K. Swirski, V. Koteliansky, R. Langer, R. Weissleder, D. G. Anderson and M. Nahrendorf, Nat. Biotechnol., 2011, 29, 1005–1010 CrossRef CAS PubMed.
- A. Bershteyn, M. C. Hanson, M. P. Crespo, J. J. Moon, A. V. Li, H. Suh and D. J. Irvine, J. Controlled Release, 2012, 157, 354–365 CrossRef CAS PubMed.
- M. Look, E. Stern, Q. A. Wang, L. D. DiPlacido, M. Kashgarian, J. Craft and T. M. Fahmy, J. Clin. Invest., 2013, 123, 1741–1749 CAS.
- P. von Hoegen, Adv. Drug Delivery Rev., 2001, 51, 113–125 CrossRef CAS.
- T. A. Wynn, A. Chawla and J. W. Pollard, Nature, 2013, 496, 445–455 CrossRef CAS PubMed.
- C. M. J. Hu, S. Kaushal, H. S. T. Cao, S. Aryal, M. Sartor, S. Esener, M. Bouvet and L. F. Zhang, Mol. Pharmaceutics, 2010, 7, 914–920 CrossRef CAS PubMed.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. W. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. M. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS PubMed.
- Z. Wang and P. C. Ho, Small, 2010, 6, 2576–2583 CrossRef CAS PubMed.
- A. Z. Wang, K. Yuet, L. F. Zhang, F. X. Gu, M. Huynh-Le, A. F. Radovic-Moreno, P. W. Kantoff, N. H. Bander, R. Langer and O. C. Farokhzad, Nanomedicine, 2010, 5, 361–368 CrossRef CAS PubMed.
- L. F. Zhang, J. M. Chan, F. X. Gu, J. W. Rhee, A. Z. Wang, A. F. Radovic-Moreno, F. Alexis, R. Langer and O. C. Farokhzad, ACS Nano, 2008, 2, 1696–1702 CrossRef CAS PubMed.
- Z. Y. Xiao, E. Levy-Nissenbaum, F. Alexis, A. Luptak, B. A. Teply, J. M. Chan, J. J. Shi, E. Digga, J. Cheng, R. Langer and O. C. Farokhzad, ACS Nano, 2012, 6, 696–704 CrossRef CAS PubMed.
- G. Sahay, W. Querbes, C. Alabi, A. Eltoukhy, S. Sarkar, C. Zurenko, E. Karagiannis, K. Love, D. Chen, R. Zoncu, Y. Buganim, A. Schroeder, R. Langer and D. G. Anderson, Nat. Biotechnol., 2013, 31, 653–658 CrossRef CAS PubMed.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- J. Gilleron, W. Querbes, A. Zeigerer, A. Borodovsky, G. Marsico, U. Schubert, K. Manygoats, S. Seifert, C. Andree, M. Stoter, H. Epstein-Barash, L. G. Zhang, V. Koteliansky, K. Fitzgerald, E. Fava, M. Bickle, Y. Kalaidzidis, A. Akinc, M. Maier and M. Zerial, Nat. Biotechnol., 2013, 31, 638-U102 CrossRef PubMed.
- J. Nguyen and F. C. Szoka, Acc. Chem. Res., 2012, 45, 1153–1162 CrossRef CAS PubMed.
- L. Treuel, X. E. Jiang and G. U. Nienhaus, J. R. Soc. Interface, 2013, 10, DOI:10.1098/rsif.2012.0939.
- R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef PubMed.
- J. K. Vasir and V. Labhasetwar, Adv. Drug Delivery Rev., 2007, 59, 718–728 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Preat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- J. M. Anderson and M. S. Shive, Adv. Drug Delivery Rev., 1997, 28, 5–24 CrossRef CAS.
- J. M. Chan, L. F. Zhang, R. Tong, D. Ghosh, W. W. Gao, G. Liao, K. P. Yuet, D. Gray, J. W. Rhee, J. J. Cheng, G. Golomb, P. Libby, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2213–2218 CrossRef CAS PubMed.
- S. Sengupta, D. Eavarone, I. Capila, G. L. Zhao, N. Watson, T. Kiziltepe and R. Sasisekharan, Nature, 2005, 436, 568–572 CrossRef CAS PubMed.
- E. M. Enlow, J. C. Luft, M. E. Napier and J. M. DeSimone, Nano Lett., 2011, 11, 808–813 CrossRef CAS PubMed.
- W. Hasan, K. Chu, A. Gullapalli, S. S. Dunn, E. M. Enlow, J. C. Luft, S. M. Tian, M. E. Napier, P. D. Pohlhaus, J. P. Rolland and J. M. DeSimone, Nano Lett., 2012, 12, 287–292 CrossRef CAS PubMed.
- J. L. Perry, K. P. Herlihy, M. E. Napier and J. M. DeSimone, Acc. Chem. Res., 2011, 44, 990–998 CrossRef CAS PubMed.
- J. Zhou, T. R. Patel, M. Fu, J. P. Bertram and W. M. Saltzman, Biomaterials, 2012, 33, 583–591 CrossRef CAS PubMed.
- C. M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. F. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- J. Thevenot, A. L. Troutier, L. David, T. Delair and C. Ladaviere, Biomacromolecules, 2007, 8, 3651–3660 CrossRef CAS PubMed.
- J. Thevenot, A. L. Troutier, J. L. Putaux, T. Delair and C. Ladaviere, J. Phys. Chem. B, 2008, 112, 13812–13822 CrossRef CAS PubMed.
- A. L. Troutier, T. Delair, C. Pichot and C. Ladaviere, Langmuir, 2005, 21, 1305–1313 CrossRef CAS PubMed.
- H. J. Wang, P. Q. Zhao, W. Y. Su, S. Wang, Z. Y. Liao, R. F. Niu and J. Chang, Biomaterials, 2010, 31, 8741–8748 CrossRef CAS PubMed.
- Z. Y. Liao, H. J. Wang, X. D. Wang, P. Q. Zhao, S. Wang, W. Y. Su and J. Chang, Adv. Funct. Mater., 2011, 21, 1179–1186 CrossRef CAS.
- S. M. D'Addio and R. K. Prud'homme, Adv. Drug Delivery Rev., 2011, 63, 417–426 CrossRef CAS PubMed.
- Y. Liu, J. Pan and S. S. Feng, Int. J. Pharm., 2010, 395, 243–250 CrossRef CAS PubMed.
- C. H. Chu, Y. C. Wang, H. Y. Huang, L. C. Wu and C. S. Yang, Nanotechnology, 2011, 22, 185601 CrossRef PubMed.
- J. M. Chan, L. F. Zhang, K. P. Yuet, G. Liao, J. W. Rhee, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 1627–1634 CrossRef CAS PubMed.
- X. Z. Yang, S. Dou, Y. C. Wang, H. Y. Long, M. H. Xiong, C. Q. Mao, Y. D. Yao and J. Wang, ACS Nano, 2012, 6, 4955–4965 CrossRef CAS PubMed.
- A. Bershteyn, J. Chaparro, R. Yau, M. Kim, E. Reinherz, L. Ferreira-Moita and D. J. Irvine, Soft Matter, 2008, 4, 1787–1791 RSC.
- M. T. Stephan, J. J. Moon, S. H. Um, A. Bershteyn and D. J. Irvine, Nat. Med., 2010, 16, 1035–1041 CrossRef CAS PubMed.
- R. H. Fang, S. Aryal, C. M. J. Hu and L. F. Zhang, Langmuir, 2010, 26, 16958–16962 CrossRef CAS PubMed.
- R. H. Fang, K. N. H. Chen, S. Aryal, C. M. J. Hu, K. Zhang and L. F. Zhang, Langmuir, 2012, 28, 13824–13829 CrossRef CAS PubMed.
- P. M. Valencia, O. C. Farokhzad, R. Karnik and R. Langer, Nat. Nanotechnol., 2012, 7, 623–629 CrossRef CAS PubMed.
- B. K. Johnson and R. K. Prud'homme, Phys. Rev. Lett., 2003, 91, 118302 CrossRef.
- L. Capretto, W. Cheng, M. Hill and X. L. Zhang, in Microfluidics: Technologies and Applications, ed. B. C. Lin, Springer-Verlag, Berlin, 2011, vol. 304, pp. 27–68 Search PubMed.
- P. M. Valencia, P. A. Basto, L. F. Zhang, M. Rhee, R. Langer, O. C. Farokhzad and R. Karnik, ACS Nano, 2010, 4, 1671–1679 CrossRef CAS PubMed.
- Y. T. Kim, B. L. Chung, M. M. Ma, W. J. M. Mulder, Z. A. Fayad, O. C. Farokhzad and R. Langer, Nano Lett., 2012, 12, 3587–3591 CrossRef CAS PubMed.
- C. S. Kim, G. Y. Tonga, D. Solfiell and V. M. Rotello, Adv. Drug Delivery Rev., 2013, 65, 93–99 CrossRef CAS PubMed.
- R. Richter, A. Mukhopadhyay and A. Brisson, Biophys. J., 2003, 85, 3035–3047 CrossRef CAS.
- S. Mornet, O. Lambert, E. Duguet and A. Brisson, Nano Lett., 2005, 5, 281–285 CrossRef CAS PubMed.
- R. Rapuano and A. M. Carmona-Ribeiro, J. Colloid Interface Sci., 1997, 193, 104–111 CrossRef CAS.
- R. Rapuano and A. M. Carmona-Ribeiro, J. Colloid Interface Sci., 2000, 226, 299–307 CrossRef CAS.
- F. Q. Tang, L. L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- Y. Wan and D. Y. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed.
- D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- M. Vallet-Regi, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S. Lin, Small, 2010, 6, 1952–1967 CrossRef CAS PubMed.
- J. W. Liu, A. Stace-Naughton, X. M. Jiang and C. J. Brinker, J. Am. Chem. Soc., 2009, 131, 1354–1355 CrossRef CAS PubMed.
- L. S. Wang, L. C. Wu, S. Y. Lu, L. L. Chang, I. T. Teng, C. M. Yang and J. A. A. Ho, ACS Nano, 2010, 4, 4371–4379 CrossRef CAS PubMed.
- J. Liu, X. Jiang, C. Ashley and C. J. Brinker, J. Am. Chem. Soc., 2009, 131, 7567–7569 CrossRef CAS PubMed.
- J. Liu, A. Stace-Naughton and C. J. Brinker, Chem. Commun., 2009, 5100–5102 RSC.
- C. E. Ashley, E. C. Carnes, K. E. Epler, D. P. Padilla, G. K. Phillips, R. E. Castillo, D. C. Wilkinson, B. S. Wilkinson, C. A. Burgard, R. M. Kalinich, J. L. Townson, B. Chackerian, C. L. Willman, D. S. Peabody, W. Wharton and C. J. Brinker, ACS Nano, 2012, 6, 2174–2188 CrossRef CAS PubMed.
- K. Epler, D. Padilla, G. Phillips, P. Crowder, R. Castillo, D. Wilkinson, B. Wilkinson, C. Burgard, R. Kalinich, J. Townson, B. Chackerian, C. Willman, D. Peabody, W. Wharton, C. J. Brinker, C. Ashley and E. Carnes, Adv. Healthcare Mater., 2012, 1, 348–353 CrossRef CAS PubMed.
- H. T. Lv, S. B. Zhang, B. Wang, S. H. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- S. A. Audouy, L. F. de Leij, D. Hoekstra and G. Molema, Pharm. Res., 2002, 19, 1599–1605 CrossRef CAS.
- S. J. Soenen, A. R. Brisson and M. De Cuyper, Biomaterials, 2009, 30, 3691–3701 CrossRef CAS PubMed.
- I. T. Teng, Y. J. Chang, L. S. Wang, H. Y. Lu, L. C. Wu, C. M. Yang, C. C. Chiu, C. H. Yang, S. L. Hsu and J. A. Ho, Biomaterials, 2013, 34, 7462–7470 CrossRef CAS PubMed.
- R. A. Roggers, V. S. Y. Lin and B. G. Trewyn, Mol. Pharmaceutics, 2012, 9, 2770–2777 CrossRef CAS PubMed.
- V. Cauda, H. Engelke, A. Sauer, D. Arcizet, C. Brauchle, J. Radler and T. Bein, Nano Lett., 2010, 10, 2484–2492 CrossRef CAS PubMed.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2002, 54, 13–36 CrossRef CAS.
- O. Wichterle and D. Lim, Nature, 1960, 185, 117–118 CrossRef.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- K. Raemdonck, J. Demeester and S. De Smedt, Soft Matter, 2009, 5, 707–715 RSC.
- R. T. Chacko, J. Ventura, J. M. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836–851 CrossRef CAS PubMed.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS PubMed.
- S. Buck, P. S. Pennefather, H. Y. Xue, J. Grant, Y. L. Cheng and C. J. Allen, Biomacromolecules, 2004, 5, 2230–2237 CrossRef CAS PubMed.
- B. G. De Geest, S. De Koker, J. Demeester, S. C. De Smedt and W. E. Hennink, Polym. Chem., 2010, 1, 137–148 RSC.
- T. Jin, P. Pennefather and P. I. Lee, FEBS Lett., 1996, 397, 70–74 CrossRef CAS.
- P. F. Kiser, G. Wilson and D. Needham, Nature, 1998, 394, 459–462 CrossRef CAS PubMed.
- P. F. Kiser, G. Wilson and D. Needham, J. Controlled Release, 2000, 68, 9–22 CrossRef CAS.
- N. MacKinnon, G. Guerin, B. X. Liu, C. C. Gradinaru, J. L. Rubinstein and P. M. Macdonald, Langmuir, 2010, 26, 1081–1089 CrossRef CAS PubMed.
- Q. Saleem, B. X. Liu, C. C. Gradinaru and P. M. Macdonald, Biomacromolecules, 2011, 12, 2364–2374 CrossRef CAS PubMed.
- A. Jesorka, M. Markstrom, M. Karlsson and O. Orwar, J. Phys. Chem. B, 2005, 109, 14759–14763 CrossRef CAS PubMed.
- M. Peyrot, A. M. Sautereau, J. M. Rabanel, F. Nguyen, J. F. Tocanne and D. Samain, Int. J. Pharm., 1994, 102, 25–33 CrossRef CAS.
- I. De Miguel, L. Imbertie, V. Rieumajou, M. Major, R. Kravtzoff and D. Betbeder, Pharm. Res., 2000, 17, 817–824 CrossRef CAS.
- L. Fenart, A. Casanova, B. Dehouck, C. Duhem, S. Slupek, R. Cecchelli and D. Betbeder, J. Pharmacol. Exp. Ther., 1999, 291, 1017–1022 CAS.
- A. Erten, W. Wrasidlo, M. Scadeng, S. Esener, R. M. Hoffman, M. Bouvet and M. Makale, Nanomedicine, 2010, 6, 797–807 CrossRef CAS PubMed.
- T. G. Van Thienen, B. Lucas, F. M. Flesch, C. F. van Nostrum, J. Demeester and S. C. De Smedt, Macromolecules, 2005, 38, 8503–8511 CrossRef CAS.
- T. G. Van Thienen, K. Raemdonck, J. Demeester and S. C. De Smedt, Langmuir, 2007, 23, 9794–9801 CrossRef CAS PubMed.
- T. G. Van Thienen, J. Demeester and S. C. De Smedt, Int. J. Pharm., 2008, 351, 174–185 CrossRef CAS PubMed.
- S. Kazakov, M. Kaholek, I. Teraoka and K. Levon, Macromolecules, 2002, 35, 1911–1920 CrossRef CAS.
- S. Kazakov, M. Kaholek, D. Kudasheva, I. Teraoka, M. K. Cowman and K. Levon, Langmuir, 2003, 19, 8086–8093 CrossRef CAS.
- J. P. Schillemans, F. M. Flesch, W. E. Hennink and C. F. van Nostrum, Macromolecules, 2006, 39, 5885–5890 CrossRef CAS.
- J. Park, S. H. Wrzesinski, E. Stern, M. Look, J. Criscione, R. Ragheb, S. M. Jay, S. L. Demento, A. Agawu, L. P. Licona, A. F. Ferrandino, D. Gonzalez, A. Habermann, R. A. Flavell and T. M. Fahmy, Nat. Mater., 2012, 11, 895–905 CrossRef CAS PubMed.
- J. S. Hong, W. N. Vreeland, S. H. Paoli Lacerda, L. E. Locascio, M. Gaitan and S. R. Raghavan, Langmuir, 2008, 24, 4092–4096 CrossRef CAS PubMed.
- X. F. Su, J. Fricke, D. G. Kavanagh and D. J. Irvine, Pharmaceutics, 2011, 8, 774–787 CrossRef CAS PubMed.
- T. Ishida, D. L. Iden and T. M. Allen, FEBS Lett., 1999, 460, 129–133 CrossRef CAS.
- P. S. Uster, T. M. Allen, B. E. Daniel, C. J. Mendez, M. S. Newman and G. Z. Zhu, FEBS Lett., 1996, 386, 243–246 CrossRef CAS.
- B. Romberg, W. E. Hennink and G. Storm, Pharm. Res., 2008, 25, 55–71 CrossRef CAS PubMed.
- M. E. Werner, S. Karve, R. Sukumar, N. D. Cummings, J. A. Copp, R. C. Chen, T. Zhang and A. Z. Wang, Biomaterials, 2011, 32, 8548–8554 CrossRef CAS PubMed.
- M. E. Werner, J. A. Copp, S. Karve, N. D. Cummings, R. Sukumar, C. X. Li, M. E. Napier, R. C. Chen, A. D. Cox and A. Z. Wang, ACS Nano, 2011, 5, 8990–8998 CrossRef CAS PubMed.
- Y. Liu, K. Li, J. Pan, B. Liu and S. S. Feng, Biomaterials, 2010, 31, 330–338 CrossRef CAS PubMed.
- Y. Zheng, B. Yu, W. Weecharangsan, L. Piao, M. Darby, Y. Mao, R. Koynova, X. Yang, H. Li, S. Xu, L. J. Lee, Y. Sugimoto, R. W. Brueggemeier and R. J. Lee, Int. J. Pharm., 2010, 390, 234–241 CrossRef CAS PubMed.
- P. R. Desai, S. Marepally, A. R. Patel, C. Voshavar, A. Chaudhuri and M. Singh, J. Controlled Release, 2013, 170, 51–63 CrossRef CAS PubMed.
- J. J. Shi, Z. Y. Xiao, A. R. Votruba, C. Vilos and O. C. Farokhzad, Angew. Chem., Int. Ed., 2011, 50, 7027–7031 CrossRef CAS PubMed.
- X. Z. Yang, S. Dou, T. M. Sun, C. Q. Mao, H. X. Wang and J. Wang, J. Controlled Release, 2011, 156, 203–211 CrossRef CAS PubMed.
- J. M. Chan, J. W. Rhee, C. L. Drum, R. T. Bronson, G. Golomb, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19347–19352 CrossRef CAS PubMed.
- C. M. J. Hu and L. F. Zhang, Biochem. Pharmacol., 2012, 83, 1104–1111 CrossRef CAS PubMed.
- S. Aryal, C. M. J. Hu and L. F. Zhang, Small, 2010, 6, 1442–1448 CrossRef CAS PubMed.
- S. Aryal, C. M. J. Hu, V. Fu and L. F. Zhang, J. Mater. Chem., 2012, 22, 994–999 RSC.
- V. P. Chauhan, T. Stylianopoulos, J. D. Martin, Z. Popovic, O. Chen, W. S. Kamoun, M. G. Bawendi, D. Fukumura and R. K. Jain, Nat. Nanotechnol., 2012, 7, 383–388 CrossRef CAS PubMed.
- R. M. Schiffelers, A. Ansari, J. Xu, Q. Zhou, Q. Tang, G. Storm, G. Molema, P. Y. Lu, P. V. Scaria and M. C. Woodle, Nucleic Acids Res., 2004, 32, e149 CrossRef PubMed.
- R. Manchanda, A. Fernandez-Fernandez, A. Nagesetti and A. J. McGoron, Colloids Surf., B, 2010, 75, 260–267 CrossRef CAS PubMed.
- C. F. Zheng, M. B. Zheng, P. Gong, D. X. Jia, P. F. Zhang, B. H. Shi, Z. H. Sheng, Y. F. Ma and L. T. Cai, Biomaterials, 2012, 33, 5603–5609 CrossRef CAS PubMed.
- P. Wust, B. Hildebrandt, G. Sreenivasa, B. Rau, J. Gellermann, H. Riess, R. Felix and P. M. Schlag, Lancet Oncol., 2002, 3, 487–497 CrossRef CAS.
- M. B. Zheng, C. X. Yue, Y. F. Ma, P. Gong, P. F. Zhao, C. F. Zheng, Z. H. Sheng, P. F. Zhang, Z. H. Wang and L. T. Cai, ACS Nano, 2013, 7, 2056–2067 CrossRef CAS PubMed.
- R. Cheng, F. Meng, C. Deng, H. A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS PubMed.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- S. D. Kong, M. Sartor, C. M. J. Hu, W. Z. Zhang, L. F. Zhang and S. H. Jin, Acta Biomater., 2013, 9, 5447–5452 CrossRef PubMed.
- C. Clawson, L. Ton, S. Aryal, V. Fu, S. Esener and L. F. Zhang, Langmuir, 2011, 27, 10556–10561 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Preat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- M. L. De Temmerman, H. Dewitte, R. E. Vandenbroucke, B. Lucas, C. Libert, J. Demeester, S. C. De Smedt, I. Lentacker and J. Rejman, Biomaterials, 2011, 32, 9128–9135 CrossRef CAS PubMed.
- K. A. Woodrow, Y. Cu, C. J. Booth, J. K. Saucier-Sawyer, M. J. Wood and W. M. Saltzman, Nat. Mater., 2009, 8, 526–533 CrossRef CAS PubMed.
- A. Alshamsan, A. Haddadi, S. Hamdy, J. Samuel, A. O. El-Kadi, H. Uludag and A. Lavasanifar, Mol. Pharmaceutics, 2010, 7, 1643–1654 CrossRef CAS PubMed.
- N. Murata, Y. Takashima, K. Toyoshima, M. Yamamoto and H. Okada, J. Controlled Release, 2008, 126, 246–254 CrossRef CAS PubMed.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- S. Muro, C. Garnacho, J. A. Champion, J. Leferovich, C. Gajewski, E. H. Schuchman, S. Mitragotri and V. R. Muzykantov, Mol. Ther., 2008, 16, 1450–1458 CrossRef CAS PubMed.
- D. J. Irvine, Nat. Mater., 2011, 10, 342–343 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- N. J. Carroll, S. Pylypenko, P. B. Atanassov and D. N. Petsev, Langmuir, 2009, 25, 13540–13544 CrossRef CAS PubMed.
- A. Deshpande, P. Sicinski and P. W. Hinds, Oncogene, 2005, 24, 2909–2915 CrossRef CAS PubMed.
- M. de Virgilio, A. Lombardi, R. Caliandro and M. S. Fabbrini, Toxins, 2010, 2, 2699–2737 CrossRef CAS PubMed.
- A. Schloßbauer, A. M. Sauer, V. Cauda, A. Schmidt, H. Engelke, U. Rothbauer, K. Zolghadr, H. Leonhardt, C. Brauchle and T. Bein, Adv. Healthcare Mater., 2012, 1, 316–320 CrossRef PubMed.
- K. Berg, M. Folini, L. Prasmickaite, P. K. Selbo, A. Bonsted, B. O. Engesaeter, N. Zaffaroni, A. Weyergang, A. Dietze, G. M. Maelandsmo, E. Wagner, O. J. Norum and A. Hogset, Curr. Pharm. Biotechnol., 2007, 8, 362–372 CAS.
- A. Hogset, L. Prasmickaite, P. K. Selbo, M. Hellum, B. O. Engesaeter, A. Bonsted and K. Berg, Adv. Drug Delivery Rev., 2004, 56, 95–115 CrossRef CAS PubMed.
- K. Berg, P. K. Selbo, L. Prasmickaite, T. E. Tjelle, K. Sandvig, D. Moan, G. Gaudernack, O. Fodstad, S. Kjolsrud, H. Anholt, G. H. Rodal, S. K. Rodal and A. Hogset, Cancer Res., 1999, 59, 1180–1183 CAS.
- P. K. Selbo, A. Weyergang, A. Hogset, O. J. Norum, M. B. Berstad, M. Vikdal and K. Berg, J. Controlled Release, 2010, 148, 2–12 CrossRef CAS PubMed.
- K. Raemdonck, B. Naeye, K. Buyens, R. E. Vandenbroucke, A. Hogset, J. Demeester and S. C. De Smedt, Adv. Funct. Mater., 2009, 19, 1406–1415 CrossRef CAS.
- K. Raemdonck, B. Naeye, A. Hogset, J. Demeester and S. C. De Smedt, J. Controlled Release, 2010, 145, 281–288 CrossRef CAS PubMed.
- E. Bringas, O. Koysuren, D. V. Quach, M. Mahmoudi, E. Aznar, J. D. Roehling, M. D. Marcos, R. Martinez-Manez and P. Stroeve, Chem. Commun., 2012, 48, 5647–5649 RSC.
- B. T. Luk, R. H. Fang and L. F. Zhang, Theranostics, 2012, 2, 1117–1126 CrossRef CAS PubMed.
- B. G. De Geest, B. G. Stubbe, A. M. Jonas, T. Van Thienen, W. L. J. Hinrichs, J. Demeester and S. C. De Smedt, Biomacromolecules, 2006, 7, 373–379 CrossRef PubMed.
- C. Allal, S. Sixou, R. Kravtzoff, N. Soulet, G. Soula and G. Favre, Br. J. Cancer, 1998, 77, 1448–1453 CrossRef CAS.
- M. Vanneman and G. Dranoff, Nat. Rev. Cancer, 2012, 12, 237–251 CrossRef CAS PubMed.
- D. M. Pardoll, Nat. Rev. Cancer, 2012, 12, 252–264 CrossRef CAS PubMed.
- D. I. Gabrilovich, S. Ostrand-Rosenberg and V. Bronte, Nat. Rev. Immunol., 2012, 12, 253–268 CrossRef CAS PubMed.
- R. Juliano, Nat. Rev. Drug Discovery, 2013, 12, 171–172 CrossRef CAS PubMed.
- S. C. Balmert and S. R. Little, Adv. Mater., 2012, 24, 3757–3778 CrossRef CAS PubMed.
- J. W. Yoo, D. J. Irvine, D. E. Discher and S. Mitragotri, Nat. Rev. Drug Discovery, 2011, 10, 521–535 CrossRef CAS PubMed.
- M. G. Damiano, R. K. Mutharasan, S. Tripathy, K. M. McMahon and C. S. Thaxton, Adv. Drug Delivery Rev., 2013, 65, 649–662 CrossRef CAS PubMed.
- D. P. Cormode, T. Skajaa, M. M. van Schooneveld, R. Koole, P. Jarzyna, M. E. Lobatto, C. Calcagno, A. Barazza, R. E. Gordon, P. Zanzonico, E. A. Fisher, Z. A. Fayad and W. J. M. Mulder, Nano Lett., 2008, 8, 3715–3723 CrossRef CAS PubMed.
- S. El Andaloussi, I. Mager, X. O. Breakefield and M. J. Wood, Nat. Rev. Drug Discovery, 2013, 12, 347–357 CrossRef CAS PubMed.
- C. M. Hu, R. H. Fang and L. Zhang, Adv. Healthcare Mater., 2012, 1, 537–547 CrossRef CAS PubMed.
- P. L. Rodriguez, T. Harada, D. A. Christian, D. A. Pantano, R. K. Tsai and D. E. Discher, Science, 2013, 339, 971–975 CrossRef CAS PubMed.
- R. K. Tsai, P. L. Rodriguez and D. E. Discher, Blood Cells, Mol. Dis., 2010, 45, 67–74 CrossRef CAS PubMed.
- A. Judge, K. McClintock, J. R. Phelps and I. MacLachlan, Mol. Ther., 2006, 13, 328–337 CrossRef CAS PubMed.
- M. Cavadas, A. Gonzalez-Fernandez and R. Franco, Nanomedicine, 2011, 7, 730–743 CrossRef CAS PubMed.
- T. Ishida, M. Ichihara, X. Wang, K. Yamamoto, J. Kimura, E. Majima and H. Kiwada, J. Controlled Release, 2006, 112, 15–25 CrossRef CAS PubMed.
- S. M. Moghimi, A. J. Andersen, S. H. Hashemi, B. Lettiero, D. Ahmadvand, A. C. Hunter, T. L. Andresen, I. Hamad and J. Szebeni, J. Controlled Release, 2010, 146, 175–181 CrossRef CAS PubMed.
- C. Salvador-Morales, L. F. Zhang, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 2231–2240 CrossRef CAS PubMed.
- C. M. Hu, R. H. Fang, J. Copp, B. T. Luk and L. Zhang, Nat. Nanotechnol., 2013, 8, 336–340 CrossRef CAS PubMed.
- L. De Backer, K. Braeckmans, J. Demeester, S. C. De Smedt and K. Raemdonck, Nanomedicine, 2013, 8, 1625–1638 CrossRef CAS PubMed.
- O. M. Merkel and T. Kissel, Acc. Chem. Res., 2012, 45, 961–970 CrossRef CAS PubMed.
- M. Benfer and T. Kissel, Eur. J. Pharm. Biopharm., 2012, 80, 247–256 CrossRef CAS PubMed.
- J. Nguyen, R. Reul, T. Betz, E. Dayyoub, T. Schmehl, T. Gessler, U. Bakowsky, W. Seeger and T. Kissel, J. Controlled Release, 2009, 140, 47–54 CrossRef CAS PubMed.
- M. L. De Temmerman, J. Rejman, R. E. Vandenbroucke, S. De Koker, C. Libert, J. Grooten, J. Demeester, B. Gander and S. C. De Smedt, J. Controlled Release, 2012, 158, 233–239 CrossRef CAS PubMed.
- W. L. Jiang, R. K. Gupta, M. C. Deshpande and S. P. Schwendeman, Adv. Drug Delivery Rev., 2005, 57, 391–410 CrossRef CAS PubMed.
- S. L. Demento, W. G. Cui, J. M. Criscione, E. Stern, J. Tulipan, S. M. Kaech and T. M. Fahmy, Biomaterials, 2012, 33, 4957–4964 CrossRef CAS PubMed.
- S. L. Demento, A. L. Siefert, A. Bandyopadhyay, F. A. Sharp and T. M. Fahmy, Trends Biotechnol., 2011, 29, 294–306 CrossRef CAS PubMed.
- V. Cerundolo, J. D. Silk, S. H. Masri and M. Salio, Nat. Rev. Immunol., 2009, 9, 28–38 CrossRef CAS PubMed.
- F. Joris, B. B. Manshian, K. Peynshaert, S. C. De Smedt, K. Braeckmans and S. J. Soenen, Chem. Soc. Rev., 2013 10.1039/C3CS60145E.
- A. Nel, T. Xia, L. Madler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- B. Pelaz, G. Charron, C. Pfeiffer, Y. L. Zhao, J. M. de la Fuente, X. J. Liang, W. J. Parak and P. del Pino, Small, 2013, 9, 1573–1584 CrossRef CAS PubMed.
- S. J. Soenen, P. Rivera-Gil, J. M. Montenegro, W. J. Parak, S. C. De Smedt and K. Braeckmans, Nano Today, 2011, 6, 446–465 CrossRef CAS PubMed.
- A. Nel, T. Xia, H. Meng, X. Wang, S. Lin, Z. Ji and H. Zhang, Acc. Chem. Res., 2013, 46, 607–621 CrossRef CAS PubMed.
- C. D. Walkey and W. C. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- J. Szebeni, F. Muggia, A. Gabizon and Y. Barenholz, Adv. Drug Delivery Rev., 2011, 63, 1020–1030 CrossRef CAS PubMed.
- R. Kedmi, N. Ben-Arie and D. Peer, Biomaterials, 2010, 31, 6867–6875 CrossRef CAS PubMed.
- S. M. Moghimi, A. J. Andersen, D. Ahmadvand, P. P. Wibroe, T. L. Andresen and A. C. Hunter, Adv. Drug Delivery Rev., 2011, 63, 1000–1007 CrossRef CAS PubMed.
- S. M. Moghimi, I. Hamad, T. L. Andresen, K. Jorgensen and J. Szebeni, FASEB J., 2006, 20, 2591–2593 CrossRef CAS PubMed.
- J. Szebeni, P. Bedocs, Z. Rozsnyay, Z. Weiszhar, R. Urbanics, L. Rosivall, R. Cohen, O. Garbuzenko, G. Bathori, M. Toth, R. Bunger and Y. Barenholz, Nanomedicine, 2012, 8, 176–184 CrossRef CAS PubMed.
- T. Ishida, X. Wang, T. Shimizu, K. Nawata and H. Kiwada, J. Controlled Release, 2007, 122, 349–355 CrossRef CAS PubMed.
- X. Y. Wang, T. Ishida and H. Kiwada, J. Controlled Release, 2007, 119, 236–244 CrossRef CAS PubMed.
- S. Abu Lila, H. Kiwada and T. Ishida, J. Controlled Release, 2013, 172, 38–47 CrossRef PubMed.
- S. J. Soenen, L. De Backer, B. Manshian, S. Doak, K. Raemdonck, J. Demeester, K. Braeckmans and S. C. De Smedt, Nanomedicine, 2013 DOI:10.2217/nnm.12.208.
- I. I. Slowing, C. W. Wu, J. L. Vivero-Escoto and V. S. Lin, Small, 2009, 5, 57–62 CrossRef CAS PubMed.
- T. Yu, A. Malugin and H. Ghandehari, ACS Nano, 2011, 5, 5717–5728 CrossRef CAS PubMed.
- V. Hornung, F. Bauernfeind, A. Halle, E. O. Samstad, H. Kono, K. L. Rock, K. A. Fitzgerald and E. Latz, Nat. Immunol., 2008, 9, 847–856 CrossRef CAS PubMed.
- B. Sun, X. Wang, Z. Ji, R. Li and T. Xia, Small, 2013, 9, 1595–1607 CrossRef CAS PubMed.
- J. G. van den Boorn, M. Schlee, C. Coch and G. Hartmann, Nat. Biotechnol., 2011, 29, 325–326 CrossRef CAS PubMed.
- C. Lonez, M. Vandenbranden and J. M. Ruysschaert, Adv. Drug Delivery Rev., 2012, 64, 1749–1758 CrossRef CAS PubMed.
- A. K. Larsen, D. Malinska, I. Koszela-Piotrowska, L. Parhamifar, A. C. Hunter and S. M. Moghimi, Mitochondrion, 2012, 12, 162–168 CrossRef CAS PubMed.
- J. S. Hong, S. M. Stavis, S. H. DePaoli Lacerda, L. E. Locascio, S. R. Raghavan and M. Gaitan, Langmuir, 2010, 26, 11581–11588 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |