Chemical reduction of graphene oxide: a synthetic chemistry viewpoint
Chun Kiang
Chua
and
Martin
Pumera
*
Nanyang Technological University – Chemistry and Biological, Chemistry, Singapore 637371, Singapore. E-mail: pumera@ntu.edu.sg
First published on 11th October 2013
Abstract
The chemical reduction of graphene oxide is a promising route towards the large scale production of graphene for commercial applications. The current state-of-the-art in graphene oxide reduction, consisting of more than 50 types of reducing agent, will be reviewed from a synthetic chemistry point of view. Emphasis is placed on the techniques, reaction mechanisms and the quality of the produced graphene. The reducing agents are reviewed under two major categories: (i) those which function according to well-supported mechanisms and (ii) those which function according to proposed mechanisms based on knowledge of organic chemistry. This review will serve as a valuable platform to understand the efficiency of these reducing agents for the reduction of graphene oxide.
 Chun Kiang Chua | Chun Kiang Chua obtained his BSc (Hons.) in Chemistry & Biological Chemistry from the Nanyang Technological University, Singapore, in 2010. He received his PhD degree from the same university in 2013. He pursued his PhD with Prof. Martin Pumera on a thesis focused on the synthesis and electrochemistry of graphene materials obtained by the top-down approach. His research interests revolve around the organic chemistry modifications and electrochemistry of graphene materials. |
 Martin Pumera | Prof. Martin Pumera has been a faculty member at Nanyang Technological University, Singapore, since 2010. He received his PhD from Charles University, Czech Republic, in 2001. After two postdoctoral stays (in the USA and Spain), he joined the National Institute for Materials Science, Japan, in 2006 on a tenure-track arrangement and stayed there until spring 2008 when he accepted a tenured position at NIMS. In 2009, Prof. Pumera received an ERC-StG award. Prof. Pumera has a broad interest in nanomaterials and microsystems, in the specific areas of electrochemistry and synthetic chemistry of carbon nanomaterials, nanomotors, nanotoxicity, and energy storage devices. He is associate editor of Science and Technology of Advanced Materials, member of Editorial board of Electrophoresis, Electroanalysis, The Chemical Records, ChemElectroChem and eight other journals. He has published over 200 peer-reviewed articles and has an h-index of 41. |
1. Introduction
The key to success in new materials' commercial applications lies in the ability to produce high quality materials on an industrially viable scale. There is no doubt that the 21st century is the era of carbon nanomaterials, from graphite to carbon nanotubes and now with intense focus on graphene. Graphene belongs to a new class of carbon nanomaterials – 2-D materials made up entirely of conjugated sp2 carbons arranged in a honeycomb structure. Ever since the 2004 isolation of free-standing graphene and subsequent 2010 Nobel Prize in Physics awarded to both Andre Geim and Konstantin Novoselov, graphene research has experienced a phenomenal growth. The main reason behind the fast-paced development of graphene research is due to the unique properties of graphene. Several experimentally measured properties have already surpassed those observed in other types of materials. With enhanced electrical conductivity, high mechanical strength, high thermal conductivity, high impermeability to gases and optical transparency, graphene holds great promise as the next wonder material.1–4 Such an ideal is, however, achievable only if the large scale production of high quality graphene is attained.The current state of graphene production is divided between two approaches – the bottom-up and top-down. The bottom-up approach seeks to build graphene sheets from scratch, starting with simple carbon molecules such as methane and ethanol. On the other hand, the top-down approach relies on the fundamental idea of extracting layers of graphene from graphite. Both methods provide graphene of contrasting quality and yield. However, in terms of high yield and low cost, the top-down approach via chemical oxidation and reduction is the most convenient method to date. Moreover, graphene obtained via such chemical treatment is important for a large portion of the graphene community that endeavours to use graphene in applications such as composites, coatings, paint/ink, transparent conductive layers, bioapplications and energy storage.5–7 Interested readers are recommended several review articles for more information on other production methods of graphene.3,8–10
There is currently a wide range of reducing agents for graphene oxide in the literature. As the list continues to expand, the graphene community could be easily overwhelmed by the collection of this vast knowledge. Several key concerns of the graphene community are with regard to achieving the highest reduction capability, healing the defective graphene oxide, selectively removing a single type of oxygen moiety, improving the dispersion stability of the resulting graphene, as well as applying environmentally friendly and affordable reducing agents. The pursuit in search of a reducing agent which could achieve most of these has resulted in the introduction of several new chemicals/compounds to function as reducing agents. As such, it is critical, at this juncture, to pause and review the general progress of this field.
This review will focus on the fundamental core issues of graphene oxide reduction (of oxygen functionalities) from a synthetic chemistry point of view. Although the term ‘reduction’ is most often defined as the gain of electrons or decrease in oxidation number by inorganic chemists, organic chemists often view such a process as a loss of oxygen or gain of hydrogen. In fact, the practice of organic chemistry qualitatively defined reduction as conversion of a functional group in a molecule from one category to a lower one.11 Such conversion can occur based on several mechanisms, such as direct electron transfer, hydride transfer or hydrogen atom transfer. While the exact mechanism of a reduction process is often difficult to deduce, it is perhaps only appropriate to apply reducing agents that have been previously demonstrated on smaller organic molecules, onto graphene oxide. This can hopefully provide a higher confidence for further modifications of the obtained graphene.
As such, this review covers the chemical reduction of graphene oxide, including brief discussions on the preparation and structure of graphite oxide, its precursor. The chemical reduction approaches are classified into methods with (i) well-supported and (ii) proposed mechanisms. Reducing agents found in the first category are known to specifically reduce or remove carbon–oxygen moieties based on well-accepted mechanisms or are at least widely applied in organic chemistry. On the other hand, the second category includes unconventional reducing agents with unproven/unknown reducing capabilities for carbon–oxygen functionalities in accordance with organic chemistry.
2. Discovery and preparation of graphite oxide
Graphite oxide was discovered much earlier than graphene. In 1859, when an abundance of graphite from Ceylon (now known as Sri Lanka) arrived in Britain, Professor Brodie from the University of Oxford attempted to measure the molecular weight of the graphite.12 A mixture of graphite and fuming nitric acid in the presence of potassium chlorate was heated at 60 °C for three to four days. The material was then washed, and re-oxidised under the same conditions, up to four repetitions, to finally give a light yellow solid upon drying at 100 °C. The C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H:O ratio of the material was determined to be 61.04:1.85:37.11. Since the material was found to disperse in pure or alkaline water, but not in acidic media, Brodie termed the material “graphic acid”. That was the very first sample of graphite oxide prepared experimentally.
:H:O ratio of the material was determined to be 61.04:1.85:37.11. Since the material was found to disperse in pure or alkaline water, but not in acidic media, Brodie termed the material “graphic acid”. That was the very first sample of graphite oxide prepared experimentally.
In 1898, Staudenmaier improved the oxidation method by adding potassium chlorate in small portions and further acidifying the mixture with concentrated sulphuric acid.13 The material obtained by Staudenmaier gave almost similar C/O ratio as that prepared by Brodie. However, this method was practically more convenient since it does not require four repetitions of oxidation.
A few decades later in 1937, Hofmann replaced the usage of fuming nitric acid with non-fuming nitric acid in his oxidation method.14 In 1958, Hummers and Offeman presented a safer approach by utilizing potassium permanganate as oxidant in a mixture of concentrated sulphuric acid and sodium nitrate.15 This method was safer since it produced nitric acid in situ, and thus the usage of highly corrosive fuming nitric acid could be avoided. Despite the absence of fuming nitric acid, the combination of potassium permanganate and sodium nitrate resulted in a more heavily oxygenated form of graphite oxide. As such, the Hummers method has been very well received and adopted by many researchers. However, all these procedures resulted in the production of toxic gases, mainly NO2, N2O4 and explosive ClO2 (Staudenmaier–Hofmann).
In an effort to improve the oxidation method, Tour in 2010 replaced in situ production of nitric acid with less corrosive phosphoric acid.16 The method claimed to provide a more oxidised form of graphite oxide and highlighted a more intact graphitic basal-plane. Moreover, the possibility for large scale production of graphite oxide seems likely since the method does not involve a large exotherm or release of toxic gases.
Over the span of 150 years, improvements over current synthesis methods of graphite oxide are constantly pursued to provide safer and more effective alternatives. The effectiveness of an oxidation process is often evaluated by the magnitude of carbon/oxygen ratios of the graphene. In fact, graphite oxides obtained from these oxidation methods were previously shown to differ substantially in the structural and electrochemical properties.17
3. Structure of graphite oxide
Understanding the structure of graphite oxide is important and crucial prior to any subsequent chemical modifications of the material. Graphite oxide is primarily composed of carbon, oxygen and hydrogen atoms. It is common for graphite oxide to retain a carbon–oxygen ratio of approximately 1.5 to 2.5. Although the synthesis method of graphite oxide has been known for nearly 150 years, the exact chemical structure of graphite oxide remained elusive to the graphene community. Several structures have been proposed over the years, such as the Hofmann, Ruess, Scholz–Boehm, Nakajima–Matsuo, Lerf–Klinowski, Dékány and Ajayan models (Fig. 1).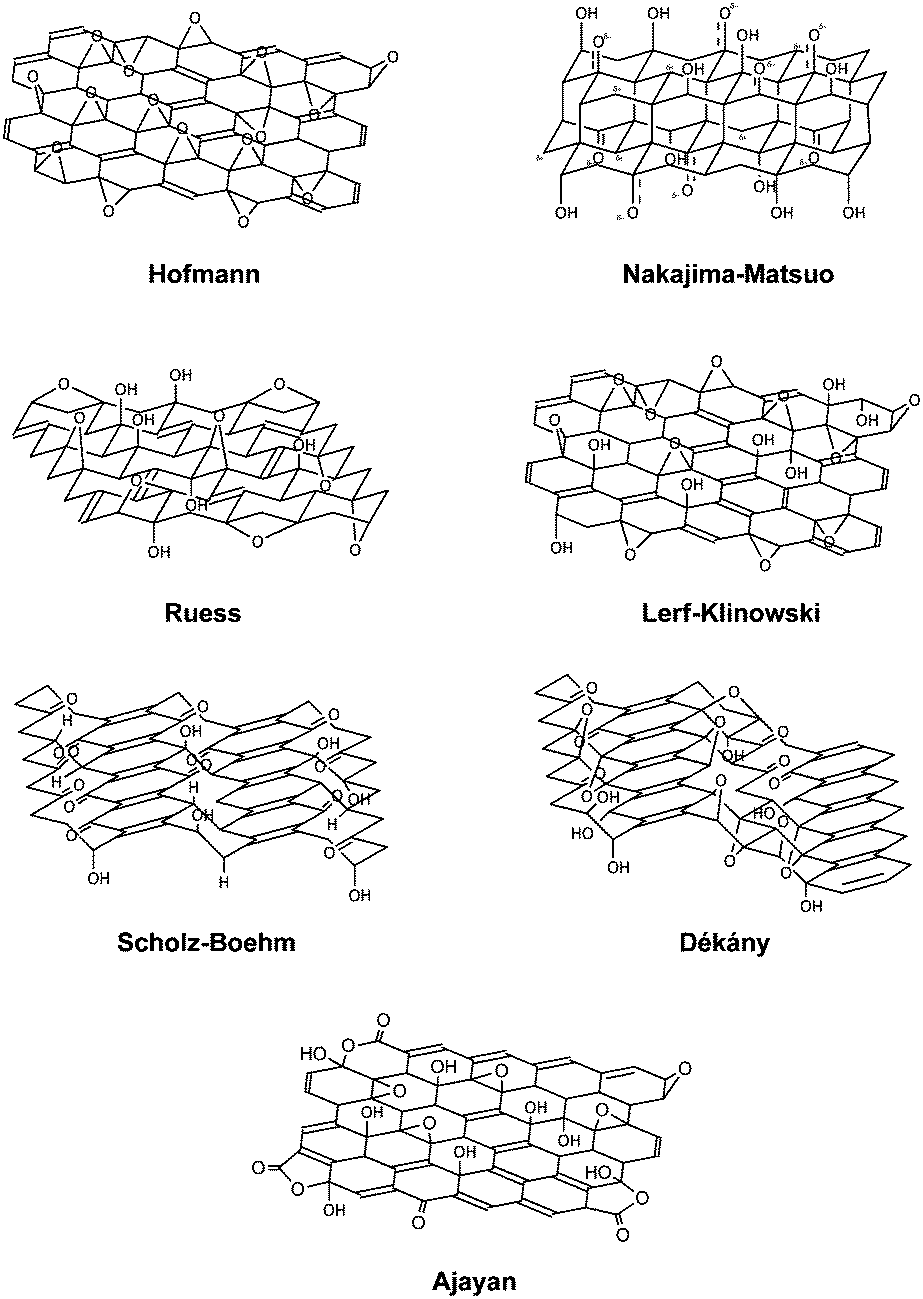 | ||
| Fig. 1 Proposed structures of graphite oxide. | ||
The very first model of graphite oxide was proposed by Hofmann and Holst.18 The suggested structure contained repeating units of 1,2-epoxides on the entire basal-planes of graphene. In 1946, Ruess proposed a new model made up of sp3 hybridised basal-planes as opposed to the sp2 hybridised system of Hofmann and Holst.19 The model consisted of 1,3-epoxide and hydroxyl groups to account for the presence of hydrogen atoms found in graphite oxide. Twenty years later in 1969, Scholz and Boehm proposed a new model which was made up of only hydroxyl and ketone groups.20 The subsequent model introduced by Nakajima and Matsuo gave rise to a structure which was very similar to a graphite intercalation compound.21 The aforementioned models were mostly deduced based on elemental compositions, chemical reactivity and X-ray diffraction (XRD) studies.
Subsequent models derived from 13C and 1H magic-angle spinning nuclear magnetic resonance (MAS NMR) studies performed by Lerf and Klinowski concluded on two contrasting regions of graphite oxide.22–24 Graphite oxide was postulated to consist of an aromatic region with unoxidised benzene rings and another region with aliphatic six-membered rings. Oxygen functionalities such as 1,2-epoxides and hydroxyl groups populated the basal-plane while the edge-plane contained mostly carboxyl and hydroxyl groups. In fact, a very recent observation of graphite oxide using high-resolution transmission electron microscopy (TEM) has indicated the presence of such features on the graphite oxide sheets, in support of the Lerf–Klinowski model.25
Another model of interest was proposed by Dékány based on elemental analysis, X-ray photoelectron spectroscopy, diffuse reflectance infrared Fourier transform spectroscopy, electron spin resonance, TEM, XRD and 13C MAS NMR.26 Building on the Ruess and Scholz–Boehm models, Dékány's depiction of graphite oxide comprised of two distinct domains containing trans linked cyclohexane chairs and corrugated hexagon ribbons. The cyclohexane chairs contained 1,3-epoxide and tertiary hydroxyl groups while the hexagon ribbons were populated with cyclic ketones and quinones. Furthermore, phenolic groups were introduced into the model to explain the acidity of graphite oxide.
In a more recent attempt to determine the detailed structure of graphite oxide, Ruoff and co-workers synthesized a 100% 13C-labelled graphite oxide for 13C MAS NMR analysis.27 Most of the hydroxyl and epoxide carbons were found to be bonded to each other. Moreover, the study highlighted the spatial separation of carbonyl groups from the majority of the sp2, hydroxyl and epoxide groups. More importantly, signals detected near 100 ppm were indicative of the presence of non-protonated carbons, but were not specifically assigned to any functionality. Despite that, the data obtained from the NMR analyses narrowed down the most possible structure of graphite oxide to both the Lerf–Klinowski and Dékány models.
A subsequent follow-up work by Ajayan and co-workers suggested the signal at 100 ppm of 13C MAS NMR to originate from the presence of lactols, specifically 2-hydroxynaphthalic anhydrides or 1,3-dihydroxyxanthones.28 The relative ratios of functional groups on graphite oxide were proposed to be 115 (hydroxyl and epoxide): 3(lactol O–C–O): 63(graphitic sp2 carbon): 10(lactol + ester + acid carbonyl): 9(ketone carbonyl).
Tour and co-workers took a different approach in determining the structural details of graphite oxide. By introducing alcohol instead of water during the work-up step of graphite oxide synthesis, they were able to produce ‘pristine graphite oxide’ which was dominated by epoxide groups.29 A small amount of covalent sulphates and hydroxyl groups readily reacted with water through numerous chemical transformations to effect the acidic properties of graphite oxide. In fact, the acidic properties were brought about by edge-plane ketones which were in equilibrium with their hydrated forms or incompletely hydrolysed covalent sulphates. This was in contrast to classical interpretation which attributed the acidic properties to the presence of carboxyl groups. Moreover, the signal at 100 ppm of 13C MAS NMR was suggested to originate from hemiacetal moieties. Extended investigations of the acidic properties of graphite oxide by the group subsequently highlighted the fact that acidic functional groups (i.e. vinylogous acid) on graphite oxide were gradually generated via interaction with water.30 It was also suggested that graphite oxide does not exist as a static structure with a definite set of functional groups, but constantly evolves in the presence of water. Such a proposed model was termed the ‘dynamic structural model’.
4. From graphite oxide to graphene
The scientific discoveries revolving around graphite oxide span a long period of time. It should not be forgotten that, above all, graphite oxide is a very important intermediate material towards the production of graphene (Fig. 2). The oxidative treatment of graphite helps to increase the interlayer distance between graphene sheets in graphite for an easy exfoliation, since the sheets are usually held by strong van der Waals forces. This is with reference to the top-down approach, which is the most direct route, and it is also one which provides the opportunity for the large scale production of graphene.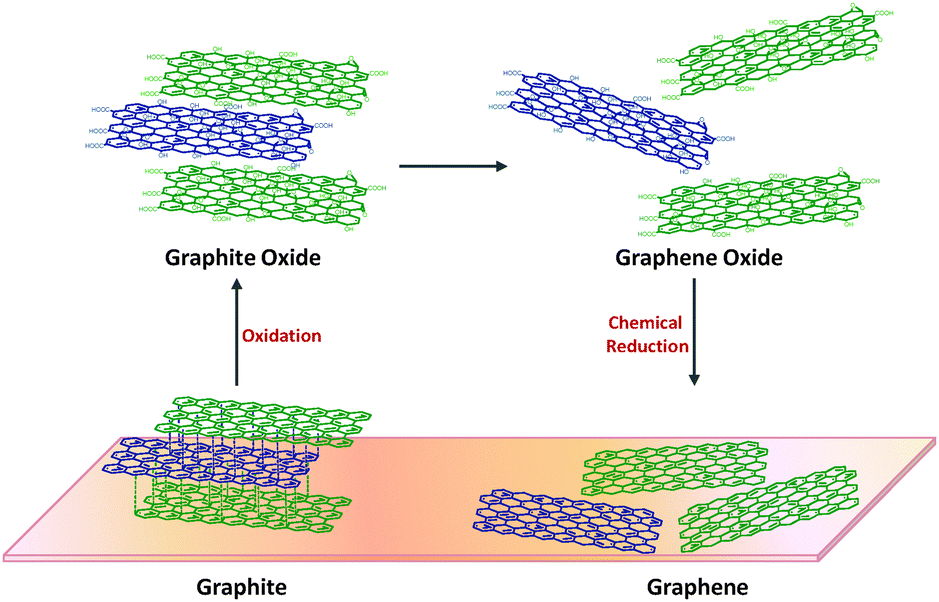 | ||
| Fig. 2 Synthesis of graphene from graphite. | ||
Graphite oxide is seen as a highly oxidised form of graphite, which retains the lamellar structure (multilayer) of its precursor albeit with a higher interlayer spacing due to the presence of oxygen functionalities. Based on the widely accepted Lerf–Klinowski model of graphite oxide, the oxygen functionalities encompass moieties such as hydroxyl, epoxide, carbonyl and carboxyl groups. The basal-plane of the graphite oxide is highly populated with hydroxyls and epoxides while the edge-plane mainly consists of carboxyl and carbonyl groups. These oxygen containing groups account for the structural defects that lead graphite oxide to deviate from the state of pristine graphene.
The corresponding exfoliated form, graphene oxide, is seen as another important intermediate between graphite and graphene. Graphene oxide is structurally different but chemically similar to graphite oxide. It retains the oxygen functionalities of its precursor, but largely exists as mono-, bi- or few-layer graphene sheets. Graphene oxide is usually achieved via mechanical stirring or ultrasonication methods in a polar organic solvent or aqueous media. Although the ultrasonication method ensures a more efficient and faster exfoliation of the stacked graphite oxide sheets, it often entails structural damages and results in the breaking of graphene oxide sheets into smaller fragments.31
In order to achieve the conversion of graphene oxide to graphene, thermal, electrochemical and chemical reduction methods are often applied. The reduction methods are envisaged to facilitate the removal of the oxygen functionalities on graphite/graphene oxide. These three different methods would result in graphene of varying performances in terms of electronic, structural, physical and surface morphological properties.32 Although these graphene materials consist of defective sheets of the sp2 carbon network, they share close resemblance to pristine graphene and are highly suitable for applications that require a large amount of graphene materials.
5. Chemical reduction of graphene oxide
While the preparation of graphite oxide was first carried out 150 years ago, attempts to remove the oxygen functionalities on the surface of graphite oxide via chemical means did not occur until approximately another 80 years later. A record by Brauer in 1963 noted the treatment of graphite oxide with hydrazine, hydroxylamine, hydroiodic acid, iron(II) and tin(II) ions for reduction.33 Fast forward to today. This collection of knowledge has been transferred onto graphene oxide. The chemical composition of graphene oxide does not differ much from that of graphite oxide at all. They are only structurally different in terms of the number of stacked graphene layers.The transformation of graphene oxide to graphene is, by experimental observations, often indicated by a colour change of the reaction mixture from brown (of graphene oxide) to black (of graphene), and an increase of hydrophobicity/aggregation of the material as a result of the removal of oxygen containing groups. Based on more detailed analyses, the decrease in elemental oxygen contents (often represented by an increase of C/O ratio) and the increase in the current conductivity of the graphene materials often signify the efficiency of a particular reduction method.
Numerous methods of chemical reduction of graphite/graphene oxide have surfaced or resurfaced in recent years. Despite the wide arrays of chemical reduction strategies, a number of reducing agents known to date may not be supported by mechanisms. For discussion purposes, reduction methods involving these reducing agents are grouped into two categories: reduction methods with ‘well-supported’ mechanisms and reduction methods with ‘proposed’ mechanisms (Table 1). Reducing agents under the category of ‘well-supported’ mechanisms consist of reducing agents which have been traditionally applied in synthetic chemistry and have demonstrated definite modes of reaction towards specific oxygen functional groups (i.e. metal hydrides are reactive towards carbonyl functional groups). On the other hand, reducing agents under the category of ‘proposed’ mechanisms include reducing agents that have not been previously applied in synthetic chemistry as reducing agents and do not have any definite modes of reaction towards specific oxygen moieties. These reducing agents are not widely applied in synthetic chemistry for the removal/reduction of oxygen functionalities. In fact, most of these are usually known to have non-specific antioxidant or oxygen scavenging properties.
| Reducing agents | Conductivitya (S m−1) | C/O ratio | Doping | Conditionsb | Ref. |
|---|---|---|---|---|---|
| a Conductivity obtained from as-produced graphene without annealing. b Reduction carried out in aqueous medium or pure solution of the reducing agent unless stated otherwise. c X-ray photoelectron spectroscopy. d Elemental analysis. e Energy dispersive spectroscopy. | |||||
| Reduction methods with ‘well-supported’ mechanisms | |||||
| Borohydrides | |||||
| NaBH4 | 17 | — | — | 80 °C, 1 h | 35 |
| 82 | 4.8c | — | 80 °C, 1 h | 28 | |
| 45 | 8.6c | — | RT, 2 h | 36 | |
| — | 2.5c | — | MeOH, 70 °C, 2 h | 37 | |
| NaBH3(CN) | — | 2.5c | — | MeOH, 70 °C, 2 h | 37 |
| NaBH(OAc)3 | — | 2.2c | — | MeOH, 70 °C, 2 h | 37 |
| NH3BH3 | 19300 |
14.2c | B/N-doped | 80 °C, 12 h | 38 |
| 20300 |
9.8c | B/N-doped | THF, 66 °C, 12 h | 38 | |
| Aluminium hydride | |||||
| LiAlH4 | — | 12c | — | THF, 70 °C, 24 h | 39 |
| Hydrohalic acid | |||||
| HI/AcOH | 30400 |
11.5d | — | 40 °C, 40 h | 40 |
| HI/TFA | — | 12.5d | — | −10 °C, 40 h | 41 |
| HI | 29800 |
12c | I-doped | 100 °C, 1 h | 42 |
| HBr | 2.3 × 10−2 | 3.9c | Br-doped | 110 °C, 24 h | 43 |
| HBr–KOtBu | — | 7.1c | — | THF, 66 °C, 0.5 h | 44 |
| Sulphur-containing reducing agents | |||||
| Thiourea dioxide/NaOH | — | 14.5c | — | EtOH/H2O, 90 °C, 1 h | 46 |
| Thiourea dioxide/NaOH/cholate | 3205 | 5.8c | Adsorbed | 80 °C, 0.5 h | 47 |
| Thiourea dioxide/NH3 | 290 | 6.0c | — | RT, 1 h | 48 |
| Ethanethiol/AlCl3 | — | 4.7c | — | THF, 70 °C, 5 h | 49 |
| Lawesson's reagent | 4760 | — | S-doped | Toluene, 110 °C, 24 h | 53 |
| Reduction methods with ‘proposed’ mechanisms | |||||
| Nitrogen-containing reducing agents | |||||
| Hydrazine | 2420 | 10.3d | N-doped | 100 °C, 24 h | 54 |
| 1700 | 11d | N-doped | DMF/H2O, 80 °C, 12 h | 58 | |
| Phenylhydrazine | 4700 | 9.5d | N-doped | RT, 24 h | 59 |
| Hydroxylamine/NH3 | 1122 | 9.7c | — | 90 °C, 1 h | 61 |
| Hydroxylamine | — | 1.5c | N-doped | 80 °C, 30 h | 62 |
| Pyrrole | — | 7.7c | Adsorbed | 95 °C, 12 h | 63 |
| Benzylamine | — | 4.7c | Adsorbed | 90 °C, 1.5 h | 65 |
| p-Phenylene diamine | 15000 |
7.4c | Adsorbed | DMF/H2O, 90 °C, 24 h | 66 |
| Ethylenediamine | 220 | 7.8c | N-doped | DMF, 80 °C, 8 h | 67 |
| Urea/NH3 | 43 | 4.5c | Adsorbed | 95 °C, 30 h | 68 |
| Dimethyl ketoxime/NH3 | 100 | 6.5c | Adsorbed | 100 °C, 3 h | 69 |
| Hexamethylenetetramine | — | — | Adsorbed | 100 °C, 12 h | 70 |
| Polyelectrolyte | — | — | Adsorbed | 90 °C, 5 h | 71 |
| Poly(amido amine) | — | 8.1c | Covalent | 90 °C, 1 h | 72 |
| Oxygen-containing reducing agents | |||||
| Methanol | 3.2 × 10−5 | 4.0d | — | 100 °C, 5 days | 73 |
| Ethanol | 1.8 × 10−4 | 6.0d | — | 100 °C, 5 days | 73 |
| Isopropyl alcohol | 1019 | 6.9d | — | 100 °C, 5 days | 73 |
| Benzyl alcohol | 4600 | 30d | — | 100 °C, 5 days | 73 |
| Hydroquinone | — | — | — | RT, 20 h | 75 |
| L-Ascorbic acid/L-tryptophan/NaOH | 14 | — | Adsorbed | 80 °C, 24 h | 76 |
| L-Ascorbic acid | 800 | — | Adsorbed | RT, 48 h | 77 |
| L-Ascorbic acid/NH3 | 7700 | 12.5c | — | 95 °C, 15 min | 78 |
| Glucose/NH3 | — | — | Adsorbed | 95 °C, 1 h | 79 |
| Dextran/NH3 | 1.1 | — | Adsorbed | 95 °C, 3 h | 80 |
| Gallic acid | 36 | 5.3c | Adsorbed | 95 °C, 6 h | 81 |
| Sulphur-containing reducing agents | |||||
| NaHSO3 | 6500 | 7.9d | S-doped | 95 °C, 3 h | 82 |
| Na2S2O4/NaOH | 1377 | — | — | 60 °C, 15 min | 83 |
| Thiourea | 635 | 5.6c | Adsorbed | 95 °C, 8 h | 84 |
| Thiophene | — | 10.9c | Adsorbed | 80 °C, 24 h | 85 |
| Metal–acid | |||||
| Al/HCl | 2100 | 18.6c | — | RT, 30 min | 86 |
| Fe/HCl | 2300 | 7.9c | Fe-doped | RT, 6 h | 87 |
| Zn/HCl | 15000 |
33.5c | — | RT, 1 min | 88 |
| 650 | 8.2c | — | RT, 30 min | 89 | |
| Zn/H2SO4 | 3416 | 21.2c | — | RT, 2 h | 90 |
| Sn(II)/HCl | — | 7.6c | — | RT, 7 h | 91 |
| Al foil/HCl | 12530 |
21.1d | — | RT, 20 min | 92 |
| Mg/HCl | 10 | 3.9e | — | RT, 5 min | 93 |
| Metal–alkaline | |||||
| Zn/NH3 | — | 8.6c | — | RT, 10 min | 94 |
| Zn/NaOH | — | 5.7d | — | RT, 6 h | 95 |
| 7540 | 17.9d | — | RT, 6 h | 92 | |
| Al foil/NaOH | 1120 | 5.3d | — | RT, 20 min | 92 |
| Na/NH3 | — | 16.6c | N-doped | −78 °C, 30 min | 96 |
| Amino acid | |||||
| L-Cysteine | 1.2 × 10−1 | — | — | RT, 72 h | 97 |
| Glycine | — | 11.2c | N-doped | 95 °C, 36 h | 98 |
| L-Lysine | — | 8.5c | N-doped | 90 °C, 9 h | 99 |
| L-Glutathione | — | — | Adsorbed | 50 °C, 6 h | 100 |
| Plant extracts | |||||
| Green tea | 53 | — | Adsorbed | 90 °C, 2.5 h | 101 |
| C. esculenta leaf | 4006 | 7.1d | Adsorbed | RT | 102 |
| M. ferrea Linn. leaf | 3185 | 6.1d | Adsorbed | RT | 102 |
| C. sinensis peel | 3033 | 6.0d | Adsorbed | RT | 102 |
| R. damascena | — | — | — | 95 °C, 5.5 h | 103 |
| Microorganisms | |||||
| Shewanella | — | — | — | Anaerobic, 72 h | 104 |
| — | 3.1c | — | Aerobic, 60 h | 105 | |
| E. coli culture | — | — | — | 37 °C, 48 h | 106 |
| E. coli biomass | — | — | — | 37 °C, 72 h | 107 |
| Baker's yeast/NADPH | 43 | 5.9c | Adsorbed | 35–40 °C, 72 h | 108 |
| Wild carrot roots | — | 11.9c | Adsorbed | 25 °C, 72 h | 109 |
| Proteins | |||||
| Bovine serum albumin/NaOH | — | — | Adsorbed | 55–90 °C, 3–24 h | 110 |
| Hormones | |||||
| Melatonin/NH3 | — | — | Adsorbed | 80 °C, 3 h | 112 |
5.1 Reduction methods with ‘well-supported’ mechanisms
The usage of NaBH4 as a reducing agent for graphene oxide was first reported by Kamat and co-workers to achieve the physisorption of gold nanoparticles on a graphene-octadecylamine material.34 At about the same time, Si and Samulski performed the reduction of graphene oxide with NaBH4 as the first of a three-step synthesis towards a total reduction effort to obtain a sulphonated graphene which dispersed well in aqueous and organic solvents.35 The pre-reduced sulphonated graphene showed a conductivity of 17 S m−1. A similar reduction method with NaBH4 was also carried out by Ajayan and co-workers in their investigations related to the total reduction of graphene oxide.28 Following that, Lee and co-workers conducted a study on the effect of NaBH4 concentration on the electrical properties of the resulting graphene.36 A conductivity of 45 S m−1 and a C/O ratio of 8.6 were measured on the graphene prepared by dipping a strip of graphene oxide into a 150 mM solution of NaBH4. The electrical resistance of the graphene was measured to be lower than that of hydrazine-reduced graphene, possibly due to the absence of heteroatoms. Surprisingly, the presence of boron oxide complexes was only observed on graphene strips subjected to a low concentration of 15 mM NaBH4.
On another note, a study was recently carried out on the reducing capabilities of sodium borohydride, sodium triacetoxy-borohydride (NaBH(OAc)3), and sodium cyanoborohydride (NaBH3(CN)) in an effort to tailor the types of oxygen functionalities on graphene oxide.37 Unlike previous studies, the reactions were carried out in methanol solvent. The resulting graphene showed variable C/O ratios in the range of ∼2.2–2.5. Subsequent electrochemical impedance spectroscopy study highlighted charge transfer resistances of NaBH4–, NaBH3(CN)– and NaBH(OAc)3–graphene at 1.64, 1.72 and 4.92 kΩ, respectively (graphite oxide: 2.27 kΩ, glassy carbon: 0.6 kΩ).
Chung and co-workers recently introduced ammonia borane (NH3BH3) in aqueous and organic solvents to reduce graphene oxide.38 Ammonia borane is a mild reducing agent with a similar reactivity to sodium borohydride and exhibits potential as a material for hydrogen storage. The resulting graphene highlighted traces of boron and nitrogen doping in the form of BC2 and pyrrolic N, with the reaction in organic solvent showing a higher extent of doping up to 0.98 at%. While the reaction in organic solvent showed a lower C/O ratio than the reaction in aqueous solution, its measured conductivity was higher than the latter. Moreover, the graphene possessed specific capacitance in the range of 100–130 F g−1.
Although hydrohalic acid has been previously applied for the reduction of graphite oxide, it has resurfaced for the reduction of graphene oxide in recent years, as achieved by Lee and co-workers.40 The group was inspired by a prior report on the conversion of polycyclic quinones and phenols into the corresponding arenes with a mixture of hydroiodic acid and acetic acid. When the reduction of graphene oxide was carried out in two different phases, the liquid phase reaction performed much efficiently than the vapour phase reaction. The resulting graphene from the liquid phase reaction provided a C/O ratio of 11.5 and a measured conductivity of 30400 S m−1. Residual I− or I2 were absent in the graphene based on combustion ion chromatography analysis. The reduction steps of epoxide, hydroxyl and carbonyl moieties were suggested to follow the mechanisms as shown in Scheme 1. These included the iodination of alcohols, cleavage of ether, reduction of aromatic iodides and partial reduction of the carbonyl moiety.
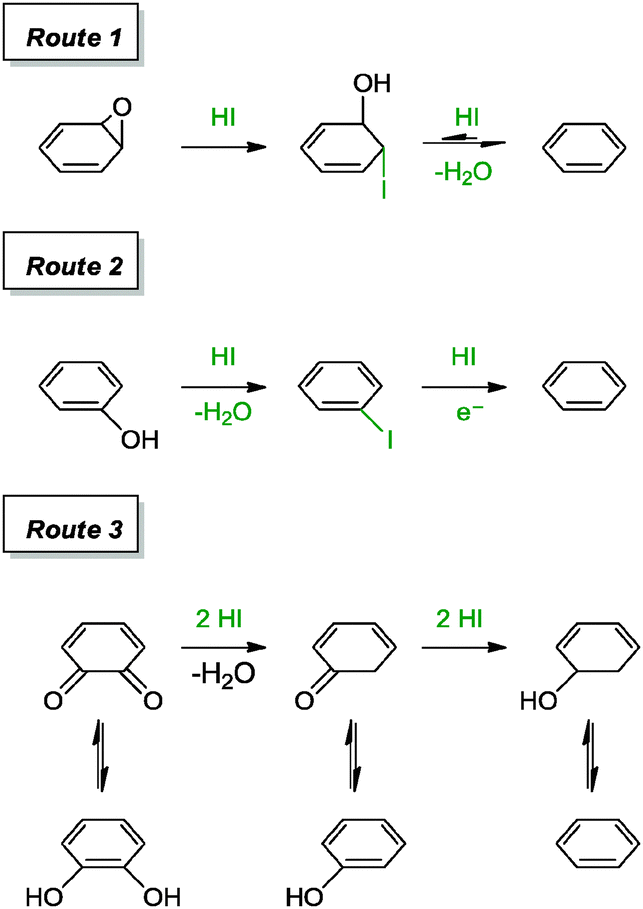 | ||
| Scheme 1 Suggested mechanisms for the reduction of epoxide, hydroxyl and diketone groups with HI/AcOH. Adapted with permission from ref. 40. | ||
The group further examined a reducing mixture of hydroiodic acid and trifluoroacetic acid, which could perform under subzero temperature of −10 °C.41 Trifluoroacetic acid was expected to exceed the performance of acetic acid (in previous study) due to its 100000-fold increase in acidity and a lower freezing point of −15.4 °C. The mixture provided highly graphitized graphene with a C/O ratio of 12.5 and a sheet resistance of 2 Ω sq−1.
In another similar study by Cheng and co-workers, graphene oxide was subjected to reduction with only hydroiodic acid at 100 °C to result in graphene with a C/O ratio of 12 and a measured conductivity of 29800 S m−1.42 Traces of residual I− or I2 were detected with X-ray photoelectron spectroscopy analysis despite a thermal annealing process at 400 °C for 2 h. The reaction was expected to proceed via a ring opening of epoxide groups by iodides and subsequent elimination due to the weak binding energy of the carbon–iodide bond, which was similar to that suggested by Lee and co-workers.
Apart from that, Ma and co-workers achieved a reduction of graphene oxide via treatment with hydrobromic acid to provide graphene with a measured conductivity of 0.023 S m−1.43 The group postulated that the low conductivity could have resulted from the presence of residual bromides. The presence of such residual bromides was consequently exploited to regenerate the sp2 carbon network of graphene which was previously populated with sp3 carbons.44 Treatment of the brominated graphene with potassium tert-butoxide led to a dehydrobromination reaction which provided graphene with an improved conductivity. Measurements of the conductivities with an interdigitated gold electrode showed a measured conductivity of 51 mS for the dehydrobrominated graphene, 39 mS for the brominated graphene and 34 mS for graphene reduced with hydrazine. A simultaneous control study that excluded the possibility of a dehydrobromination step was carried out to successfully ascertain the feasibility of the transformation.
5.1.4.1 Thiourea dioxide. While the usage of conventional reducing agents for graphene oxide may be preferred, unconventional reducing agents could potentially provide comparable efficiencies as well. Thiourea dioxide has long been applied as a strong reducing agent in textile printing, paper, photographic and leather processing industries. More importantly, thiourea dioxide has been reported to demonstrate excellent deoxygenation of α,β-epoxy ketones as well as reduction of ketones, aromatic nitro, azo, azoxy, hydrazo and organosulfur compounds. The reduction with thiourea dioxide in alkaline conditions was suggested to proceed via a one electron transfer process to provide urea and sodium sulphite as end products.45 On top of that, thiourea dioxide may provide an opportunity for the large scale manufacturing of graphene due to its low cost and the production of only common industrial wastes from the reaction, i.e. urea and sodium sulphite.
Such properties of thiourea dioxide (formamidinesulfinic acid) in an alkaline (i.e. NaOH) mixture has led to its usage as a reducing agent for graphene oxide.46 The reaction was screened up to 20 h and a 1 h reaction time provided graphene with the most favourable structural and electrochemical properties. The corresponding graphene exhibited a C/O ratio of 14.5 and remained insoluble in aqueous solution. Moreover, a low charge transfer resistance of 0.11 kΩ (graphite oxide: 5.43 kΩ, bare glassy carbon: 0.76 kΩ) based on electrochemical impedance spectroscopy was observed.
Fugetsu and co-workers reported similar reduction capability of thiourea in alkaline (i.e. NaOH) conditions.47 Sodium cholate was also added to stabilize the graphene sheets. The graphene showed a C/O ratio of 5.89 and a conductivity of 3205 S m−1. A parallel comparison conducted with graphene oxide reduced with sodium hydrosulfite and L-ascorbic acid highlighted the superior reduction capability of thiourea dioxide.
Consequently, Guo and co-workers reported the usage of thiourea dioxide as a reducing agent for graphene oxide while replacing NaOH with NH3 to achieve an alkaline condition.48 The resulting graphene showed a C/O ratio of 6 and a measured conductivity of 290 S m−1. A reduction time of 1 h was optimised with UV-vis analysis and this coincided with the result obtained previously, as aforementioned.
5.1.4.2 Ethanethiol–aluminium chloride. A recent effort on the reduction of graphene oxide has been achieved with the utilization of ethanethiol–aluminium chloride complexes.49 Convinced by the fact that most reducing agents such as sodium borohydride and lithium aluminium could only reduce oxygen-containing groups to hydroxyl moieties, an attempt to selectively remove the hydroxyl groups was pursued. This method was highly inspired by the work of Fujita and co-workers which showed the effective defunctionalization of alkoxyl, peroxy, hydroxyl, alkylthio and halo groups from the parent aromatic moieties based on a one electron transfer pathway.50 Graphene obtained from the reducing mixture showed a C/O ratio of 4.71. Control experiments which carefully excluded one active reactant at a time ascertained the success and efficiency of the reaction based on XPS and FTIR analyses. Furthermore, the resulting graphene showed dispersion stability in aqueous media, possibly due to the presence of carboxyl groups which were not reduced. Subsequent analyses based on electrochemical impedance spectroscopy indicated a low charge transfer resistance of 0.09 kΩ (graphite oxide: 4.56 kΩ, bare glassy carbon: 0.55 kΩ). Conductivity study with an interdigitated gold electrode on the graphene highlighted a twenty-fold increment in conductivity at 75 mS as compared to 2–3 mS for the remaining graphene materials obtained from the control experiments.
5.1.4.3 Lawesson's reagent. Lawesson's reagent (2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane-2,4-dithione) is a common thionation agent consisting of a four membered ring of alternating phosphorus atoms. It was used for the conversion of carbonyl moieties in amide or ester functionalities to thiocarbonyl groups.11 Moreover, the olefination of furandiones via a dithietane intermediate was achieved with Lawesson's reagent.51 The reactivity of Lawesson's reagent is governed by its reactive intermediates – dithiophosphine ylides. Thionation is driven by the formation of a stable P
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond via a thioxaphosphetane intermediate, analogous to the Wittig reaction. On top of that, the thiolation of hydroxyl groups has been achieved with Lawesson's reagent as well.52
O bond via a thioxaphosphetane intermediate, analogous to the Wittig reaction. On top of that, the thiolation of hydroxyl groups has been achieved with Lawesson's reagent as well.52
Zhu and co-workers have reported the reduction of graphene oxide based on Lawesson's reagent.53 By subjecting graphene oxide sheets or films to Lawesson's reagent, graphene with a measured conductivity of 4760 S m−1 was obtained (Scheme 2). Traces of sulphur was observed based on XPS analysis at 1.03 at% (graphene oxide: 0.94 at%). The group suggested that the nucleophilic sulphur atom of the reactive dithiophosphine ylides could possibly establish a strong interaction with oxygen groups on the graphene oxide to result in the ring opening of epoxide groups. The hydroxyl groups were eliminated via a direct nucleophilic attack by the dithiophosphine ylides. More importantly, the group attributed the high conductivity of the resulting graphene to the regeneration of the sp2 conjugation on graphene oxide via the olefination between two neighbouring carbonyl groups. Further annealing of the graphene at 300 °C provided a measured conductivity of 30900 S m−1 (graphene oxide annealed at 300 °C: 2260 S m−1).
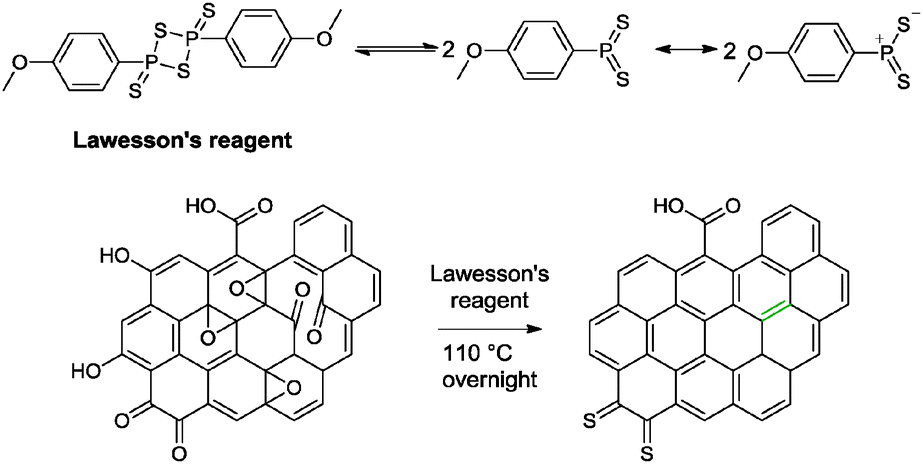 | ||
| Scheme 2 The reactive species of Lawesson's reagent and its reactivity with graphene oxide. Adapted with permission from ref. 53. | ||
5.2 Reduction methods with ‘proposed’ mechanisms
5.2.1.1 Hydrazine. Hydrazine, N2H4, is typically known to form hydrazone with carbonyl groups. It is also important for applications in the well-known Wolff–Kishner reduction and for the preparation of heterocyclic compounds. Being a common antioxidant, it scavenges oxygen while it is broken down to nitrogen and water. Despite being widely used in the industry, hydrazine is highly toxic and dangerous.
Following the usage of hydrazine for the reduction of graphite oxide, an effort to obtain individual sheets of graphene has thus led to its application in the reduction of graphene oxide. This was first demonstrated by Ruoff and co-workers in their work which highlighted graphene with a C/O ratio of 10.3 and a measured conductivity of 2420 S m−1 (graphite: 2500 S m−1).54 As a result of the loss of oxygen containing groups, the obtained graphene was highly hydrophobic and consisted of aggregated layers of graphene sheets. While the graphene showed a low amount of oxygen content, traces of atomic nitrogen were discovered in the sample. Since graphite oxide consists mostly of hydroxyl and epoxide groups based on the Lerf–Klinowski model, the group proposed a reduction pathway for the epoxide moiety. The initial mechanism of reduction was postulated to proceed via a direct nucleophilic attack of hydrazine on an epoxide group to result in a hydrazine alcohol moiety, which released a water molecule towards the formation of an aminoaziridine and finally underwent a thermal elimination of diimide to form a double bond as shown in Scheme 3A. Subsequent calculations based on density functional theory (DFT) suggested a H abstraction of the epoxide group from hydrazine to result in the ring opening process.55 Moreover, the presence of residual nitrogen atoms was explained by the possible formation of hydrazides and hydrazones, owing to the reaction between hydrazine and lactones, anhydrides or quinones found on graphene oxide.
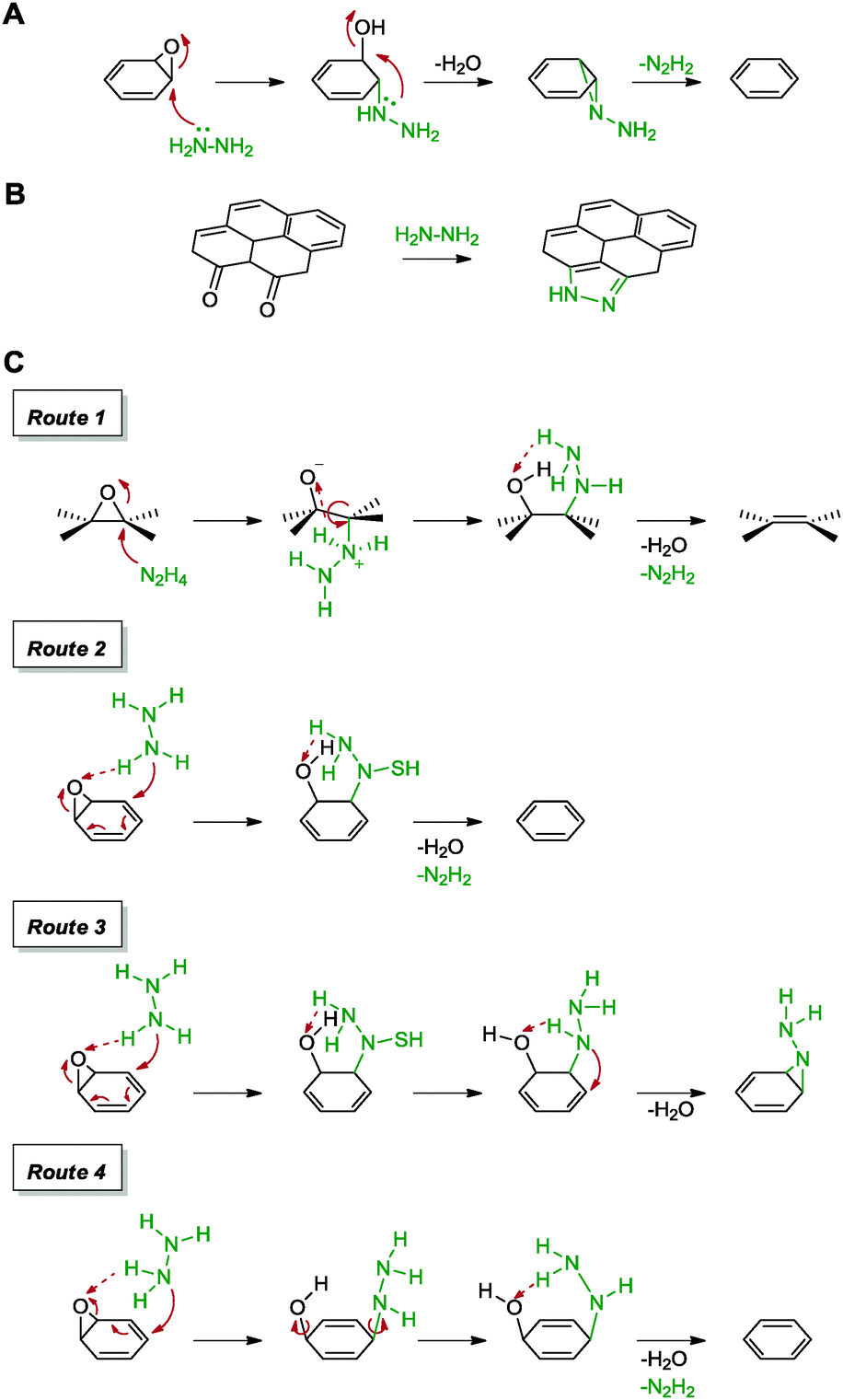 | ||
| Scheme 3 (A) Proposed mechanism for the reduction of the epoxide group with hydrazine. (B) Formation of the pyrazole ring upon usage of hydrazine as a reducing agent. (C) Proposed mechanisms for the reduction of epoxide groups with hydrazine via four different routes. Adapted with permission from ref. 54–57. | ||
In order to probe the structure of graphite oxide and to verify their initial claims, the group reported a detailed study based on solid state NMR and XPS analyses of 13C- and 15N-labelled graphene obtained via hydrazine monohydrate treatment.56 Although the mechanisms of reduction remained questionable, the study revealed the formation of five-membered pyrazole or pyrazoline rings at the edges of the graphene sheet (Scheme 3B). Chemical substitutions occurred preferentially at the edges of the graphene sheets where diketone groups were the most abundant. Further efforts to unravel the mechanisms of reduction were pursued by Nagase and co-workers with DFT calculations, as shown in Scheme 3C.57
On another note, a colloidal sample of graphene, without any surfactants or stabilizers, was also obtained by the group via reduction with hydrazine hydrate in DMF/H2O medium.58 Attempts at reduction in acetone, THF, Et2O, toluene, DCB, DMSO, ethanol, NMP and acetonitrile were unfavourable. The resulting graphene showed a conductivity of 1700 S m−1 (16000 S m−1 after drying at 150 °C). Thereafter, Chung and co-workers applied phenylhydrazine in the hope of improving the dispersion stability of the graphene.59 The obtained graphene was stable in DMAc, DMF, NMP and propylene carbonate for at least 2 months. Such stability was credited to the covalently bonded phenylhydrazine which conferred steric effects thereby preventing aggregation of graphene sheets. The graphene showed a conductivity of 4700 S m−1 (21000 S m−1 after drying at 150 °C).
Realizing that the usage of hydrazine in aqueous solvents often provided aggregated sheets of graphene, Kaner and co-workers attempted a solution-based approach towards the production of large scale mono-layer graphene.60 This was achieved via the usage of pure hydrazine on graphene oxide films. The resulting graphene was easily dispersed, most probably due to the presence of N2H4+ counter-ions. Moreover, the graphene could be dried and re-suspended in DMSO or DMF solvents for months. The group managed to obtain mono-layer graphene sheets of 20 × 40 μm in size with this method.
5.2.1.2 Hydroxylamine. Hydroxylamine, NH2OH, is a common precursor of functional groups such as oximes and hydroxamic acids. It is applied in the production of Nylon 6® and the reduction of metal ions such as Cu2+, Pd2+, Ag+ and Au3+. Moreover, hydroxylamine could function as a free radical scavenger and as a peroxide decomposer. Such antioxidant properties and weak reactivity with water have thus prompted its usage as a reducing agent for graphene oxide.
Guo and co-workers were the first to introduce hydroxylamine as a reducing agent for graphene oxide.61 Based on their approach, hydroxylamine was generated in situ from a mixture of hydroxylamine hydrochloride and ammonia. The resulting graphene provided a C/O ratio of 9.7 and a measured conductivity of 1122 S m−1. Due to the alkaline nature of the reaction mixture, the graphene formed a stable suspension in aqueous solution, possibly due to the repulsive negatively charged graphene surfaces. The group proposed a series of mechanisms for the reduction of graphene oxide as shown in Scheme 4. For the reduction of the epoxide group, hydroxylamine resulted in a ring opening followed by a proton transfer and elimination of the water molecule to arrive at an N-hydroxylaziridine intermediate. A similar intermediate was derived for the removal of hydroxyl groups. Finally, each N-hydroxylaziridine intermediate was then converted into a conjugated vinyl while accompanied by the release of unstable species of nitrogen oxide and water molecule. Carbonyl groups were unlikely to be reduced with hydroxylamine.
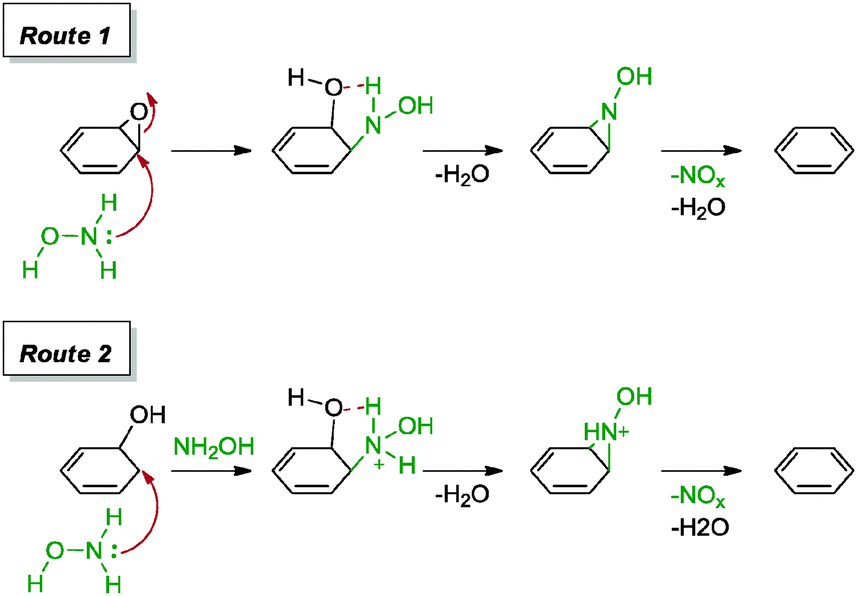 | ||
| Scheme 4 Proposed mechanisms for the reduction of epoxide and hydroxyl groups with hydroxylamine. Adapted with permission from ref. 61. | ||
At about the same time, Chen and co-workers demonstrated the treatment of graphene oxide with hydroxylamine hydrochloride to provide graphene with a C/O ratio of 1.5.62 The C/O ratio increased up to 2.5 upon thermal annealing at 200 °C. Moreover, high resolution XPS analysis revealed the presence of nitrogen doping on the graphene material. The graphene showed good dispersion stability in DMF up to one week. Furthermore, a direct-current measurement indicated a resistance of ∼105 Ω (graphite oxide: ∼1010 Ω) for the graphene and ∼102 Ω after thermal annealing. In terms of the reduction mechanism, the group proposed a release of electrons from the N− moiety in hydroxylamine to remove oxygen containing groups in graphene oxide with the presence of H+ in the reaction mixture. The resulting graphene showed a p-type semiconductor behaviour under ambient conditions with a Dirac point beyond +40 V when analysed for its FET characteristics. When tested as a gas sensor, the fabricated graphene exhibited fast response and high sensitivity to NO2 (100 ppm) and NH3 (1%) diluted in dry air.
5.2.1.3 Pyrrole. Pyrrole is a weak base and a general precursor to the synthesis of many natural products. The usage of pyrrole as a reducing agent and stabilizer was first demonstrated by Lee and co-workers.63 The group was inspired by the work of Sakura and Nagasaki which utilized pyrrole derivatives as reducing agents for the preparation of gold colloids.64 Graphene obtained from the reduction with pyrrole showed a C/O ratio of 7.7 and displayed good dispersion stabilities in several organic solvents such as ethanol, isopropanol, DMF, DMSO, NMP, THF and acetone. Such stability was thought to be conferred by the adsorbed oxidation product of pyrrole on the graphene surfaces. As for the reduction mechanism, it was postulated to be driven by the transfer of electrons from pyrrole to graphene oxide while pyrrole itself was oxidised to form oligomers.
5.2.1.4 Benzylamine. Benzylamine was applied by Sun and co-workers as a reducing agent for graphene oxide and also as a stabilizer for the resulting graphene material.65 Graphene oxide treated with benzylamine for 1.5 h gave graphene with a C/O ratio of 4.7 while a longer reaction time up to 2 h increased the C/O ratio to 10.2. Furthermore, the resulting graphene provided a measured resistance of 5 × 108 Ω and showed good dispersion stability in aqueous solution. Such stability was associated with the presence of basic polar amine groups of adsorbed benzylamine molecules and negatively charged carboxyl groups on graphene. The reduction process was attributed to the direct redox reaction between the –NH2 group of benzylamine and graphene oxide. Subsequently, the graphene was hybridised with AgNPs to demonstrate a high sensitivity for the electrochemical reduction of H2O2.
5.2.1.5 p-Phenylene diamine. Ma and co-workers established a new reduction method using p-phenylene diamine (PPD), which acted as both a reducing agent and a stabilizer.66 The resulting graphene was positively charged as a result of the adsorbed oxidised PPDs that contained –N+ residuals. This prevented π–π stacking interactions and agglomeration among the graphene sheets. As such, the graphene showed enhanced dispersion stabilities in ethanol, glycol and NMP (it showed only short-term stability in DMF). Furthermore, the resulting graphene gave a measured conductivity of 15
000 S m−1 in the plane of the graphene film. The reaction mechanism remained unknown.
5.2.1.6 Ethylenediamine. Another reduction method of graphene oxide with diamine compounds, specifically ethylenediamine, was observed by Xiao and co-workers.67 While the group intended to functionalize graphene oxide with ethylenediamine, the reduction of graphene oxide was unexpectedly achieved instead. The resulting graphene was N-doped based on XPS analysis and showed a C/O ratio of 7.87 as well as a measured conductivity of 220 S m−1 (graphite oxide: 0.02 S m−1, hydrazine reduced: 1300 S m−1). The obtained graphene formed a stable dispersion in DMF, most likely due to the increased compatibility of the graphene with DMF and the presence of amide groups on the graphene surfaces. The reduction was suggested to result from the presence of the diamine moiety since a control experiment with monoamine n-butylamine showed a less extensive reduction. A cyclization-removal pathway was postulated as the dominant reaction mechanism (Scheme 5). Upon a ring opening addition of ethylenediamine with an epoxide, cyclization occurred with the adjacent hydroxyl group to give a piperazine moiety which was subsequently removed to release the tension of the ring structure. Apart from that, a direct removal of the ethylenediamine with a neighbouring hydroxyl group in the form of hydroxylamine was concurrently proposed.
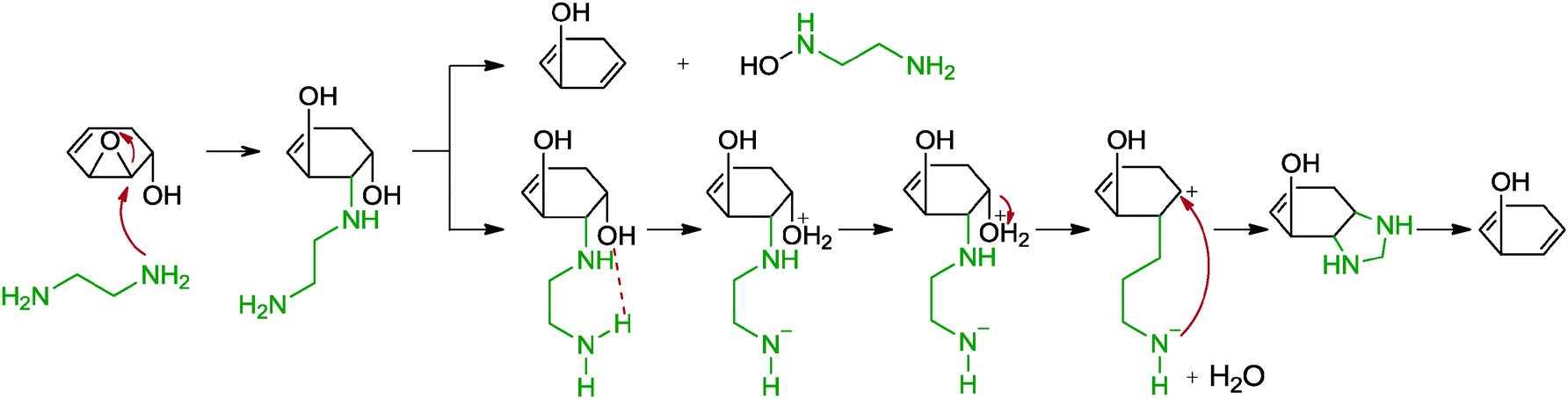 | ||
| Scheme 5 Proposed mechanisms for the reduction of epoxide with ethylenediamine. Adapted with permission from ref. 67. | ||
5.2.1.7 Urea. Urea, a simple chemical with a long stretch of history in synthetic chemistry, was introduced as a reducing agent towards the production of graphene by Zhao and co-workers.68 The group was encouraged by previous applications of urea as a reducing agent to prepare graphene analogues of BN in the presence of B2O3 at 900 °C and as an expansion–reduction agent towards graphene. Graphene obtained from reduction with urea in alkaline conditions showed excellent dispersion stabilities in water, ethylene glycol and DMF for up to 36 h. While ethanol and 2-propanol were less suitable for preparing dispersions of graphene, chloroform was not able to disperse the graphene at all. The graphene exhibited a C/O ratio of 4.5 and a measured conductivity of 43 S m−1 (graphite oxide: 0.0012 S m−1). Subsequent thermal annealing at 800 °C provided graphene with a C/O ratio of 19.7 and a measured conductivity of 4520 S m−1. Traces of nitrogen were observed at 1.2 at% based on high resolution XPS analysis, which indicated the presence of pyridine and quaternary types of nitrogen moieties. Following the application of the graphene and annealed graphene as electrode materials for supercapacitors, the materials exhibited specific capacitance of 255 and 172 F g−1, respectively. The reduction mechanism remained unknown.
5.2.1.8 Dimethyl ketoxime. Graphene oxide was reduced by dimethyl ketoxime, a common oxygen scavenger in boiler water, as applied by Ning and co-workers.69 An alkaline aqueous suspension of graphene oxide treated with dimethyl ketoxime provided graphene with a C/O ratio of 6.53 and a measured conductivity of 100 S m−1 (graphite oxide: <0.01 S m−1). The graphene displayed excellent dispersion stability in aqueous solution up to 6 months. Apart from that, the resulting graphene was N-doped at 3.67 at% based on high resolution XPS analysis, which included pyridinic, pyrrolic and graphitic nitrogen moieties. While the exact mechanism was still in question, the active reducing agent was proposed to be hydroxylammonium species generated in situ, alongside acetone from the hydrolysis of dimethyl ketoxime under alkaline conditions. Furthermore, the graphene provided a specific capacitance of 141 F g−1.
5.2.1.9 Hexamethylenetetramine. Hexamethylenetetramine (HMTA), a heterocyclic cage-like compound similar to adamantine, was utilized by Zhu and co-workers since HMTA was previously observed to reduce water-soluble metal nanoparticles.70 Formaldehyde and ammonia are the main hydrolysed products of HMTA and the former was suggested to promote the reduction of graphene oxide. The obtained graphene displayed high dispersion stability in aqueous solution as a result of electrostatic stabilization of negatively charged carboxyl groups and positively charged adsorbed HMTA molecules.
5.2.1.10 Polyelectrolyte. In addition to small nitrogen-containing compounds, a nitrogenous polymer could also reduce graphene oxide, as highlighted by Lin and co-workers with the application of poly(diallyldimethylammonium chloride) (PDDA).71 PDDA was initially intended to aid the dispersion of graphene in aqueous solution since this was previously achieved with carbon nanotubes. PDDA could adsorb through π–π and electrostatic interactions to result in electrostatic repulsion between the nanotubes and prevented aggregation in aqueous suspensions. However, when graphene oxide was exposed to PDDA, a colour change from brown to black was observed, which thus prompted the investigation of PDDA as both a reducing agent and a stabilizer. The resulting graphene formed homogeneous colloidal suspensions in water, ethanol, glycol and DMF. The group postulated that the positively charged PDDA, which contained inherent N+ groups, would attack the oxygen atom of epoxide in graphene oxide to induce a ring-opening reaction (Scheme 6). This resulted in a single electron on the α-carbon followed by a subsequent elimination of the nitroso group to provide an olefin. The hybrid graphene material further prompted the uniform deposition of metal nanoparticles to result in a high activity towards formic acid oxidation.
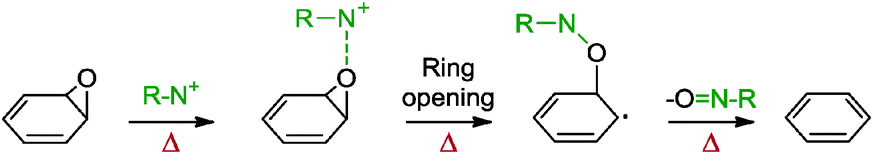 | ||
| Scheme 6 Proposed mechanism for the reduction of epoxide groups with polyelectrolyte. Adapted with permission from ref. 71. | ||
5.2.1.11 Poly(amido amine). A recent work by Liu and co-workers examined the reduction of graphene oxide with a third generation poly(amido amine) dendrimer (G3-PAMAM).72 The reduction provided graphene with a C/O ratio of 8.10 and high dispersion stabilities in DMF and DMSO for at least two months. The dispersing ability in aqueous solution was however short-lived, at only one day. The group suggested that, similar to other reducing agents containing amino functionalities, the G3-PAMAM underwent a similar mechanism of reduction with its terminal amido groups upon anchoring on the graphene oxide surface via formation of amide bonds, Schiff bases or hydrogen bonding (Scheme 7). The G3-PAMAM–graphene hybrid was subsequently applied as a catalyst for the Knoevenagel condensation between benzaldehyde and dimethyl malonate with up to 78% yield (G6-PAMAM–graphene gave 99% yield).
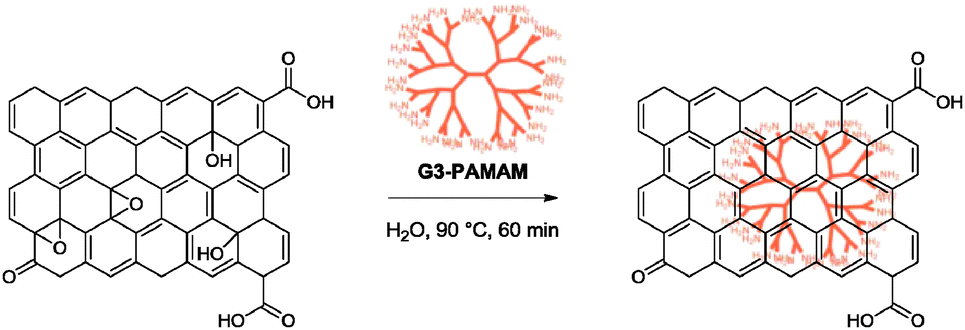 | ||
| Scheme 7 Reduction of graphene oxide with G3-PAMAM. Adapted with permission from ref. 72. | ||
5.2.2.1 Alcohol. Simple alcohols, namely MeOH, EtOH, iPrOH, and BnOH, showed interesting reducing capability towards graphite oxide as demonstrated by Bielawski and co-workers.73 While BnOH provided the highest extent of reduction among the rest, specifically with a C/O ratio of 30 and a conductivity of 4600 S m−1, the reduced graphite oxide retained much of its lamellar structure. The reduction process possibly proceeded via a thermally mediated process. In addition, the presence of benzaldehyde and benzoic acid in the reduced graphite oxide solution suggested the direct involvement of BnOH in the reduction process. The reduced sample showed a specific capacitance of 35 F g−1. On another note, ethanol vapour was able to reduce graphene oxide films at 600 °C.74
5.2.2.2 Hydroquinone. In another study, Yao and co-workers introduced the reduction of graphene oxide with hydroquinone in an aqueous ethanolic mixture.75 Hydroquinone was postulated to lose either one H+ to form a monophenolate ion or two H+ to form a diphenolate ion (quinone). The graphene was hydrophobic and could only be dispersed in water for a few hours.
5.2.2.3 L-Ascorbic acid. Ascorbic acid, also known as vitamin C, is a common essential nutrient that exhibits antioxidant properties. This property has inspired Zhang and co-workers to apply it as a reducing agent in conjunction with L-tryptophan for graphene oxide.76 In this work, L-ascorbic acid acted as a reducing agent while L-tryptophan functioned to stabilize the resulting graphene by adsorbing onto the graphene sheet via π–π interactions. The stabilization effect of L-tryptophan acted synergistically with the remaining negatively charged carboxylic acid residuals to provide electrostatic repulsions between individual graphene sheets. The obtained graphene provided a measured conductivity of 14.1 S m−1 (graphite oxide: 5.72 × 10−6 S m−1). The postulated reduction mechanism proceeded via a two-step SN2 nucleophilic reaction followed by another one-step thermal elimination as shown in Scheme 8. Prior to the first part, the electron withdrawing side chain of the five-membered ring of L-ascorbic acid increased the acidity of the β- and γ-hydroxyl groups, resulting in the dissociation of two protons to form an oxygen anion of L-ascorbic acid (HOAO−). Following that, the anion underwent a back-side SN2 nucleophilic attack, twice on the epoxide and diol groups of graphene oxide, to arrive at an intermediate. Finally, the intermediate was thermally eliminated at which ascorbic acid was oxidised to dehydroascorbic acid. This resulted in the successful conversion of graphene oxide to graphene.
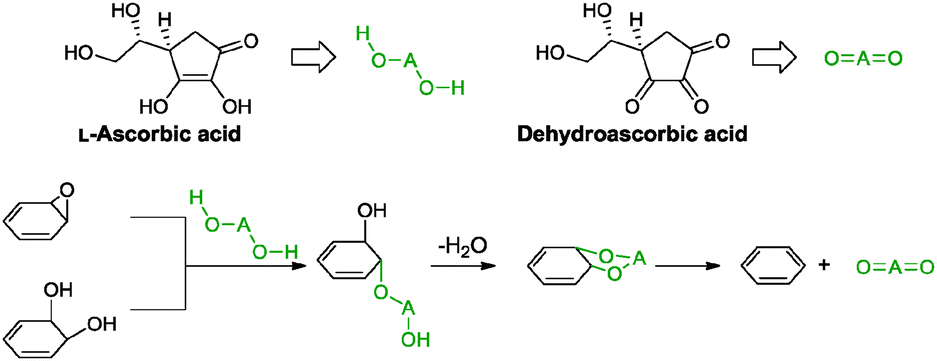 | ||
| Scheme 8 Proposed mechanisms for the reduction of epoxide and di-hydroxyl groups with L-ascorbic acid. Adapted with permission from ref. 76. | ||
At about the same time, Guo and co-workers reported the reduction of graphene oxide using only L-ascorbic acid to provide graphene with a measured conductivity of 800 S m−1.77 While the entire procedure took 48 h, a higher concentration of L-ascorbic acid was able to shorten the reaction time. The resulting graphene displayed good dispersion stability in aqueous solution. This observation was attributed to the presence of oxalic and guluronic acids, which were generated from the breakdown of dehydroascorbic acid. These acids could form hydrogen bonding with residual oxygen functionalities found on graphene surfaces to prevent agglomeration and π–π interaction of the graphene sheets.
In a comparative study by Tascón and co-workers, graphene oxide reduced by L-ascorbic acid in the presence of NH3 was shown to disperse well in organic solvents such as DMF and NMP.78 On top of that, the measured conductivity of graphene obtained from reduction with L-ascorbic acid/NH3 (2 mM) was 7700 S m−1 as compared to that with hydrazine (21 mM) treatment at 9960 S m−1 (graphene oxide/NH3 (206 mM) = 0.32 S m−1). The measured C/O ratio of the former was 12.5.
5.2.2.4 Saccharides. Another essential nutrient that has been introduced in this area of research is saccharides. Generally divided into groups such as mono-, di-, oligo- and polysaccharides, their chemical structures are populated with oxygen functionalities. Monosaccharides such as glucose and fructose are common reducing sugars that are capable of reducing Tollen's reagent or Fehling's solution due to the presence of free carbonyl and hydroxyl groups. Such reducing capability operating via a redox pathway is highly dependent on the ability of the saccharides to form open-chain structures, which is usually not observed with non-reducing sugars such as sucrose.
Dong and co-workers demonstrated the usage of glucose, fructose and sucrose in aqueous ammonia solution for the reduction of graphene oxide.79 The presence of ammonia was highly critical to achieve a full extent of reduction since it contributed to a synergistic augmentation of the reaction rate and deoxygenation process. The resulting graphene displayed excellent dispersion stabilities in aqueous solutions for more than one month. Furthermore, the mechanism of the reduction process was examined by the group to show that glucose, an aldohexose, was first oxidised to aldonic acid by graphene oxide in the presence of ammonia solution. The aldonic acid was then further converted to lactone. The mixture of aldonic acid and lactone, which were rich in hydroxyl and carboxyl groups, could form hydrogen bonding with residual oxygen functionalities on graphene surfaces to prevent aggregation of sheets by π–π stacking. On the other hand, fructose also displayed reducing capability towards graphene oxide since it was capable of undergoing a keto–enol tautomerism under alkaline conditions. As for sucrose, despite being a non-reducing sugar, it could readily break down to fructose and glucose upon hydrolysis in an alkaline environment to effect the reduction of graphene oxide. However, the reducing capability was lower than that of glucose and fructose. Furthermore, the resulting graphene obtained from glucose treatment highlighted good electrocatalytic activity towards dopamine, epinephrine and norepinephrine.
In another work, dextran (polysaccharide) in aqueous ammonia solution was employed by Min and co-workers.80 Similar to the synergistic effect of ammonia observed as mentioned before, the reduction step was shortened to 3 h instead of 3 days with the addition of ammonia. The mechanism of the reduction was postulated to go through a similar route as that of glucose. Moreover, the obtained graphene showed a measured conductivity of 1.1 S m−1 (on thermal annealing at 500 °C in Ar atmosphere: 10000 S m−1). Based on the toxicity study with MTT and CCK-8 assays on HeLa cells after 24 h exposure to graphene of concentration 450 μg mL−1, the cell viability stood at 82 and 80%, respectively (with graphene oxide: 88 and 73%, respectively). The graphene was subsequently applied as a template for the direct synthesis of gold nanoparticles on its surface.
5.2.2.5 Gallic acid. More recently, the application of gallic acid for the reduction of graphene oxide was introduced by Xiao and co-workers.81 Gallic acid was selected as it has been widely adopted as an effective antioxidant in the area of food packaging, food processing, and toiletries for the purpose of preventing rotting induced by lipid peroxidation and spoilage. The three hydroxyl groups on gallic acid were postulated to induce the reduction process of graphene oxide. In their work, an aqueous dispersion of graphene oxide was subjected to treatment with gallic acid and ammonia solution under two different conditions. The first set of conditions was based on stirring the mixture at room temperature for 24 h, while the second set of conditions was based on stirring the mixture at 95 °C for 6 h. The graphene materials from the two conditions gave different values of measured conductivities, with the former exhibiting 0.96 S m−1 while the latter showed 36 S m−1. Moreover, the C/O ratio of the latter was 5.3. Both graphene materials were readily dispersed in deionized water (1.2 mg mL−1), NMP (2 mg mL−1), DMSO (4 mg mL−1), DMF (1.5 mg mL−1) and methanol (1.5 mg mL−1) due to the presence of adsorbed gallic acid. The reaction mechanism remained unknown.
5.2.3.1 Sulphur-containing compounds. Sulphur-containing compounds, namely NaHSO3, Na2SO3, Na2S2O3, Na2S·9H2O, SOCl2 and SO2, were applied by Bangal and co-workers as reducing agents for graphene oxide.82 The reduction was carried out in a mixture of graphene oxide in DMAc/H2O medium as DMAc was previously shown to promote the reduction of graphene oxide. Among the sulphur-containing compounds, SOCl2 provided the best extent of reduction at which the resulting graphene has a C/O ratio of 8.48. However, due to the exothermic nature of SOCl2 upon contact with aqueous solution, NaHSO3, the second best reducing agent, was utilized. The obtained graphene showed a C/O ratio of 7.89 while graphene reduced with hydrazine provided a C/O ratio of 11.4. Although the disparities between the C/O ratios were rather wide, the measured conductivity varied by only a small extent: the NaHSO3–graphene gave a measured conductivity of 5100 S m−1 while hydrazine–graphene gave a measured conductivity of 6500 S m−1. Despite that, traces of sulphur atoms were detected from the elemental analyses of the graphene materials. On top of that, the group proposed a possible mechanism for NaHSO3 as a reducing agent based on a two-step SN2 nucleophilic reaction followed by a thermal elimination (Scheme 9). In short, HSO3− was oxidised to SO42−. The group supported this based on a positive qualitative test whereby Ba2+ cation formed a precipitate of BaSO4 in an aqueous solution of the graphene. As for SOCl2, the HSO3− anion was readily produced upon contact with water based on eqn (1)–(3). Moreover, the group also suggested that the Na2S, Na2SO3 and Na2S2O3 reducing agents proceeded through the HSO3− anion route somewhere along their reaction pathways.
| SOCl2 + 2H2O → H2SO3 + 2HCl | (1) |
| H2SO3 → HSO3− + H+ | (2) |
| HSO3− + GO → Graphene | (3) |
5.2.3.2 Sodium hydrosulphite. Inspired by the usage of sodium hydrosulphite, Na2S2O4, as an ancient reducing agent in the textile industry, Fu and co-workers applied it for the reduction of graphene oxide.83 The group subjected an alkaline suspension of graphene oxide to Na2S2O4 for 15 min to obtain graphene with a measured conductivity of 1377 S m−1 (graphite oxide: 3 × 10−4 S m−1). Sodium hydroxide was present in the reaction mixture to act as a catalyst and stabilizer. The low electrode potential of Na2S2O4 (E0 SO32−/S2O42− = −1.12 V) in alkaline solution was postulated to promote the reduction process. Accordingly, S2O42− nucleophilically attacked the epoxide or hydroxyl groups on graphene oxide, followed by a consequent release of H2O molecule to result in the formation of an intermediate. A subsequent thermal elimination of the intermediate led to the formation of graphene, while S2O42− was oxidised to SO32−.
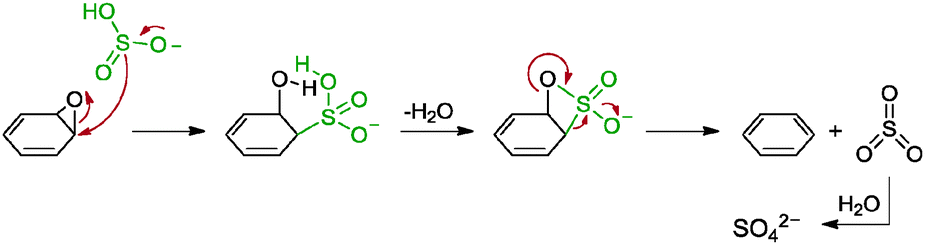 | ||
| Scheme 9 Proposed mechanisms for the reduction of epoxide groups with HSO3− ions. Adapted with permission from ref. 82. | ||
5.2.3.3 Thiourea. In another study, Wang and co-workers attempted to establish a large scale production of graphene with thiourea.84 An 8 h reaction resulted in graphene with a C/O ratio of 5.6, measured conductivity of 635 S m−1 and good dispersion stabilities in ethanol and DMF solvents. The group postulated the activation of diamine bonds of thiourea by the inherent C
S bond, which consequently reacted with the epoxide, carboxyl and hydroxyl groups on graphene oxide. More elaborately, the diamine reacted with epoxide groups via a cyclization-removal reaction, with carboxyl groups via grafting and subsequent deoxygenation, and with hydroxyl groups via a hydrogen bonding interaction with consequent deoxidation.
5.2.3.4 Thiophene. The application of thiophene as both reducing and healing agent was recently introduced by Lee and co-workers (Fig. 3).85 Graphene obtained from a direct treatment of thiophene for 24 h showed a C/O ratio of 10.9 (N2H4–graphene: 14) and a measured sheet resistance of 59 Ω sq−1 (N2H4–graphene: 26 Ω sq−1). At this juncture, thiophene was oxidised and polymerized while the released electrons served to reduce graphene oxide. The polymerization of thiophene coupled with the release of SO2 gas generated a π-conjugated polyhydrocarbon which adsorbed onto graphene surfaces. Subsequent thermal annealing at 800 °C removed the adsorbed polyhydrocarbon resulting in an improvement in C/O ratio to 16.8 and a measured sheet resistance of 9 Ω sq−1. Moreover, an observed lower ID/IG ratio of the annealed graphene at 0.83 in comparison to graphite oxide at 0.91 suggested a certain healing ability conferred by the polyhydrocarbon (ID/IG ratios: thiophene–graphene: 0.41, N2H4–graphene: 1.2). Traces of sulphur and nitrogen were not detected on the resulted graphene.
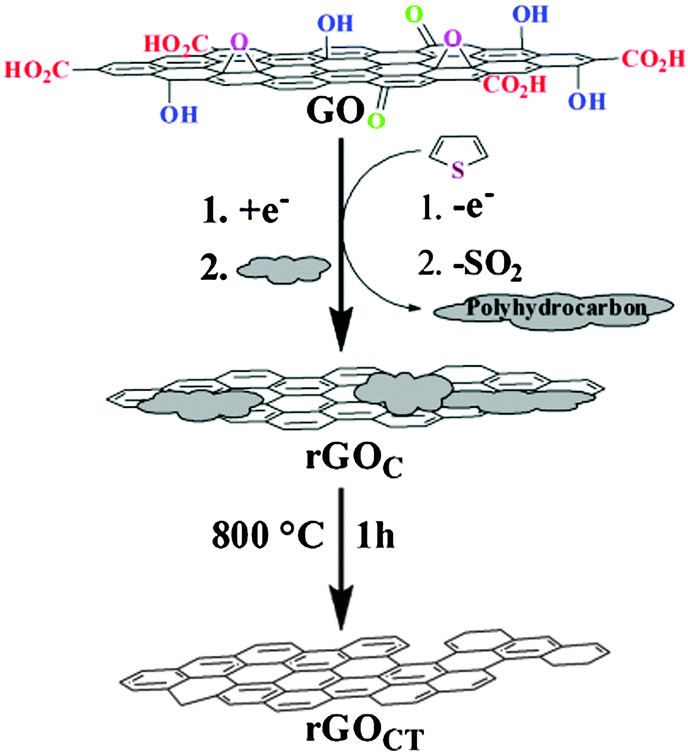 | ||
| Fig. 3 Reduction of graphene oxide with thiophene followed by thermal annealing. Reproduced with permission from ref. 85. | ||
The earliest piece of work based on the first concept was reported by Fan and co-workers, who achieved the reduction of graphene oxide using an aluminium powder (10 μm) and hydrochloric acid mixture in 30 min.86 The obtained graphene provided a C/O ratio of 18.6 and a measured conductivity of 2100 S m−1 (graphite: 3200 S m−1). Aluminium powder was selected since the standard reduction potential of aluminium was considerably more negative than that of sodium borohydride and hydrazine, which stood at −1.68, −1.24 and −1.16 V respectively. Despite that, aluminium failed to reduce graphene oxide in the absence of hydrochloric acid. It was thus deduced that hydrochloric acid aided in dissolving a passive Al2O3 on the surface of aluminium and improved the reduction potential. The close contact between the electrostatically attracted graphene sheets and aluminium could have driven the electron transfer process as well. The involvement of hydrogen gas as the key reducing agent was denounced based on a control experiment which observed no traces of reduction in a dispersion of graphene oxide bubbled with pure hydrogen gas.
In a follow-up work by the group, iron powder (10 μm) in the presence of hydrochloric acid was applied as a reducing agent instead.87 The graphene sample obtained after 6 h showed a C/O ratio of 7.9, a measured conductivity of 2300 S m−1 and a ID/IG ratio of 0.32 (graphite oxide: 0.86). The lower ID/IG ratio suggested a restoration effect of the sp2 network on graphene. Similar to their previous work, the group observed the wrapping of iron particles by graphene oxide sheets, possibly due to the electrostatic attraction between Fe2+ on iron surfaces and negatively charged graphene oxide. The close contact, as such, facilitated the fast electron transport from Fe/Fe2+ to the negatively charged graphene oxide sheets, based on eqn (4). Similarly, the reduction of graphene oxide was not observed in the absence of hydrochloric acid.
| GO + aH+ + be− → Graphene + cH2O | (4) |
In another study, Mei and Ouyang showed that zinc powder in hydrochloric acid was able to reduce graphene oxide within 1 min under ultrasonication.88 At a zinc to graphene oxide weight ratio of 2, the obtained graphene exhibited a C/O ratio of 33.5 and a measured conductivity of 15000 S m−1. The group postulated that the spontaneous reduction process was driven by a wide gap between the standard reduction potentials of Zn2+/Zn and graphene oxide (at pH 4) at −0.76 V and −0.40 V, respectively. Similar work carried out by Wang and co-workers with Zn/HCl resulted in graphene with a measured conductivity of 650 S m−1 and a C/O ratio of 8.2.89
In a more recent report by Panigrahi and co-workers, the group achieved the reduction of graphene oxide with solid zinc filings and sulphuric acid after a reaction time of 2 h.90 The graphene highlighted a C/O ratio of 21.2 and a measured conductivity of 3416 S m−1 (graphite oxide: <3 S m−1). The reduction of graphene oxide failed to reach completion within 2 h in the event when hydrochloric acid was replaced with sulphuric acid or acetic acid. The group also proposed a series of mechanisms that facilitated the reduction process, as shown in Scheme 10. In short, the reduction of carbonyl and epoxide groups followed through a series of steps towards the formation of hydroxyl groups, which ultimately underwent dehydration to afford olefins (Routes 1 and 2). The dehydration process was believed to be catalysed by the presence of ZnSO4. As for the carboxyl group, the CC bond conjugated to CO was protonated at the α-position, with subsequent decarboxylation and regeneration of the CC bond (Route 3).
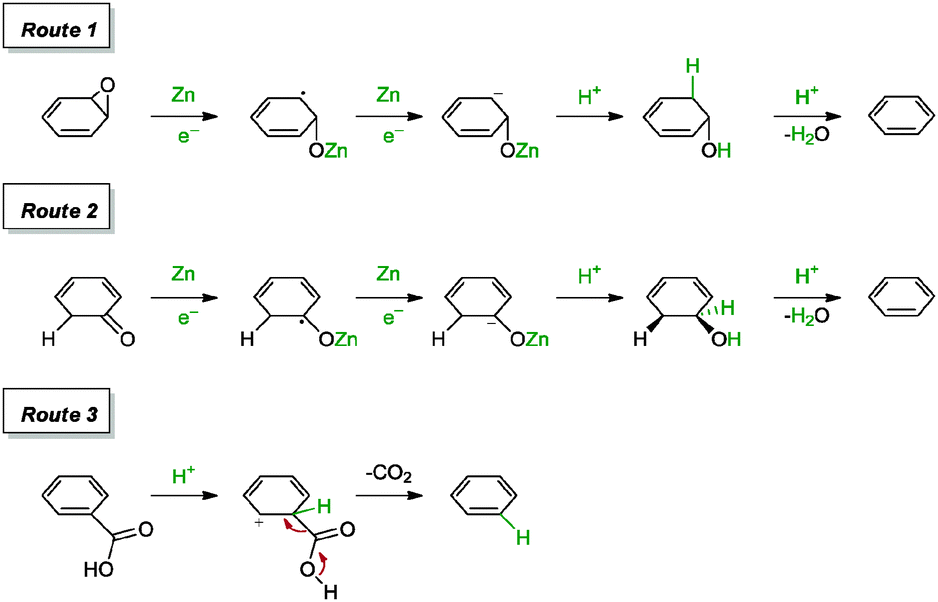 | ||
| Scheme 10 Proposed mechanisms for the reduction of epoxide, carbonyl and carboxyl groups with Zn/H2SO4. Adapted with permission from ref. 90. | ||
An effort by Dubios and co-workers to produce graphene free of paramagnetic metallic impurities has seen the usage of tin(II) chloride in hydrochloric acid for the reduction of graphene oxide (note that Zn and Zn2+are diamagnetic species too).91 An increment of C/O ratio from 2.6 for graphene oxide to 7.6 for graphene was observed. The group credited the success of the reduction process to previous reports on the reducing capability of Sn(II) towards nitro groups and the opening of epoxide groups under acidic conditions. Moreover, the oppositely charged Sn(II) and graphene oxide could facilitate the electron transfer according to eqn (5). It was further mentioned that the Sn(IV)–GO–graphene complexes could be easily reduced since the adsorbed Sn(IV) ions could act as an electron mediator and enhance additional electron transfer from the remaining Sn(II) in the bulk solution. Subsequent X-band EPR, magnetisation and Brillouin analyses highlighted the absence of magnetic impurities in the graphene sample.
| GO + aH+ + bSn(II) → bSn(IV) + cH2O + Graphene | (5) |
On the other hand, the second concept of reduction with metal in acidic medium was supported by the work of Chung and co-workers, whereby aluminium foil was added into a dispersion of graphene oxide containing hydrochloric acid.92 A 20 min reaction provided graphene with a C/O ratio of 21.1, a measured conductivity of 12530 S m−1 and a sheet resistance of 144700 Ω sq−1. Additional experiments were carried out with aluminium foil and Zn in alkaline environments (vide infra). Since the phenomenon of graphene oxide enclosing metal surfaces was not observed in their work, the group attributed the success of the reduction to the generation of in situ nascent hydrogen, instead of direct electron transfer between metal and surfaces. This claim was further supported by a control experiment in which a graphene oxide film kept away from direct contact with zinc metal was observed to undergo a reduction process. Following that, the group proposed several mechanisms which could have led to the reduction of graphene oxide (Scheme 11).
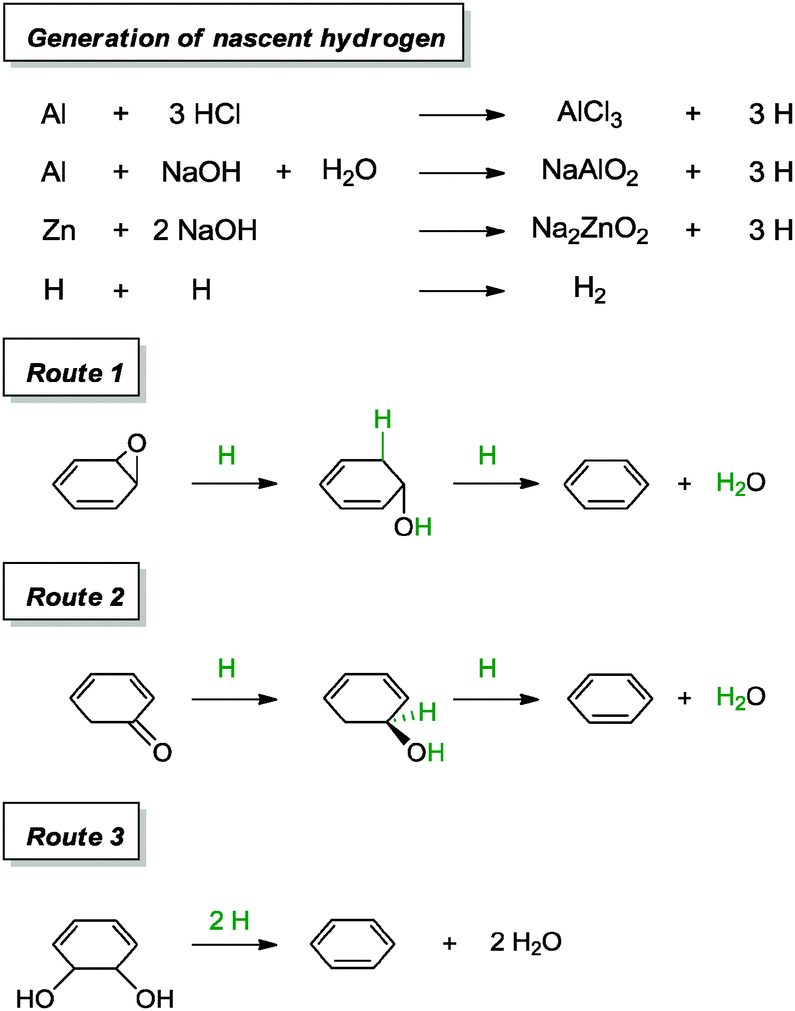 | ||
| Scheme 11 Proposed mechanisms for the reduction of epoxide, carbonyl and diol with nascent hydrogen. Adapted with permission from ref. 92. | ||
The active role of nascent hydrogen as the main reducing agent of graphene oxide was also observed by Nanda and co-workers from a mixture of magnesium in 2 M hydrochloric acid.93 The reduction was completed within 5 min and could be improved to within 1 min with a higher concentration of hydrochloric acid at 10 M. The graphene provided a C/O ratio of 3.9 and a measured conductivity of 10 S m−1. The evolution of nascent hydrogen was measured with Ag–guar gum based hydrogen sensors to show a higher intensity of nascent hydrogen produced from magnesium than zinc in acidic media.
While the general mechanisms of the metal–acid approach remained debatable, this category of reducing agents provided one of the shortest reaction times to obtain graphene, albeit with a slight probability of trace metal doping.
| Zn − 2e− → Zn2+ | (6) |
| NH4+ + e− → NH3 + H | (7) |
| Zn2+ + 4NH3 → [Zn(NH3)4]2+ | (8) |
| GO + 2H → Graphene + H2O | (9) |
In the work of Chung and co-workers as mentioned above (see Section 5.2.4), the reduction of graphene oxide was also achieved with aluminium foil and zinc powder in sodium hydroxide solution after reaction times of 20 min and 6 h, respectively.92 The difference in the reaction time could be due to the contrasting reduction potential of aluminium and zinc in alkaline solutions, at −2.33 and −1.2 V. As such, zinc took a longer time than aluminium to dissolve. Interestingly, the long reaction time of zinc possibly released nascent hydrogen (Scheme 11) gradually to result in a better reduction. Graphene obtained from zinc reduction showed a C/O ratio of 17.96 and a conductivity of 7540 S m−1 as compared to that from aluminium reduction at a C/O ratio of 5.35 and a conductivity of 1120 S m−1.
A recent work by Yang and co-workers demonstrated the cooperative behaviour of zinc and alkaline solution in the reduction of graphene oxide.95 By adding zinc powder into an alkaline (i.e. NaOH) dispersion of graphene oxide for 6 h at room temperature, the group obtained graphene with a C/O ratio of 5.73. When carried out at 100 °C, the C/O ratio increased up to 7.19 due to changes in the redox potential and occurrence of deoxygenation under high temperature. The group postulated the reduction as due to the transfer of electrons from zinc to graphene oxide (eqn (10)). Specifically, the reaction condition improved the extent of reduction via a dual approach. Firstly, the redox potential of graphene oxide/graphene was enhanced upon ionic exchange of Na+ in solution with the H+ of carboxyl groups in graphene oxide. Secondly, the reduction process was further driven by the formation of Zn(OH)42− species under alkaline conditions, resulting in an increased standard reduction potential of Zn2+/Zn (−0.76 V) compared to Zn(OH)42−/Zn (−1.25 V).
| GO + aZn + bOH− → Graphene + aZn(OH)42− + cH2O | (10) |
Li and co-workers examined the reduction of graphene oxide using the sodium–ammonia (Na–NH3) system.96 Pieces of sodium metal were added into a dispersion of graphene oxide in liquid NH3 and the mixture was kept in a dry ice–acetone bath for 30 min to afford a highly reduced graphene. The resulting graphene highlighted a C/O ratio of 16.61 and a measured sheet resistance of 350 Ω sq−1. The graphene was N-doped at 0.86 at%. The group postulated that the dissolution of sodium metal in liquid ammonia provided ionized sodium cations and solvated electrons (es−) that were strongly associated with the ammonia solvent. The solvated electrons were possibly capable of cleaving the carbon–oxygen bond via four routes to form a carbon radical or a radical anion on the surface of graphene oxide (Scheme 12). These included the removal of epoxide (Route 1), hydroxyl (Route 2), carbonyl (Route 3) and carboxyl (Route 4) groups. Thereafter, the presence of solvated electrons and partially delocalized π-conjugation on graphene oxide were proposed to stabilize the carbon radical and subsequently lead to the formation of π bonds and restoration of defects on graphene oxide upon rearranging into the lowest energy state. Moreover, the graphene highlighted a specific capacitance of 263 F g−1 and a hole mobility of 123 cm2 V−1s−1 on a FET system.
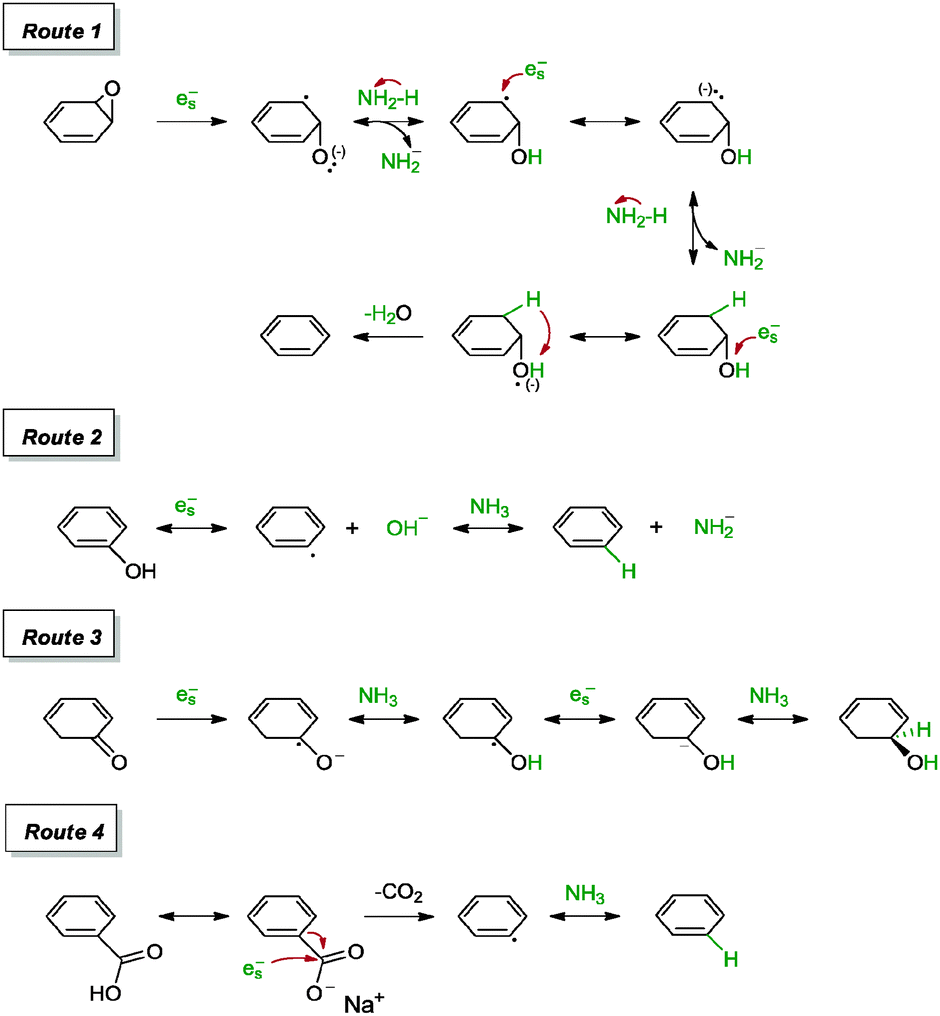 | ||
| Scheme 12 Proposed reduction mechanisms of epoxide, hydroxyl, carbonyl and carboxyl groups with solvated electrons. Adapted with permission from ref. 96. | ||
5.2.6.1 L-Cysteine. The first amino acid applied as a reducing agent for graphene oxide was L-cysteine, as demonstrated by Guo and co-workers.97 Generally known for its antioxidant properties, the thiol group of L-cysteine is susceptible to oxidation towards the formation of a disulphide derivative L-cystine. Graphene obtained upon 72 h of reduction with L-cysteine showed a measured conductivity of 0.124 S m−1 (graphite oxide: 7.97 × 10−7 S m−1). The low conductivity was deduced to result from an ineffective reduction of carboxyl groups on graphene oxide, which on the hindsight granted the ability to form a homogeneous aqueous dispersion of graphene at pH 10. In addition, the reduction mechanism was proposed to go through a two-step SN2 nucleophilic reaction on epoxide and hydroxyl groups, followed by thermal eliminations as shown in Scheme 13. The initial nucleophilic attack was driven by the release of proton from a thiol group to provide a nucleophilic thiol. The free proton had high affinity towards oxygen atoms on hydroxyl groups and departed as water molecules. Finally, the L-cysteine was oxidised to L-cystine while graphene was obtained.
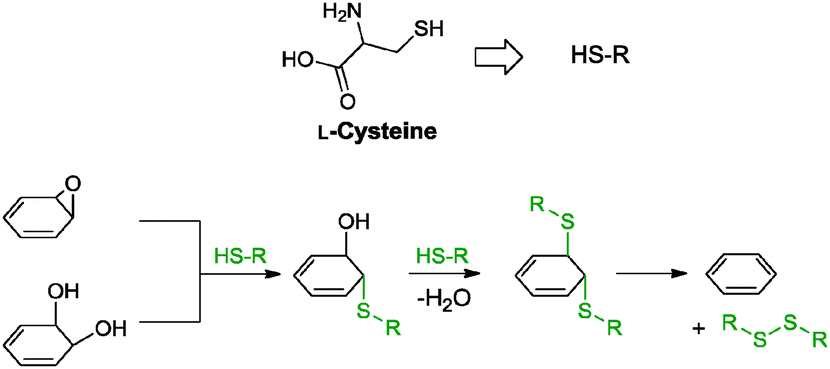 | ||
| Scheme 13 Proposed mechanisms for the reduction of epoxide and di-hydroxyl groups with L-cysteine. Adapted with permission from ref. 97. | ||
5.2.6.2 Glycine. Thereafter, the reduction of graphene oxide with glycine by Lee and co-workers provided graphene with a C/O ratio of 11.2 after 36 h of reaction time.98 A higher amount of glycine was observed to prevent aggregation of graphene sheets during the reduction process. The reduction was postulated to undergo a two-step process. In the first part, the amine functionality of glycine nucleophilically attacked the epoxide and the β-carbon atom of the hydroxyl groups. Upon such functionalization, the reduction was subsequently promoted by the catalytic action of glycine at high temperature. The functionalized graphene would then form certain intermediates and consequently release unwanted species while the π-conjugated network of graphene oxide was replenished. The mechanism of reduction was still under consideration.
5.2.6.3 L-Lysine. Gao and co-workers applied L-lysine as a reducing agent in the presence of carboxymethyl starch (CMS) as a dispersant to stabilize the resulting graphene in aqueous solution.99 The graphene–CMS hybrid indeed exhibited a high dispersion stability in aqueous solution and provided a C/O ratio of 8.5. A trace of nitrogen content at 3.8 at% was detected. The hybrid material was then crosslinked and freeze-dried to provide porous graphene–CMS foams which facilitated the effective absorption of metal ions such as Cu2+.
5.2.6.4 L-Glutathione. L-Glutathione reduced (GSH) is a natural antioxidant in cells capable of reducing many reactive oxygen species. Jeong and co-workers exploited this property of GSH to obtain graphene which showed good dispersion stabilities in aqueous and polar aprotic solvents such as THF, DMF and DMSO.100 Stabilization and prevention of agglomeration were achieved as GSH functioned as a capping agent. Such stabilization effects were attributed to the formation of hydrogen bonding between oxidised products of GSH and residual oxygen functionalities on graphene surfaces. The presence of residual carboxyl groups could possibly enhance the effect as well. Based on the proposed mechanism, GSH was able to release a proton and undergo intermolecular reactions with another GSH to form a glutathione disulphide (Scheme 14). The released protons would then bind to the oxygen groups present on the surfaces of graphene oxide, and subsequently be released as water molecules.
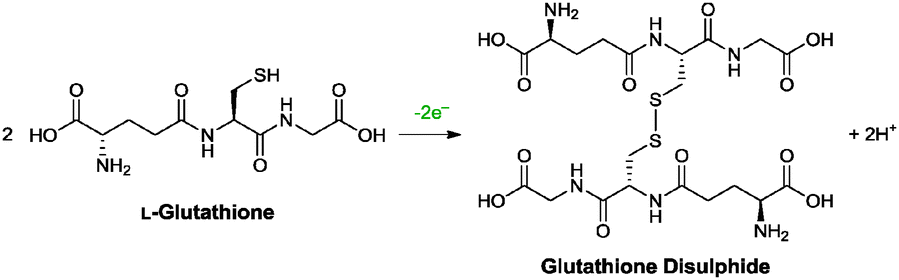 | ||
| Scheme 14 Proposed mechanisms for formation of glutathione disulphide with the simultaneous release of electrons for the reduction of graphene oxide. Adapted with permission from ref. 100. | ||
Yin and co-workers demonstrated the reduction of graphene oxide with green tea since the multiple pyrogallol and catechol moieties of polyphenolic compounds found in tea exerted excellent antioxidant properties and reactivity towards oxygen species.101 These phenolic groups were often converted to the corresponding quinone forms upon oxidation. In fact, the reduction capabilities of polyphenols were previously observed in the synthesis and stabilization of Au, Ag, Pd and Fe microparticles. Graphene obtained from green tea extract showed a conductivity of 53 S m−1 and good dispersion stabilities in polar solvents such as ethanol, methanol, acetone, DMF, NMP, DMAc and DMSO.
Apart from that, phytochemicals obtained from the leaves of Colocasia esculenta, Mesua ferrea Linn., and the peels of Citrus sinensis were used by Thakur and Karak to achieve similar reduction properties.102 The common phytochemicals found in these leaves and peels are pectins, flavonoids, ascorbic acid, apigenin, luteolin and various flavones which would convert to the respective quinones upon oxidation. Graphene obtained from the reduction with C. esculenta, M. ferrea Linn., and C. sinensis extracts provided C/O ratios of 7.11, 6.09 and 5.97, respectively. The conductivities were measured as 4006, 3185 and 3033 S m−1, respectively. In comparison, the precursor graphene oxide highlighted a C/O ratio of 2.68 and a measured conductivity of 104 S m−1. Furthermore, the specific capacitances achieved were 17, 18 and 21 F g−1, respectively. The mechanisms suggested by the group were similar to that of L-ascorbic acid as aforementioned.
In the work of Haghighi and Tabrizi, Rosa damascena (more commonly known as rose) was applied as a reducing agent since rose water contains natural antioxidants such as phenolic compounds and flavanol glycosides.103 The resulting graphene could disperse well in aqueous solution for more than one month. Subsequent electrochemical impedance spectroscopy highlighted a low charge transfer resistance of the graphene at 0.18 kΩ (graphite oxide: 28 kΩ, bare glassy carbon: 0.40 kΩ). Moreover, electrocatalytic activities towards catechol, NADH and immobilized glucose oxidase were demonstrated by the graphene.
Apart from that, the usage of Escherichia coli (E. coli) bacteria utilizing a mixed-acid fermentation pathway under anaerobic conditions for the reduction of graphene oxide was highlighted by Akhavan and Ghaderi.106 The graphene oxide sample and E. coli were incubated for 48 h to provide graphene with a sheet resistance of 3.4 × 109 Ω sq−1 (graphene oxide: 2.9 × 1011 Ω sq−1). The metabolically generated electrons and hydrogen from the glycolysis process were postulated to be involved in the mechanism of graphene oxide reduction. Moreover, graphene oxide was found to provide biocompatible sites for the adsorption and proliferation of E. coli whilst the resulting graphene inhibited further proliferation. In a subsequent work by Kim and co-workers, similar reduction capability of E. coli was demonstrated by stirring a mixture of E. coli biomass with graphene oxide at 37 °C for 72 h.107
Another reduction method with microorganisms based on baker's yeast containing nicotinamide adenine dinucleotide phosphate (NADPH) was described by Lee and co-workers.108 The group was motivated by previous reports on the ability of baker's yeast in reducing prochiral ketones and α,β-epoxy ketones as well as the reducing capability of NADPH towards organic ketones. Graphene obtained after 72 h of treatment showed a C/O ratio of 5.9 and a measured conductivity of 43 S m−1 (graphite oxide: 0.002 S m−1). The graphene was simultaneously bio-functionalized as NADP/NADPH remained attached to the surface of the graphene.
Moreover, Lee and co-workers highlighted the application of wild carrot root for the reduction of graphene oxide.109 This was pursued based on a previous report on the reduction capability of wild carrot root towards aliphatic and aromatic ketones, cyclic ketones, β-keto esters, and azidoketones. The reduction process was attributed to the reducing capabilities of endophytic microorganisms present in wild carrot root. By treating an aqueous dispersion of graphene oxide with slices of wild carrot roots for 48 h and another 24 h under reflux conditions, graphene with a C/O ratio of 11.9 was obtained.
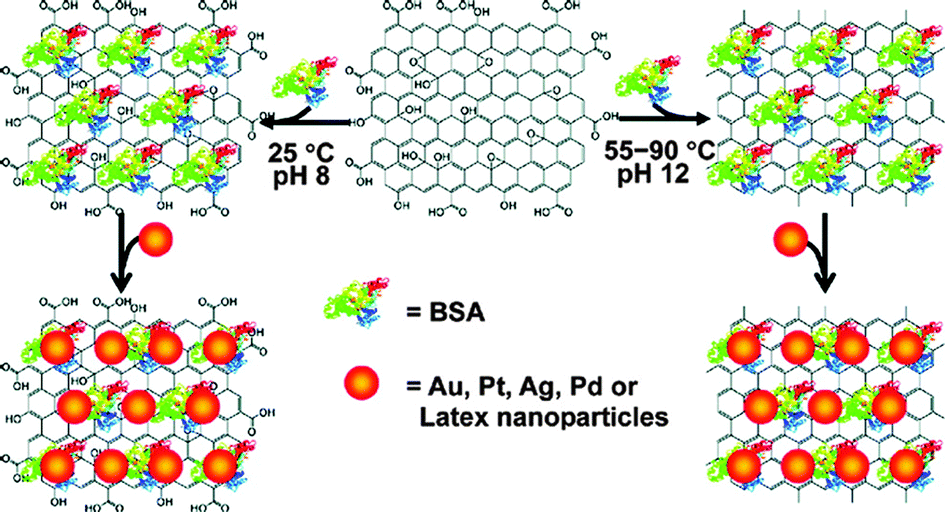 | ||
| Scheme 15 Effects of BSA on graphene oxide under different incubation conditions. Reproduced with permission from ref. 110. | ||
The group proposed a series of mechanisms for the reduction whereby the epoxide groups on the graphene oxide surfaces were nucleophilically attacked by melatonin in two different manners (Scheme 16, Routes 1 and 2). Based on Route 1, a covalent C–C bond was formed and a subsequent hydrogen transfer step from another melatonin molecule led to the reduction of hydroxyl groups via a dehydration process. The dimer was subsequently adsorbed on the surface of the graphene sheet. As for Route 2, the nucleophilic attack of the nitrogen on the amide group led to the formation of a C–N covalent bond. On the other hand, the hydroxyl groups on graphene oxide surfaces were eliminated based on Routes 3 and 4. In Route 3, the transfer of electrons and hydrogen resulted in the formation of an indolyl cation radical, while the hydroxyl group was removed through a dehydration process. In Route 4, hydroxyl and graphene radicals were generated under heat treatment in an alkaline condition. The hydroxyl radical was then scavenged by a melatonin cation radical to produce a hydroxymelatonin compound, which subsequently scavenged a second hydroxyl radical to form a dehydroxymelatonin compound.
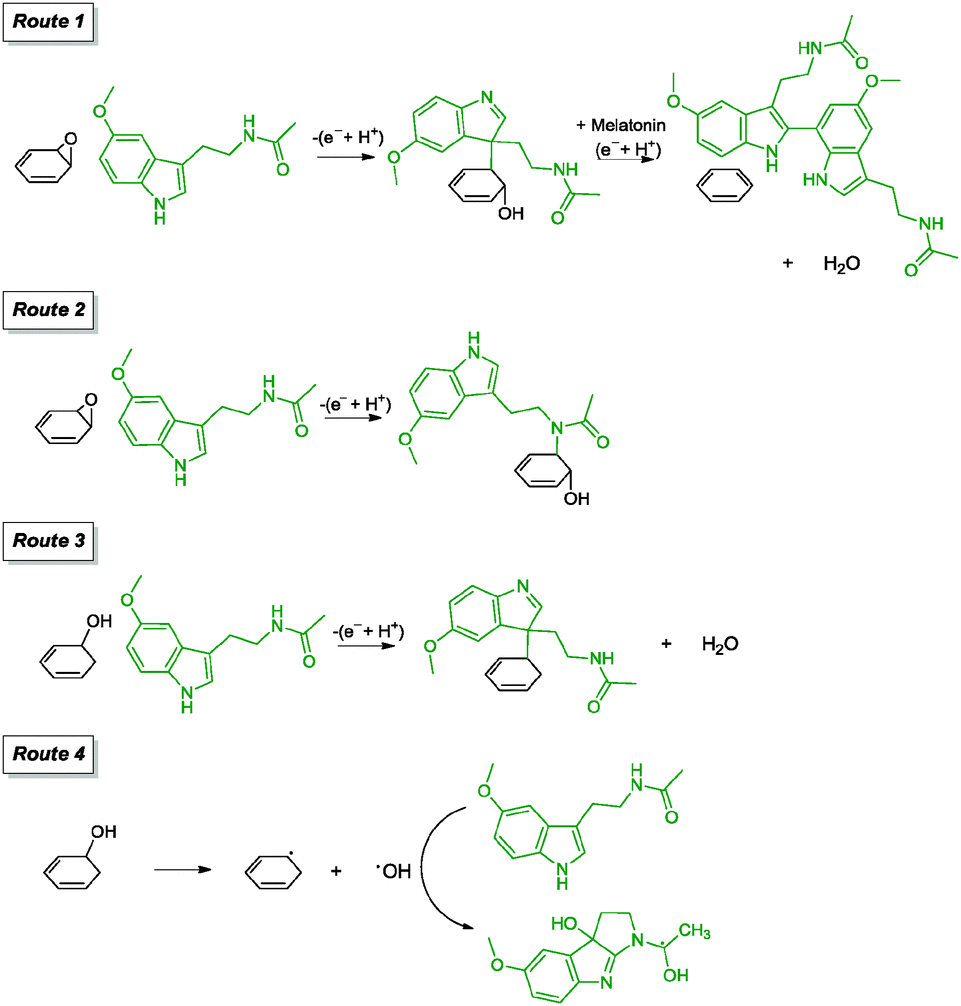 | ||
| Scheme 16 Proposed mechanisms for the reduction of epoxide and hydroxyl groups with melatonin. Adapted with permission from ref. 112. | ||
6. Conclusion and outlook
The amount of scientific research dedicated to the reduction methods of graphene oxide is tremendous. The reduction strategies covered in this review are by no means exhaustive as new approaches are constantly being explored. Although we may have a basic understanding of the efficiencies of each reducing agent based on the available material characterization techniques, much remains to be done to elucidate the mechanisms of reduction. This is of utmost importance since two important questions may plague future experimentalists interested in producing graphene via the chemical methods, mainly whether a reducing agent of interest may (i) specifically target designated oxygen functionalities in a certain mode of reaction or (ii) undergo a random mode of reaction accredited to the antioxidant and oxygen scavenging properties of the reducing agents. We have to tread lightly on this. Given past experiences on how grades of commodity plastics are determined by the structures of monomers, it seems that only by understanding the chemical modifications occurring at the molecular level can the exact nature of a bulk material be ascertained with a high certainty. After all, it is of interest to chemists to determine the exact chemical composition and constitution of compounds. Graphene obtained via chemical means, which is made up of basic carbon and oxygen atoms, is nevertheless related to or governed by the knowledge of organic chemistry. Despite that, given the current worldwide concern on a greener chemistry, alternative reducing agents could be valuable as well. It could potentially be beneficial for the graphene community if such alternative agents are capable of demonstrating reducing capabilities towards oxygenated polycyclic aromatic hydrocarbons (PAH).At the current state of graphene research, the synthesis of graphene via chemical methods is the most promising approach towards large scale production. Although the resulting graphene is imperfect as compared to graphene obtained from other methods of synthesis, it has shown potential applications ranging from transparent conductive layers, nanoelectronics, sensors, bioapplications, coatings, paint/ink, composites and energy storage. It is thus envisaged that a cheaper cost of production and the ability to fine-tune the quality of graphene could result in a faster development of these technologies and subsequent introduction into the consumer market. More importantly, rational synthetic modifications of the graphene materials could potentially provide high quality graphene sheets or even novel graphene-based materials for future practical applications. In all likelihood, the progress of commercial applications of graphene is still in its infant stage. It is thus critical to apply the most appropriate reduction method of graphene oxide to ensure the quality for commercial applications. Collective efforts from the graphene community are of utmost importance towards the realization of graphene as the ‘wonder-material’.
Notes and references
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- Y. W. Zhu, S. Murali, W. W. Cai, X. S. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- M. Pumera, Energy Environ. Sci., 2011, 4, 668–674 CAS.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876–1902 CrossRef CAS PubMed.
- P. Avouris and C. Dimitrakopoulos, Mater. Today, 2012, 15, 86–97 CrossRef CAS.
- Y. Zhang, L. Zhang and C. Zhou, Acc. Chem. Res., 2013 DOI:10.1021/ar300203n.
- R. S. Edwards and K. S. Coleman, Nanoscale, 2013, 5, 38–51 RSC.
- M. B. Smith and J. March, March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, New Jersey, 6th edn, 2007 Search PubMed.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 149, 249–259 CrossRef.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481–1487 CrossRef CAS.
- U. Hofmann and E. Konig, Z. Anorg. Allg. Chem., 1937, 234, 311–336 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- C. K. Chua, Z. Sofer and M. Pumera, Chem.–Eur. J., 2012, 18, 13453–13459 CrossRef CAS PubMed.
- U. Hofmann and R. Holst, Ber. Dtsch. Chem. Ges., 1939, 72, 754–771 CrossRef.
- G. Ruess, Monatsh. Chem., 1946, 76, 381–417 CrossRef.
- W. Scholz and H. P. Boehm, Z. Anorg. Allg. Chem., 1969, 369, 327–340 CrossRef CAS.
- T. Nakajima and Y. Matsuo, Carbon, 1994, 32, 469–475 CrossRef CAS.
- H. He, T. Riedl, A. Lerf and J. Klinowski, J. Phys. Chem., 1996, 100, 19954–19958 CrossRef CAS.
- A. Lerf, H. Y. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- A. Lerf, H. He, T. Riedl, M. Forster and J. Klinowski, Solid State Ionics, 1997, 101–103(Part 2), 857–862 CrossRef CAS.
- K. Erickson, R. Erni, Z. Lee, N. Alem, W. Gannett and A. Zettl, Adv. Mater., 2010, 22, 4467–4472 CrossRef CAS PubMed.
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef.
- W. W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. X. Yang, A. Velamakanni, S. J. An, M. Stoller, J. H. An, D. M. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS PubMed.
- W. Gao, L. B. Alemany, L. J. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed.
- A. Dimiev, D. V. Kosynkin, L. B. Alemany, P. Chaguine and J. M. Tour, J. Am. Chem. Soc., 2012, 134, 2815–2822 CrossRef CAS PubMed.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2012, 7, 576–588 CrossRef PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- A. Ambrosi, A. Bonanni, Z. Sofer, J. S. Cross and M. Pumera, Chem.–Eur. J., 2011, 17, 10763–10770 CrossRef CAS PubMed.
- G. Brauer, Handbook of Preparative Inorganic Chemistry, Academic Press, 1963 Search PubMed.
- R. Muszynski, B. Seger and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 5263–5266 CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- H.-J. Shin, K. K. Kim, A. Benayad, S.-M. Yoon, H. K. Park, I.-S. Jung, M. H. Jin, H.-K. Jeong, J. M. Kim, J.-Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- C. K. Chua and M. Pumera, J. Mater. Chem. A, 2013, 1, 1892–1898 CAS.
- V. H. Pham, S. H. Hur, E. J. Kim, B. S. Kim and J. S. Chung, Chem. Commun., 2013, 49, 6665–6667 RSC.
- A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Chem. Mater., 2012, 24, 2292–2298 CrossRef CAS.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 73 CrossRef PubMed.
- P. Cui, J. Lee, E. Hwang and H. Lee, Chem. Commun., 2011, 47, 12370–12372 RSC.
- S. F. Pei, J. P. Zhao, J. H. Du, W. C. Ren and H. M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS PubMed.
- Y. Chen, X. Zhang, D. Zhang, P. Yu and Y. Ma, Carbon, 2011, 49, 573–580 CrossRef CAS PubMed.
- C. K. Chua and M. Pumera, J. Mater. Chem., 2012, 22, 23227–23231 RSC.
- K. Nakagawa and K. Minami, Tetrahedron Lett., 1972, 13, 343–346 CrossRef.
- C. K. Chua, A. Ambrosi and M. Pumera, J. Mater. Chem., 2012, 22, 11054–11061 RSC.
- Y. Wang, L. Sun and B. Fugetsu, Bull. Chem. Soc. Jpn., 2012, 85, 1339–1344 CrossRef CAS.
- Q. Ma, J. Song, C. Jin, Z. Li, J. Liu, S. Meng, J. Zhao and Y. Guo, Carbon, 2013, 54, 36–41 CrossRef CAS PubMed.
- C. K. Chua and M. Pumera, Chemistry, 2013, 19, 2005–2011 CrossRef CAS PubMed.
- M. Node, K. Nishide, K. Ohta and E. Fujita, Tetrahedron Lett., 1982, 23, 689–692 CrossRef CAS.
- G. Kollenz, G. Penn, R. Theuer, W. M. F. Fabian, H. A. A. ElNabi, X. Zhang, K. Peters, E. M. Peters and H. G. vonSchnering, Tetrahedron, 1996, 52, 5427–5440 CrossRef CAS.
- T. Nishio, J. Chem. Soc., Chem. Commun., 1989, 205–206 RSC.
- H. Liu, L. Zhang, Y. Guo, C. Cheng, L. Yang, L. Jiang, G. Yu, W. Hu, Y. Liu and D. Zhu, J. Mater. Chem. C, 2013, 1, 3104–3109 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- M. C. Kim, G. S. Hwang and R. S. Ruoff, J. Chem. Phys., 2009, 131, 064704 CrossRef PubMed.
- S. Park, Y. Hu, J. O. Hwang, E.-S. Lee, L. B. Casabianca, W. Cai, J. R. Potts, H.-W. Ha, S. Chen, J. Oh, S. O. Kim, Y.-H. Kim, Y. Ishii and R. S. Ruoff, Nat. Commun., 2012, 3, 638 CrossRef PubMed.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2009, 114, 832–842 Search PubMed.
- S. Park, J. An, I. Jung, R. D. Piner, S. J. An, X. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS PubMed.
- V. H. Pham, T. V. Cuong, T.-D. Nguyen-Phan, H. D. Pham, E. J. Kim, S. H. Hur, E. W. Shin, S. Kim and J. S. Chung, Chem. Commun., 2010, 46, 4375–4377 RSC.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2009, 4, 25–29 CrossRef CAS PubMed.
- X. Zhou, J. Zhang, H. Wu, H. Yang, J. Zhang and S. Guo, J. Phys. Chem. C, 2011, 115, 11957–11961 CAS.
- S. Mao, K. Yu, S. Cui, Z. Bo, G. Lu and J. Chen, Nanoscale, 2011, 3, 2849–2853 RSC.
- C. A. Amarnath, C. E. Hong, N. H. Kim, B.-C. Ku, T. Kuila and J. H. Lee, Carbon, 2011, 49, 3497–3502 CrossRef CAS PubMed.
- T. Sakura and Y. Nagasaki, Colloid Polym. Sci., 2007, 285, 1407–1410 CAS.
- S. Liu, J. Q. Tian, L. Wang and X. P. Sun, Carbon, 2011, 49, 3158–3164 CrossRef CAS PubMed.
- Y. Chen, X. Zhang, P. Yu and Y. Ma, Chem. Commun., 2009, 4527–4529 RSC.
- J. F. Che, L. Y. Shen and Y. H. Xiao, J. Mater. Chem., 2010, 20, 1722–1727 RSC.
- Z. Lei, L. Lu and X. S. Zhao, Energy Environ. Sci., 2012, 5, 6391–6399 CAS.
- P. Su, H.-L. Guo, L. Tian and S.-K. Ning, Carbon, 2012, 50, 5351–5358 CrossRef CAS PubMed.
- X. P. Shen, L. Jiang, Z. Y. Ji, J. L. Wu, H. Zhou and G. X. Zhu, J. Colloid Interface Sci., 2011, 354, 493–497 CrossRef CAS PubMed.
- S. Zhang, Y. Shao, H. Liao, M. H. Engelhard, G. Yin and Y. Lin, ACS Nano, 2011, 5, 1785–1791 CrossRef CAS PubMed.
- T. Wu, X. Wang, H. Qiu, J. Gao, W. Wang and Y. Liu, J. Mater. Chem., 2012, 22, 4772–4779 RSC.
- D. R. Dreyer, S. Murali, Y. Zhu, R. S. Ruoff and C. W. Bielawski, J. Mater. Chem., 2011, 21, 3443–3447 RSC.
- C.-Y. Su, Y. Xu, W. Zhang, J. Zhao, A. Liu, X. Tang, C.-H. Tsai, Y. Huang and L.-J. Li, ACS Nano, 2010, 4, 5285–5292 CrossRef CAS PubMed.
- G. Wang, J. Yang, J. Park, X. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192–8195 CAS.
- J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- M. J. Fernández-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2010, 114, 6426–6432 Search PubMed.
- C. Zhu, S. Guo, Y. Fang and S. Dong, ACS Nano, 2010, 4, 2429–2437 CrossRef CAS PubMed.
- Y.-K. Kim, M.-H. Kim and D.-H. Min, Chem. Commun., 2011, 47, 3195–3197 RSC.
- J. Li, G. Xiao, C. Chen, R. Li and D. Yan, J. Mater. Chem. A, 2013, 1, 1481–1487 CAS.
- W. Chen, L. Yan and P. R. Bangal, J. Phys. Chem. C, 2010, 114, 19885–19890 CAS.
- T. Zhou, F. Chen, K. Liu, H. Deng, Q. Zhang, J. Feng and Q. Fu, Nanotechnology, 2011, 22, 045704 CrossRef PubMed.
- Y. Liu, Y. Li, Y. Yang, Y. Wen and M. Wang, J. Nanosci. Nanotechnol., 2011, 11, 10082–10086 CrossRef CAS PubMed.
- S. Some, Y. Kim, Y. Yoon, H. Yoo, S. Lee, Y. Park and H. Lee, Sci. Rep., 2013, 3, 1929 Search PubMed.
- Z. Fan, K. Wang, T. Wei, J. Yan, L. Song and B. Shao, Carbon, 2010, 48, 1686–1689 CrossRef CAS PubMed.
- Z.-J. Fan, W. Kai, J. Yan, T. Wei, L.-J. Zhi, J. Feng, Y.-m. Ren, L.-P. Song and F. Wei, ACS Nano, 2010, 5, 191–198 CrossRef PubMed.
- X. Mei and J. Ouyang, Carbon, 2011, 49, 5389–5397 CrossRef CAS PubMed.
- P. B. Liu, Y. Huang and L. Wang, Mater. Lett., 2013, 91, 125–128 CrossRef CAS PubMed.
- R. S. Dey, S. Hajra, R. K. Sahu, C. R. Raj and M. K. Panigrahi, Chem. Commun., 2012, 48, 1787–1789 RSC.
- N. A. Kumar, S. Gambarelli, F. Duclairoir, G. Bidan and L. Dubois, J. Mater. Chem. A, 2013, 1, 2789–2794 CAS.
- V. H. Pham, H. D. Pham, T. T. Dang, S. H. Hur, E. J. Kim, B. S. Kong, S. Kim and J. S. Chung, J. Mater. Chem., 2012, 22, 10530–10536 RSC.
- B. K. Barman, P. Mahanandia and K. K. Nanda, RSC Adv., 2013, 3, 12621–12624 RSC.
- Y. Liu, Y. Li, M. Zhong, Y. Yang, W. Yuefang and M. Wang, J. Mater. Chem., 2011, 21, 15449–15455 RSC.
- S. Yang, W. Yue, D. Huang, C. Chen, H. Lin and X. Yang, RSC Adv., 2012, 2, 8827–8832 RSC.
- H. Feng, R. Cheng, X. Zhao, X. Duan and J. Li, Nat. Commun., 2013, 4, 1539 CrossRef PubMed.
- D. Chen, L. Li and L. Guo, Nanotechnology, 2011, 22, 325601 CrossRef PubMed.
- S. Bose, T. Kuila, A. K. Mishra, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 9696–9703 RSC.
- J. K. Ma, X. R. Wang, Y. Liu, T. Wu, Y. Liu, Y. Q. Guo, R. Q. Li, X. Y. Sun, F. Wu, C. B. Li and J. P. Gao, J. Mater. Chem. A, 2013, 1, 2192–2201 CAS.
- T. A. Pham, J. S. Kim, J. S. Kim and Y. T. Jeong, Colloids Surf., A, 2011, 384, 543–548 CrossRef CAS PubMed.
- Y. Wang, Z. Shi and J. Yin, ACS Appl. Mater. Interfaces, 2011, 3, 1127–1133 CAS.
- S. Thakur and N. Karak, Carbon, 2012, 50, 5331–5339 CrossRef CAS PubMed.
- B. Haghighi and M. Amouzadeh Tabrizi, RSC Adv., 2013, 3, 13365–13371 RSC.
- E. C. Salas, Z. Sun, A. Lüttge and J. M. Tour, ACS Nano, 2010, 4, 4852–4856 CrossRef CAS PubMed.
- G. Wang, F. Qian, C. Saltikov, Y. Jiao and Y. Li, Nano Res., 2011, 4, 563–570 CrossRef CAS PubMed.
- O. Akhavan and E. Ghaderi, Carbon, 2012, 50, 1853–1860 CrossRef CAS PubMed.
- S. Gurunathan, J. W. Han, V. Eppakayala and J. H. Kim, Colloids Surf., B, 2013, 102, 772–777 CrossRef CAS PubMed.
- P. Khanra, T. Kuila, N. H. Kim, S. H. Bae, D.-S. Yu and J. H. Lee, Chem. Eng. J., 2012, 183, 526–533 CrossRef CAS PubMed.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Carbon, 2012, 50, 914–921 CrossRef CAS PubMed.
- J. Liu, S. Fu, B. Yuan, Y. Li and Z. Deng, J. Am. Chem. Soc., 2010, 132, 7279–7281 CrossRef CAS PubMed.
- J. P. Rourke, P. A. Pandey, J. J. Moore, M. Bates, I. A. Kinloch, R. J. Young and N. R. Wilson, Angew. Chem., 2011, 123, 3231–3235 CrossRef.
- A. Esfandiar, O. Akhavan and A. Irajizad, J. Mater. Chem., 2011, 21, 10907–10914 RSC.
| This journal is © The Royal Society of Chemistry 2014 |