 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards cancer cell-specific phototoxic organometallic rhenium(I) complexes†
Anna
Leonidova‡
a,
Vanessa
Pierroz‡
ab,
Riccardo
Rubbiani
a,
Jakob
Heier
c,
Stefano
Ferrari
b and
Gilles
Gasser
*a
aInstitute of Inorganic Chemistry, University of Zurich, Winterthurerstrasse 190, CH 8057 Zurich, Switzerland. E-mail: gilles.gasser@aci.uzh.ch; Fax: +41 44 635 46 03; Tel: +41 44 635 46 30
bInstitute of Molecular Cancer Research, Winterthurerstrasse 190, CH 8057 Zurich, Switzerland
cLaboratory for Functional Polymers, Empa. Swiss Federal Laboratories for Material Science and Technology, Uberlandstrasse 129, CH 8600, Dübendorf, Switzerland
First published on 1st August 2013
Abstract
Over the recent years, several Re(I) organometallic compounds have been shown to be toxic to various cancer cell lines. However, these compounds lacked sufficient selectivity towards cancer tissues to be used as novel chemotherapeutic agents. In this study, we probe the potential of two known N,N-bis(quinolinoyl) Re(I) tricarbonyl complex derivatives, namely Re(I) tricarbonyl [N,N-bis(quinolin-2-ylmethyl)amino]-4-butane-1-amine (Re–NH2) and Re(I) tricarbonyl [N,N-bis(quinolin-2-ylmethyl)amino]-5-valeric acid (Re–COOH), as photodynamic therapy (PDT) photosensitizers. Re–NH2 and Re–COOH proved to be excellent singlet oxygen generators in a lipophilic environment with quantum yields of about 75%. Furthermore, we envisaged to improve the selectivity of Re–COOHvia conjugation to two types of peptides, namely a nuclear localization signal (NLS) and a derivative of the neuropeptide bombesin, to form Re–NLS and Re–Bombesin, respectively. Fluorescent microscopy on cervical cancer cells (HeLa) showed that the conjugation of Re–COOH to NLS significantly enhanced the compound's accumulation into the cell nucleus and more specifically into its nucleoli. Importantly, in view of PDT applications, the cytotoxicity of the Re complexes and their bioconjugates increased significantly upon light irradiation. In particular, Re–Bombesin was found to be at least 20-fold more toxic after light irradiation. DNA photo-cleavage studies demonstrated that all compounds damaged DNA via singlet oxygen and, to a minor extent, superoxide production.
Introduction
Over the last few years, an impressive amount of organometallic compounds has been screened for cytotoxicity to various cancer cell lines.1–6 Among them, several organometallic rhenium complexes have been reported to possess an anti-proliferative activity comparable to or even exceeding that of cisplatin.7–13 However, to avoid the severe side-effects afflicting patients during chemotherapy, high cytotoxicity is not enough – an improved selectivity towards cancer cells is required.14 One of the possible strategies to overcome this important problem is to identify a target specific to cancer cells – e.g. an overexpressed receptor – and to conjugate the active compound to a moiety recognized by that target. Rhenium compounds such as Re(I) tricarbonyl N,N-bis(quinolinoyl) complex derivatives can easily be coupled to potential targeting vectors, as previously reported by Valliant, Zubieta, Doyle, Metzler-Nolte and our group.15–23 Due to their long lifetimes, polarized emission and large Stokes shifts, these organometallic complexes have been initially developed as luminescent probes24–28 and proven themselves particularly useful to study the behaviour of bioconjugates in cells. As mentioned earlier, these probes have been coupled to numerous biologically relevant molecules, such as formyl peptide receptor targeting peptide,15 biotin,16 glucose,17 β-breaker peptide derivatives,18 folate,19 cell permeation peptides,20 vitamin B12,21 and peptide nucleic acids.22,23 In addition to their imaging properties, some of the conjugates have shown therapeutic potential. For instance, the Re–β-breaker peptides inhibit amyloid plaque formation associated with Alzheimer's disease.18 The Re–B12 derivative showed only a moderate cytotoxicity to the choriocarcinoma cell line (BeWo),21 but the Re–folate conjugate was found to be strongly toxic to human multidrug-resistant ovarian carcinoma (A2780/AD) and Chinese hamster ovary (CHO) cell lines.19 Of note, the Re(I) complexes used in these studies also possess a certain anti-proliferative activity on their own. Importantly, due to the isostructurality of Re(I) with Tc(I), the 99mTc bioconjugate analogues could be prepared in a few of these studies allowing radioimaging to be performed.15,16,18,29Another approach to specifically kill cancer cells is to use an external trigger to select the area where an initially inactive compound can be activated. A good example of such an approach is photodynamic therapy (PDT). This medical technique is based on the ability of certain molecules called photosensitizers (PS) to generate highly reactive singlet oxygen (1O2) upon light irradiation. Although some PS, such as certain porphyrin derivatives, tend to accumulate more in tumour tissues,30 other PS often lack selectivity for cancer cells. This can lead to accumulation of PS in healthy tissues rendering patients photosensitive, in some cases, up to several weeks after the end of the treatment.31 Although many Re complexes are known to be efficient triplet PS,32–35 so far, most of them have been applied in photocatalysis and, to the best of our knowledge, the first anti-cancer application of rhenium(I) complexes with light was only recently reported by the group of Meggers.36 This is very surprising since, given their high activity as triplet PS in photocatalysis, such compounds should be excellent 1O2 generators in cells.
In this article, we present the characterization of 1O2 production of two Re(I) organometallic complexes, the preparation of nuclear and bombesin receptor targeting peptide conjugates of a Re(I) tricarbonyl N,N-bis(quinolinoyl) complex, as well as the biological activity to human cancer cells (HeLa) and fibroblasts (MRC-5) of the Re(I) complexes and conjugates thereof.
Results and discussion
Synthesis and characterization
Re(I) tricarbonyl [N,N-bis(quinolin-2-ylmethyl)amino]-4-butane-1-amine (Re–NH2) and Re(I) tricarbonyl [N,N-bis(quinolin-2-ylmethyl)amino]-5-valeric acid (Re–COOH) complexes (Fig. 1) were prepared via reductive amination of quinoline-2-carboxyaldehyde with the corresponding amine, followed by complexation with [Re(CO)3Br3][NEt3], as previously reported.19,21Re–COOH was then successfully coupled to two different targeting peptides on the solid phase using a similar experiment described by Metzler-Nolte et al.37,38 to obtain the bioconjugates Re–NLS and Re–Bombesin, respectively (Fig. 1). Re–NLS contains a short nuclear localization signal (NLS) peptide39 in order to bring the Re complex to the nucleus in close proximity of DNA. Most approved PS localize in other organelles rather than the nucleus (e.g. membranes, mitochondria, lysosomes, endoplasmic reticulum and Golgi apparatus).40 However, many PS have long been known to damage DNA upon light irradiation.41,42 The second conjugate, Re–Bombesin, holds a derivative of the neuropeptide bombesin, known for its excellent in vivo stability and biodistribution.43,44 The purpose of this peptide is to selectively transport the complex to cancer cells (over healthy cells), as it targets a receptor overexpressed in certain types of cancer. Both Re–NLS and Re–Bombesin were purified by preparative reverse phase HPLC and lyophilized to obtain light-yellow powders. The presence of the two expected bioconjugates was ascertained by HR ESI-MS and MALDI-TOF (Fig. S8, S10, S12, S13†). Several charge states, namely at m/z 342.69 [M + 3H]4+, 456.85 [M + 2H]3+ and 684.78 [M + H]2+, were observed by HR ESI-MS for Re–NLS, as can be expected for such a highly positively charged peptide conjugate. By contrast, only charge state 929.88 [M + H]2+ was detected for the less charged Re–Bombesin conjugate. Characteristic Re isotopic pattern was present in both HR ESI-MS and MALDI-TOF spectra. The purity of Re–NLS and Re–Bombesin was further confirmed by LC-MS (Fig. S9 and S14†) and elemental analysis.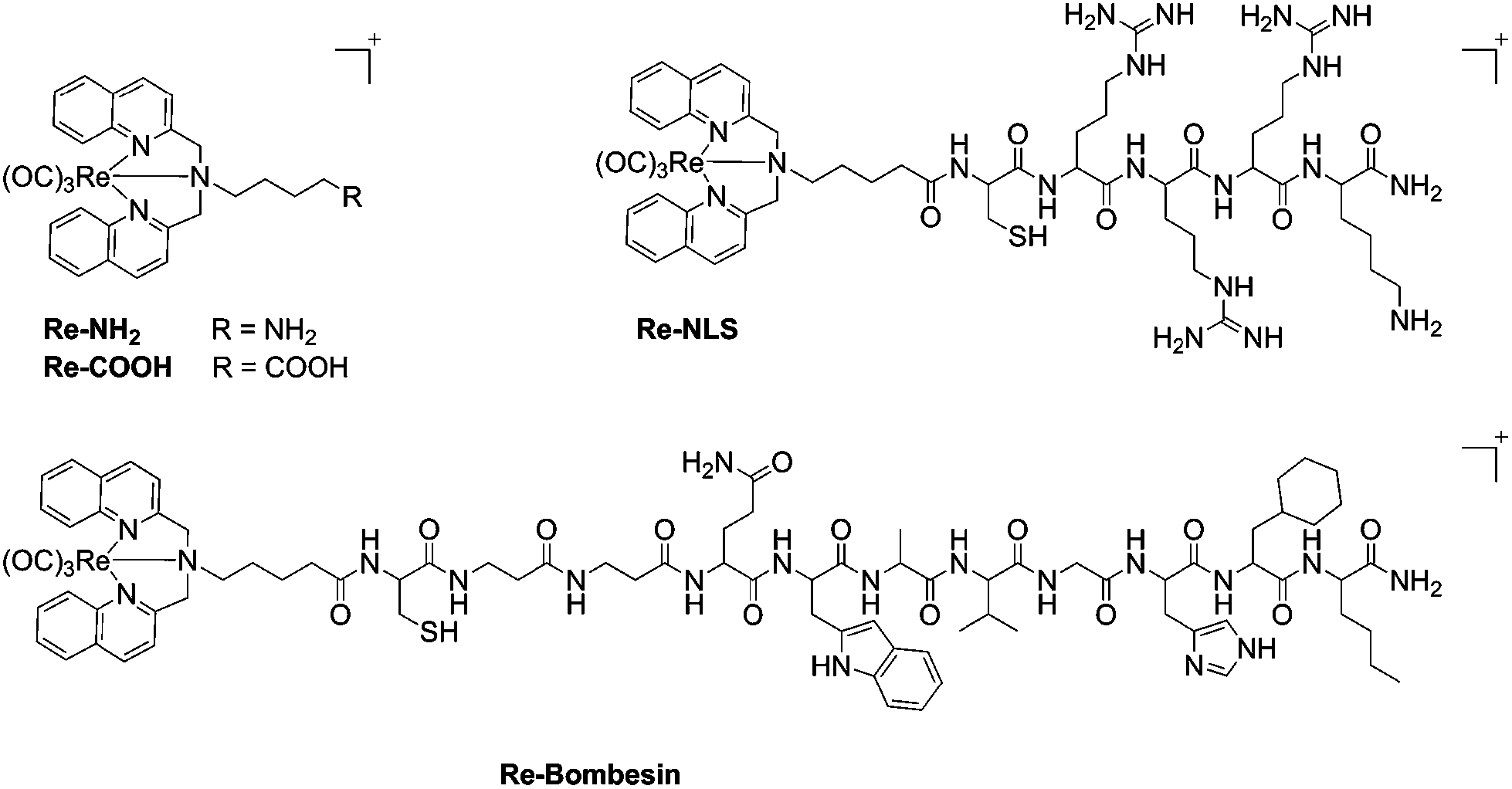 | ||
| Fig. 1 Structures of Re complexes and their derivatives. | ||
Singlet oxygen production
In order to assess the phototoxic potential of our Re(I) organometallic complexes, Re–NH2 and Re–COOH were assayed in water and acetonitrile for 1O2 production using both an indirect method – an N,N-dimethyl-4-nitrosoaniline (RNO)/histidine test45 – and a direct detection by near-infrared luminescence.46 In the first assay, histidine quenches 1O2 and the resulting trans-annular peroxide bleaches RNO absorbance at 440 nm (Fig. S15 and S16†). In acetonitrile, imidazole instead of histidine was used due to the poor solubility of histidine in this solvent. To verify the indirect method results, direct measurements of characteristic 1O2 near-IR luminescence at 1270 nm were performed (Fig. S17†). For both methods, 1O2 quantum yields were then calculated using phenalenone as the reference.47–49 As can be seen in Table 1, the two methods yielded similar results. The nature of the solvent strongly affected the 1O2 quantum yields of the Re complexes. Although they are mediocre PS in water (quantum yields of about 25%), these compounds are excellent 1O2 producers in more lipophilic solvents such as acetonitrile with quantum yields of about 75%. Since cells provide not only polar, but also lipophilic environments, our compounds could prove to be efficient PS in vitro as well.Intracellular localization
To understand the behaviour of the organometallic complexes Re–NH2 and Re–COOH and of the bioconjugates Re–NLS and Re–Bombesin, their cellular localization was evaluated in HeLa cells. For this purpose, the cells were incubated with the compounds, fixed with formaldehyde and imaged by fluorescence microscopy. As shown in Fig. 2, Re–NH2 alone is observed exclusively in the cytoplasm and Re–COOH is homogeneously distributed throughout the cell. Conjugation to an NLS peptide greatly enhances Re complex accumulation in the nucleoli. These subnuclear domains mainly house ribosomal RNA transcription, processing and assembly, but they have other important functions, such as cell cycle and mitosis regulation, stress response and assembly of non-ribosomal ribonucleoprotein particles.50 Multifunctional, nucleoli contain both DNA (mostly ribosomal) and RNA, as well as proteins, which are either dynamic or immobilized. Re–NLS could potentially interact with one or several of these components. While NLS sequences have been well characterized, nucleolar localisation signals (NoLS) have been far less defined.51 They do seem to share structural motifs with NLS, such as the presence of Arg and Lys residues, and are even sometimes part of NLS.52–54 Hence, it is also possible that the combination of NLS with the Re complex is recognized as a NoLS. Of note, a Ru complex conjugated to an octaarginine peptide with fluorescein has been found to be mainly localized in nucleoli.55 On the other hand, some proteins are only captured by nucleoli as part of the cellular stress response that can be caused by toxins.56 Thus, it cannot be excluded that the compound initially interacts with one of these proteins and is later transported with them to nucleoli. Overall, given the importance of nucleoli in proper cell function, the production of singlet oxygen by Re–NLS should severely affect cell health. Since it has been reported that the nucleoli localization could sometimes be observed as an artefact of cell fixation,12 the results were confirmed in living cells (Fig. S22†). Re–Bombesin gave a very weak luminescence signal and therefore could not be localized. This could either be due to its poor uptake by cells or the quenching of the fluorescence inside the cell. Indeed, the luminescence of similar complexes has already been reported to be quenched by vitamin B12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 57 or in living cells.22
57 or in living cells.22
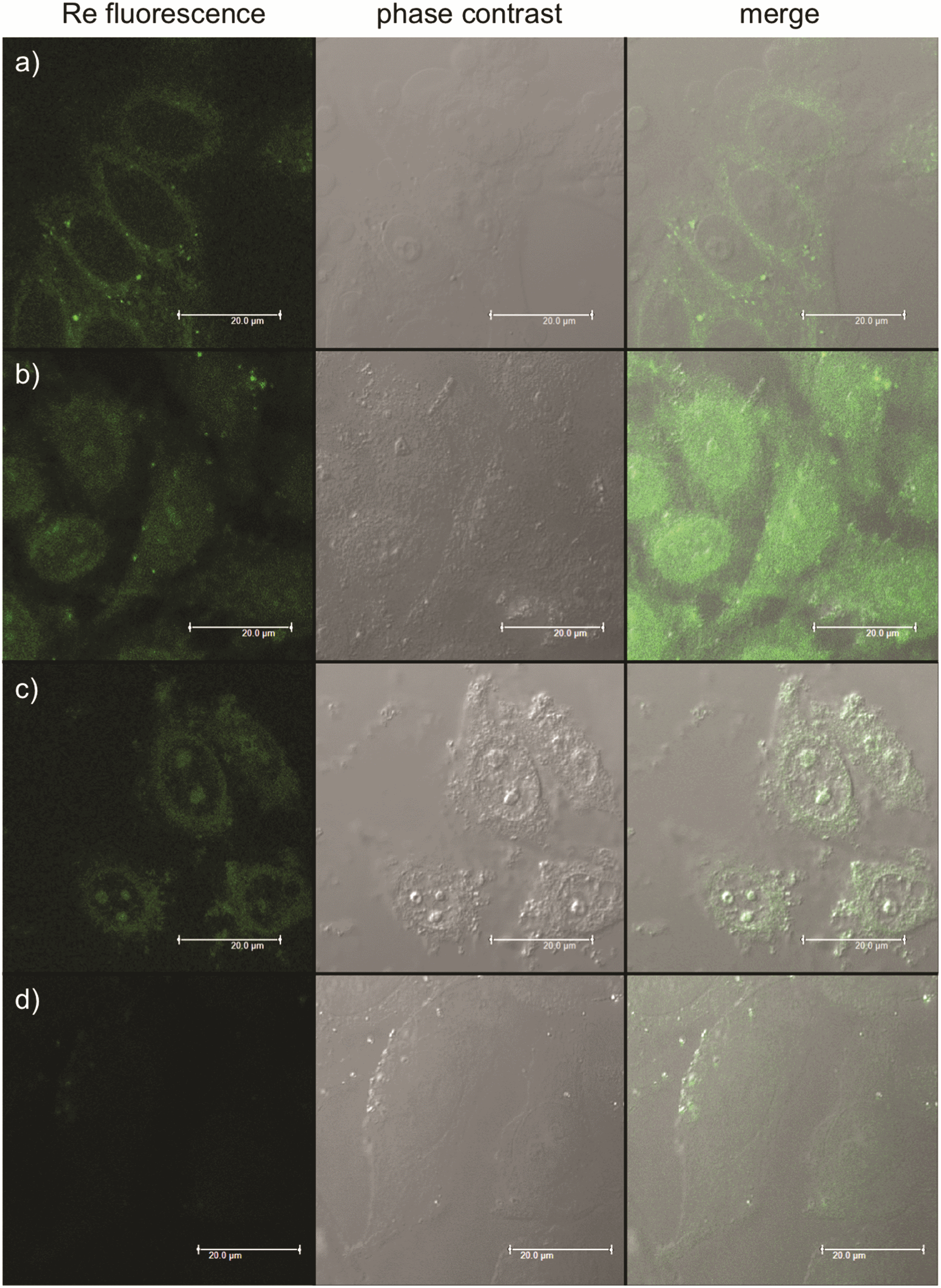 | ||
| Fig. 2 Fluorescence microscopy images showing HeLa cells fixed after treatment with: (a) Re–NH2 100 μM 2 h; (b) Re–COOH 2.5 μM 2 h; (c) Re–NLS 100 μM 1 h; (d) Re–Bombesin 50 μM 2 h. | ||
Cytotoxicity studies
With this information in hand, we then assessed the cytotoxicity of the Re complexes Re–NH2 and Re–COOH and of bioconjugates Re–NLS and Re–Bombesin on two different cell lines, namely the cervical cancer (HeLa) and human fibroblast (MRC-5) cell lines. As shown in Table 2, Re–NH2 and Re–COOH alone only affected HeLa and MRC-5 cells at concentrations higher than 100 μM. Conjugation of the bombesin derivative to Re–COOH did not affect its toxicity to HeLa cells, but made Re–Bombesin moderately toxic to MRC-5 cells. The latter have already been reported to be slightly affected by bombesin derivatives, albeit less than the cancer cells.58 The difference in Re–Bombesin cytotoxicity to HeLa and MRC-5 could also be due to the fact that the bombesin derivative used in this study has been optimized on the androgen-independent human prostate cancer (PC-3) cell line.43,44 PC-3 cells mainly overexpress gastrin-releasing peptide (GRP) receptor of the bombesin receptor family.59 HeLa, on the other hand, mostly express bombesin receptor subtype 3 (BRS-3). Although BRS-3 can interact with the same ligands as GRP receptor, its affinity for them is lower.59 In any case, the cytotoxicity of our Re–Bombesin to MRC-5 is lower compared to bombesin-mitochondria-disrupting peptide derivative.58 Coupling of the NLS peptide increased dark toxicity of Re–NLS to both HeLa and MRC-5, probably due to the compound's interactions with nucleoli. Of note, both peptides, namely NLS and Bombesin alone, were found to be non-toxic to cells at concentrations up to 100 μM. To probe the light toxicity, HeLa cells incubated with our compounds were irradiated with UVA light (emission maximum at 350 nm, 2.58 J cm−2). Little changes were observed for Re–NLS that was already cytotoxic in the dark. All other compounds became more cytotoxic upon light irradiation. The most pronounced increase (20-fold) was observed for the Re–Bombesin derivative that had a cytotoxicity comparable to cisplatin after a 2.58 J cm−2 irradiation dose – an amount smaller than or comparable to that normally used for UVA activated compounds, such as certain platinum complexes.60–65| Compound | HeLa dark IC50 (μM) | HeLa UV IC50 (μM) | MRC-5 dark IC50 (μM) |
|---|---|---|---|
| a IC50 after 48 h treatment incubation. | |||
| NLS | >100a | — | >100a |
| Bombesin | >100a | — | >100a |
| Re–NH2 | 187.1 ± 17.9a | 17.3 ± 2.9 | >100a |
| Re–COOH | >100 | 9.3 ± 2.2 | >100a |
| Re–NLS | 35.1 ± 1.8 | 18.3 ± 1.4 | 17.8 ± 1.8a |
| Re–Bombesin | >100 | 5.3 ± 1.0 | 44.1 ± 9.9a |
| Cisplatin | 9.2 ± 0.6a | — | 10.5 ± 2.8a |
DNA photo-cleavage
Encouraged by the promising results obtained on cancer cells, we decided to obtain more insight into the potential mode of action of our compounds by evaluating the possible damage induced by Re–NH2, Re–NLS, Re–COOH and Re–Bombesin to nucleic acids. DNA photo-cleavage experiments were performed on closed circular plasmid DNA (pcDNA3) as previously reported by Barton et al.66 pcDNA3 was incubated with various concentrations of our compounds and irradiated for 10 min (2.58 J cm−2) at 350 nm. Upon light irradiation, all compounds converted the supercoiled DNA form to circular/nicked forms, albeit at different concentrations (Fig. S18 and S20†). While Re–NH2, Re–COOH and Re–Bombesin displayed an effect for high concentration (50 μM–100 μM), Re–NLS, for instance, already extensively damaged DNA at a concentration of 5 μM (Fig. 3), 5–10-fold lower than the other compounds. Such a strong effect is possibly due to its high positive charge (5+) at physiological pH that allows a stronger interaction of Re–NLS with the negatively charged DNA backbone. Interestingly though, Re–NLS has the lowest photo-toxicity of all compounds. Further investigation on pcDNA3 treated with Re–NLS and reactive oxygen species quenchers, namely KI (superoxide: O2−), NaN3 (1O2) and mannitol (hydroxyl radical: OH˙), revealed a major involvement of 1O2 and, to a lesser extent, O2− in DNA photo-cleavage (Fig. S19†). DNA incubated with our compounds in the dark did not show a significant effect up to the highest concentration used.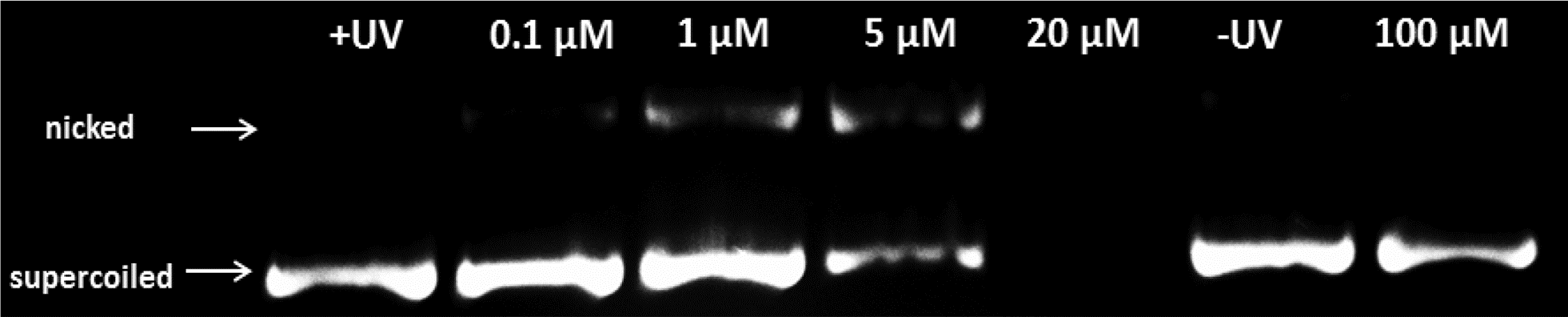 | ||
| Fig. 3 Electrophoresis experiment of DNA photo-cleavage of pcDNA3 plasmid treated with different concentrations of the complex Re–NLS; +UV = untreated DNA irradiated (lane 1); probes irradiated for 10 minutes at 350 nm (lanes 2–5); −UV = untreated DNA not irradiated (lane 6); probes not irradiated (lane 7). | ||
Conclusions
In this article, we present one of the first studies on the use of Re(I) organometallic complexes as photosensitizers for PDT purposes. We demonstrated that Re(I) tricarbonyl N,N-bis(quinolinoyl) complexes could efficiently produce 1O2 in lipophilic environments and induce DNA damage. Importantly, the localization or cellular uptake of the organometallic complexes could be controlled by attachment to targeting molecules. Although the irradiation wavelengths used in this study are not ideal since they only slightly penetrate through the skin, this work opens new avenues for the search for metal-containing photosensitizers. Indeed, of the 14 PDT agents accepted by the FDA, two already contain a metal ion (lutetium and tin) and another one containing a palladium ion (Tookad) is in clinical trial phase III.40,67 We believe that the specific physico-chemical properties of metal complexes and in particular of organometallic complexes will have an important role to play in this field of research. In addition, since 99mTc analogues of Re(I) tricarbonyl N,N-bis(quinolinoyl) complexes can be easily prepared, in vivo imaging studies could be performed, shedding light on important biodistribution data.Experimental section
Materials
Chemicals and solvents were of reagent grade or better and were purchased from commercial suppliers. They were used without further purification unless otherwise specified. The plasmid pcDNA3 was obtained from Invitrogen.Instrumentation and methods
1H and 13C NMR spectra were recorded using Bruker 400 and 500 spectrometers. Elemental microanalyses were obtained on a LecoCHNS-932 elemental analyser. High-resolution accurate mass spectra were performed on a Bruker maXis QTOF high-resolution mass spectrometer (Bruker Daltonics, Bremen, Germany). UV spectra were recorded using a Carry 50 Scan Varian spectrophotometer. ESI-MS and LC-MS spectra were obtained using a Bruker Daltonics HCT 6000 mass spectrometer. LC-MS spectra were measured using an Acquity™ from Waters system equipped with a PDA detector and an auto sampler using an Agilent Zorbax 300SB-C18 analytical column (3.5 μm particle size, 300 Å pore size, 150 × 4.6 mm). The LC run (flow rate: 0.5 mL min−1) was performed with a linear gradient of A (double distilled water containing 0.1% v/v formic acid) and B (acetonitrile containing 0.1% v/v formic acid); t = 0 min, 5% B; t = 3 min, 5% B; t = 17 min, 100% B; t = 20 min, 100% B; t = 25 min, 5% B. HPLC purification was performed using a Varian ProStar system and an Agilent Zorbax 300 SB-C18 prep column (5 μm particle size, 300 Å pore size, 150 × 21.1 mm; flow rate: 20 mL min−1). The runs were carried out with a linear gradient of A (double distilled water containing 0.1% v/v TFA) and B (acetonitrile containing 0.1% v/v TFA, Sigma-Aldrich HPLC-grade). Preparative runs: t = 0 min, 5% B; t = 25 min, 100% B; t = 30 min, 100% B; t = 32 min, 5% B. For singlet oxygen indirect detection and cell culture irradiation, a Rayonet RPR-200 photochemical reactor with 6 bulbs (14 W each) with maximum intensity output at 350 nm was used. Samples were irradiated in a fluorescence quartz cuvette (width 1 cm) placed at the centre of the reactor. The light intensity on that spot measured with an X11 optometer (Gigahertz-Optik) was 42 W m−2. The temperature inside the reactor was 30 °C. Singlet oxygen near-IR luminescence was recorded using a Fluorolog-3 spectrofluorometer (Jobin Yvon Horiba, model FL3-11) with a 450 W xenon lamp light source and single-grating excitation and emission spectrometers. To reach high beam intensity, the excitation slits were set to a maximum value of 29.4 nm. The emission was recorded at right angle to the excitation path with an IR-sensitive liquid nitrogen cooled germanium diode detector (bias −160 V, Edinburgh Instruments, model EI-L). The signal-to-noise ratio of the Ge-diode was improved with a lock-in amplifier (Stanford Research Systems, model SR510) referenced to the chopper frequency of 126 Hz. Data-acquisition was done using DataMax.Synthesis and characterization
Peptide synthesis
The peptide synthesis was performed similarly to a procedure reported by Metzler-Nolte.37 More specifically, the peptides with C-terminal amide were prepared manually in one-way polypropylene syringes (5 or 10 mL) equipped with a frit using a standard solid-phase peptide synthesis procedure. All reactions were carried out using a mechanical shaker. Polystyrene resin beads of TentaGel S RAM Lys(Boc)Fmoc (capacity 0.23 mmol g−1) or TentaGel S RAM (capacity 0.24 mmol g−1) were swollen in DMF for 1 hour before use. The resin was deprotected with piperidine (20% v/v in DMF) for 2 + 10 min. The resin was then washed with DMF (5×), DCM (5×), and DMF (5×). A Fmoc-protected amino acid was first pre-activated (4.5 equiv. amino acid, 4 equiv. TBTU, 8 equiv. DIPEA in DMF) for 2 min and then reacted with the resin for 30 min. Afterwards, the resin beads were washed with DMF (5×) and DCM (5×), and a Kaiser test was performed to check if the coupling was complete. Prior to cleavage, the resin was shrunk in methanol and dried. The peptides were cleaved using a mixture of trifluoroacetic acid–water–triisopropylsilane, 38:1:1 v/v/v (3 × 1 mL, for 1.5 h each). The collected solutions were combined and dried and the crude peptides were precipitated with cold ether. They were then purified by preparative HPLC.
Re–NLS and Re–Bombesin
Re–COOH was coupled to NLS or bombesin derivative peptide by solid-phase synthesis prior to peptide cleavage as described above. Characterization data forRe–NLS. ESI-MS m/z 342.9 [M + 3H]4+, 456.8 [M + 2H]3+, 684.6 [M + H]2+. HR ESI-MS found m/z 342.8947, 456.8573, 684.7820, calcd for [C55H79N19O9ReS] m/z 342.8956, 456.8582, 684.7832. MALDI-TOF m/z 1368.4 [M]+. Characterization data forRe–Bombesin: ESI-MS m/z 620.3 [M + 2H]3+, 929.8 [M + H]2+. HR ESI-MS found m/z 929.8817, calcd for [C84H109N19O16ReS] m/z 929.8825. MALDI-TOF m/z 1858.04 [M]+. Anal. Found: C, 49.41; H, 5.09; N, 12.03. Calc. for [C90H109F9N19O22ReS]: C, 49.17; H, 5.00; N, 12.11.Singlet oxygen production
where S is the slope of the absorbance vs. irradiation time and I is the rate of light absorption calculated as the overlap of the lamp emission spectra and the absorption spectra of the compound according to the following formula:
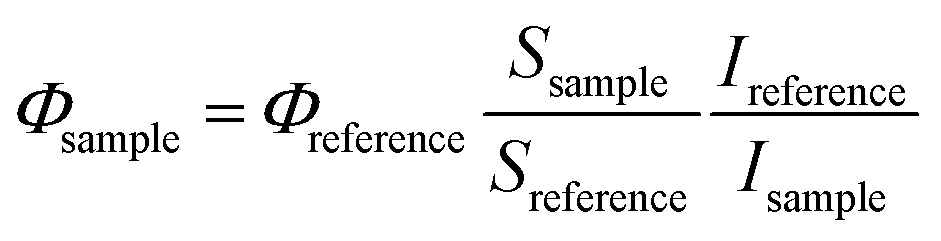
where I0 is the light-flux intensity of the lamp and A is the absorbance of the compound.
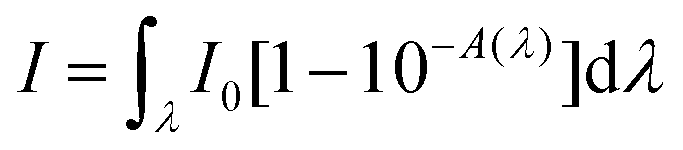
Near-infrared luminescence
A 20 mM stock solution in DMSO of the compound to measure was diluted in D2O or acetonitrile to reach approximately 0.2 absorbance at the irradiation wavelength. This solution was then irradiated in fluorescence quartz cuvettes (width 1 cm) using a UV lamp (350 nm, slit 29.4 nm). Singlet oxygen near-IR luminescence at 1271 nm was measured by recording spectra from 1200 to 1350 nm (emission slit 5 nm, detector sensitivity 100, integration 3 (1)). The intensity of irradiation was varied via neutral density filters. Singlet oxygen luminescence peaks at different irradiation intensities were integrated and the resulting areas were plotted vs. irradiation intensities. The quantum yields were then calculated by applying the same formulas as those for the N,N-dimethyl-4-nitrosoaniline/histidine assay.DNA photo-cleavage
The DNA photo-cleavage effect provoked by complexes Re–NH2, Re–NLS, Re–COOH and Re–Bombesin was investigated by electrophoresis. Supercoiled pcDNA3 plasmid (0.10 μg) was treated with increasing concentrations of the rhenium compound in a buffer (50 mM Tris/HCl, 18 mM NaCl, pH 7.2), incubated 20 minutes at 25 °C and irradiated at 350 nm for 10 minutes (Rayonet Chamber Reactor Complex, 2.58 J cm−2). Moreover, a further series of positive controls of pcDNA3 plasmid treated with 2 in the presence of mannitol (15 mM, to quench ˙OH), NaN3 (15 mM, to quench 1O2) and KI (15 mM, to quench O2−) was also performed (see Fig. S19†). A series of negative controls of the plasmid treated with different concentrations of Re–NH2, Re–NLS, Re–COOH and Re–Bombesin in the dark was used for comparative purposes. The cleavage of the target plasmid not photo-mediated was also studied at different incubation temperatures and in the presence of the restriction enzyme BstXI (1 h incubation at 37 °C) which linearized pcDNA3 (see Fig. S20†). Upon irradiation, loading buffer (2.5% Bromophenol Blue, 1% SDS, 0.1 M EDTA, 20% Ficoll 400 in 100 mL of H2O) was added to samples and they were analyzed by electrophoresis in 0.8% agarose in TBE 1× (diluted from a 10× solution of 108 g of Tris/HCl, and 55 g of H3BO3 in 900 mL of H2O) at 70 V (BioRad Powerpack 1000, BioRad) for 2 h. The gel was prestained with 0.5 μg mL−1 ethidium bromide, photographed and worked out with an AlphaDigiDoc 1000 CCD camera (Buchner Biotec AG) and AlphaImager software.Cell culture
Human cervical carcinoma cells (HeLa) were cultured in DMEM (Gibco) with 5% fetal calf serum (FCS, Gibco), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C and 5% CO2. Normal lung fibroblasts (MRC-5) were maintained in F-10 medium (Gibco) supplemented with 10% fetal calf serum (FCS, Gibco), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C and 5% CO2.Cytotoxicity studies
The cytotoxicity of the rhenium complexes and their bioconjugates non-UV-irradiated or UV-irradiated to HeLa cells was measured by a fluorometric cell viability assay using Resazurin (Promocell GmbH). Cells were seeded in triplicates in 96-well plates at a density of 4 × 103 cells per well in 100 μL 24 h prior to treatment. To assess the cytotoxicity, cells were treated with increasing concentrations of compounds for 48 h. For phototoxicity studies, cells were treated with increasing concentrations of the compounds for 4 h only. After that, the medium was removed and replaced by complete medium prior to 10 min UV-A irradiation (30 kJ m−2). Cells were then returned to the incubator for 48 h. After incubation, the medium was replaced by 100 μL complete medium containing resazurin (0.2 mg mL−1 final concentration). Upon 4 h of incubation at 37 °C, the fluorescence of the highly red fluorescent resorufin product was quantified at 590 nm emission with 540 nm excitation wavelength using a SpectraMax M5 microplate reader.In vitro fluorescence evaluation
Cellular localization of the luminescent rhenium complexes and bioconjugates was assessed by fluorescence microscopy. HeLa cells were grown on 18 mm Menzel-Glaser coverslips in 2 mL complete medium at a density of 1 × 105 cells per mL and incubated for 1 or 2 h with Rhenium complexes at their IC50 or at 100 μM for nontoxic complexes. Cells were fixed in a 4% formaldehyde solution in PBS and mounted on slides for viewing by confocal microscopy using a CLSM Leica SP5 microscope. The rhenium complexes were excited at 405 nm and the emission above 420 nm was recorded. For living cell imaging, cells were grown on 35 mm Cellview glass bottom dishes (Greiner), washed and kept in 1× PBS prior to imaging using an Olympus IX 81 motorized inverted microscope (Olympus, Hamburg, Germany) equipped with a 60× oil-immersion lens and a digital camera. The Rhenium complex was visualized using the Cy3 filter set of the Olympus microscope (ex., 550 nm; em., 570 nm).Acknowledgements
This work was supported by the Swiss National Science Foundation (professorship no. PP00P2_133568 and research grant no. 200021_129910 to G.G.), the University of Zurich (G.G. and S.F.), the Stiftung für Wissenschaftliche Forschung of the University of Zurich (G.G. and S.F.), the Novartis Jubilee Foundation (G.G. and R.R.), the Stiftung zur Krebsbekämpfung (S.F.), the Huggenberger-Bischoff Stiftung (S.F.) and the University of Zurich Priority Program (S.F.).Notes and references
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2010, 54, 3–25 CrossRef PubMed
and references therein.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS
- P. C. A. Bruijincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef PubMed
- G. Gasser and N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 16, 84–91 CrossRef CAS PubMed
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC
-
G. Jaouen and N. Metzler-Nolte, Medicinal Organometallic Chemistry, Springer-Verlag, 2010 Search PubMed
- J. Zhang, J. J. Vittal, W. Henderson, J. R. Wheaton, I. H. Hall, T. S. A. Hor and Y. K. Yan, J. Organomet. Chem., 2002, 650, 123–132 CrossRef CAS
- W. Wang, Y. K. Yan, T. S. A. Hor, J. J. Vittal, J. R. Wheaton and I. H. Hall, Polyhedron, 2002, 21, 1991–1999 CrossRef CAS
- M. D. Bartholoma, A. R. Vortherms, S. Hillier, B. Ploier, J. Joyal, J. Babich, R. P. Doyle and J. Zubieta, ChemMedChem, 2010, 5, 1513–1529 CrossRef PubMed
- D. Can, H. W. Peindy N'Dongo, B. Spingler, P. Schmutz, P. Raposinho, I. Santos and R. Alberto, Chem. Biodiversity, 2012, 9, 1849–1866 CAS
- A. W.-T. Choi, M.-W. Louie, S. P.-Y. Li, H.-W. Liu, B. T.-N. Chan, T. C.-Y. Lam, A. C.-C. Lin, S.-H. Cheng and K. K.-W. Lo, Inorg. Chem., 2012, 51, 13289–13302 CrossRef CAS PubMed
- M.-W. Louie, A. W.-T. Choi, H.-W. Liu, B. T.-N. Chan and K. K.-W. Lo, Organometallics, 2012, 31, 5844–5855 CrossRef CAS
- J. Ho, W. Y. Lee, K. J. T. Koh, P. P. F. Lee and Y.-K. Yan, J. Inorg. Biochem., 2013, 119, 10–20 CrossRef CAS PubMed
-
S. Dhar and S. J. Lippard, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2011, pp. 79–95 Search PubMed
- K. A. Stephenson, S. R. Banerjee, T. Besanger, O. O. Sogbein, M. K. Levadala, N. McFarlane, J. A. Lemon, D. R. Boreham, K. P. Maresca, J. D. Brennan, J. W. Babich, J. Zubieta and J. F. Valliant, J. Am. Chem. Soc., 2004, 126, 8598–8599 CrossRef CAS PubMed
- S. James, K. P. Maresca, J. W. Babich, J. F. Valliant, L. Doering and J. Zubieta, Bioconjugate Chem., 2006, 17, 590–596 CrossRef CAS PubMed
- S. R. Banerjee, J. W. Babich and J. Zubieta, Inorg. Chim. Acta, 2006, 359, 1603–1612 CrossRef CAS PubMed
- K. A. Stephenson, L. C. Reid, J. Zubieta, J. W. Babich, M.-P. Kung, H. F. Kung and J. F. Valliant, Bioconjugate Chem., 2008, 19, 1087–1094 CrossRef CAS PubMed
- N. Viola-Villegas, A. E. Rabideau, J. Cesnavicious, J. Zubieta and R. P. Doyle, ChemMedChem, 2008, 3, 1387–1394 CrossRef CAS PubMed
- P. Schaffer, J. A. Gleave, J. A. Lemon, L. C. Reid, L. K. K. Pacey, T. H. Farncombe, D. R. Boreham, J. Zubieta, J. W. Babich, L. C. Doering and J. F. Valliant, Nucl. Med. Biol., 2008, 35, 159–169 CrossRef CAS PubMed
- N. Viola-Villegas, A. E. Rabideau, M. Bartholoma, J. Zubieta and R. P. Doyle, J. Med. Chem., 2009, 52, 5253–5261 CrossRef CAS PubMed
- G. Gasser, A. Pinto, S. Neumann, A. M. Sosniak, M. Seitz, K. Merz, R. Heumann and N. Metzler-Nolte, Dalton Trans., 2012, 41, 2304–2313 RSC
- G. Gasser, S. Neumann, I. Ott, M. Seitz, R. Heumann and N. Metzler-Nolte, Eur. J. Inorg. Chem., 2011, 36, 5471–5478 CrossRef
- A. J. Amoroso, M. P. Coogan, J. E. Dunne, V. Fernandez-Moreira, J. B. Hess, A. J. Hayes, D. Lloyd, C. Millet, S. J. A. Pope and C. Williams, Chem. Commun., 2007, 3066–3068 RSC
- K. K.-W. Lo, K. Y. Zhang and S. P.-Y. Li, Eur. J. Inorg. Chem., 2011, 3551–3568 CrossRef CAS
- K. K.-W. Lo, A. W.-T. Choi and W. H.-T. Law, Dalton Trans., 2012, 41, 6021–6047 RSC
- M. Patra and G. Gasser, ChemBioChem, 2012, 13, 1232–1252 CrossRef CAS PubMed
- F. L. Thorp-Greenwood, R. G. Balasingham and M. P. Coogan, J. Organomet. Chem., 2012, 714, 12–21 CrossRef CAS PubMed
- S. R. Banerjee, P. Schaffer, J. W. Babich, J. F. Valliant and J. Zubieta, Dalton Trans., 2005, 3886–3897 RSC
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Photochem. Photobiol., 2009, 85, 1053–1074 CrossRef CAS PubMed
- Y. Gao, S. Sun and K. Han, Spectrochim. Acta, Part A, 2009, 71, 2016–2022 CrossRef PubMed
- J. Liu and W. Jiang, Dalton Trans., 2012, 41, 9700–9707 RSC
- Y. Zhao, G. M. Roberts, S. E. Greenough, N. J. Farrer, M. J. Paterson, W. H. Powell, V. G. Stavros and P. J. Sadler, Angew. Chem., Int. Ed., 2012, 51, 11263–11266 CrossRef CAS PubMed
- A. A. Abdel-Shafi, J. L. Bourdelandeb and S. S. Ali, Dalton Trans., 2007, 2510–2516 RSC
- A. Kastl, S. Dieckmann, K. Wähler, T. Völker, L. Kastl, A. L. Merkel, A. Vultur, B. Shannan, K. Harms, M. Ocker, W. J. Parak, M. Herlyn and E. Meggers, ChemMedChem, 2013, 8, 924–927 CrossRef CAS PubMed
- L. Raszeja, A. Maghnouj, S. Hahn and N. Metzler-Nolte, ChemBioChem, 2011, 12, 371–376 CrossRef CAS PubMed
- A cysteine amino acid was added to the N-terminus of the peptide to allow further coupling with different biomolecules. This work will be reported in due course. Importantly, no S–S bridge formation was observed by LC-MS in this study.
- D. Kalderon, B. L. Kalderon, W. Roberts, A. Richardson and D. Smith, Cell, 1984, 39, 499–509 CrossRef CAS
- R. R. Allison and K. Moghissi, Photodiagn. Photodyn. Ther., 2013 DOI:10.1016/j.pdpdt.2013.1003.1011
- R. J. Fiel, N. Datta-Gupta, E. H. Mark and J. C. Howard, Cancer Res., 1981, 41, 3543–3545 CAS
- J. Cadet, M. Berger, C. Decarroz, J. R. Wagner, J. E. van Lier, Y. M. Ginot and P. Vigny, Biochimie, 1986, 68, 813–834 CrossRef CAS
- E. Garcìa Garayoa, D. Rüegg, P. Bläuenstein, M. Zwimpfer, I. U. Khan, V. Maes, A. Blanc, A. G. Beck-Sickinger, D. A. Tourwé and P. A. Schubiger, Nucl. Med. Biol., 2007, 34, 17–28 CrossRef PubMed
- E. Garcia Garayoa, C. Schweinsberg, V. Maes, D. Rüegg, A. Blanc, P. Bläuenstein, D. A. Tourwé, A. G. Beck-Sickinger and P. A. Schubiger, J. Nucl. Med. Mol. Imaging, 2007, 51, 42–50 CAS
- I. Kraljić and S. E. Mohsni, Photochem. Photobiol., 1978, 28, 577–581 CrossRef
- A. A. Krasnovsky Jr., Membr. Cell. Biol., 1998, 12, 665–690 CAS
- E. Oliveros, P. Suardi Murasecco, T. Aminian-Saghafi, A. M. Braun and H.-J. Hansen, Helv. Chim. Acta, 1991, 74, 79–90 CrossRef CAS
- S. Nonell, Afinidad, 1993, 50, 445–448 CAS
- C. Marti, O. Jürgens, O. Cuenca, M. Casals and S. Nonell, J. Photochem. Photobiol., A, 1996, 97, 11–18 CrossRef CAS
- F.-M. Boisvert, J. van Koningsbruggen, A. Navascués and A. Lamond, Nat. Rev. Mol. Cell Biol., 2007, 8, 574–585 CrossRef CAS PubMed
- E. Emmott and J. A. Hiscox, EMBO Rep., 2009, 10, 231–238 CrossRef CAS PubMed
- Z. Sheng, J. A. Lewis and W. J. Chirico, J. Biol. Chem., 2004, 279, 40153–40160 CrossRef CAS PubMed
- J. Boyne and A. Whitehouse, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15190–15195 CrossRef CAS PubMed
- M. A. Hahn and D. J. Marsh, FEBS Lett., 2007, 581, 5070–5074 CrossRef CAS PubMed
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2009, 131, 8738–8739 CrossRef CAS PubMed
- S. Boulon, B. J. Westman, S. Hutten, F. M. Boisvert and A. I. Lamond, Mol. Cell, 2010, 40, 216–227 CrossRef CAS PubMed
- A. R. Vortherms, A. R. Kahkoska, A. E. Rabideau, J. Zubieta, L. L. Andersen, M. Madsen and R. P. Doyle, Chem. Commun., 2011, 47, 9792–9794 RSC
- H. Yang, H. Cai, L. Wan, S. Liu, S. Li, J. Cheng and X. Lu, PLoS One, 2013, 8, e57358 CAS
- R. T. Jensen, J. F. Battey, E. R. Spindel and R. V. Benya, Pharmacol. Rev., 2008, 60, 1–60 CrossRef CAS PubMed
- P. Bednarski, R. Bednarski, M. Grünert, A. Zielzki, F. Wellner, P. Mackay and P. Sadler, Chem. Biol., 2006, 13, 61–67 CrossRef CAS PubMed
- P. Heringova, J. Woods, F. S. Mackay, J. Kasparkova, P. J. Sadler and V. Brabec, J. Med. Chem., 2006, 49, 7792–7798 CrossRef CAS PubMed
- N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. Mackay and P. J. Sadler, Angew. Chem., Int. Ed., 2010, 49, 8905–8908 CrossRef CAS PubMed
- K. L. Ciesienski, L. M. Hyman, D. T. Yang, K. L. Haas, M. G. Dickens, R. J. Holbrook and K. J. Franz, Eur. J. Inorg. Chem., 2010, 2010, 2224–2228 CrossRef
- J. Mlcouskova, J. Stepankova and V. Brabec, J. Biol. Inorg. Chem., 2012, 17, 891–898 CrossRef CAS PubMed
- J. S. Butler, J. A. Woods, N. J. Farrer, M. E. Newton and P. J. Sadler, J. Am. Chem. Soc., 2012, 134, 16508–16511 CrossRef CAS PubMed
- J. K. Barton and A. L. Raphael, J. Am. Chem. Soc., 1984, 106, 2466–2468 CrossRef CAS
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed
-
I. E. Kochevar and R. W. Redmond, in Methods in Enzymology, ed. L. Packer and H. Sies, Academic Press, 2000, vol. 319, ch. 2, pp. 20–28 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3dt51817e |
| ‡ These authors have contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2014 |