 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Narcissistic self-sorting vs. statistic ligand shuffling within a series of phenothiazine-based coordination cages†
Marina
Frank
,
Lennard
Krause
,
Regine
Herbst-Irmer
,
Dietmar
Stalke
and
Guido H.
Clever
*
Institut für Anorganische and Chemie, Georg-August Universität Göttingen, Tammannstr. 4, 37077, Göttingen, Germany. E-mail: gclever@gwdg.de
First published on 23rd January 2014
Abstract
Previously, we introduced a series of anion-binding interpenetrated double-cages based on phenothiazine and its mono- and di-S-oxygenated derivatives. Here, we complete the structural comparison of the three related assemblies by an X-ray single crystal analysis of the sulfone derivative. We further show that the three palladium cages coexist in solution upon post-assembly mixing due to the very slow ligand exchange whereas treatment of binary mixtures of the corresponding ligands with Pd(II) leads to the formation of mixed cages comprising a statistical ligand distribution. In contrast, mixtures of one of these ligands with a shorter ligand derivative lead to narcissistic self-assembly into a double-cage and a coexisting small monomeric cage, regardless of the order of mixing and Pd(II) addition.
Introduction
Self-assembled coordination cages1 have found application in various areas such as selective guest binding,2 stabilisation of reactive compounds,3 catalysis,4 redoxactive5 and light switchable materials.6 We have recently reported on a number of interpenetrated double-cages [Pd4L8] containing concave bis-monodentate pyridyl ligands L showing an allosteric anion binding behaviour.7 We further showed that small structural changes modulate the binding affinity of these cages for different halide anions8 whereas more extensive structural changes lead to a selectivity switch towards larger oxoanions such as perrhenate.9Among the reported structures, three closely related double-cage species based on the heterocycle phenothiazine and its mono- and di-oxygenated derivatives were shown to have a redox chemistry that makes them promising candidates for application in molecular electronics and organic photovoltaics. The molecular structures of the first two cages were reported by us before.10
Here, we deliver the single-crystal structure of the di-S-oxygenated double-cage, thus allowing for a comprehensive comparison of the structure–function relationships of the three cage derivatives. Furthermore, we extended our examination of the cage assembly process to mixtures of phenothiazine-based ligands and their corresponding cages in order to study the role of kinetic effects11 on the double-cage assembly. In addition, we show a phenomenon of narcissistic self-sorting12 when a shorter ligand derivative, not capable of forming a double-cage, is brought into the system.
Results and discussion
The chemical structures of the long ligands L1–3 and the shorter derivative L4 are depicted in Fig. 1a. All three long ligands were previously shown to quantitatively yield interpenetrated double-cages [Pd4L1–38] upon treatment with the metal source [Pd(CH3CN)4](BF4)2 in acetonitrile solution.10 The structures were found to contain BF4− counter anions encapsulated in their two outer pockets and one inside their central cavity. We further showed that the two outer anions could be replaced by tightly binding halides in an allosteric fashion.8 In contrast, the shorter ligand L4 was shown to assemble into a monomeric cage [Pd2L44] that cannot undergo dimerization for steric reasons.10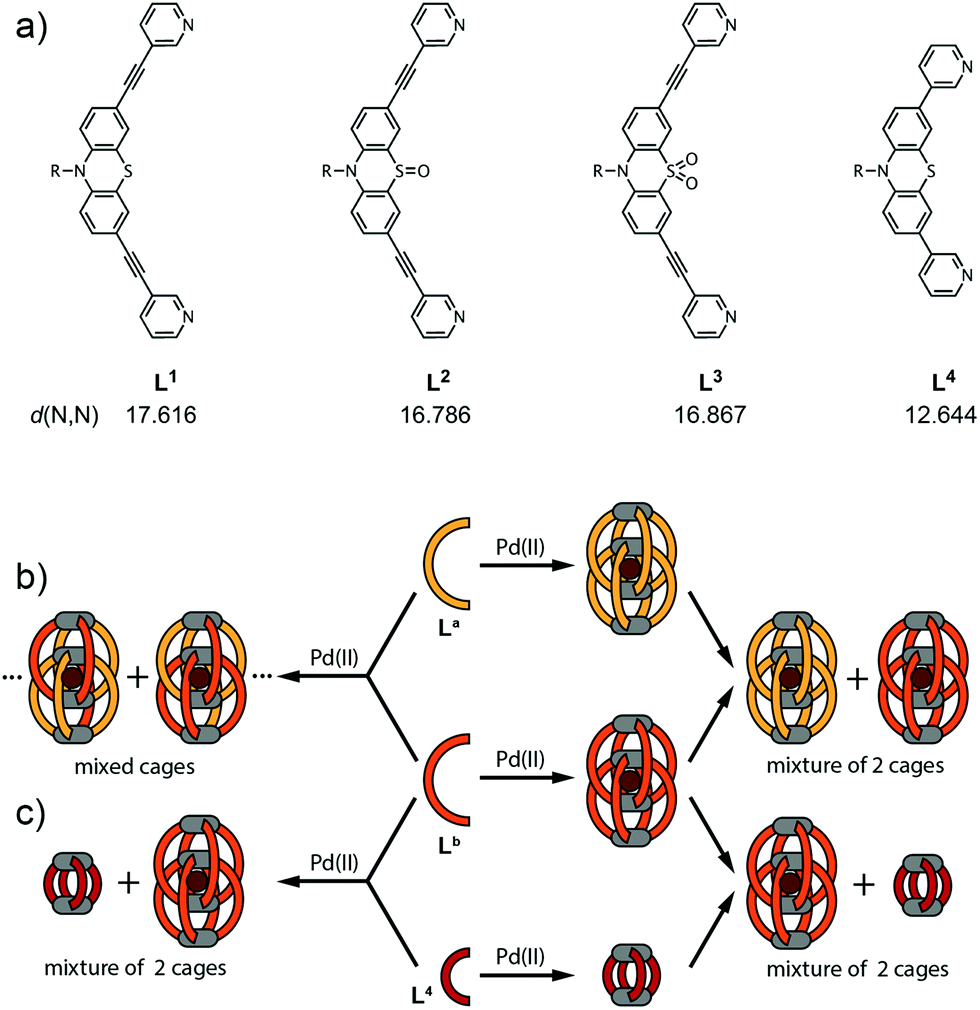 | ||
| Fig. 1 (a) Chemical structures of the cage-forming ligands L1–4. The given N,N-distances are based on double-cage X-ray structures for L1–3 and a semiempirical PM6 model for the monomeric cage based on ligand L4. (b) For binary systems containing two different long ligands La + Lb (La,Lb = L1–3; La ≠ Lb) the outcome of the self-assembly gives mixed cages showing a statistical ligand distribution when the ligands are mixed prior to the addition of palladium. In contrast, combining two preassembled double-cages [Pd4La8] + [Pd4Lb8] leads to a mixture of coexisting homogeneous structures between which ligand exchange is tremendously slowed down. (c) For binary systems containing one long ligand (Lb = L1–3) and the short ligand L4, the outcome of the self-assembly is independent of the order of mixing of the components. In both cases, this system shows narcissistic self-sorting behaviour to give [Pd4Lb8] + [Pd2L44]. | ||
The X-ray structural data for coordination cages [Pd4L18] and [Pd4L28] showed that both are interpenetrated dimers sharing a common topology but differing in their shape with regard to the Pd–Pd distances, ligand bending and pocket sizes.10 Here, we report on the single crystal X-ray structure of the third member of the three related cages, [Pd4L38], thus filling a gap in the systematic structural comparison. Table 1 lists the crystallographic parameters for the [Pd4L38] double cage. After refinement of the cage scaffold, all necessary BF4− and Cl− anions could be found in the Fourier difference map. The BF4− anions are located on special positions. All but one are exhibiting disorder and are not obeying the crystallographic symmetry. They were modeled using distance restraints with lowered standard deviations. Diffuse residual electron density was observed in the crystal voids. Heavily disordered lattice solvent (ethanol) could be modeled into these voids but a ‘squeezed' model was superior. (see the ESI†).
| CCDC number | 972251 |
| Empirical formula | C512H432B14Cl2F56N48O32Pd8S16 |
| Formula weight | 8 × 1302.43 g mol−1 |
| Temperature | 100(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Tetragonal |
| Space group | P4/nnc |
| Unit cell dimensions | a = b = 21.991(2) Å |
| c = 31.800(3) Å | |
| Volume | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 378(2) Å3 378(2) Å3 |
| Z | 8 |
| Density (calculated) | 1.125 Mg m−3 |
| Absorption coeff. | 0.363 mm−1 |
| F(000) | 5328 |
| Crystal size | 0.09 × 0.08 × 0.06 mm3 |
| Theta range for data collection | 1.126 to 23.837°. |
| Index ranges | −25 ≤ h ≤ 24, −24 ≤ k ≤ 24, −36 ≤ l ≤ 36 |
| Reflections collected | 166252 |
| Independent refl. | 5937 [Rint = 0.0583] |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 5937/1104/682 |
| Goodness-of-fit on F2 | 1.058 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0797, wR2 = 0.2565 |
| R indices (all data) | R 1 = 0.1049, wR2 = 0.3011 |
| Largest diff. peak and hole | 1.429 and −0.662 e Å−3 |
Fig. 2a and b show the double cage structure from two different perspectives. The topology of this cage assembly is in agreement with the structures reported for [Pd4L18] and [Pd4L28]. It is interesting to note that the examined crystals of [Pd4L38] were found to contain one equivalent of chloride counter anions per double-cage, positioned on the Pdn-axis between the double-cages, which was not observed in the previously reported solid states structures of [Pd4L18] and [Pd4L28]. Particularly surprising is the observation that the chloride anions (most probably being the result of a contamination) were not found in the outer two pockets of the double-cage structure but in the space outside the linearly aligned cages, which is in clear contrast to the solution behaviour.8 A plausible explanation is given as follows: in solution, the chloride anions are tightly bound inside the outer two pockets by electrostatic interactions from both Pd(pyridine)4 planes lining the cavities. As we have demonstrated previously,7,8 chloride binding is accompanied by a compression of the double cage structure along the Pd4-axis thus bringing the Pd centres of the outer pockets closer together. This allosteric effect helps to accommodate the chloride anions in the outer pockets because they are smaller than the tetrafluoroborate counter anions. As a consequence of this structural change, however, an energetic penalty has to be paid due to the enlargement of the central pocket because here the cationic metal centres are moved away from the contained tetrafluoroborate anion. Still, the incorporation of the chloride inside the outer pockets is the favoured process in solution, whereas chloride binding to the outer faces of the double cage is occurring with much lower affinity. The situation seems to change in the solid state structure due to the linear alignment of the double cages. Here, the cages can adopt an optimal spacing for the tight binding of a chloride anion between them (6.54 Å). Most importantly, binding of the chloride anion between the cages does not come with the mentioned energetic drawback of having to enlarge the cage's central cavity. For a graphical comparison of the solid state packings of the three cage derivatives see the ESI.†
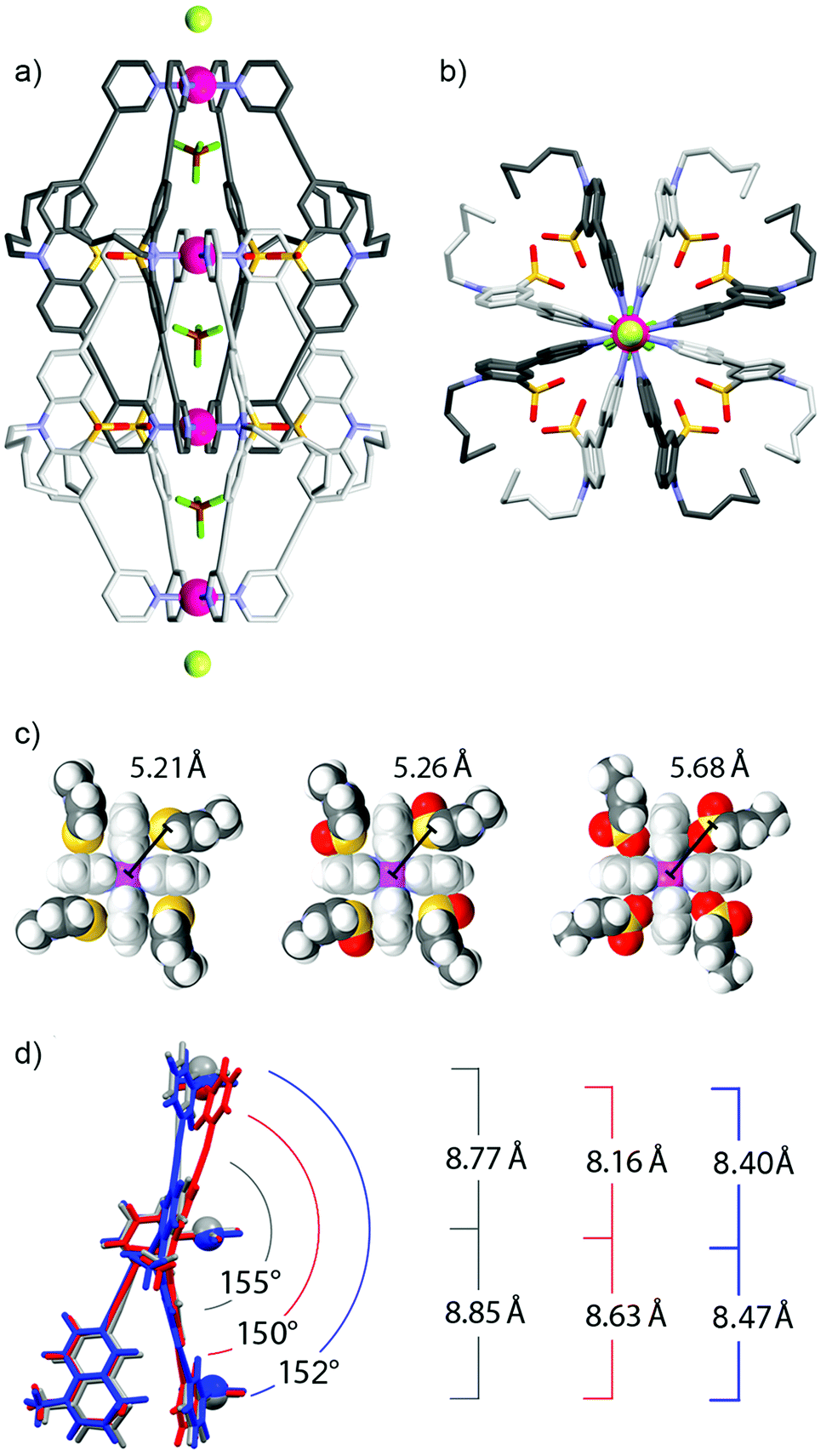 | ||
| Fig. 2 (a) Side view and (b) view along the Pd4-axis of the single crystal X-ray structure of the interpenetrated double-cage [Pd4L38] (only the three internal BF4− and the two external Cl− anions are depicted. Other anions and solvent molecules are omitted for clarity. C: grey, N: blue, O: red, S: yellow, B: brown, F: dark green, Cl: light green, Pd: purple). (c) Comparison of the space-filling top views of one of the inner Pd(pyridine)4 planes of double-cages [Pd4L18] (left), [Pd4L28] (middle) and [Pd4L38] (right). (d) Partial overlay of sub-structures of [Pd4L18] (grey), [Pd4L28] (red) and [Pd4L38] (blue) indicating the changes in the ligand structures and Pd–Pd distances. | ||
A comparison of all three X-ray structures reveals how the sulfur atoms (and the attached oxygen atoms) of the interpenetrating ligands of one cage subunit are positioned relative to the inner Pd(pyridine)4-plane of the other cage subunit (Fig. 2c). Compared to the situation observed for [Pd4L18] (left) and [Pd4L28] (middle), the extra oxygen of [Pd4L38] (right) has to squeeze between the sulfur atom and the gap between two neighbouring pyridine rings, which moves the sulfur atom further away from the palladium centre. A further structural feature, which is interesting to compare between the three cage derivatives, is the ligand bending (as defined by the angle between both of the phenothiazines’ benzene rings). Fig. 2d shows that the attachment of one oxygen substituent to the sulfur decreases the bending angle from 155° (in [Pd4L18]) to 150° (in [Pd4L28]) and back to 152° (in [Pd4L38]) which can be explained by the steric demands of the lone pairs and oxygen substituents, respectively.
Since no single crystals suitable for X-ray crystallography were obtained for the monomeric cage [Pd2L44], its structure was calculated by an unconstrained geometry optimization on the PM6 semiempirical level of theory (charge: +4; spin-multiplicity: 1) in Gaussian 09.13Fig. 3 shows the resulting optimized structure.
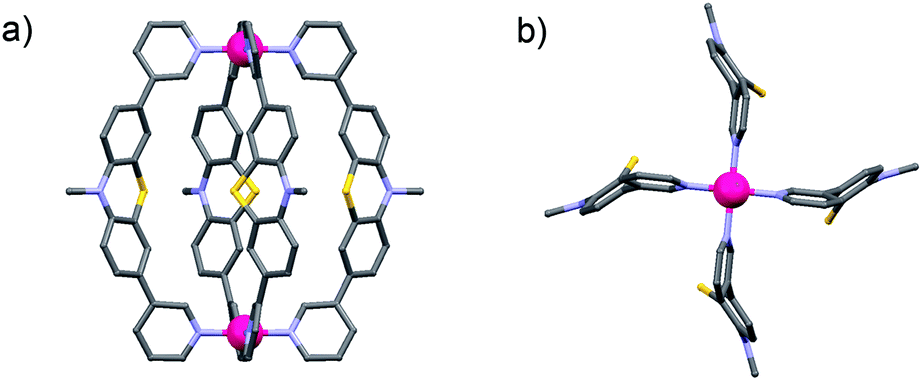 | ||
| Fig. 3 (a) Side view and (b) view along the Pd2-axis of a PM6 model of the monomeric cage [Pd2L44] (alkyl chains have been truncated and anions omitted). | ||
Having all three structures in hand, we next turned our focus to the examination of mixtures of the ligands and their corresponding cages. When two of the long ligands (named La and Lb in Fig. 1 with La,Lb = L1–3; La ≠ Lb) were mixed in equimolar ratio, a stoichiometric amount of Pd(II) cations added and the mixture heated to 70 °C overnight, a statistical mixture of interpenetrated double-cages [Pd4LamLb8−m] (m = 0–8) was obtained containing both ligands according to a binomial distribution. In contrast, when both cages [Pd4La8] and [Pd4Lb8] were preformed in separate vessels and combined subsequently, the reassembly into mixed-ligand cages was found to be extremely slow.
Fig. 4a shows the 1H NMR spectroscopic results of this experiment for ligands L1 and L3. The signals of both ligands (and corresponding double cages) are clearly distinguishable. The spectrum obtained after Pd(II) treatment of the ligand mixture unambiguously shows the pattern expected for an interpenetrated double-cage; it is however broadened as compared to the spectrum obtained for the mixture of the preformed cages.
 | ||
| Fig. 4 Comparison of 1H NMR spectra (300 MHz, 298 K, CD3CN) of the experiments described in Fig. 1 with a combination of (a) ligands L1 + L3 and (b) ligands L1 + L4. | ||
The latter spectrum does not undergo substantial changes upon prolonged heating of the sample. The ESI mass spectrometric analysis of both mixtures nicely reveals that Pd(II) addition after ligand mixing indeed leads to a binomial distribution (Fig. 5a) whereas mixing of the preformed double-cages does not (Fig. 5b). Apart from some peaks resulting from contaminating counter anions, the observed peak pattern of this sample is in very good agreement with the simulated pattern for a 1:1 mixture of [Pd4L18] + [Pd4L38].
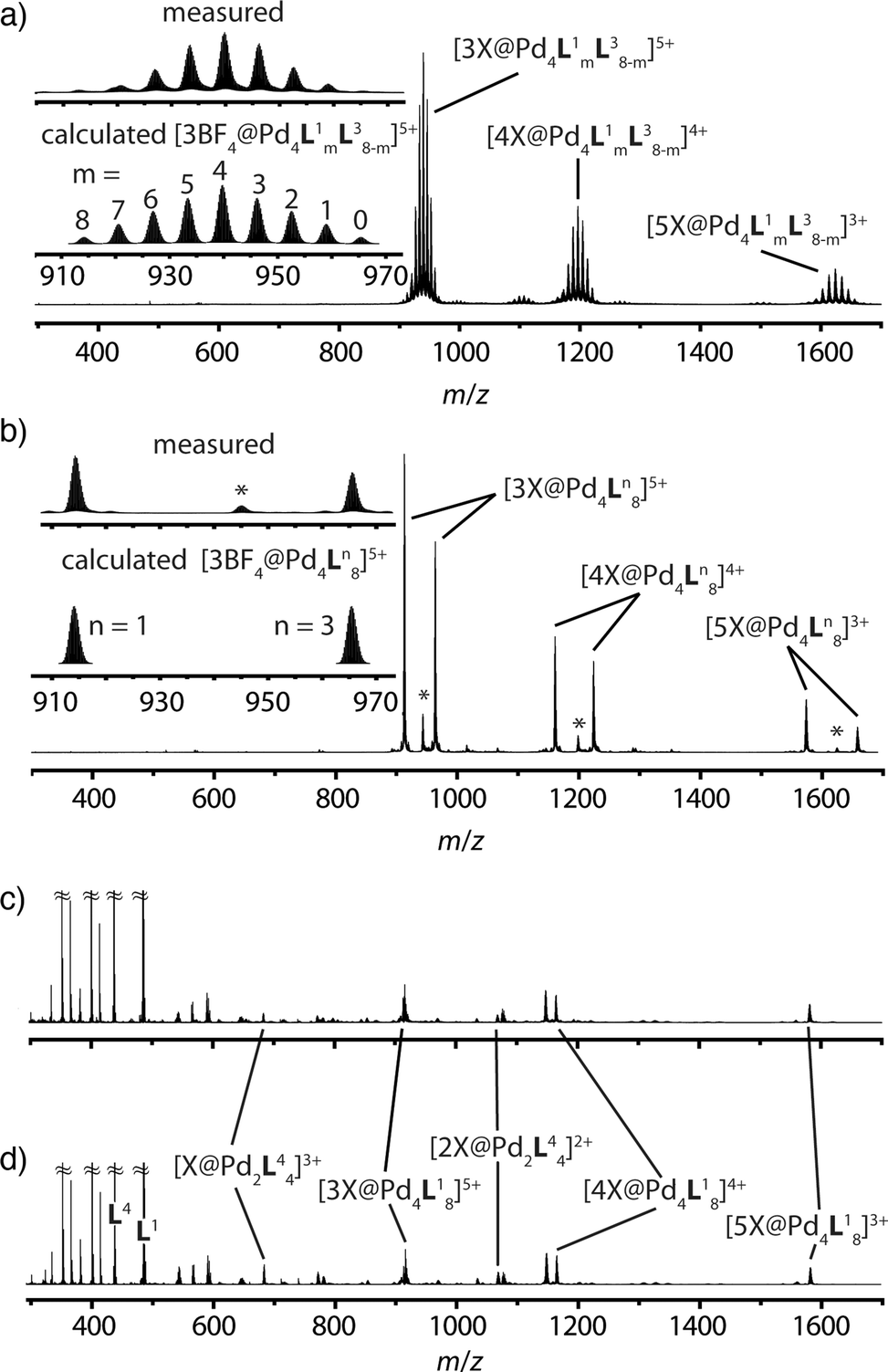 | ||
| Fig. 5 Comparison of ESI-TOF mass spectra (positive mode, CH3CN) of the experiments described in Fig. 1 showing (a) a statistical double-cage composition for the mixture of ligands L1 + L3 and subsequent Pd(II) addition and (b) non-exchanging species [Pd4L18] + [Pd4L38] for the mixture of preassembled double-cages (*corresponding double-cages containing other anions as contaminants; X = BF4− and/or other small anions). In contrast, the system L1 + L4 delivers essentially superimposable spectra showing a mixture of non-exchanging cages [Pd4L18] + [Pd2L44], both (c) for the mixture of ligands L1 + L4 and subsequent Pd(II) addition and (d) for the mixture of preassembled cages. | ||
Heating the double-cage mixture at 70 °C overnight did not result in significant formation of mixed-ligand cages.11a Prolonged heating, however, led to the slow occurrence of new peaks with low intensity in the ESI mass spectra showing the formation of the one-ligand-exchanged species [Pd4La7Lb1] and [Pd4La1Lb7] after 279 hours (Fig. 6). Even further heating for up to 26 days led to the rise of the species where more than one ligand is exchanged, thus indicating a very slow ligand exchange. In addition the intensity of the signals for the homogeneous double cages [Pd4L18] and [Pd4L38] were found to decrease after this time. Since the same procedure conducted with the mixed-ligand double-cage samples did not result in any significant deviation from the statistical distribution, we conclude that the latter state is the global thermodynamic minimum of the system.
 | ||
| Fig. 6 (Left) ESI-TOF mass spectra in positive mode of the solution of premixed ligands L1 and L3 (250 μL of a 2.8 mM solution for each ligand) after addition of 0.5 eq. [Pd(CH3CN)4](BF4)2 and heating in CD3CN at 70 °C. The spectra show that the statistical distribution of the ligands forming the double-cages [Pd4L1mL38−m] with m = 8–0 is not changing over time. (Right) The ESI-TOF mass spectra in positive mode of the solution after mixing the double cages [Pd4L18] and [Pd4L38] (250 μL of a 0.35 mM solution for each cage) and heating at 70 °C indicate a very slow, progressive ligand exchange process. Reaction times: (a) 22 h, (b) 114 h and (c) 279 h and (d) 615 h. *Denotes anion combinations with 2Cl− + BF4−; □ denotes anion combinations with NO3− + 2BF4−. | ||
Similar results were obtained for mixtures containing L1 + L2 and L2 + L3, although the NMR spectroscopic analyses involving ligand L2 were somewhat hampered by the previously reported signal broadening in the spectra of [Pd4L28] (see the ESI†).10
The picture changes when the short ligand L4 is brought into the system. Experiments involving one of the long ligands L1–3 plus L4 were found to always result in a mixture of the homogeneous double-cage [Pd4L1–38] and the small, monomeric cage [Pd2L44], regardless of the order of component addition (premixing of the ligands followed by Pd(II) vs. mixing of preassembled cages). This narcissistic self-sorting behaviour can be both seen in the 1H NMR spectroscopic examinations (Fig. 4b) and the ESI mass spectrometric analyses (Fig. 5c and d) for the combination L1 + L4 (for results of the other ligand combinations see the ESI†).
Conclusions
In summary, the successful X-ray structure determination of [Pd4L38] allows now for a systematic comparison between all three members of the family of interpenetrated cages based on phenothiazine. The effect of the extra oxygen substituent attached to each of the eight sulfur atoms in [Pd4L38] has essentially two structural implications: first, it leads to a displacement of the sulfur atoms of the interpenetrating ligands away from the inner Pd atom of the other cage substructure, and second, it influences the bending angle of the ligands with respect to the non-flat phenothiazine component. Both factors also have implications on the pocket sizes (Pd–Pd distances) and overall double-cage shapes.We further showed that binary mixtures of the long ligands L1–3 give a statistical distribution of mixed-ligand cages upon treatment with Pd(II). This is remarkable given the fact that all three ligands differ in their lengths, bending angle and steric situation around the sulfur atoms. Apparently, these discrepancies between the ligands are tolerated in the mixed cages, showing again the versatility of the [Pd4L8] structural motif. Heating a mixture of preassembled double-cages, however, only resulted in extremely slow ligand shuffling, thus indicating a strong kinetic barrier of ligand exchange once the completely assembled double-cages have been obtained. The substantial structural differences between the long ligands L1–3 and the much shorter derivative L4, however, led to a narcissistic self-sorting behaviour in binary mixtures giving the corresponding double-cage [Pd4L1–38] and the monomeric cage [Pd2L44].
Together, these results strengthen the knowledge about the structural features that govern the formation of self-assembled coordination cages from concave bispyridyl ligands. Furthermore, the observed signs of a strong kinetic influence on the double-cage assembly mechanism allow us to study self-assembled host–guest systems away from thermodynamic equilibrium in our following investigations.
Experimental
1H NMR spectra were recorded on a Bruker Avance-300 spectrometer (300 MHz) in CD3CN. ESI mass spectra were obtained using a Bruker micrOTOF mass spectrometer in positive mode. Samples were injected as acetonitrile solutions. All observed isotope patterns were in good agreement with the calculated ones.Synthesis of the cages10 and preparation of the solutions for the mixing experiments: cage compounds [Pd4L1–38] and [Pd2L44] were synthesized in quantitative yield by heating a mixture of the ligand L1–4 (2.8 μmol) in 930 μL CD3CN and a solution of [Pd(CH3CN)4](BF4)2 (1.4 μmol, 93 μL of a 15 mM solution in CD3CN) at 70 °C for 6 h to give a 0.35 mM solution of [Pd4L1–38] or a 0.7 mM solution of [Pd2L44], respectively. Mixing experiments (Fig. 1b, left side): A binary solution of two ligands was prepared by mixing the solution of ligand La (250 μL, 2.8 mM) and Lb (250 μL, 2.8 mM) in CD3CN. To this solution, [Pd(CH3CN)4](BF4)2 (0.75 μmol, 50 μL of a 15 mM solution in CD3CN) was added and the mixture was heated at 70 °C to give [Pd4LamLb8−m] with La,Lb = L1–3; La ≠ Lb; and m = 1–8.
Mixing experiment (Fig. 1b, right side): A binary solution of preassembled double cages was prepared in CD3CN by mixing 250 μL of each cage solution and heated at 70 °C.
X-ray crystallography: Yellow block shaped crystals of [Pd4L38] were grown from the solution of the acetonitrile by diffusion of ethanol. A suitable crystal of size 0.09 × 0.08 × 0.06 mm3 was selected from a batch of crystals using the X-TEMP 2 device.14 The crystal was mounted on top of glass fiber using inert perfluorinated polyether oil and placed in the cold gas stream of a Bruker Kryoflex 2 low-temperature device with a cold stream at 100(2) K on a Bruker D8 three circle diffractometer. The diffractometer was equipped with a SMART APEX II CCD detector and a rotating anode source (Mo-Kα; λ = 0.71073 Å). Data were collected in omega-scan mode at different detector 2θ angles ranging between 0° and 90°. The scan width was set to 0.5°.
Data integration was done with SAINT 8.30C.15 Each run with constant 2θ and φ settings was integrated using a separate orientation matrix. Data scaling and absorption correction were done with SADABS 2012/1.16 The space group was determined using XPREP.17 The structure was solved by direct methods using SHELXS-97.18 Refinement by full-matrix least-squares procedures was done with SHELXL-201319 within the SHELXLe-GUI.20 The hydrogen atoms were refined isotropically at calculated positions using a riding model with their Uiso values constrained to 1.5Ueq of their pivot atoms for terminal sp3 carbon atoms and 1.2 times for all other carbon atoms. All non-hydrogen atoms were refined with anisotropic displacement parameters. For the severely disordered solvent region the SQUEEZE routine of the PLATON program package was utilized (see ESI†).
Acknowledgements
M.F. thanks the Evonik Foundation for a Ph.D. fellowship. We thank the DFG (CL 489/2-1) and the Danish National Research Foundation (DNRF93) funded Center for Materials Crystallography (CMC) for financial support. We thank Dr Michael John for help with the NMR measurements and Dr Holm Frauendorf for the ESI measurements.References
- (a) M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161 RSC; (b) S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825 CrossRef CAS PubMed; (c) D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136 CrossRef CAS PubMed; (d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (e) T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476 RSC.
- (a) N. Gimeno and R. Vilar, Coord. Chem. Rev., 2006, 250, 3161 CrossRef CAS PubMed; (b) J. L. Sessler, P. Gale, W.-S. Cho and S. J. Rowan, Anion Receptor Chemistry (Monographs in Supramolecular Chemistry), Royal Society of Chemistry, Cambridge, 2006 Search PubMed; (c) S. O. Kang, J. M. Llinares, V. W. Day and K. Bowman-James, Chem. Soc. Rev., 2010, 39, 3980 RSC.
- (a) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed; (b) D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2006, 45, 745 CrossRef CAS PubMed; (c) M. Kawano, Y. Kobayashi, T. Ozeki and M. Fujita, J. Am. Chem. Soc., 2006, 128, 6558 CrossRef CAS PubMed.
- (a) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed; (b) Molecular Encapsulation: Organic Reactions in Constrained Systems, ed. U. H. Brinker, J. Mieusset, Wiley, Hoboken, 2010 Search PubMed.
- S. Bivaud, J.-Y. Balandier, M. Chas, M. Allain, S. Goeb and M. Sallé, J. Am. Chem. Soc., 2012, 134, 11968 CrossRef CAS PubMed.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319 CrossRef CAS PubMed.
- (a) S. Freye, J. Hey, A. Torras-Galán, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191 CrossRef CAS PubMed; (b) S. Freye, D. M. Engelhard, M. John and G. H. Clever, Chem.–Eur. J., 2013, 19, 2114 CrossRef CAS PubMed; (c) J. M. Dieterich, G. H. Clever and R. A. Mata, Phys. Chem. Chem. Phys., 2012, 14, 12746 RSC.
- M. Frank, J. M. Dieterich, S. Freye, R. A. Mata and G. H. Clever, Dalton Trans., 2013, 42, 15906 RSC.
- S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476 CrossRef CAS PubMed.
- M. Frank, J. Hey, I. Balcioglu, Y.-S. Chen, D. Stalke, T. Suenobu, S. Fukuzumi, H. Frauendorf and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 10102 CrossRef CAS PubMed.
- For other examples of kinetic effects in the self-assembly of coordination cages see: (a) S. Sato, Y. Ishido and M. Fujita, J. Am. Chem. Soc., 2009, 131, 6064 CrossRef CAS PubMed; (b) Y. Sakata, S. Hiraoka and M. Shionoya, Chem.–Eur. J., 2010, 16, 3318 CrossRef CAS PubMed; (c) O. Chepelin, J. Ujma, P. E. Barran and P. J. Lusby, Angew. Chem., Int. Ed., 2012, 51, 4194 CrossRef CAS PubMed; (d) H. Kumari, A. V. Mossine, S. R. Kline, C. L. Dennis, D. A. Fowler, S. J. Teat, C. L. Barnes, C. A. Deakyne and J. L. Atwood, Angew. Chem., Int. Ed., 2012, 51, 1452 CrossRef CAS PubMed.
- For reviews and selected examples see: (a) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784 CrossRef CAS PubMed; (b) M. Lal Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651 RSC; (c) R. Krämer, J.-M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394 CrossRef; (d) D. L. Caulder and K. N. Raymond, Angew. Chem., Int. Ed. Engl., 1997, 36, 1440 CrossRef CAS; (e) R. Pinalli, V. Cristini, V. Sottili, S. Geremia, M. Campagnolo, A. Caneschi and E. Dalcanale, J. Am. Chem. Soc., 2004, 126, 6516 CrossRef CAS PubMed; (f) B. Brusilowskij, E. V. Dzyuba, R. W. Troff and C. A. Schalley, Chem. Commun., 2011, 47, 1830 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef.
- Bruker SAINT v8.30C, Madison, 2013 Search PubMed.
- G. M. Sheldrick, SADABS 2012/1, Göttingen, 2012 Search PubMed.
- G. M. Sheldrick, XPREP Version 2012/1 for Windows, Göttingen, 2005 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 972251. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt53243g |
| This journal is © The Royal Society of Chemistry 2014 |