
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modified bibenzimidazole ligands as spectator ligands in photoactive molecular functional Ru-polypyridine units? Implications from spectroscopy†
Julia
Meyer-Ilse
ab,
Stefan
Bauroth
ab,
Maximilian
Bräutigam
ab,
Michael
Schmitt
a,
Jürgen
Popp
ab,
Rainer
Beckert
c,
Nils
Rockstroh
d,
T. David
Pilz
d,
Katharina
Monczak
e,
Frank W.
Heinemann
d,
Sven
Rau
de and
Benjamin
Dietzek
*ab
aInstitute of Physical Chemistry and Jena Center for Soft Matter, Friedrich-Schiller University Jena, Helmholtzweg 4, 07743 Jena, Germany. E-mail: benjamin.dietzek@uni-jena.de; Fax: +49 (0)3641 206399; Tel: +49 (0)3641 206332
bLeibniz Institute of Photonic Technology (IPHT), Albert-Einstein-Straße 9, 07745 Jena, Germany. E-mail: benjamin.dietzek@ipht-jena.de; Fax: +49 (0)3641 206399; Tel: +49 (0)3641 206332
cInstitute of Organic Chemistry and Macromolecular Chemistry, Humboldtstr. 8, 07743 Jena, Germany
dDepartment of Chemistry and Pharmacy, Friedrich-Alexander University Erlangen-Nuremberg, Egerlandstr. 3, 91058 Erlangen, Germany
eInstitute of Inorganic Chemistry, University Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany
First published on 22nd August 2014
Abstract
The photophysical properties of Ruthenium-bipyridine complexes bearing a bibenzimidazole ligand were investigated. The nitrogens on the bibenzimidazole-ligand were protected, by adding either a phenylene group or a 1,2-ethandiyl group, to remove the photophysical dependence of the complex on the protonation state of the bibenzimidazole ligand. This protection results in the bibenzimidazole ligand contributing to the MLCT transition, which is experimentally evidenced by (resonance) Raman scattering in concert with DFT calculations for a detailed mode assignment in the (resonance) Raman spectra.
Introduction
Bibenzimidazoles (bbims) have been proven to be versatile structures in the construction of environmentally sensitive photoactive Ru-complexes.1–6 One attractive aspect pointed out in the context of designing molecular photocatalysts and DNA-intercalating luminescent complexes is the fact that bbims are non-electron accepting ligands. That is, when coordinated to, e.g., ruthenium ions together with, e.g., bipyridine, phenanthroline or related ligands, the bbim ligands do not partake in the metal-to-ligand charge transfer transition (MLCT).7–11 This feature renders bbim ligands interesting units in photoactive molecular devices, in which absorption of light initiates a vectorial electron transfer across the molecular structure. Here, the use of bbims as terminal ligands, i.e., ligands directed to the opposite direction of the envisioned electron transfer acceptor, offers a promising option to enhance the directionality and efficiency of intramolecular electron transfer from the photoactive metal-unit to an acceptor unit. However, the non-coordinating nitrogen atoms of the bbim can be protonated/deprotonated, which in turn changes the photophysical properties of the complexes, despite the fact that the latter are dominated by the non-bbim ligands.7 In this context, literature reports show that upon complete deprotonation of bbim-Ru-complexes,12 a loss of luminescence and a bathochromic shift of absorption maxima by up to 100 nm can occur.13 While this behaviour is essential for molecular sensors, e.g., molecular devices, whose photophysical properties are altered upon a change in the pH-value of the solution, it is unwanted in molecular photocatalysts as, e.g., the pH-value of a reaction solution can change quite drastically during the course of a catalytic reaction, which in turn would significantly impact the photoinduced molecular reaction steps determining the function of the catalyst itself. Therefore, bbim-structures have been designed in which the non-coordinating nitrogens are protected either by phenylene14 or an 1,2-ethandiyl group. To investigate the prospects of using these ligands as environmentally non-responsive spectator ligands in photoactive complexes, heteroleptic ruthenium complexes incorporating these new ligands and well-established tert-butyl-2,2′-bipyridine (tbbpy) ligands were synthesized and characterized spectroscopically. This study focuses specifically on complexes based on the (tbbpy)2Ru(tmbbimH2)(PF6)2 (RuBib, tbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine, tmbbimH2 = 5,5′,6,6′-tetramethyl-2,2′-bibenzimidazole). In particular, the work at hand focuses on the spectroscopic characterization of [Ru(tbbpy)2(dmbbimPh)](PF6)2 (RuBibPh), in which a phenylene group is attached to the bbim moiety, and [Ru(tbbpy)2(tmbbimEt)](PF6)2 (RuBibEt), in which an additional 1,2-ethandiyl group protects the imidazole nitrogens (see Fig. 1a and b, respectively). Thus, protection is accomplished by an aromatic group for RuBibPh and an aliphatic group in RuBibEt.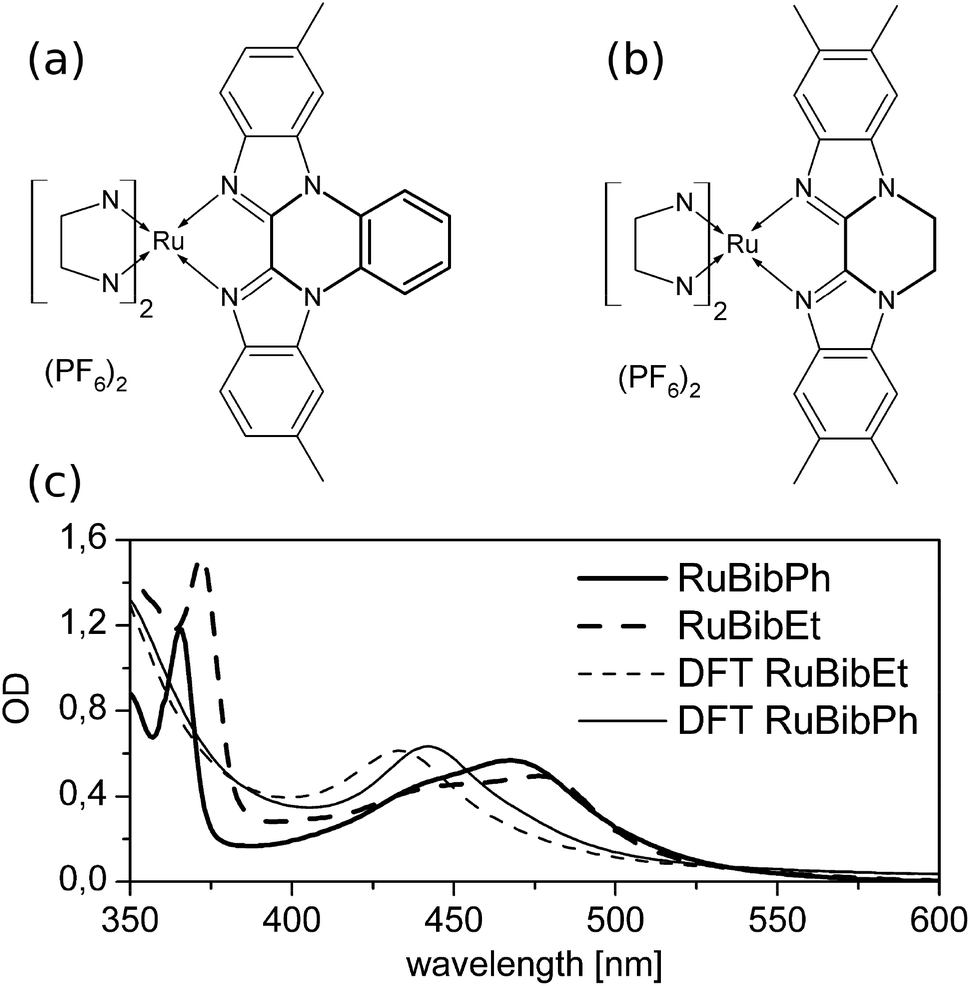 | ||
| Fig. 1 Molecular structures of the two ruthenium complexes with protected bbim ligands where the NN ligand represents the tbbpy: (a) RuBibPh and (b) RuBibEt. (c) UV-Vis spectra of the two complexes measured in DCM and their calculated absorption spectra. | ||
Experimental
Synthesis and characterization
Ruthenium trichloride and tert-butyl-pyridine were of reagent grade and used as supplied. Ru(4,4′-di-tert-butyl-2,2′-bipyridine)2Cl2,15 4,4′-di-tert-butyl-2,2′-bipyridine,16,17 1,1′-diphenylen-6,6′-dimethyl-2,2′-bibenzimidazole,14 5,5′,6,6′-tetramethyl-2,2′-bibenzimidazole18 and 1,1′-dimethylen-5,5′,6,6′-tetramethyl-2,2′-bibenzimidazole19 were prepared according to literature procedures.The NMR spectra were recorded on a Bruker 400 MHz spectrometer. The mass spectra were recorded with a SSQ 710 spectrometer (Finnigan MAT). Elemental analyses were performed on a Carlo Erba EA 1108. Steady state absorption spectra were obtained using a JASCO Spectrometer V-670. The CVs were recorded on a μAutolab Type 3 Potentiostat/Galvanostat.‡ Intensity data for the compounds were collected on a Bruker Smart APEX2 diffractometer using graphite-monochromated MoKα radiation (λ = 0.71073 Å). Data were corrected for Lorentz and polarization effects, and a semiempirical absorption correction based on multiple scans using SADABS20 was applied (Tmin = 0.676, Tmax = 0.746). Crystallographic data deposited at the Cambridge Crystallographic Data Centre under CCDC 1002151 for RuBibPh contain the supplementary crystallographic data excluding structure factors.
1H-NMR (400 MHz, CD2Cl2): δ = 8.61 (m, 2H, H8′′ or H9′′), 8.27 (d, 4J(H3–H5) = 2.0 Hz, 2H, H3 or H3′), 8.23 (s, 2H, H7′′), 8.21 (d, 4J(H3′–H5′) = 1.8 Hz, 2H, H3′ or H3), 8.01 (d, 3J(H5–H6) = 6.0 Hz, 2H, H6 or H6′), 7.98 (d, 3J(H5′–H6′) = 6.2 Hz, 2H, H6′ or H6), 7.88 (m, 2H, H9′′ or H8′′), 7.53 (dd, 3J(H5–H6) = 6.1 Hz, 4J(H3–H5) = 2.1 Hz, 2H, H5 or H5′), 7.35(dd, 3J(H5′–H6′) = 6.0 Hz, 4J(H3′–H5′) = 2.0 Hz, 2H, H5′ or H5), 7.22 (d, 3J(H4′′–H5′′) = 8.6 Hz, 2H, H5′′), 5.94 (d, 3J(H4′′–H5′′) = 8.6 Hz, 2H, H4′′), 2.62 (s, 6H, HMe′′), 1.49 (s, 18H, HtBu or HtBu′), 1.35 (s, 18H, HtBu′ or HtBu) ppm (for assignment of the hydrogen atoms see ESI†). 13C-NMR (100 MHz, CD2Cl2): δ = 162.7, 162.3, 159.6, 157.2, 153.1, 152.3, 141.0, 139.8, 138.3, 132.5, 128.9, 128.2, 127.1, 125.8, 124.4; 120.2, 120.2, 118.5, 116.5, 115.1, 35.6, 35.4, 30.2, 30.0, 22.0 ppm. MS Micro-ESI (MeCN/MeOH): m/z = 1119.3 ([M − PF6]+). EA: calculated for 4·C58H64N8RuP2F12·HPF6: (C = 53.56%, H = 4.98%, N = 8.62%); found: (C = 53.96%, H = 4.98%, N = 8.39%). Photophysical data: Abs. λmax (MeCN): 465 nm, λmax (MeOH): 465 nm, λmax (CH2Cl2): 467 nm. CV (MeCN, WE: glassy carbon, CE: Pt, RE: Pt, internal standard: Fc/Fc+, supporting electrolyte: 0.1 M TBAPF6, 20 mV s−1: 0.69 V, −1.84 V, −2.01 V, −2.24 V. Crystal data: C128H154F24N16O5P4Ru2, Mr = 2778.69 g mol−1, size 0.18 × 0.16 × 0.08 mm3, monoclinic space group P21, a = 15.632(3) Å, b = 22.082(4) Å, c = 19.317(4) (3) Å, α = 90°, β = 96.899(3)°, γ = 90°, V = 6620(2) Å3, T = 150(2) K, Z = 2, ρcalcd. = 1.394 g cm−3, no. of reflections (measured): 104![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 592, no. of reflections (unique): 27331, no. of parameters 1803/611, GooF = 1.081, R(reflection) = 0.0520 (23909), wR2(reflection) = 0.1238 (27331), largest difference peak and hole: 0.71/−0.61 e Å−3.
592, no. of reflections (unique): 27331, no. of parameters 1803/611, GooF = 1.081, R(reflection) = 0.0520 (23909), wR2(reflection) = 0.1238 (27331), largest difference peak and hole: 0.71/−0.61 e Å−3.
1H-NMR (400 MHz, 0.5 ml DMSO + 0.2 ml CD2Cl2): δ = 8.82 (d, 4J(H3–H5) = 1.8 Hz, 2H, H3 or H3′), 8.75 (d, 4J(H3′–H5′) = 1.8 Hz, 2H, H3′ or H3), 8.03 (d, 3J(H5′–H6′) = 6.0 Hz, 2H, H6′ or H6), 7.90 (d, 3J(H5–H6) = 6.2 Hz, 2H, H6 or H6′), 7.67 (s, 2H, H7′′), 7.63 (dd, 3J(H5–H6) = 6.1 Hz, 4J(H3–H5) = 2.1 Hz, 2H, H5 or H5′), 7.54 (dd, 3J(H5′–H6′) = 6.2 Hz, 4J(H3′–H5′) = 2.0 Hz, 2H, H5′ or H5), 5.50 (s, 2H, H4′′), 4.98 (m, 4H, H8′′), 2.33 (s, 6H, HMe6′′), 2.06 (s, 6H, HMe5′′), 1.47 (s, 18H, HtBu or HtBu′), 1.37 (s, 18H, HtBu′ or HtBu) ppm (for assignment of the hydrogen atoms see ESI†). 13C-NMR (100 MHz, 0.5 ml DMSO + 0.2 ml CD2Cl2): δ = 162.2, 161.4, 160.0, 157.6, 153.2, 143.1, 141.0, 135.6, 134.4, 133.8, 125.2, 124.3, 121.7, 115.9, 113.6, 79.7, 43.8, 36.0, 35.9, 30.7, 30.6, 20.7, 20.6 ppm. MS Micro-ESI (MeCN/MeOH: m/z = 1099.3 ([M − PF6]+). EA: calculated for C56H68N8RuP2F12: (C = 54.06%, H = 5.51%, N = 9.01%); found: (C = 53.82%; H = 5.51%; N = 8.86%.). Photophysical data: Abs. λmax (MeCN): 469 nm, λmax (MeOH): 463 nm, λmax (CH2Cl2): 477 nm. CV (MeCN, WE: glassy carbon, CE: Pt, RE: Pt, internal standard: Fc/Fc+, supporting electrolyte: 0.1 M TBAPF6, 20 mV s−1: 0.63 V, −1.913 V, −2.10 V, −2.36 V.
Sample preparation
For resonance Raman experiments the complexes were dissolved in DCM with an optical density of 0.5 at 475 nm. To ensure sample integrity throughout the measurements, UV-Vis spectra (Jasco 530) were measured before and after each resonance Raman measurement.Resonance Raman scattering
The details of the experimental set-up can be found elsewhere.7,21,22 In short, the excitation laser (Argon-Ion Laser 2018, Spectra-Physics) delivering the excitation light was focused into a rotating-cell cuvette.23 The scattered light was measured in 90° geometry and focused onto the entrance slit of the spectrometer (Princeton Instruments/Acton SpectraPro 2750-0.750 Triple Grating Monochromator/Spectrograph). For the measurements a grating with 2400 lines per mm was used.Measurements were performed at the excitation wavelengths of 458, 476, and 488 nm. To compare the spectra a baseline was subtracted from the data and the spectra were subsequently normalized with respect to the solvent peak of dichloromethane at 1423 cm−1.
Non-resonant Raman scattering
The Raman measurements were performed on a Bruker FT-Raman spectrometer (Bruker MultiRAM). For this the Ru-complexes, in the solid-state, were inserted into the spectrometer and illuminated with the Nd:YAG laser at a wavelength of 1064 nm.Quantum mechanical calculations
Quantum mechanical calculations were performed with the GAUSSIAN0924 program to obtain the structural and electronic data of the ruthenium complexes with protected bibenzimidazole ligands. For this, density functional theory (DFT) with the XC functional B3LYP25,26 was used to calculate the geometry, vibrational frequencies and respective normal modes of the electronic ground state. The 28-electron relativistic effective core potential MWB2827 was used with its basis set for the ruthenium atom; that is, 4s, 4p, 4d and 5s electrons are treated explicitly, whereas the three first inner shells are described by the core pseudopotential. The 6-31G(d) double-ζ basis set28 was employed for the ligands. In order to unravel the nature of the excited states contributing to the absorption spectra, the first 120 singlet excited states have been calculated at the time-dependent DFT (TDDFT) level of theory using the same XC functional, pseudopotential and basis set. Effects of solvent interaction (dichloromethane) have been taken into account on the equilibrium geometries, vibrational frequencies, excitation energies, transition dipole moments and analytical Cartesian energy derivatives of the excited states by applying the integral equation formalism of the Polarizable Continuum Model (IEFPCM).29Resonance Raman spectra were calculated using the sum-over-states approach for the bright states in the range of excitation wavelength of 476 nm, namely S3, S4, S6, and S7 (RuBibPh) as well as S3, S5, S6, and S7 (RuBibEt).12,30 In order to reproduce the experimental absorption in the region of the excitation wavelength, the excitation energies of the S7 (RuBibPh) and the S6 (RuBibEt) states have been bathochromically shifted by 2000 cm−1 and the resulting absorption was broadened by Lorentzians with a FWHM of 3000 cm−1 (equivalent to a damping factor of 1500 cm−1). Detailed information concerning the computation of resonance Raman intensities can be found in the literature.12 To correct for the lack of anharmonicity and the approximate treatment of electron correlation,31 the harmonic frequencies were scaled by a factor of 0.97. To compare the calculated bands with the measured data, a Lorentzian function with a FWHM of 5 cm−1 was added to broaden the peaks.
Results and discussion
The synthesis of all ruthenium complexes was carried out according to known procedures. Structural characterization was performed with 1H, 13C, ESI-mass spectrometry and elemental analysis. For RuBibPh it was possible to grow X-ray suitable crystals. The derived solid state structure from acetone–water is depicted in ESI Fig. 1† together with relevant distances and angles (see ESI Table 1†). The ruthenium centre is surrounded by nitrogen donor atoms in a distorted octahedral coordination geometry and, as for similar ruthenium complexes,32,33 the bond lengths (2.034(4)–2.059(4) Å) are shorter for the peripheral tbbpy as for the bibenzimidazole ligand (2.149(5) and (2.162(5) Å). Noticeably the bond to the phenylene-bridged bibenzimidazole is longer than that for the bibenzimidazole in the parent compound RuBib (see ESI Table 1†). A possible explanation is the rigidity of the bibenzimidazole induced by the phenylene moiety whereby the ligand is not able to bend around the ruthenium centre. Mononuclear ruthenium bibenzimidazole complexes show a characteristic bending of the bibenzimidazole ligand around the ruthenium centre resulting in an angle of 167.1°.32 In dinuclear complexes no bending is observed, i.e. an angle of 180° can be measured.33 The same angle of 180° is observed for RuBibPh.The UV-Vis data of RuBibPh and RuBibEt dissolved in dichloromethane and the calculated absorption spectra are displayed in Fig. 1. The spectra of the two complexes show very similar structures: in the UV region a strong transition at about 370 nm is observed, which is assigned to a ππ*-transition. The characteristic MLCT bands are observed in the visible part of the spectrum between 400 and 550 nm. The ππ*- and the MLCT-absorption maxima of RuBibEt are bathochromically shifted when compared to the absorption of RuBibPh by 441 and 405 cm−1, respectively.
In order to investigate the protection effect, the influence of the pH-value on the UV-Vis transition was investigated. The absorption properties of both complexes are within experimental errors not significantly affected by increasing the solvent pH-value (see ESI Fig. 3†), i.e. the sensitivity of the photophysical properties of the complexes to the pH value of the solution is drastically reduced when compared to the non-protected parent complex RuBib.2
Investigations on the emission in DCM properties of both complexes show that for RuBibPh no emission can be observed and for RuBibEt some – albeit very weak – luminescence is visible at approximately 610 nm.
In order to rationalise this finding, electrochemical studies were performed. The parent RuBib can be reversibly oxidised at 0.6 V vs. Fc/Fc+. However, due to irreversible electrochemical processes during the electrochemical measurements, no information is available for the reduction potentials of the coordinated ligands. For the complexes investigated here, the situation is changed: with respect to oxidation of the metal centre both complexes behave very similarly to the parent complex, i.e. the ruthenium centre is oxidized at potentials very similar to RuBib, i.e. at 0.69 and 0.63 V vs. Fc/Fc+ for RuBibPh and RuBibEt, respectively. However, three reversible reduction processes can be observed at potentials comparable to the potentials observed for Ru(tbbpy)32+ (see Table 1 and ESI Fig. 4†). Interestingly for a structurally very similar complex, [Ru(tbbpy)2(bibzim)]0 (bibzim = 2,2′-bibenzimidazolate dianion), a fully reversible reduction under electrochemical conditions was observed.33 This observation supports the notion that the irreversibility of reduction processes in bibenzimidazole ruthenium complexes correlates with the presence of relatively acidic protons. Apparently the introduction of the protection groups changes the electrochemical behaviour significantly: The observed fully reversible behaviour could be explained by an accessible state localised on the modified Bib-ligand scaffold or by a multiple reduction process of the bound bipyridine ligands. In order to clarify this question, resonance Raman spectroscopy is employed to characterize the localization of the initial electronic transition.12,34–36
| Complex | E ox (V) | E red1 (V) | E red2 (V) | E red3 (V) |
|---|---|---|---|---|
| RuBibPh | 0.69 | −1.84 | −2.01 | −2.24 |
| RuBibEt | 0.63 | −1.91 | −2.10 | −2.36 |
| RuBib | 0.60 | — | — | — |
| [Ru(tbbpy)3][PF6]2 | 0.90 | −1.73 | −1.92 | −2.17 |
Resonance Raman spectra of both complexes were recorded upon excitation at 458, 476 and 488 nm and the resulting spectra were compared to those recorded for the homoleptic reference compound Ru(tbbpy)32+ (Rutbbpy). The results are summarized in Fig. 2. In particular, Fig. 2(a–c) shows the resonance Raman data recorded at 476 nm for all three complexes (see ESI Fig. 5 and 6† for resonance Raman spectra excited at different excitation wavelengths). For easier comparison all spectra are baseline corrected and normalized to the DCM peak at 1424 cm−1. Most bands in the resonance Raman spectra of the protected bbim ruthenium complexes can be associated with those of the reference complex Rutbbpy. For example, the symmetric ring-breathing modes of the tbbpy-ligand located at 1031 and 1040 cm−1 can be found in all three spectra.37 In the RuBibPh (RuBibEt) there exist bands at 1081 and 1692 cm−1 (1681 cm−1) that are unique to the protected bbim complexes investigated here. They are found neither in the spectra of Rutbbpy nor in literature reports on related complexes bearing unprotected bbim ligands.7,11,37 To evaluate the molecular origin of these bands, non-resonant Raman measurements of RuBibPh and RuBibEt have been performed using solid powder samples of the complexes (Fig. 2d–e). These experiments are complemented by density functional theory (DFT) calculations (see Fig. 3) aimed at a detailed band assignment of the bands characteristic of the protected bbim structures. Based on the DFT calculations it can be shown that the band measured at 1081 cm−1 in RuBibPh is a ring breathing mode of the imidazole moieties on the protected bbim-ligand (see Fig. 4B). The weak band at 1499 cm−1 corresponds to ring stretching of the imidazole rings of the bbim ligand (see Fig. 4C) and the prominent band at 1692 cm−1 is a symmetric stretching mode located on the protected bbim-ligand with maximal displacement along the C–C bond between the imidazole moieties (see Fig. 4D). For RuBibEt the band at 1681 cm−1 is associated with a symmetric stretching mode of the bbim ligand with maximal displacement along the C–C bond between the imidazole moieties and strong coupling to the stretching of the C-atoms of the 1,2-ethandiyl bridge connecting the two benzimidazoles (see Fig. 4A). A complete mode assignment is listed in Table 2.
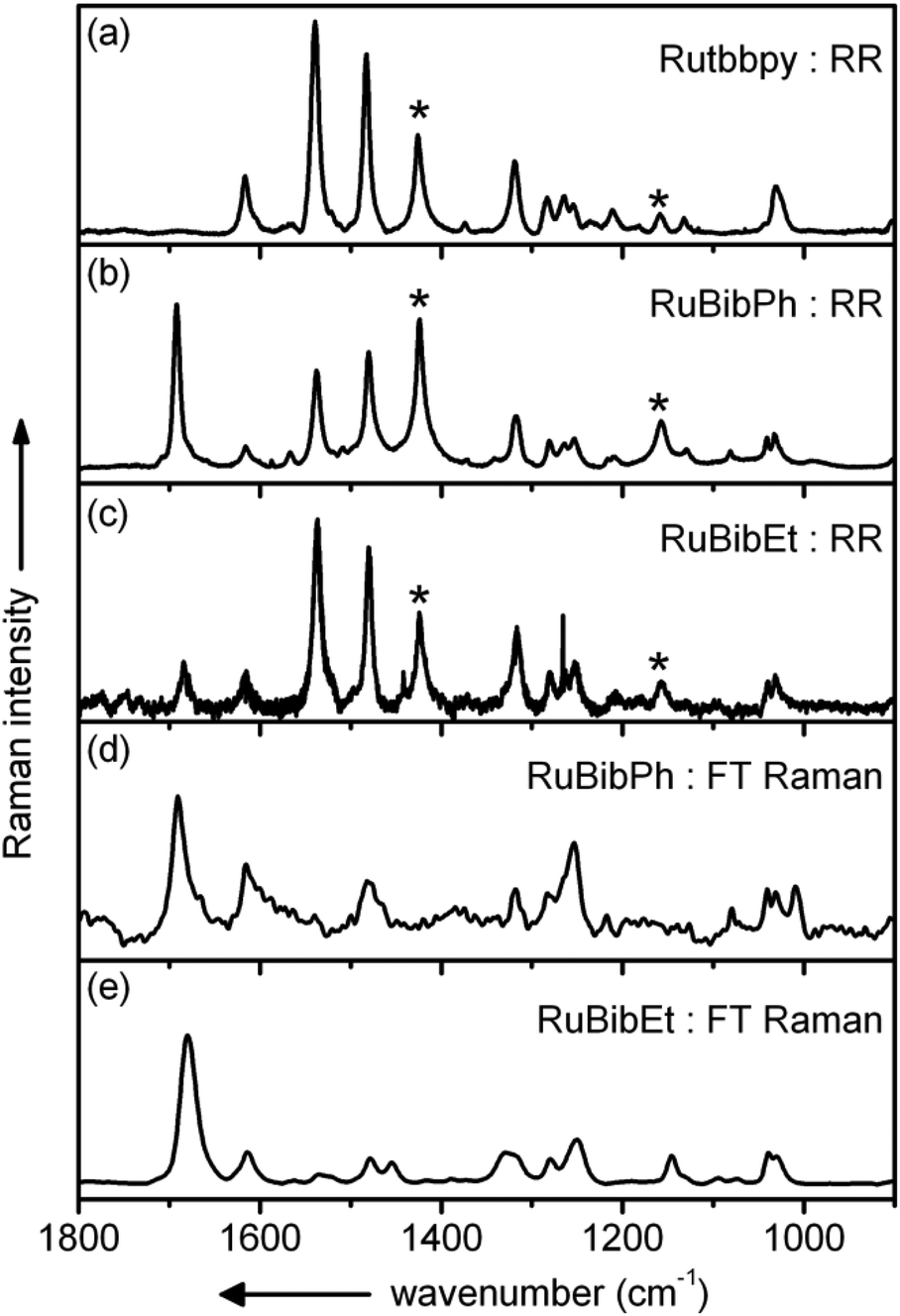 | ||
| Fig. 2 (a–c) Experimental resonance Raman spectra of the protected bbim complexes in resonance with the absorption band in the visible range (λex = 476 nm). The experimental resonance Raman spectrum of the homoleptic reference complex Rutbbpy (a) is given for the assignment of the tbbpy vibrations. The DCM solvent bands are indicated by asterisks. (d, e) Experimental non-resonant Raman spectra of the protected bbim complexes (λex = 1064 nm). | ||
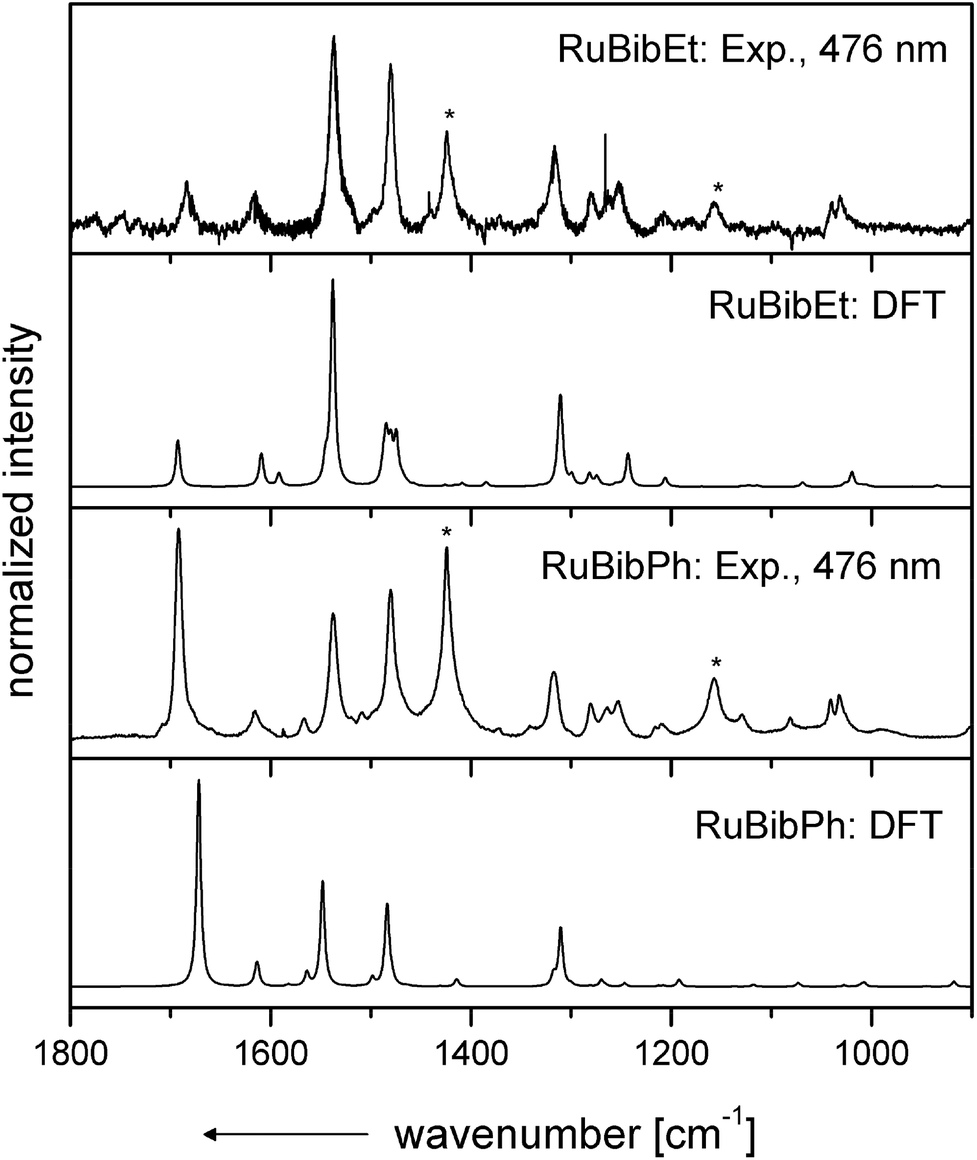 | ||
| Fig. 3 Comparison of the experimental and calculated resonance Raman spectra of both RuBibPh and RuBibEt. The calculated spectra clearly evidence the contribution of the bbim ligands in the excited state (see bands at >1670 cm−1). | ||
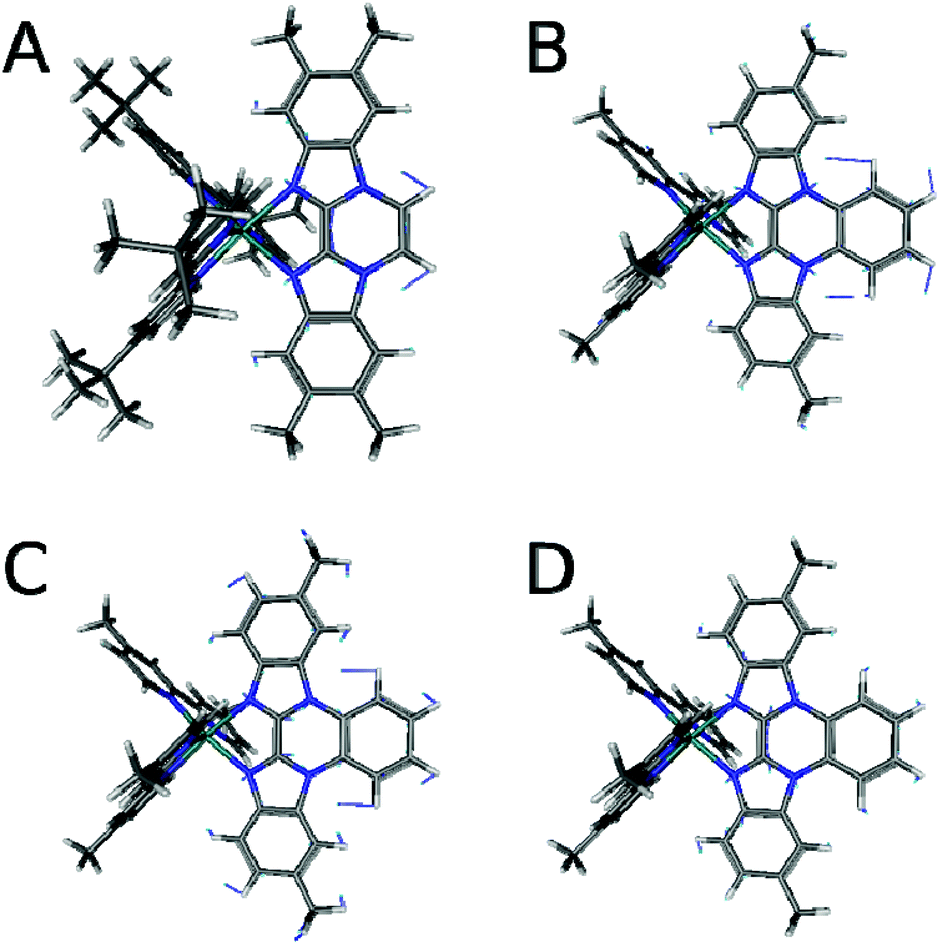 | ||
| Fig. 4 Displacement vectors of the bbim vibrations from DFT calculations of the RuBib complexes. (A) RuBibEt at 1693 cm−1, (B) RuBibPh at 1073, (C) 1499, and (D) 1672 cm−1. | ||
| RuBibPh | RuBibEt | Rutbbpy | Description | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DFT | 456 nm | 476 nm | 488 nm | 1064 nm | DFT | 458 nm | 476 nm | 488 nm | 1064 nm | ||
| cm−1 | cm−1 | cm−1 | cm−1 | cm−1 | cm−1 | cm−1 | cm−1 | cm−1 | cm−1 | cm−1 | |
| a Raman-active vibrational modes unique to the protected bibenzimidazole ligand. | |||||||||||
| 1007 | 1031 | 1033 | 1032 | 1031 | 1019 | 1030 | 1032 | 1030 | 1030 | 1030 | tbbpy, ring-breathingsym |
| 1040 | 1041 | 1041 | 1041 | 1039 | 1039 | 1039 | |||||
| 1073 | 1081 | 1081 | 1081 | 1079 | δ-ring, bbim | ||||||
| 1118 | 1129 | 1129 | 1130 | 1146 | 1132 | δ-CH, tbbpy | |||||
| 1208 | 1210 | 1210 | 1210 | 1217 | 1206 | 1206 | 1208 | 1206 | 1212 | tbbpy, tert-butyl | |
| 1247 | 1254 | 1253 | 1253 | 1243 | 1255 | 1253 | 1251 | 1250 | 1254 | ν-CC tbbpy | |
| 1270 | 1280 | 1281 | 1281 | 1283 | 1274, 1282 | 1278 | 1280 | 1280 | 1279 | 1283 | tbbpy, νasym-ring |
| 1310 | 1317 | 1318 | 1320 | 1317 | 1310, 1312 | 1315 | 1317 | 1315 | 1329 | 1319 | tbbpy, νasym-ring |
| 1484 | 1480 | 1480 | 1481 | 1481 | 1485 | 1479 | 1480 | 1479 | 1478 | 1483 | tbbpy, νasym-ring |
| 1499 | 1509 | 1509 | 1509 | ν sym-ring, bibzimPh | |||||||
| 1548 | 1538 | 1538 | 1537 | 1538 | 1534 | 1537 | 1535 | 1535 | 1539 | tbbpy, δ-ring, bbim | |
| 1564 | 1567 | 1566 | 1567 | 1540 | 1561 | 1566 | ν sym,asym-ring tbbpy, bbim | ||||
| 1614 | 1616 | 1618 | 1616 | 1615 | 1609, 1610 | 1615 | 1615 | 1613 | 1613 | 1617 | tbbpy, νasym-ring |
| 1672 | 1692 | 1692 | 1692 | 1691 | 1693 | 1681 | 1684 | 1682 | 1680 | ν-CC bbim | |
These data reveal that – in contrast to unprotected bbim ligands – the protected ligands do participate in the MLCT-transition. Hence, the beneficial feature of the unprotected bbim ligands, i.e., acting as non-electron accepting spectator ligands on the photophysical properties of thus-derived Ru-complexes, is lost.
The resonance Raman spectrum of RuBibEt reveals a reduced signal-to-noise ratio when compared to the spectrum of RuBibPh. This is due to the residual luminescence observed in the former case, which is entirely absent in the latter complex. The observation of a complete loss of luminescence for RuBibPh is potentially related to the extended π-electron scaffold in the protected Bib-ligand. The effect of an extended π-scaffold is thoroughly investigated and well understood for Ru-complexes bearing phenanthroline (phen) and dipyridophenazine (dppz) ligands.38,39 Here for instance, the phen-complexes show relatively strong emission in water with quantum yields in the range of 0.032–0.072,40–42 while dppz complexes in water barely emit. Sophisticated insights into the dynamics in dppz complexes were achieved by using a combination of time-resolved resonance Raman, time-resolved infrared spectroscopy and DFT calculations.43 Thus, it is speculated that protection of the bbim-structure in RuBibPh creates an electronic situation comparable to the dppz ligand, which efficiently quenches the – anyhow weak – luminescence of RuBibEt. In analogy to dppz complexes this quenching is speculated to be achieved by ultrafast picosecond removal of electron density from the proximity of the coordinated Ru-ion to orbitals spatially separated38,43–51 – a decay channel, which is apparently more prominently accessible in RuBibPh compared to RuBibEt.
Conclusion
The study presented here reveals that protecting the non-coordinating nitrogen atoms in bbim-ligands affects the photophysical properties of thereof derived Ru-complexes. It is shown that the protection route suggested here leads to a loss of the highly-advantageous feature of unprotected bbim ligands, i.e., the ability to act as non-electron accepting spectator ligands on Ru-polypyridine complexes. Furthermore, indications are reported that the protection of the non-coordinating nitrogens leads to dppz-like electronic structures in the ligand and resulting photophysical properties. The results presented suggest that the strategies pursued to date, to protect the uncoordinated nitrogen atoms of the bbim moiety against protonation/deprotonation and – at the same time – preserve the nature of the ligand as a non-electron accepting spectator ligand, do not succeed. One feasible alternative might be to use conventional Bib type ligands and form corresponding metal complexes with zinc centres as they render the compound pH-value insensitive while at the same time retaining luminescence.7Acknowledgements
Support by the COST Action CM1202 Perspect-H2O is highly acknowledged. The authors are thankful to Dr Maria Wächtler for fruitful discussions.Notes and references
- M.-A. Haga, Inorg. Chim. Acta, 1983, 75, 29–35 CrossRef CAS.
- A. M. Bond and M. Haga, Inorg. Chem., 1986, 25, 4507–4514 CrossRef CAS.
- M. Haga, T. Matsumura-Inoue and S. Yamabe, Inorg. Chem., 1987, 26, 4148–4154 CrossRef CAS.
- M.-A. Haga, Inorg. Chim. Acta, 1980, 45, L183–L184 CrossRef CAS.
- O. Takeshi, N. Koichi, I. Noriaki and H. Masa-aki, in Electron Transfer in Inorganic, Organic, and Biological Systems, American Chemical Society, 1991, vol. 228, pp. 215–228 Search PubMed.
- T. Ohno, K. Nozaki and M. Haga, Inorg. Chem., 1992, 31, 548–555 CrossRef CAS.
- M. Bräutigam, M. Wächtler, S. Rau, J. Popp and B. Dietzek, J. Phys. Chem. C, 2011, 116, 1274–1281 Search PubMed.
- B. Dietzek, W. Kiefer, J. Blumhoff, L. Böttcher, S. Rau, D. Walther, U. Uhlemann, M. Schmitt and J. Popp, Chem. – Eur. J., 2006, 12, 5105–5115 CrossRef CAS PubMed.
- H.-J. Mo, Y.-L. Niu, M. Zhang, Z.-P. Qiao and B.-H. Ye, Dalton Trans., 2011, 40, 8218–8225 RSC.
- D. P. Rillema, R. Sahai, P. Matthews, A. K. Edwards, R. J. Shaver and L. Morgan, Inorg. Chem., 1990, 29, 167–175 CrossRef CAS.
- C. Herrmann, J. Neugebauer, M. Presselt, U. Uhlemann, M. Schmitt, S. Rau, J. Popp and M. Reiher, J. Phys. Chem. B, 2007, 111, 6078–6087 CrossRef CAS PubMed.
- M. Wächtler, J. Guthmuller, L. González and B. Dietzek, Coord. Chem. Rev., 2012, 256, 1479–1508 CrossRef PubMed.
- S. Rau, T. Büttner, C. Temme, M. Ruben, H. Görls, D. Walther, M. Duati, S. Fanni and J. G. Vos, Inorg. Chem., 2000, 39, 1621–1624 CrossRef CAS.
- J. Preßler, R. Beckert, S. Rau, R. Menzel, E. Birckner, W. Günther and H. Görls, Z. Naturforsch., B: Chem. Sci., 2012, 67, 367–372 CrossRef PubMed.
- S. Rau, B. Schäfer, A. Grüßing, S. Schebesta, K. Lamm, J. Vieth, H. Görls, D. Walther, M. Rudolph, U. W. Grummt and E. Birkner, Inorg. Chim. Acta, 2004, 357, 4496–4503 CrossRef CAS PubMed.
- T. B. Hadda and H. Le Bozec, Polyhedron, 1988, 7, 575–577 CrossRef.
- P. Belser and A. V. Zelewsky, Helv. Chim. Acta, 1980, 63, 1675–1702 CrossRef CAS PubMed.
- E. Muller, G. Bernardinelli and J. Reedijk, Inorg. Chem., 1995, 34, 5979–5988 CrossRef CAS.
- N. Schoenberger, E. Schinzel, T. Martini and G. Roesch, Bridged Quaternary Benzimidazolyl-Benzimidazole Derivatives, Process for Their Preparation and Their Use, EP0027897 (A1), May 6, 1981.
- Bruker AXS Inc., SADABS 2008/1, Madison WI, U.S.A., 2008 Search PubMed.
- S. Tschierlei, B. Dietzek, M. Karnahl, S. Rau, F. M. MacDonnell, M. Schmitt and J. Popp, J. Raman Spectrosc., 2008, 39, 557–559 CrossRef CAS PubMed.
- M. Wächtler, S. Kupfer, J. Guthmuller, J. Popp, L. González and B. Dietzek, J. Phys. Chem. C, 2011, 115, 24004–24012 Search PubMed.
- W. Kiefer, Appl. Spectrosc., 1973, 27, 253–257 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- S. Kupfer, M. Wächtler, J. Guthmuller, J. Popp, B. Dietzek and L. González, J. Phys. Chem. C, 2012, 116, 19968–19977 CAS.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 11683–11700 CrossRef CAS PubMed.
- N. Rockstroh, K. Peuntinger, H. Görls, D. M. Guldi, F. W. Heinemann, B. Schäfer and S. Rau, Z. Naturforsch., B: Chem. Sci., 2010, 65, 281–290 CAS.
- S. Rau, M. Ruben, T. Büttner, C. Temme, S. Dautz, H. Görls, M. Rudolph, D. Walther, A. Brodkorb, M. Duati, C. O'Connor and J. G. Vos, J. Chem. Soc., Dalton Trans., 2000, 3649–3657 RSC.
- W. R. Browne, P. Passaniti, M. T. Gandolfi, R. Ballardini, W. Henry, A. Guckian, N. O'Boyle, J. J. McGarvey and J. G. Vos, Inorg. Chim. Acta, 2007, 360, 1183–1190 CrossRef CAS PubMed.
- S. L. Howell, K. C. Gordon, M. R. Waterland, K. H. Leung and D. L. Phillips, J. Phys. Chem. A, 2006, 110, 11194–11199 CrossRef CAS PubMed.
- N. J. Lundin, P. J. Walsh, S. L. Howell, J. J. McGarvey, A. G. Blackman and K. C. Gordon, Inorg. Chem., 2005, 44, 3551–3560 CrossRef CAS PubMed.
- M. Wächtler, M. Bräutigam, J. Popp and B. Dietzek, RSC Adv., 2013, 3, 5597–5606 RSC.
- E. J. C. Olson, D. Hu, A. Hörmann, A. M. Jonkman, M. R. Arkin, E. D. A. Stemp, J. K. Barton and P. F. Barbara, J. Am. Chem. Soc., 1997, 119, 11458–11467 CrossRef CAS.
- M. K. Brennaman, T. J. Meyer and J. M. Papanikolas, J. Phys. Chem. A, 2004, 108, 9938–9944 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- K. Nakamaru, Bull. Chem. Soc. Jpn., 1982, 55, 2697–2705 CrossRef CAS.
- A. Boisdenghien, C. Moucheron and A. Kirsch-De Mesmaeker, Inorg. Chem., 2005, 44, 7678–7685 CrossRef CAS PubMed.
- R. Horvath and K. C. Gordon, Inorg. Chim. Acta, 2011, 374, 10–18 CrossRef CAS PubMed.
- C. Kuhnt, M. Karnahl, S. Tschierlei, K. Griebenow, M. Schmitt, B. Schäfer, S. Krieck, H. Görls, S. Rau, B. Dietzek and J. Popp, Phys. Chem. Chem. Phys., 2010, 12, 1357–1368 RSC.
- E. Amouyal, A. Homsi, J.-C. Chambron and J.-P. Sauvage, J. Chem. Soc., Dalton Trans., 1990, 1841–1845 RSC.
- J. R. Schoonover, W. D. Bates and T. J. Meyer, Inorg. Chem., 1995, 34, 6421–6422 CrossRef CAS.
- G. Pourtois, D. Beljonne, C. Moucheron, S. Schumm, A. Kirsch-De Mesmaeker, R. Lazzaroni and J.-L. Brédas, J. Am. Chem. Soc., 2004, 126, 683–692 CrossRef CAS PubMed.
- Y. Sun, D. A. Lutterman and C. Turro, Inorg. Chem., 2008, 47, 6427–6434 CrossRef CAS PubMed.
- J. Fees, W. Kaim, M. Moscherosch, W. Matheis, J. Klima, M. Krejcik and S. Zalis, Inorg. Chem., 1993, 32, 166–174 CrossRef CAS.
- N. J. Lundin, P. J. Walsh, S. L. Howell, A. G. Blackman and K. C. Gordon, Chem. – Eur. J., 2008, 14, 11573–11583 CrossRef CAS PubMed.
- M. Atsumi, L. González and C. Daniel, J. Photochem. Photobiol., A, 2007, 190, 310–320 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and instrumentation, resonance Raman spectra recorded at excitation wavelengths of 458 and 488 nm, emission data. See DOI: 10.1039/c4dt01399a |
| ‡ CV data for dinuclear bibenzimidazolate complexes show reversible redox chemistry also in the reductive part. Here a reversible reduction of all bipyridine ligands but no reduction of the bibenzimidazolate could be observed.33 |
| This journal is © The Royal Society of Chemistry 2014 |