Electrochemical energy storage in a sustainable modern society
John B.
Goodenough
*
Texas Materials Institute, The University of Texas at Austin, 204 E. Dean Keeton, C2200, Austin, Texas 78712, USA. E-mail: john.goodenough@mail.utexas.edu; Tel: +1-512-471-1646
First published on 7th October 2013
Abstract
The storage of electrical energy in a rechargeable battery is subject to the limitations of reversible chemical reactions in an electrochemical cell. The limiting constraints on the design of a rechargeable battery also depend on the application of the battery. Of particular interest for a sustainable modern society are (1) powering electric vehicles that can compete with cars powered by the internal combustion engine and (2) stationary storage of electrical energy from renewable energy sources that can compete with energy stored in fossil fuels. Existing design strategies for the rechargeable battery have enabled the wireless revolution and the plug-in hybrid electric car, but they show little promise of providing safe, adequate capacity with an acceptable shelf and cycle life to compete in cost and convenience with the chemical energy stored in fossil fuels. Electric vehicles that are charged overnight (plug-in vehicles) offer a distributed energy storage, but larger battery packs are needed for stationary storage of electrical energy generated from wind or solar farms and for stand-by power. This paper outlines the limitations of existing commercial strategies and some developing strategies that may overcome these limitations.
Broader contextThis opinion highlights the limitations of Li-ion rechargeable batteries that use as cathodes an oxide host into/from which lithium is inserted/extracted reversibly. As a power source for portable electronic devices and power tools, they do not need to compete with fossil fuels and the internal combustion engine. Limits to energy density at required power outputs are expected to restrict their use in electric cars to plug-in hybrid vehicles, and then only after costs are significantly lowered and safety concerns are eliminated. Although a viable Na-ion battery appears to have been demonstrated, the Na-ion battery is not expected to compete with the Li-ion battery for powering an electric car. Storage in a rechargeable battery of electrical energy generated by variable renewable energy resources allows alternative electrochemical strategies. Those suggested require identification of a thin, mechanically robust solid Li+ and/or Na+ electrolyte membrane capable of blocking dendrites from a lithium or sodium anode and soluble redox molecules from reaching the anode from the cathode side. Al2O3/polymer electrolytes have been demonstrated. Identification of a solid O2− electrolyte having a conductivity σ0 ≃ 10−2 S cm−1 at 300 °C may allow combining a solid oxide fuel cell with an Fe/Fe2O3 storage bed. |
Introduction
Energy is most conveniently stored as chemical energy. Modern society consumes a huge quantity of energy obtained from the chemical energy stored in fossil fuels; fossil fuels can still be extracted and distributed at a relatively low apparent cost. However, the combustion of fossil fuels pollutes the air and is responsible for global warming; extraction and distribution of fossil fuels creates other environmental costs; the protection of national vulnerabilities to inadequate local fossil fuels is another expense. These societal costs can no longer be ignored. A sustainable modern society in a global economy must be able to store cleanly huge quantities of electrical energy derived from wind and radiant solar energy beyond what is available from hydroelectric power and biofuels. Electric energy can be stored in a rechargeable battery; but what are the limitations of this electrochemical technology, and how can these limitations be overcome by alternative electrochemical strategies?Traditional batteries
A traditional battery stores electrical energy as chemical energy in its two electrodes, the anode (a reductant) and the cathode (an oxidant). Its output on discharge is an electronic current Id at a voltage Vd for a time Δtd. A battery consists of one or more electrochemical cells that are connected in series to provide a desired Vd and in parallel to provide a desired Id and thus a desired electric power Pd = IdVd. The energy available from a single cell at a given discharge current Id is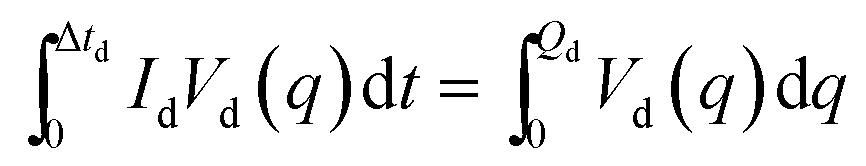 | (1) |
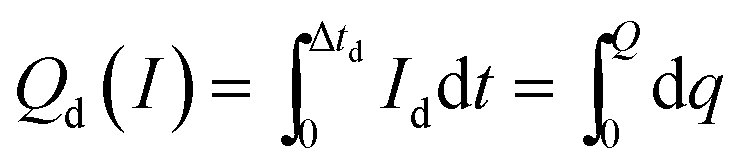 | (2) |
Of particular interest for the designer of a battery cell is its specific and/or volumetric capacity Qd(Id)/weight and/or volume and its voltage output Vd(q). For a given electrode, of interest is its voltage Vd(q) versus a reference voltage at any given Id and its capacity Q(Id). The larger the energy density of a cell for a given Id, the fewer the cells needed for a given application; fewer cells means simpler battery management and lower cost. For portable batteries, both the volume energy density and the specific energy density are important.
The chemical reaction between the two electrodes of a cell has an ionic and an electronic component. The two electrodes are separated inside a cell by a solid electrolyte or by an insulating polymer separator that is permeable to a liquid electrolyte. The electrolyte conducts the ionic component (the working ion), but it is an electronic insulator to force the electronic component of the chemical reaction into an external circuit where it can do work. The relatively low conductivity of the working ion requires fabrication of a thin electrolyte layer between the anode and cathode.
The electrolyte is a critical cell component; a thermodynamically stable cell has the electrochemical potentials EA of the anode and EC of the cathode within the electrolyte “window”. The window, Eg, of a liquid electrolyte is the energy difference between its lowest unoccupied and highest occupied molecular orbitals, i.e. its LUMO and HOMO, and of a solid electrolyte it is the energy gap between the conduction and valence bands. A discharge voltage Vd > Eg/e (e is the magnitude of the electron charge) requires development of a passivation layer on the anode if EA < LUMO or on the cathode if EC > HOMO. The passivation layer on a solid electrode with a liquid electrolyte is commonly referred to as a solid-electrolyte interphase (SEI) layer. Traditional batteries use a strongly acidic or strongly alkaline aqueous electrolyte in which H+ is the working ion with a conductivity of σH ≈ 10−1 S cm−1; but the aqueous window of 1.23 eV restricts the voltage of a traditional cell with a stable shelf life to a V ≲ 1.5 V. A kinetic stability of the electrode discharge reaction provides the additional 0.3 V.
The chemical reaction of a rechargeable cell must be reversible. Traditional strategy is to use a metal anode, an aqueous electrolyte, and as cathode a host transition-metal oxide into which the working H+ ion can be inserted reversibly over a finite solid-solution range. For example, in the 1.5 V Ni–Cd cell containing a KOH electrolyte, the reversible electrode reactions are
| Cd + 2H2O = Cd(OH) + 2H+ + 2e− (anode) |
| 2NiOOH + 2H+ + 2e− = 2Ni(OH)2 (cathode) |
The EA of the Cd anode is well matched to the H2O/H2 LUMO and the EC of the NiIII/NiII redox couple of NiOOH to the O2/H2O HOMO of the electrolyte, so it has a stable shelf life. On the other hand, the 2.0 V lead–acid cell containing an H2SO4 electrolyte and the reversible electrode reactions
| Pb + H2SO4 = PbSO4 + 2H+ + 2e− (anode) |
| PbO2 + 2H+ + 2e− = Pb(OH)2 (cathode) |
| Pb(OH)2 + H2SO4 = PbSO4 + 2H2O (cathode) |
Genesis of the Li-ion rechargeable battery
In 1967, Kummer and Webber of the Ford Motor Co. discovered fast Na+ conduction at 350 °C in the solid oxide Na2O·11Al2O3 (β-alumina). This discovery led them to invent the Na–S battery, which uses a Na+ solid electrolyte and liquid electrodes: molten sodium as the anode and molten sulfur impregnated with a carbon felt as the cathode. This invention coincided with the first energy crisis in 1970 that highlighted the national vulnerabilities resulting from dependence on imported oil. Although operation at 350 °C introduces high maintenance costs, nevertheless the Na–S battery focused minds on the possibility of using a non-aqueous electrolyte having a window of Eg > 1.23 eV in a rechargeable battery. Although non-aqueous liquids do not support a high σH, it was known that separation of Li+ from a lithium salt is sufficient in some solvents, including organic liquid carbonates or ethers, to give a Li+ conductivity σLi ≃ 10−2 S cm−1, which is adequate. The liquid dimethyl and/or diethyl carbonates (DMC/DEC) have a LUMO ca. 1.2 eV and a HOMO ca. 4.2 eV below the EA of metallic lithium, and an ethylene carbonate (EC) additive provides a passivating SEI layer on a lithium anode. With LiClO4 or LiPF6 as the salt in an organic liquid-carbonate electrolyte, a lithium primary (non-rechargeable) cell is capable of a voltage V ≈ 4.0 V. Therefore, the organic-liquid-carbonate electrolyte has become the standard Li+ liquid electrolyte used commercially in a Li-ion rechargeable battery.With insertion of H+ into layered NiO(OH) as a model rechargeable cathode, the first attempted rechargeable Li battery had a layered TiS2 cathode and a metallic-lithium anode; Li can be inserted (intercalated) reversibly between the TiS2 layers as LixTiS2 (0 ≤ x ≤ 1) at a TiIV/TiIII voltage of ca. 2.2 V versus Li0. However, in a rechargeable cell, the anode SEI layer prevents a uniform plating of lithium back onto the anode during charge; a “mossy” surface is formed on the anode. On repeated discharge/charge cycles, lithium dendrites are formed that can grow across a thin polymer separator and electrolyte layer to short-circuit the battery, which has disastrous consequences with a flammable, or even explosive, electrolyte. After a few incendiary events, initial attempts to design a rechargeable Li battery were abandoned.
Layered oxides only exist in V2O5 and MoO3 where the vanadium and molybdenum ions form vanadyl or molybdyl cations that create dipole–dipole bonding between strongly bonded oxide layers. Different vanadium oxides were investigated as cathodes of a rechargeable battery that is assembled in a charged state, but assembly in a charged state retains a metallic-lithium anode.
With the recognition that the voltage of a layered sulfide is limited by the energy of the top of the S-3p bands, an investigation of the reversible extraction of Li from a discharged LiMO2 (M = transition metal) cathode was initiated. LiCoO2, for example, has a rock-salt structure with Li+ and CoIII ordered into alternate octahedral-site (111) planes, making it analogous to LiTiS2 except for cubic rather than hexagonal stacking of the anion planes. Most battery manufacturers failed to recognize that the assembly of a rechargeable cell in a discharged state is practical for a rechargeable battery and allows consideration of alternative discharged anodes of higher voltage. Since reversible lithium insertion into graphite was being demonstrated at 0.2 V versus a lithium anode without the formation of dendrites, assembly of a discharged cell with a carbon anode and a LiCoO2 cathode was shown to have a Vd ≃ 3.8 V. The SONY Corporation licensed the LiCoO2/C technology to market the first cell telephone that launched the wireless revolution.
Although lithium is plated out on the surface of a carbon anode if the cell is charged too rapidly, this limitation is acceptable for a battery that powers an electronic device, or even a power tool or the battery of an electric vehicle where a long recharge time is tolerable. A rechargeable battery assembled in a discharged state with an anode other than metallic lithium, but with a Li+ working ion, is called a lithium-ion battery.
The challenge for Li-ion batteries
As indicated in the Introduction, two global challenges for electrochemical storage of energy are realization of (1) a commercially competitive portable store that can power an electric vehicle rivaling cars powered by the internal combustion engine and (2) an affordable, safe, stationary storage of electrical energy generated by a renewable, but variable energy source over a period of at least 24 h. Is the present strategy for the Li-ion battery up to either task?Strategies for portable power
Two approaches to powering electric vehicles are (1) Li-ion rechargeable batteries and (2) ambient-temperature fuel cells. The ambient-temperature fuel cell runs on pure hydrogen that needs to be stored, uses an aqueous electrolyte with an expensive proton-exchange membrane (PEM), and requires a more stable and cheaper catalyst for the oxygen-reduction reaction (ORR). It doesn't appear to be competitive with a rechargeable battery. Unfortunately, assembly in a discharged state of a Li-ion cell having a liquid-carbonate electrolyte creates a limitation on its energy density; if the anode has an EA ≲ 1.0 V, which is needed for a high Vd(q), formation of a Li+-permeable SEI layer on the anode during the initial charge robs Li irreversibly from the cathode of the cell, which decreases a cathode capacity already limited by the solid-solution range of the working ion in the cathode host. Consequently, the designer of a portable Li-ion power battery is forced to preform the anode SEI layer and to maximize the capacity of a stable high-voltage host cathode. Conventional strategies for Li-ion batteries are based on oxide-host cathodes and Li-alloy anodes. The cathode hosts are, so far, limited to layered Li1−xMO2, spinel Li1−x[M2]O4, or olivine Li1−xMPO4 structures, all with 0 ≤ x ≤ 1 and M = transition metal. These oxide hosts provide, respectively, 2D, 3D, and 1D Li-ion pathways for Li extraction/insertion; their structures and capacities are shown in Fig. 1 together with host voltages and those of the LUMO and HOMO of the organic liquid-carbonate electrolytes versus Li+/Li0. (Note: the sign of the voltage is opposite to that of the energy.) The room-temperature Li mobility in a close-packed oxide-ion array can approach 10−3 S cm−1, so high charge/discharge rates are possible with all the hosts of Fig. 1 provided the cathode particles have at least one nanosized dimension in the direction of a mobile-Li pathway that is accessible by the electrolyte. The Li mobility in an oxide host is not limiting. (Note: small host particles must be connected electronically to the current collector by being imbedded in a Li-permeable conductive matrix, normally a form of carbon, or attached to a conductive polymer contacting the current collector.)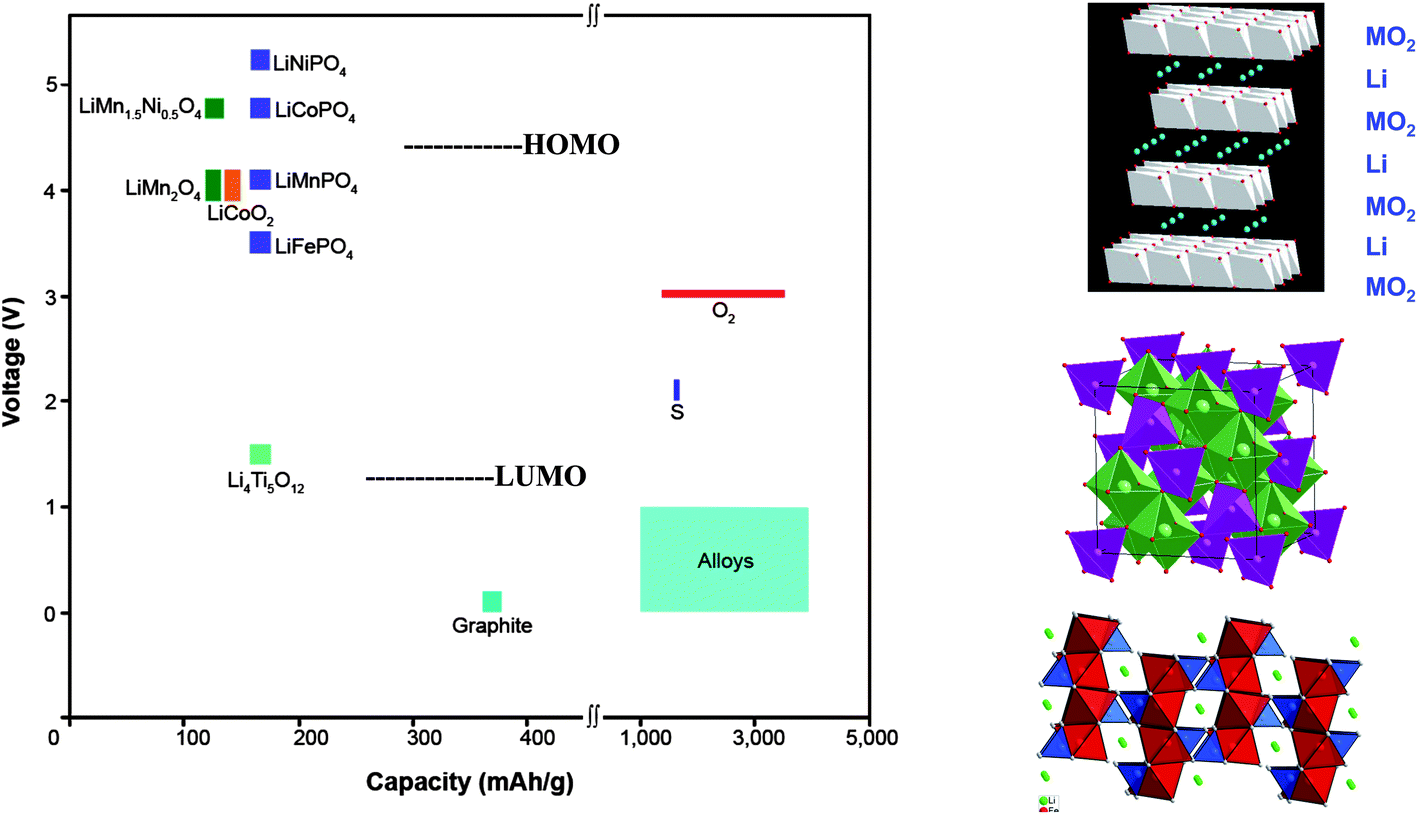 | ||
| Fig. 1 Voltage versus capacity of insertion-compound electrodes (left side), of lithium alloy anodes and sulfur or oxygen cathodes (right side) for a lithium-ion battery. The LUMO and HOMO are for organic liquid-carbonate electrolytes. Oxide Structures (top to bottom) are layered LiMO2, spinel Li[M2]O4, and olivine LiMPO4. | ||
The voltage of a host cathode may be limited by either the energy of the top of the O-2p bands or by the HOMO of the electrolyte. Where the voltage is limited by a pinning of the host redox couple at the top of the host O-2p bands, the capacity may also be limited by the loss of O2. For example, the capacities of the layered Li1−x(Ni1−yCoy)O2 compounds are limited by loss of oxygen before all the Li is extracted. In these layered compounds, the NiIV/NiIII and CoIV/CoIII redox states of d-orbital symmetry contain a large O-2p fraction that increases with oxidation because the couples are pinned at the top of the O-2p bands. Where the O-2p fraction becomes too large, the mobile holes find it energetically favorable to become trapped in surface peroxide ions (O2)2− with subsequent loss of O2. The introduction of MnIV lowers the energy of the top of the O-2p bands to allow complete oxidation of the NiIV/NiIII couple in Li1−x(Ni0.5−yCo2yMn0.5−y)O2. Of greater interest is the more stable spinel Li1−x[Ni0.5Mn1.5]O4; it shows no significant energy gap between the NiIII/NiII and NiIV/NiIII couples at about 4.7 V versus Li+/Li0, which still allows extraction of one Li per formula unit. At 4.7 V, which is about 0.5 V above the HOMO of a liquid-carbonate electrolyte, either an alternative liquid electrolyte with a more stable HOMO or an SEI layer permeable to Li+ or an intrinsic kinetic barrier to reaction with the electrolyte is needed to stabilize this high-voltage cathode. This spinel represents an upper limit to the voltage of a host-cathode for a Li-ion battery with an organic liquid-carbonate electrolyte, but a host with double the capacity may be possible. These observations indicate that with the existing strategy for a Li-ion battery, the voltage will be limited to V < 5 V and the capacity to less than one Li per host cation, i.e. 220 mA h g−1.
The anode presents another problem. Li alloys permit both increasing the capacity well-beyond that of a carbon anode and lowering the EA to allow a fast charge, albeit at the cost of the cell voltage. However, with an EA < LUMO, a Li+-permeable SEI layer needs to be formed on the surface of the anode particles. Li-alloy particles buffered by carbon or a polymer need to be small in size for the buffer to absorb the large volume changes that occur on cycling, which means the anode has a large surface area needing to be passivated. To circumvent a large irreversible loss of Li from the cathode on the initial charge, the passivating layer on the anode must be performed before cell assembly.
In order to increase the volume-capacity of an oxide cathode, the synthesis of electronically conductive mesoporous structures to which nanosized cathode particles can be chemically attached is being explored. Mesoporous, electronically conductive structures can provide both the working ion from the electrolyte and electrons from the current collector access to the cathode particles distributed over a large surface area. This cathode strategy together with a Li-alloy anode having a preformed SEI layer may prove capable of providing batteries that can power electric vehicles with acceptable driving range, power, cycle life, and safety; the electric vehicles would be charged at night during off-peak power demand to provide a distributed storage of electrical energy. The remaining problem will be to lower the cost of manufacture and recycling to where these vehicles can compete with the internal combustion engine. A “plug-in”—overnight-charge—electric car does not need a fast charge, which would allow use of a Li–Si alloy with a Vd(q) ≈ 0.4 V versus Li+/Li0; but where a fast charge is required, a Li alloy with a Vd(q) > 0.5 V would be needed to avoid plating of Li on the SEI layer.
The search for non-flammable liquid electrolytes with larger windows will continue, but to date the ionic liquids have proven to have too high a viscosity.
A desired further increase in capacity will require a cathode strategy that either uses a multivalent working ion or a reversible cathode chemical reaction other than that of a host for working-ion insertion/extraction. Although use of multivalent working cations is being explored, this approach is problematic. As shown in Fig. 1, sulfur and air cathodes offer much larger capacities than insertion compounds. Although a Li–S room-temperature battery may find application in electrical vehicles, for this application their volumetric energy density needs to be increased. Increased capacity and cost are the overriding concern for stationary storage of electrical energy; metal–sulfur batteries for this application are not restricted by the requirement of a high volumetric energy density.
Strategies for stationary storage of electrical energy
Although large Li-ion batteries having Li-insertion electrodes have been installed in Quebec and in China to store electrical energy generated by wind farms, this approach is not optimal. Alternative cell strategies of higher capacity begin with the requirement of a solid working-ion electrolyte that can be used as a separator of anode and cathode chemical reactions. The solid-electrolyte separator must be fabricated as a thin, mechanically robust membrane having a working-ion conductivity σi > 10−4 S cm−1 at room temperature; it must also be chemically stable in any liquid electrolytes used and must block any dendrites from the anode side and any soluble products from the cathode side. Two approaches are possible: (1) a composite oxide/polymer membrane having the same liquid electrolyte either side and (2) a solid oxide membrane capable of a different electrolyte at the anode and the cathode.An oxide/polymer composite electrolyte can be made thin and mechanically robust; it is porous to the liquid electrolyte and remains somewhat flexible with sufficient oxide loading and small enough pore size to block dendrites from a lithium anode. This configuration eliminates problems with a large impedance to Li+ transport across solid–liquid electrolyte interfaces. However, a thin, solid layer, which may be a mixed electron/Li+ conductor, may need to be coated on the cathode side to block soluble redox molecules in a liquid cathode. Al2O3/PEO membranes have already been shown to be capable of blocking dendrites from the anode, which allows use of a metallic-lithium anode and a cell assembly in the charged state. Two immediate targets with an oxide/polymer membrane are (1) a sulfur cathode and (2) a redox flow-through liquid cathode. (3) A Li–air cell with this membrane will probably require identification of a molecular shuttle to carry the O2−/(O2)2− reaction product away from the catalyst to a Li2O2 particle build-up as well as to oxidize the Li2O2 to ferry O2− back to the current collector to be oxidized to O2 on charge.
Sulfur is abundant and environmentally friendly; replacement of an expensive transition-metal oxide by sulfur as the cathode would lower cost and greatly improve the cathode capacity. However, a low Vd(q) ≈ 2.15 V versus Li+/Li0 requires a metallic-lithium (or metallic-sodium) anode for capacity balance and an optimized cell voltage. A Li–S cell promises to meet the target of a 500 km range for an electric vehicle as well as lower the cost of large stationary batteries for storing electrical energy obtained from wind or radiant solar energy. However, utilization of a solid sulfur cathode in an organic electrolyte faces several problems: the cyclo-S8 parent charged phase and its reaction products with lithium are all electronic insulators, and the structural changes associated with the Li2S6, Li2S4, Li2S2, and Li2S products create unstable contact with the current collector. Moreover, the intermediate Li2S2 is soluble in the electrolyte and migrates to the anode unless blocked by a membrane. Strategies for realizing the potential of a sulfur cathode have concentrated on encapsulating sulfur in binder-free, porous carbon networks such as self-intertwining curly carbon nanotubes or in hollow carbon tubes. The carbon network acts as the current collector. To block the dissolved Li2S2 species from migrating to the anode and to improve contact of the soluble species with the electronically conductive carbon network, a carbon paper can be placed between the carbon/sulfur composite cathode and the cell separator. To increase further the volumetric capacity, conductive insertion compounds of comparable voltage e.g. TiS2, may be introduced to replace some of the carbon in the encapsulating conductive matrix. Optimization of the morphology and content of the composite sulfur cathode leaves much room for imaginative chemical synthesis.
Insoluble intermediate species, Li2S6 and Li2S4, offer fast charge/discharge rates; the soluble Li2S2 in the Li2S2–Li2S range yields a slower charge/discharge rate. However, lithium polysulfides are highly soluble in some organic solvents such as tetrahydrofuran (THF), and the dissolved Li2S2 redox molecule can be used in a liquid cathode operating in a redox flow-through mode. This strategy shows excellent potential, but it has been hampered to date by migration of the redox molecule to the anode. A solid Li+ electrolyte that can block the Li2S2 molecules from migrating to the cathode should solve this problem.
In addition to the challenge of fabrication as a thin, mechanically robust membrane, a solid oxide Li+-electrolyte separator may have a large impedance for Li+ transfer across a solid–liquid electrolyte interface and also may be unstable on contact with an aqueous electrolyte. These problems have been found to be the case with the Li+ garnet-framework electrolyte Li7−xLa3Zr2−xTaxO12, which has an acceptable Li+ conductivity σLi ≳ 5 × 10−4 S cm−1 at room temperature. Coatings to reduce the interface impedance and reaction with an aqueous electrolyte need to be explored.
Replacement of Li by Na is another goal to reduce both cost and national vulnerability to the global distribution of lithium deposits. However, reversible Na insertion into an oxide framework does not appear to be a promising strategy. Nevertheless, a Na–S cell may prove viable as oxide Na+-electrolyte membranes that block Na dendrites are known with a room-temperature σNa > 10−4 S cm−1. Moreover, sodium insertion into MnFe(CN)6 and an Al2O3/polymer-membrane separator has been shown to provide a viable Na-ion battery.
Novel approaches
Completely novel configurations may be invented as new material properties are discovered. For example, electrical energy storage has been demonstrated with a solid oxide fuel cell (SOFC) connected to a reversible metal-to-oxide bed. On discharge, H2 is fed to the anode of the SOFC to produce an electrical current Id at a Vd ≃ 1 V and exhausts H2O at the anode; the H2O is fed to the metallic bed where it is reduced back to H2 by oxidation of the bed. On charge, the circulation is reversed; H2O is fed back to the anode of the SOFC where it is reduced back to H2 that, in turn, reduces the oxide bed back to the metallic state. Good cyclability has been demonstrated at 550 °C; the capacity is determined by the size of the bed. With the discovery of a new solid O2− electrolyte having σO ≃ 10−4 S cm−1 at room temperature, this scheme may be operated at 300 °C if not all the way down to room temperature, depending on the activities of the catalytic electrodes of the SOFC.If the new O2− solid electrolyte is able to generate H2 from low-temperature steam, it will provide a huge saving at commercial refineries as well as a source of pure H2 for a low-temperature fuel cell. For this application, a solid for H2 storage at modest pressure will be needed to accompany the stationary storage of electrical energy.
Not explicitly considered in the above narrative will be an on-going development of improved oxide catalysts for the oxygen-reduction and oxygen-evolution (ORR and OER) reactions.
| This journal is © The Royal Society of Chemistry 2014 |