Effects of all-trans retinoic acid, retinol, and β-carotene on murine macrophage activity†
Huey-Ming
Lo
ab,
Shih-Wei
Wang
c,
Chih-Li
Chen
a,
Pi-Hui
Wu
a and
Wen-Bin
Wu
*a
aSchool of Medicine, Fu-Jen Catholic University, Wen-Bin Wu, No.510, Zhongzheng Rd., Xinzhuang Dist., New Taipei City 24205, Taiwan. E-mail: wenbin@mail.fju.edu.tw; Fax: +886-2-29052096; Tel: +886-2-29053497
bSection of Cardiology, Department of Internal Medicine, Shin Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan
cDepartment of Medicine, Mackay Medical College, New Taipei City, Taiwan
First published on 5th November 2013
Abstract
Previous studies have demonstrated that vitamin A and carotenoids regulate immune function in lymphocytes and splenocytes, and that the carotenoid lutein regulates matrix metalloproteinase-9 (MMP-9) production in macrophages. In this study, we investigated the effects of all-trans retinoic acid (atRA, a bioactive vitamin A metabolite), retinol (vitamin A), and β-carotene (vitamin A precursor) on the activity of murine RAW264.7 and peritoneal macrophages. Our results indicated that atRA and retinol could induce GM-CSF and IL-16 expression, whereas all these tested substances enhanced MMP-9 production. Interestingly, the expression of GM-CSF, IL-16, and MMP-9 was distinctly regulated by these three substances. AtRA and retinol affected GM-CSF and IL-16 expression mainly through RA receptor β (RARβ). However, atRA induced MMP-9 production was via RARα activation and retinol and β-carotene caused MMP-9 production via RARα and β activation. These were supported by the observations that the RARα and β agonists/antagonists differentially affected MMP-9 production and that atRA and β-carotene enhanced RARE-mediated and MMP-9 promoter luciferase activity. In parallel, while the MMP-9 induction by atRA was not affected by the MAPKs inhibitors, its induction by retinol and β-carotene was repressed by the inhibitor targeting ERK1/2. Finally, we show that all the tested substances could functionally enhance macrophage phagocytosis. Taken together, we provide evidence here for the first time that atRA, retinol, and β-carotene differentially regulate GM-CSF, IL-16, and MMP-9 production in macrophages, explaining at least in part why these vitamin A-related substances are beneficial for immunity.
1. Introduction
Vitamin A (retinol) is essential for life and has important roles in embryonic development, spermatogenesis, vision and cellular differentiation.1 It is also known for its role in immunity. Vitamin A deficiency affects the mucosal barriers of several tissues, compromising the ability of macrophages and neutrophils to migrate to sites of infection, phagocytose, and kill bacteria.2,3 β-Carotene, a carotenoid which can be converted into vitamin A in human body, has also been demonstrated to prevent organ infections in vitamin A deficient rats4 and reduce ear infections in young children.5β-Carotene is one of the main dietary carotenoids and structurally belongs to the hydrocarbons of carotenoids, sharing a polyene backbone structure with vitamin A (retinol) and its bioactive metabolite, all-trans retinoic acid (atRA).6 Retinol and atRA have been known for interaction with RA-binding transcriptional regulators belonging to the nuclear receptors (NRs) superfamily. All-trans and 9-cis RA bind to the RA receptors (RARα, β, and γ isotypes), whereas 9-cis RA binds to the retinoid receptors (RXRα, β, and γ isotypes).7 Some carotenoids have been shown to affect the activity of RAR, RXR, and peroxisome proliferator activated receptors (PPARs), but it is not yet clear if the observed changes in the transcriptional activity of these nuclear receptors are related to direct interaction of carotenoid derivatives with the relevant receptors as ligands.8,9 According to the current model of gene regulation by retinoids, RARs binding to RA response elements (RAREs) can maintain chromatin in a repressed state in the absence of ligand but initiates a recruitment of the coregulator complexes with different enzymatic activities to alter the chromatin structure for recruitment of the transcription machinery upon ligand binding.10
Macrophages are highly efficient phagocytes that play an important role in the recognition and clearance of many potential pathogens. They play important roles in primary innate immunity and function as regulator and effector cells in both humoral and cell mediated immune responses.11 Macrophages produce many cytokines, chemokines, growth factors and proteases. They produce reactive-oxygen species and reactive-nitrogen metabolites which are required for pathogen clearance. They also secrete diverse matrix metalloproteinases (MMPs) in large quantities, which mediate several important functions.12
Matrix metalloproteinases (MMPs) are the principal matrix-degrading proteinases that play key roles in embryonic development, organ morphogenesis, and wound healing.13 Monocyte/macrophage MMP production has been linked to various biological functions, including ECM degradation; cell migration; and regulation of cytokines, chemokines, and other soluble proteins during inflammation.12 Recently, MMP-25 induced-vimentin cleavage was shown to be required for the phagocytic activity of macrophages,14 and MMP-9 was required for forming multinucleated giant cells, which are believed to enhance the defensive capacity of macrophages.15 Our previous study has also shown that the carotenoid lutein induced MMP-9 production, which participated in macrophage phagocytosis.16
Although vitamin A and β-carotene (with vitamin A activity) play a role in immunity, their effects on macrophages remain unclear. In the present study, we investigated the effects of atRA, retinol, and β-carotene on macrophage activity. We found that atRA, retinol, and β-carotene had differential effects on the expression of granulocyte macrophage-colony stimulating factor (GM-CSF) and interleukin-16 (IL-16). However, all of them caused MMP-9 production in murine macrophages. Their possible mechanism of action on macrophage activity was explored.
2. Materials and methods
2.1. Materials
All-trans retinoic acid (AtRA) and retinol were purchased from Sigma-Aldrich Chemical Co. (St Louis, MO, USA). β-Carotene was from Nacalai Tesque, Inc. (Kyoto, Japan). The RAR antagonists were from Tocris Bioscience (Bristol, United Kingdom). The MMP-9 antibody (Ab) against C-terminal region of MMP-9 was purchased from Abcam (Cambridge, MA, USA). The Ab against phospho-ERK1/2 was from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and the Abs for JNK, phospho-p38 and phospho-JNK were from Cell Signaling Technology, Inc. (Danvers, MA, USA). The Abs for total p38 and ERK1/2 were from R&D systems, Inc. (MN, USA). AtRA and retinol were dissolved in ethanol and β-carotene was dissolved in tetrahydrofuran (THF) containing 0.025% butylated hydroxytoluene as an antioxidant (Sigma Chemical Co).2.2. Cell cultures
RAW264.7 macrophages were obtained from the Food Industry Research and Development Institute (Hsinchu, Taiwan). They were cultured in 90% Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS supplemented with amino acids and antibiotics. Primary-cultured peritoneal macrophages were prepared following the procedure described previously.16 Briefly, ICR mice were injected i.p. with 1.0 ml of 3% (w/v) thioglycolate (BD Difco, MD, USA) in serum-free medium, 5 days prior to cell harvest. ICR mice were sacrificed by CO2 asphyxiation. The peritoneal macrophages were collected by peritoneal lavage using 10 ml serum-free medium. After several washes, cells were allowed to adhere to culture dishes and were cultured in the same medium used for RAW264.7 before further analysis. Animal experimental procedures were approved by the Laboratory Animal Use and Care Committee of Fu-Jen University.2.3. RT-PCR analysis of GM-CSF, IL-16 and MMP-9 mRNA levels
Oligonucleotide PCR primers targeting to GM-CSF, IL-16, MMP-9 and GAPDH were synthesized (MDBio Inc., Taipei, Taiwan) (Table 1). Macrophage total RNA was extracted using Trizol reagents (Invitrogen Technologies). The reverse transcription reaction was performed with aliquots of 1–2 μg of total RNA and random hexaprimers using the Superscript III First-Strand Synthesis System (Invitrogen Technologies) according to the manufacturer's instructions. PCR was performed with 30 cycles of denaturation at 94 °C for 1 min, annealing at 51–56 °C for 1 min, and elongation at 72 °C for 1.5 min using the ABI 7200 Thermal Cycler (Applied Biosystems, Foster city, CA, USA). The amplification products were analyzed by electrophoresis in 2% agarose gels.| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| GM-CSF | AGATATTCGAGCAGGGTCTAC | GGGATATCAGTCAGAAAGGTT |
| IL-16 | AACCGAGGACAGGAACCACT | CTTGAGAGATTTGCCATTGA |
| MMP-9 | TTCTGCCCTACCCGAGTGGA | CATAGTGGGAGGTGCTGTCGG |
| GAPDH | ATGGTGAAGGTCGGTGTGAACG | GTTGTCATGGATGATCTTGGCC |
2.4. Measurement of GM-CSF in culture medium by ELISA
GM-CSF release by macrophages was measured by a mouse GM-CSF ELISA Development kit (R&D Systems, Inc., MN, USA) according to the manufacturer's protocol. Briefly, macrophages were treated with vehicle, atRA, or β-carotene for 16 h. The culture media were collected, centrifuged and GM-CSF release was measured. The product of this enzymatic reaction was yellowish in color and absorbed strongly at 450 nm. The intensity of this color is proportional to the amount of GM-CSF present in the well during the incubation. The GM-CSF concentrations in the macrophage culture medium were calculated from the standard curve.2.5. Gelatin zymography and Western blot analysis of MMP-9
For measuring the MMP-9 activity in the culture media, equal numbers of RAW264.7 macrophages were seeded into 24-well culture plates, to which equal volumes of medium were added. After the cells were treated with atRA, retinol, or β-carotene, equal volumes of culture medium were removed, and MMP-9 activity was analyzed by gelatin zymography under non-reducing conditions.17 The influence of these tested substances on cell viability was monitored by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. The gelatinolytic activity of MMP-9 was detected as clear bands. Western blot analysis was used to detect MMP-9 expression and release in cell lysates or culture media. Cell lysates were prepared by washing cells with prechilled PBS followed by lysis in radioimmunoprecipitation assay buffer [20 mM Tris–HCl (pH 7.4), 137 mM NaCl, 2 mM EDTA, 1 mM sodium fluoride, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10% glycerol, 1 mM sodium orthovanadate, 1 mM PMSF, and 1 μg ml−1 aprotinin and leupeptin (freshly prepared)]. The protein content was quantified using the Pierce protein assay kit (Pierce, Rockford, IL, USA). Equal amounts of samples were analyzed by Western blotting.18 Zymographs and chemiluminescent immunoblot images were captured using the BioSpectrum Imaging System (Ultra-Violet Products Ltd., Cambridge, UK). The intensity of each band in the captured images was analyzed using the ImageQuant 5.0 software (Molecular Dynamics).2.6. Transient transfection and luciferase reporter assay
For the MMP-9 luciferase reporter assay, cells were transfected with pGL-MMP-9-Luc.16 For the pRAR-Luc reporter assay, cells were transfected with pRAR-Luc [a reporter construct containing 3 copies of the retinoic acid response element (RARE)] [Signosis Inc. (Sunnyvale, CA, USA)]. Reporter transfection was performed by seeding RAW264.7 macrophages (2.5 × 105 cells per ml) in 12-well culture plates (Costar); the cells were then transfected with 0.75 μg of total DNA using PolyJet™ in vitro DNA Transfection Reagent (SignaGen Lab, Rockville, MD, USA) for 18 h in medium according to the manufacturer's protocol. All DNAs were prepared using endotoxin-free plasmid preparation kits (Qiagen) with 0.75 μg of reporter construct or pSV-β-galactosidase (β-gal) control vector (Promega). Following transfection, cells were washed once with an endotoxin-free medium and then allowed to grow for 24 h in complete medium containing antibiotics. The reporter firefly luciferase values were obtained by analyzing 1 ml of purified cell extract according to standard instructions provided by the Luciferase Kit (Promega) in a Wallac Victor 3 1420 multilabel counter (Perkin Elmer, Turku, Finland). β-Gal activity was determined using o-nitrophenyl-β-D-galactopyranoside as a substrate.19 The relative luciferase activities of cell extracts were typically represented as firefly luciferase/β-gal values.2.7. Phagocytosis assay
The phagocytosis of yeasts by macrophages using 2′,7′ bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein acetoxymethyl ester (BCECF/AM) (Invitrogen) has been described previously.16 Briefly, macrophages were incubated with vehicle, atRA, retinol, or β-carotene for 16 h. The yeasts were then labeled with BCECF/AM (10 μg ml−1) for 30 min at 37 °C, and an equal number (1 × 107 per well) were directly added into the macrophage serum-free medium. After incubation for 1 h, cells were washed thrice with medium followed by fixation with 4% paraformaldehyde. Phagocytosis was examined and photographed under a Nikon Eclipse Ti–S inverted fluorescence microscope (Japan) equipped with a high-resolution camera. The phagocytic index was measured by the direct counting of ingested yeasts (fluorescent spots) in nine randomly selected regions at a magnification of 200× (high power field, HPF).2.8. Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM). The means of the two groups of data were compared using the unpaired, two-tailed Student's t-test.3. Results
3.1. Effect of atRA, retinol, and β-carotene on MMP-9 and cytokines production in murine macrophages
In our previous study, we had observed that the carotenoid lutein was able to induce MMP-9 release.16 Meanwhile, we also observed that lutein could cause the release and synthesis of GM-CSF and IL-16 in murine macrophages using a cytokine antibody array assay and RT-PCR (Fig. 1S†). Based on these observations, in the beginning of this study we examined the effect of atRA and β-carotene on MMP-9 mRNA expression in macrophages. In Fig. 1A, we found that both atRA and β-carotene enhanced the MMP-9 mRNA level in murine RAW264.7 macrophages. AtRA and β-carotene concentration-dependently increased MMP-9 mRNA level, 0.1 μM of atRA and 2 μM of β-carotene were sufficient to achieve such an inducing effect. Moreover, atRA and β-carotene enhanced the MMP-9 mRNA level in a time-dependent manner (Fig. 1B). We also tested whether retinol (vitamin A) had an inducing effect on MMP-9 production. Fig. 1C shows that retinol caused MMP-9 production in a concentration-dependent manner. Next, we examined whether atRA and β-carotene could induce GM-CSF and IL-16 expression in murine macrophages by RT-PCR. As shown in Fig. 2A, atRA at 0.1 μM was sufficient to increase GM-CSF and IL-16 mRNA levels in murine RAW264.7 macrophages. Surprisingly, β-carotene did not have such an effect, even at 10 μM. However, retinol also caused GM-CSF and IL-16 expression in a concentration-dependent manner (Fig. 2B). An ELISA indicated that a higher concentration of atRA was capable of inducing GM-CSF release into the culture medium (Fig. 2C).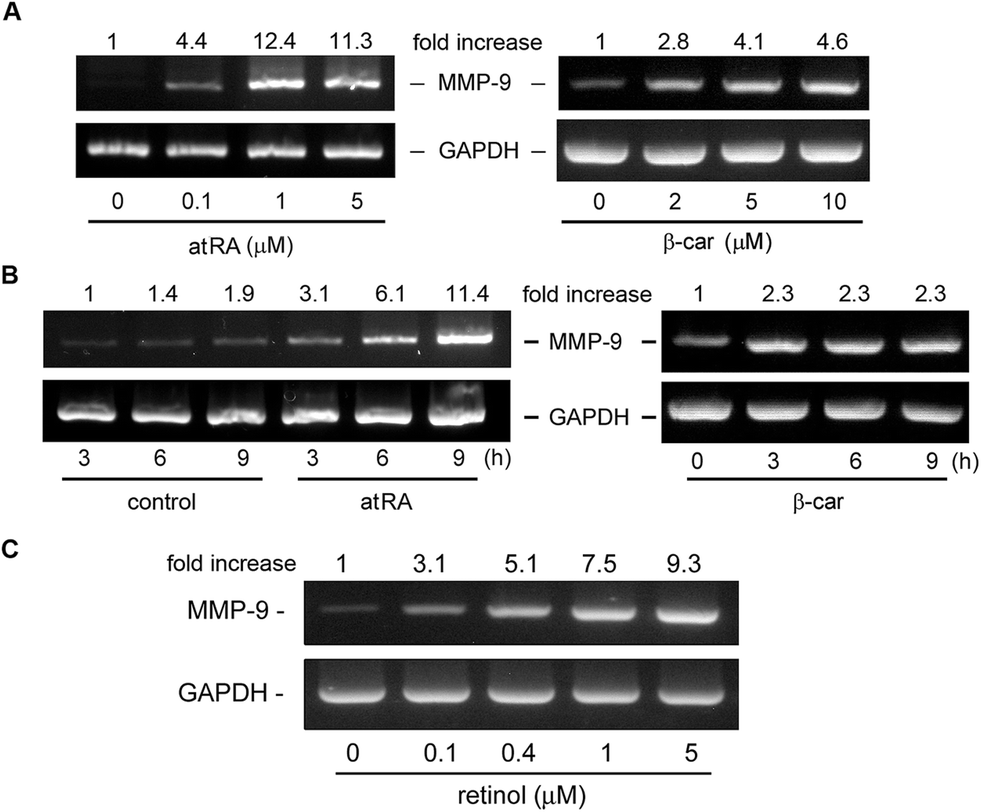 | ||
| Fig. 1 Effect of atRA, retinol, and β-carotene on the MMP-9 mRNA level. RAW264.7 macrophages were treated with (A and C) the indicated concentrations of atRA, β-carotene (β-car), or retinol for 6 h or (B) atRA (0.1 μM) and β-carotene (10 μM) for the indicated time intervals. MMP-9 and GAPDH mRNA level were analyzed by RT-PCR (n = 3). | ||
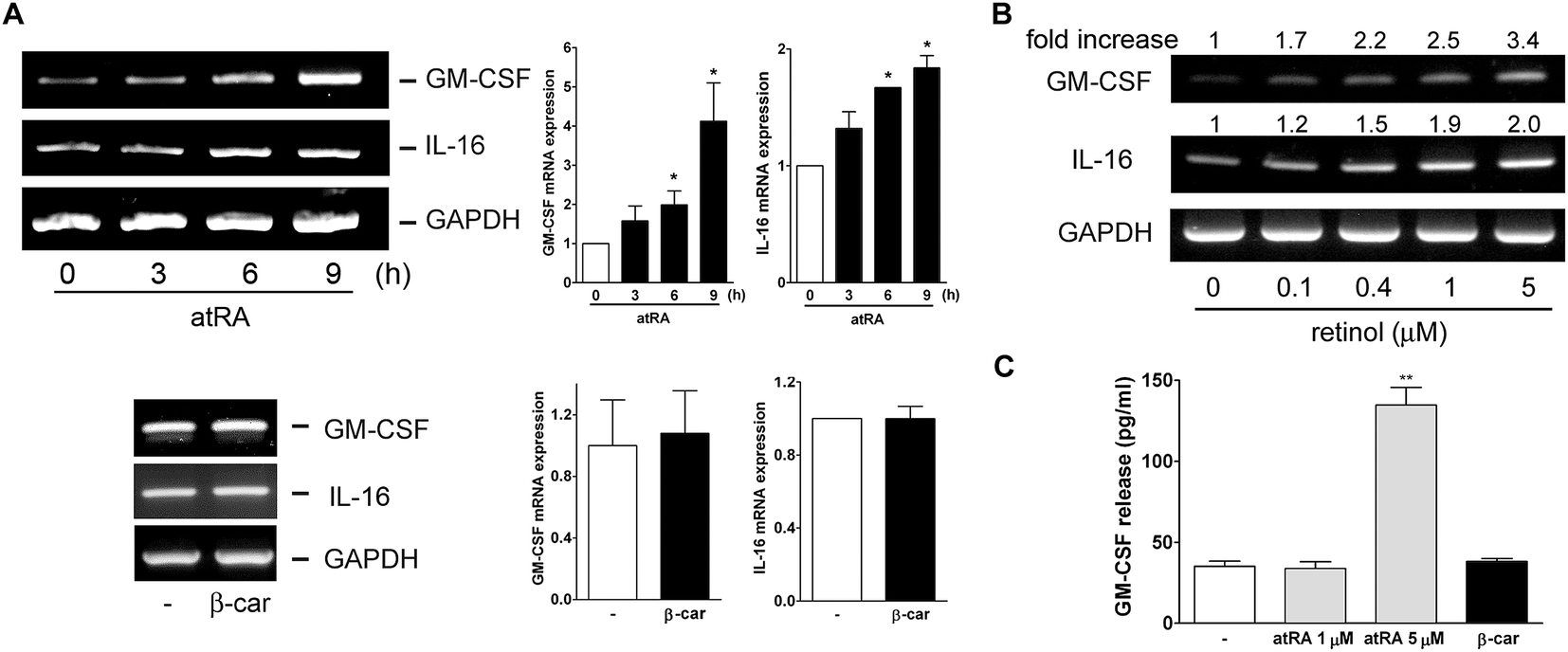 | ||
| Fig. 2 Effect of atRA, retinol, and β-carotene on GM-CSF and IL-16 expression. (A and B) RT-PCR analysis of GM-CSF and IL-16 mRNA level in RAW264.7 macrophages. Cells were treated with (A) atRA (0.1 μM) for the indicated time intervals or β-carotene (β-car, 10 μM) for 6 h or (B) retinol for 8 h. After total RNA was extracted, GM-CSF, IL-16 and GAPDH mRNA levels were analyzed by RT-PCR and was analyzed by densitometry (n = 3). (C) Macrophages were treated with atRA and β-carotene (β-car) for 16 h, and GM-CSF release into the culture medium was determined by ELISA (n = 3–4). *P < 0.05 and **P < 0.001 versus basal level. | ||
To investigate whether MMP-9 protein production was simultaneously increased by these substances, we performed gelatin zymography and Western blotting. Fig. 3A shows that atRA, retinol, and β-carotene enhanced MMP-9 production in the culture media of RAW264.7 macrophages. To confirm this observation, we examined whether these substances had a similar inducing effect on primary-cultured peritoneal macrophages. As shown in Fig. 3B, atRA, β-carotene, and retinol induced MMP-9 release in primary-cultured peritoneal macrophages in a concentration-dependent manner, 0.1 μM of atRA, 1 μM of retinol and 2 μM of β-carotene were sufficient to cause MMP-9 production. RT-PCR analysis also confirmed an increase in the MMP-9 mRNA expression by retinol in peritoneal macrophages.
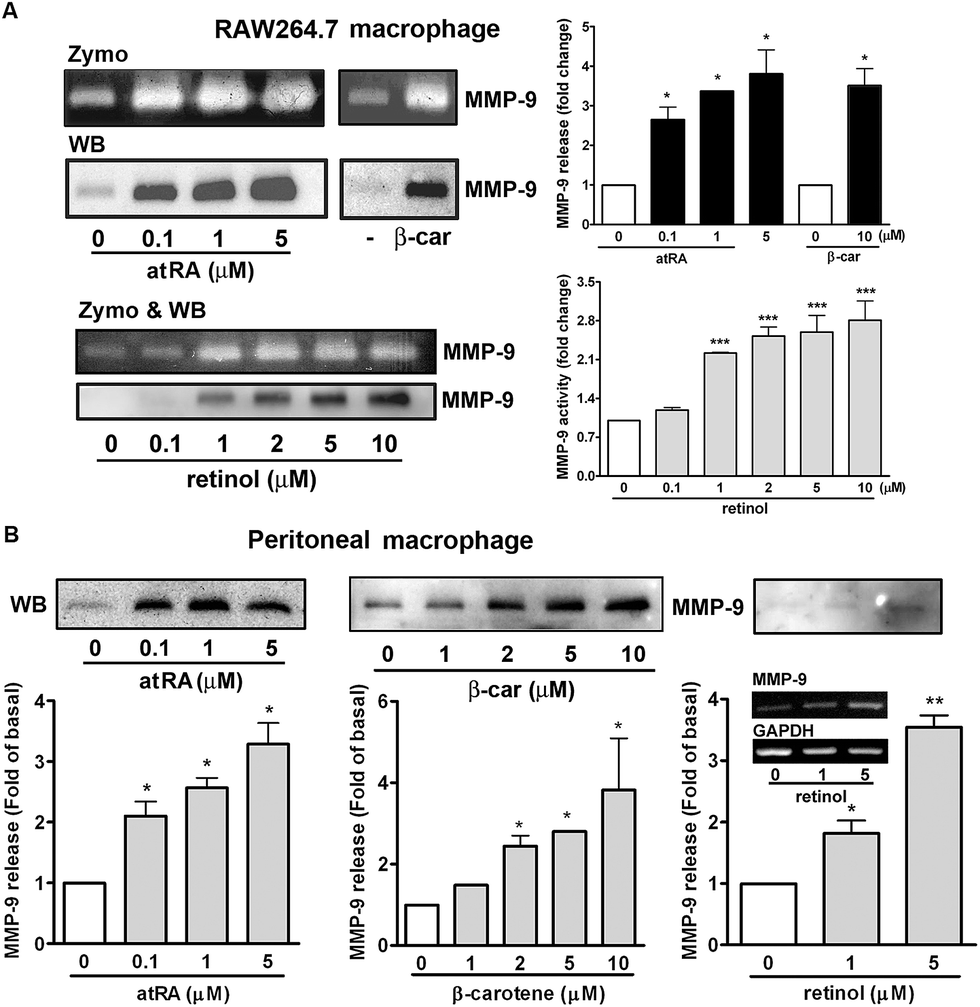 | ||
| Fig. 3 Effect of atRA, retinol, and β-carotene on MMP-9 production. (A) RAW264.7 or (B) primary-cultured peritoneal macrophages were treated with atRA, β-carotene (β-car), or retinol for 16 h. MMP-9 production in the culture media was analyzed by gelatin zymography (zymo) and/or Western blotting (WB). The MMP-9 release or activity was quantified by densitometry (n = 3–5). *P < 0.05 versus control. An inset in right panel of (B) indicates a representative RT-PCR result of MMP-9 mRNA expression in peritoneal macrophages by retinol treatment. | ||
3.2. Role of RAR in atRA-, retinol-, and β-carotene-induced GM-CSF, IL-16, and MMP-9 expression
The RARs bind atRA and 9-cis RA, whereas the RXRs only bind 9-cis RA.7 We have observed that atRA and retinol could induce GM-CSF, IL-16, and MMP-9 expression, whereas β-carotene only induced MMP-9 expression. To investigate whether they affected macrophage activity through NRs activation, specific inhibitors targeting RAR were used for pharmacological intervention. Fig. 4A shows that CD2665 (a RARβ/γ antagonist) obviously inhibited β-carotene-induced MMP-9 release but did not affect atRA-induced MMP-9 release and mRNA levels. This was confirmed by the observations that LE135 (an antagonist specific for RARβ) but not MM11253 (an antagonist specific for RARγ) abolished β-carotene-induced MMP-9 activity and expression. In Fig. 4B, we show that BMS195614 (a selective RARα antagonist) inhibited atRA- and β-carotene-induced MMP-9 production. In line with these observations, the RARα agonist (AM80) and RARβ/γ agonist (adapalene) caused a robust increase in MMP-9 activity and UVI3003 (a RXR antagonist) did not affect atRA and β-carotene-induced MMP-9 release (Fig. S2†). Interestingly, Fig. 4C shows that the inhibitory profile of retinol was similar to that of β-carotene but not to that of atRA. CD2665, LE135, and BMS195614 were able to inhibit retinol-induced MMP-9 production. These indicated differential involvement of RARβ and α in MMP-9 induction by β-carotene, retinol, and atRA.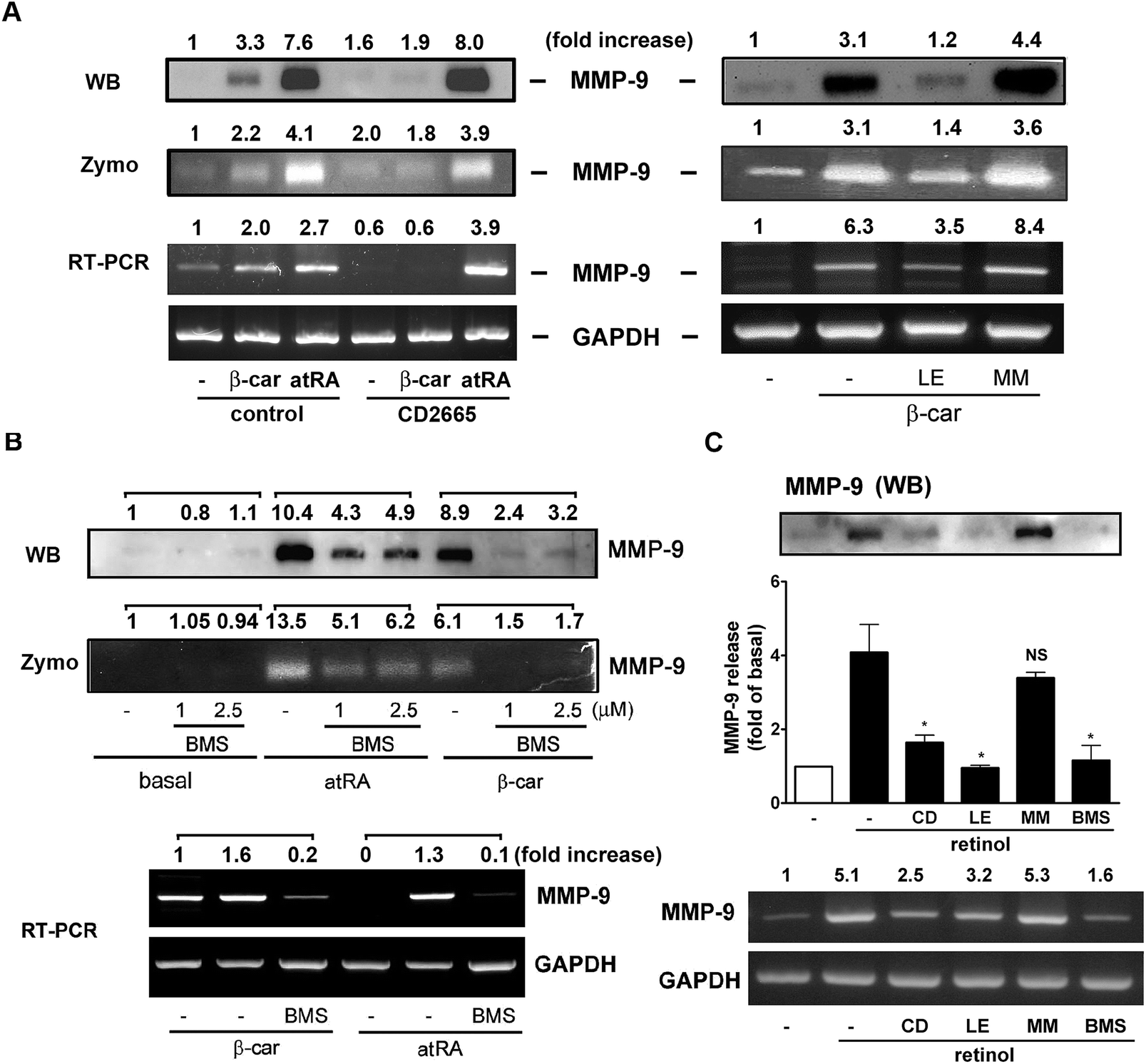 | ||
| Fig. 4 Effect of RAR antagonists on atRA-, retinol, and β-carotene-induced MMP-9 production. RAW264.7 macrophages were pretreated with (A) CD2665 (CD, 1 μM), LE135 (LE), or MM11253 (MM) (1 μM for each) or (B) BMS195614 (BMS, 1 μM) for 1 h. Cells were then incubated with atRA (0.1 μM) or β-carotene (β-car, 10 μM) for 16 h, the MMP-9 release in medium was analyzed by zymography (zymo) and Western blotting (WB), and the MMP-9 mRNA level in the cells was analyzed by RT-PCR (n = 4). (C) CD2665, LE135, and BMS195614 (1 μM for each) inhibited retinol (1 μM)-induced MMP-9 protein and mRNA production (n = 3). *P < 0.05 versus control. NS: non significant. | ||
On the other hand, whether NRs activation was responsible for GM-CSF and IL-16 expression in macrophages was investigated. As shown in Fig. 5, CD2665 and LE135 obviously inhibited atRA- and retinol-induced GM-CSF and IL-16 expression, whereas BMS195614 did not. This suggested that atRA and retinol induced GM-CSF and IL-16 expression via RARβ activation.
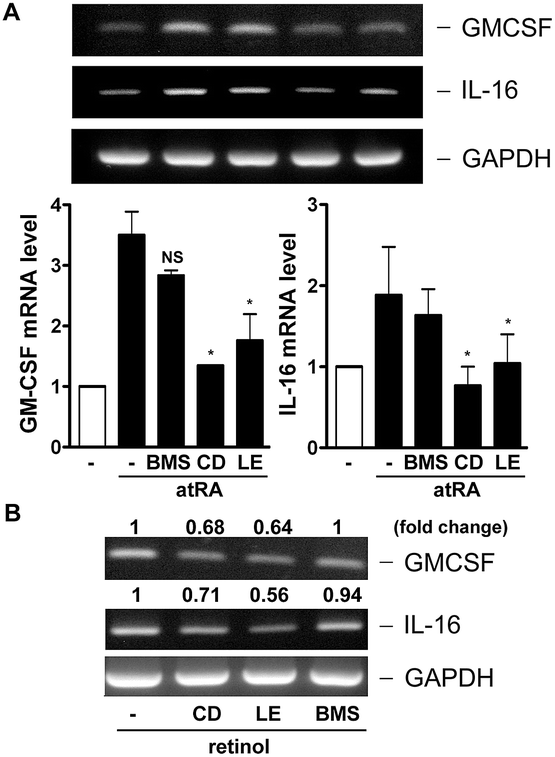 | ||
| Fig. 5 Effect of RAR antagonists on atRA- and retinol-induced GM-CSF and IL-16 expression. RAW264.7 macrophages were pretreated with BMS195614 (BMS, 1 μM), CD2665 (CD, 1 μM), LE135 (LE), or MM11253 (MM) (1 μM for each) for 1 h. Cells were then incubated with (A) atRA (0.1 μM) for 6 h or (B) retinol (1 μM) for 8 h. GM-CSF, IL-16 and GAPDH mRNA levels were analyzed by RT-PCR (n = 2–3). | ||
Next, we used a RA response element (RARE) luciferase reporter system (pRAR-Luc) to determine whether atRA and β-carotene could induce RAR activation. Cells with pRAR-Luc reporter, when treated with atRA and β-carotene markedly increased RARE-mediated luciferase activity by 2.5–7 and 1.8–3-fold, respectively (Fig. 6A), suggesting that atRA and β-carotene could induce RAR activation. In parallel, the luciferase reporter assay, using a construct with murine MMP-9 promoter region, revealed that atRA and β-carotene slightly enhanced luciferase activity (Fig. 6B).
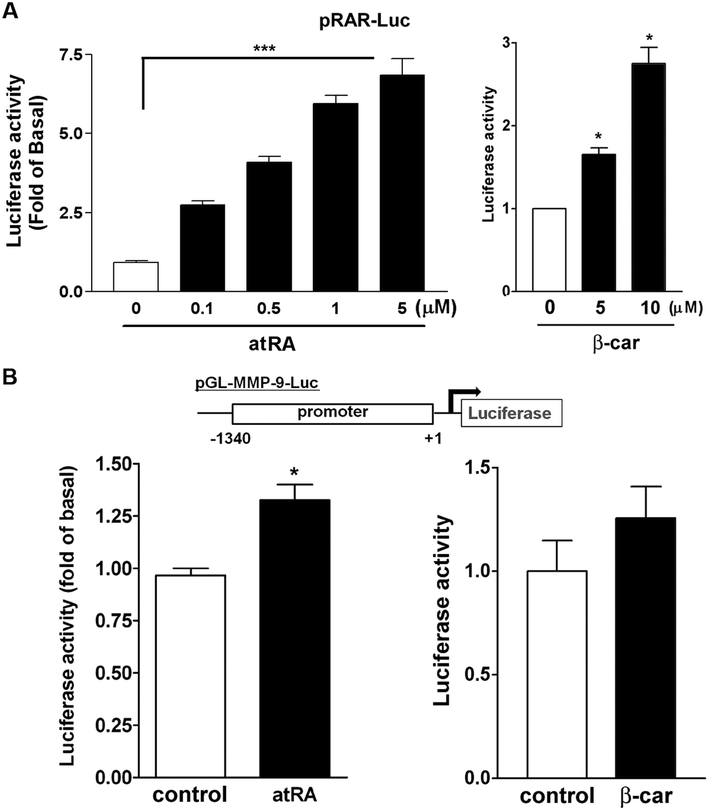 | ||
| Fig. 6 Role of RAR activation in atRA- and β-carotene-induced MMP-9 production. (A) The RARE luciferase reporter or (B) the 1340-bp MMP-9 reporter construct was transiently transfected into RAW264.7 macrophages for 18 h, followed by maintaining for 24 h in endotoxin-free medium. Cells were treated with vehicle (control), atRA (0.1 μM), or β-carotene (10 μM) for 12 h. Cells were then lyzed and luciferase activity was determined by luminometry (n = 3–4). *P < 0.05, ***P < 0.001 versus control. | ||
3.3. Role of MAPKs and PI-3K in atRA-, retinol-, and β-carotene-induced MMP-9 production
It has been reported that retinoids mediate transcription through protein kinases and mitogen-activated protein kinases (MAPKs).20 In our previous study, we also demonstrated that ERK1/2 and p38 MAPK were involved in MMP-9 regulation by lutein. To investigate the possible signaling pathway(s) responsible for induction of MMP-9 by atRA, retinol, and β-carotene, we used various inhibitors targeting cellular kinases, including PD (targets ERK1/2 upstream kinase), SB (targets p38 MAPK), SP (targets JNK), and LY (targets PI-3K). It was shown that all of these inhibitors failed to block atRA-induced MMP-9 release, and inversely, SP slightly enhanced atRA-induced MMP-9 release (Fig. 7A, a). Interestingly, PD slightly inhibited retinol-induced MMP-9 release and the mRNA level (Fig. 7A, b). Moreover, PD markedly and significantly reduced β-carotene-induced MMP-9 release (Fig. 7B, a) and the MMP-9 mRNA level (panel b). SB and LY slightly affected β-carotene-induced MMP-9 expression, but it was not statistically significant. Further, the activation status of MAPKs, PI-3K, and Akt in RAW264.7 macrophages by atRA or β-carotene was examined. Fig. 7C shows that β-carotene markedly induced ERK1/2, p38 MAPK, JNK, PI-3K, and Akt activation, whereas atRA slightly induced ERK1/2 and markedly affected p38 MAPK activation.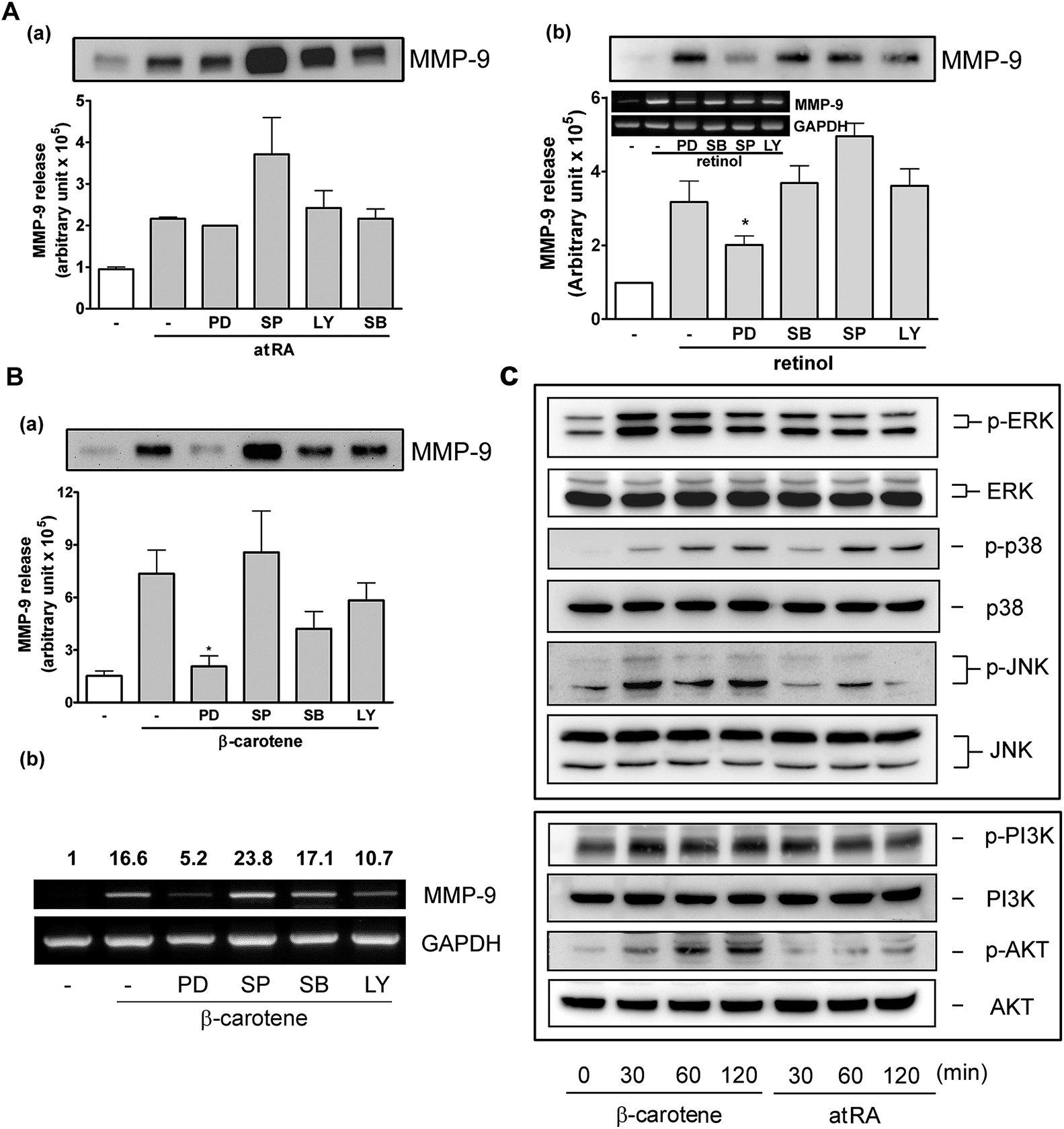 | ||
| Fig. 7 Role of MAPKs and PI-3K in atRA, retinol, and β-carotene-induced MMP-9 production. (A and B) Effect of MAPKs and PI-3K inhibitors on MMP-9 production. RAW264.7 macrophages were pretreated with DMSO or the signaling inhibitors (10 μM each) for 30 min and followed by the addition of (A) atRA (0.1 μM) or retinol (1 μM), or (B) β-carotene (10 μM) for 16 h. MMP-9 production in the culture media was analyzed by Western blotting (n = 3) and the MMP-9 mRNA level was analyzed by RT-PCR (n = 4). The fold increase of the MMP-9 mRNA level, determined by densitometry, was indicated above the panel. *P < 0.05 versus control. (C) Effect of atRA and β-carotene on MAPKs, PI-3K and Akt activation. Macrophages were treated with β-carotene (10 μM) or atRA (0.1 μM) for the indicated time intervals. The protein kinase activation and expression were analyzed by Western blotting (n = 3). | ||
3.4. AtRA, retinol, and β-carotene enhance macrophage phagocytosis
In a functional study, we examined whether atRA, retinol, and β-carotene enhanced macrophage phagocytosis. A phagocytic assay by fluorescence microscopy was performed.16 Quantitative analysis of the fluorescent spots, representing the ingested yeasts, demonstrated that the phagocytosis was increased by atRA, retinol, and β-carotene treatment but was reduced in the presence of MMP inhibitor-GM6001 (Fig. 8).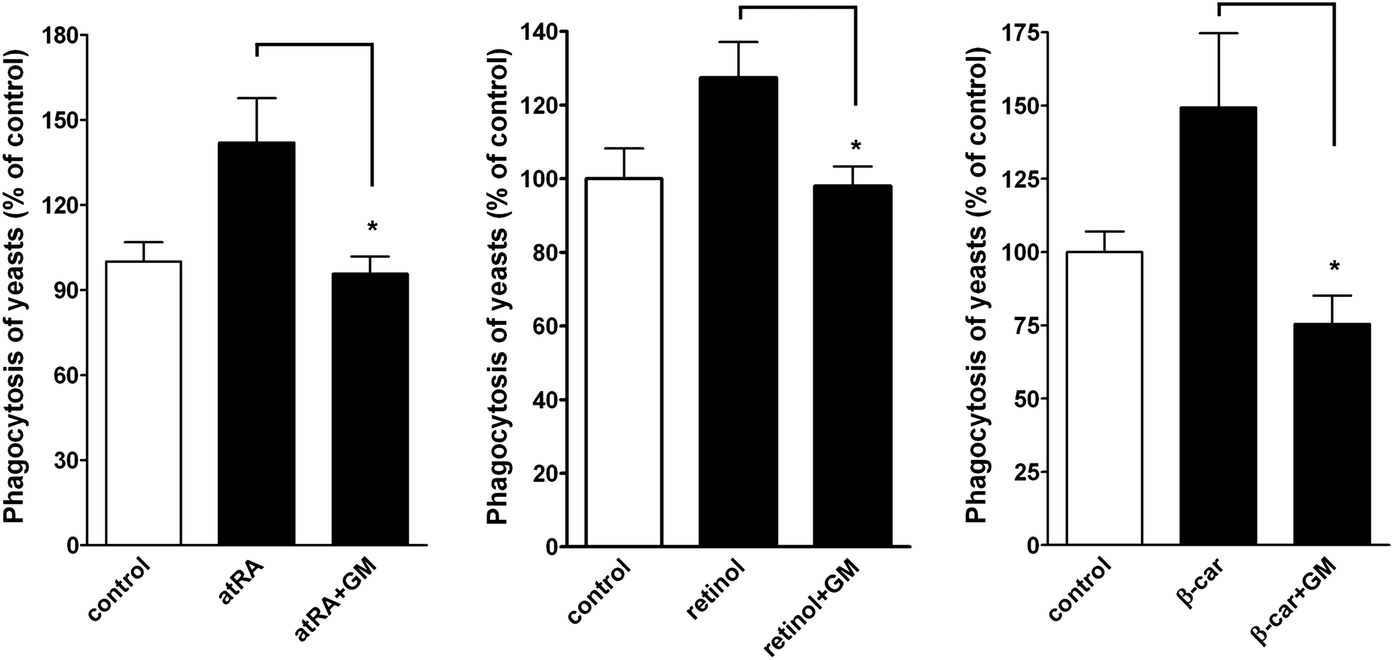 | ||
| Fig. 8 Effect of atRA, retinol, and β-carotene on phagocytosis. RAW264.7 macrophages were primed with DMSO (control), atRA (5 μM), retinol (1 μM) or β-carotene (10 μM) in the presence of vehicle or GM6001 (10 μM) for 16 h, followed by incubation with fluorescent dye-loaded yeasts for 1 h. At the end of incubation, phagocytosis was analyzed by fluorescence microscopy (n = 3–5). Data were expressed as a percentage of control. *P < 0.05. | ||
4. Discussion
In this study, we evaluated whether atRA, retinol, and β-carotene affected the production of proteases and cytokines in murine macrophages. We demonstrate here for the first time that atRA, retinol, and β-carotene have differential effects on GM-CSF and IL-16 expression in murine RAW264.7 macrophages. Both atRA and retinol could induce apparent GM-CSF and IL-16 mRNA expression at a transcriptional level and atRA induced GM-CSF protein release at a higher concentration. In contrast, β-carotene did not cause GM-CSF and IL-16 mRNA expression and release (Fig. 2). Because these three substances affected MMP-9 production, both in murine RAW264.7 and primary-cultured peritoneal macrophages, their detailed action mechanisms were investigated throughout this study. According to a previous study, the predominant carotenoids found in human plasma are lycopene, β-carotene, and lutein, and their concentrations vary from 0 to 8 μM, depending upon dietary intake.21 In the present study, 0.1 μM of atRA, 1 μM of retinol, and 2 to10 μM of β-carotene were used in most of experiments. Therefore, the concentrations used were reasonable and within physiological range.GM-CSF, IL-16, and MMP-9 induction by atRA, retinol, and/or β-carotene appears to be through a transcriptional regulation and is RAR-dependent. Although RARs and RXRs have been shown to function as RXR/RAR heterodimers, in this study RXR was unlikely to be involved in induction of these genes by atRA, retinol and β-carotene because the RXR antagonist UVI3003 did not affect MMP-9 induction (Fig. S2†). The RARE luciferase reporter assay indicated that atRA and β-carotene markedly induced RAR interaction with RARE (Fig. 6A). In contrast to lipopolysaccharide (LPS), they only weakly induced MMP-9 luciferase reporter activity in murine macrophages (data not shown). LPS has been shown to induce MMP-9 expression through the ROS/p38 kinase-dependent pathway, leading to activation of AP-1 activity in RAW264.7 macrophages.22 Using the Transcription Element Search System (http://www.cbil.upenn.edu/cgi-bin/tess/tess), we found several AP-1 binding sites but only one RARβ/γ and RXRα/β binding sequence within our 1340-bp mmp-9 promoter-luciferase reporter construct (data not shown). However, there is no RARα binding sequence within it. A previous study has shown that the promoter region upstream of the murine mmp-9 gene does not contain a canonical consensus RARE.23 However, in our previous study, the carotenoid lutein induced this mmp-9 promoter-luciferase activity about 1.5 fold than control in the same cells.16 As reported by others, the MMP-9 production in myeloid dendritic cells by atRA has been suggested to act through a nonclassical (epigenetic) transcriptional mechanism, which involves RARα promoter binding and recruitment of coregulators.24,25 Our results indicated that RARα was required for atRA-induced MMP-9 production, whereas RARβ was primarily responsible for atRA and retinol-induced GM-CSF and IL-16 expression. On the other hand, both RARα and β were essential for retinol- and β-carotene-induced MMP-9 induction (Fig. 4). A previous study also reported that β-cryptoxanthin and lutein serve as ligands for RAR but not for RXR.9 Interestingly, atRA is sufficient to cause MMP-9 production at a concentration of 0.1 μM, but only exhibited 1.5-fold luciferase activity at a concentration of 5 μM. In addition, lutein and β-carotene displayed similar MMP-9 inductory activity through RARβ activation, but exhibited differential luciferase activity. These findings need to be explored in our future studies. It is possible that these substances bind to RAR but weakly interact with mmp-9 promoter through unidentified binding sequence(s) or indirectly affect mmp-9 promoter activity through an epigenetic mechanism, as observed before that atRA increases MMP-9 production in myeloid dendritic cells through RARα.24,25
The MMP-9 activity, production, and mRNA level induced by β-carotene were markedly reduced by PD98059 and slightly suppressed by SB202190 and LY294002. In addition, the MMP-9 production and mRNA level induced by retinol was slightly affected by PD98059. However, atRA-induced MMP-9 activity and production was not affected by these inhibitors (Fig. 7A and B). It has been reported that atRA induces nuclear factor kappaB (NF-κB) activation and MMP-9 expression in human SK-N-BE 9N neuroblastoma cells.26 However, the NF-κB signaling inhibitor (BAY 11-7085, 5 μM), protein kinase (PK) A inhibitor (H-89, 5 μM), PKC inhibitor (GF109203X, 1 μM), and PPAR inhibitor (GW9662, 10 μM) all did not affect atRA- and β-carotene-induced MMP-9 activity (data not shown). These data suggest that ERK1/2 activation plays a certain role in retinol- and β-carotene-induced MMP-9 production in RAW264.7 macrophages. This is supported by the observation that β-carotene induced marked ERK1/2, PI-3K and Akt activation in parallel and atRA primarily caused p38 MAPK activation (Fig. 7C). It has been reported that retinol and atRA induce ERK1/2 phosphorylation but increase MMP-2 activity by different pathways in cultured Sertoli cells. The authors suggested that ROS contributes to the action of retinol but not to that of atRA.27,28 The various signal transduction pathways can also cross-talk with retinoid receptors transactivation through phosphorylation of the coactivators and corepressors. In addition, RARβ and γ have been reported to be phosphorylated by MAPKs.20 In this study, atRA did not cause ROS production in RAW264.7 macrophages, even at a higher concentration of 5 μM (data not shown). Therefore, we suggested that atRA drives mmp-9 transcription through direct RARα activation, whereas β-carotene and retinol may activate MAPKs and RARα or β and then drives MMP-9 transcription machinery.
AtRA or vitamin A have been shown to upregulate MMP-9 expression in some cell types29,30 but downregulate it in other cell types.31 To our knowledge, we have shown for the first time that atRA and retinol are able to induce GM-CSF and IL-16 expression and MMP-9 production in macrophages. GM-CSF is a cytokine that can stimulate stem cells to produce granulocytes and has been implicated to activate macrophage and IL-16 is a pleiotropic cytokine that functions as a chemoattractant, a modulator of T cell activation. Our previous study and the work reported here has also shown that MMP-9 was involved in macrophage phagocytosis.16 These results indicate atRA, retinol, and β-carotene with a different immunomodulatory activity. However, it still needs to be further investigated why β-carotene, with a similar structure to lutein, did not affect GM-CSF and IL-16 expression (Fig. S1†).
5. Conclusions
We provide here the first evidence that shows that atRA, retinol, and β-carotene differentially cause GM-CSF, IL-16, and MMP-9 production in murine macrophages. An appropriate increased macrophage activity leads to immunomodulation, anti-tumor activity, wound-healing and other therapeutic effects.32 Therefore, the results presented here demonstrate atRA, retinol, and β-carotene with modulatory activity on macrophages, explaining at least in part why these substances are beneficial for immunity.Acknowledgements
This work was supported by the grants of the National Science Council (NSC 98-2320-B-030 -001 -MY3) and Shin Kong Wu Ho-Su Memorial Hospital (100-SKH-FJU-06)Notes and references
- R. M. Niles, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 2004, vol. 555, pp. 97–105 Search PubMed.
- S. S. Twining, D. P. Schulte, P. M. Wilson, B. L. Fish and J. E. Moulder, J. Nutr., 1997, 127, 558–565 CAS.
- C. B. Stephensen, Annu. Rev. Nutr., 2001, 21, 167–192 CrossRef CAS PubMed.
- H. Green and E. Mallanby, Br. J. Exp. Pathol., 1930, 11, 88–89 Search PubMed.
- S. Clausen, Trans. Am. Pediatr. Soc., 1931, 43, 27–30 Search PubMed.
- A. El-Agamey, G. M. Lowe, D. J. McGarvey, A. Mortensen, D. M. Phillip, T. G. Truscott and A. J. Young, Arch. Biochem. Biophys., 2004, 430, 37–48 CrossRef CAS PubMed.
- M. Mark, N. B. Ghyselinck and P. Chambon, Annu. Rev. Pharmacol. Toxicol., 2006, 46, 451–480 CrossRef CAS PubMed.
- Y. Sharoni, K. Linnewiel-Hermoni, M. Khanin, H. Salman, A. Veprik, M. Danilenko and J. Levy, Mol. Nutr. Food Res., 2012, 56, 259–269 CAS.
- A. Matsumoto, H. Mizukami, S. Mizuno, K. Umegaki, J.-i. Nishikawa, K. Shudo, H. Kagechika and M. Inoue, Biochem. Pharmacol., 2007, 74, 256–264 CrossRef CAS PubMed.
- V. Duong and C. Rochette-Egly, Biochim. Biophys. Acta, Mol. Basis Dis., 2011, 1812, 1023–1031 CrossRef CAS PubMed.
- N. Morrissette, E. Gold and A. Aderem, Trends Cell Biol., 1999, 9, 199–201 CrossRef CAS.
- N. L. Webster and S. M. Crowe, J. Leukocyte Biol., 2006, 80, 1052–1066 CrossRef CAS PubMed.
- C. M. Overall and C. Lopez-Otin, Nat. Rev. Cancer, 2002, 2, 657–672 CrossRef CAS PubMed.
- A. E. Starr, C. L. Bellac, A. Dufour, V. Goebeler and C. M. Overall, J. Biol. Chem., 2012, 287, 13382–13395 CrossRef CAS PubMed.
- S. MacLauchlan, E. A. Skokos, N. Meznarich, D. H. Zhu, S. Raoof, J. M. Shipley, R. M. Senior, P. Bornstein and T. R. Kyriakides, J. Leukocyte Biol., 2009, 85, 617–626 CrossRef CAS PubMed.
- H.-M. Lo, C.-L. Chen, C.-M. Yang, P.-H. Wu, C.-J. Tsou, K.-W. Chiang and W.-B. Wu, J. Leukocyte Biol., 2013, 93, 723–735 CrossRef CAS PubMed.
- H. M. Lo, C. F. Hung, Y. L. Tseng, B. H. Chen, J. S. Jian and W. B. Wu, Biochem. Pharmacol., 2007, 74, 54–63 CrossRef CAS PubMed.
- C.-P. Chen, C.-F. Hung, S.-C. Lee, H.-M. Lo, P.-H. Wu and W.-B. Wu, Naunyn-Schmiedebergs Arch. Pharmacol., 2010, 381, 401–414 CrossRef CAS PubMed.
- M. R. Green and J. Sambrook, Molecular cloning: A laboratory manual, Cold Spring Habor Laboratory Press, 2012 Search PubMed.
- J. Bastien and C. Rochette-Egly, Gene, 2004, 328, 1–16 CrossRef CAS PubMed.
- A. Collins, B. Olmedilla, S. Southon, F. Granado and S. Duthie, Carcinogenesis, 1998, 19, 2159–2162 CrossRef CAS PubMed.
- C.-H. Woo, J.-H. Lim and J.-H. Kim, J. Immunol., 2004, 173, 6973–6980 CAS.
- J. E. Balmer and R. Blomhoff, J. Lipid Res., 2002, 43, 1773–1808 CrossRef CAS PubMed.
- D. E. Lackey and K. A. Hoag, J. Nutr., 2010, 140, 1502–1508 CrossRef CAS PubMed.
- D. E. Lackey, S. L. Ashley, A. L. Davis and K. A. Hoag, J. Nutr., 2008, 138, 1512–1519 CAS.
- A. R. Farina, M. P. Masciulli, A. Tacconelli, L. Cappabianca, G. De Santis, A. Gulino and A. R. Mackay, Cell Growth Differ., 2002, 13, 343–354 CAS.
- R. J. S. Dalmolin, A. Zanotto-filho, R. B. de Oliveira, R. F. Duarte, M. A. B. Pasquali and J. C. F. Moreira, Free Radical Res., 2007, 41, 1338–1347 CrossRef CAS PubMed.
- D. P. Gelain, M. Cammarota, A. Zanotto-Filho, R. B. de Oliveira, F. Dal-Pizzol, I. Izquierdo, L. R. M. Bevilaqua and J. C. F. Moreira, Cell. Signalling, 2006, 18, 1685–1694 CrossRef CAS PubMed.
- R. Zaragozá, A. Gimeno, V. J. Miralles, E. R. García-Trevijano, R. Carmena, C. García, M. Mata, I. R. Puertes, L. Torres and J. R. Viña, Am. J. Physiol.: Endocrinol. Metab., 2007, 292, E1140–E1148 CrossRef PubMed.
- A.-M. Chambaut-Guérin, S. Hérigault, P. Rouet-Benzineb, C. Rouher and C. Lafuma, J. Neurochem., 2000, 74, 508–517 CrossRef.
- M. Frankenberger, R. W. Hauck, B. Frankenberger, K. Haussinger, K. L. Maier, J. Heyder and H. W. Ziegler-Heitbrock, Mol. Med., 2001, 7, 263–270 CAS.
- I. A. Schepetkin and M. T. Quinn, Int. Immunopharmacol., 2006, 6, 317–333 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3fo60309a |
| This journal is © The Royal Society of Chemistry 2014 |