Advances in 5-hydroxymethylfurfural production from biomass in biphasic solvents
Basudeb
Saha
*ab and
Mahdi M.
Abu-Omar
*ab
aBrown Laboratory, Department of Chemistry and School of Chemical Engineering, Purdue University, 560 Oval Drive, West Lafayette, IN 47907, USA. E-mail: sahab@purdue.edu; mabuomar@purdue.edu
bThe Center for Direct Catalytic Conversion of Biomass to Biofuels (C3Bio), Discovery Park, Purdue University, West Lafayette, IN 47907, USA
First published on 24th September 2013
Abstract
Biomass derived 5-hydroxymethylfurfural (HMF) has emerged as an important platform chemical for the production of value added chemicals and liquid fuels that are currently obtained from petroleum. Although a significant amount of research has been performed over the past decade, the high production cost of HMF is still a bottleneck for its sustainable utilization for making other value added chemicals and fuels on a commercial scale. Among several factors, low product selectivity and high purification cost are major constraints. To address these drawbacks, HMF production methodology in recent years has been directed towards utilization of biphasic media for concurrent extraction of HMF into an organic phase immediately after its formation in the reactive phase. As a result, several dozens of journal and patent articles have appeared, demonstrating the benefit of biphasic media for effective HMF extraction. This review article summarizes the findings of the most recent research articles with critical discussion on the factors that enhance the performance of biphasic media. Particular emphasis has been given to the development of more effective extracting solvents and their beneficial effect in enhancing HMF yield and selectivity, improvement of partition coefficient, mechanistic role of the bi-functional acid catalysts and factors that control high HMF selectivity for solid catalysts.
 Basudeb Saha | Dr Basudeb Saha, born in Calcutta (India), graduated in chemistry at Calcutta University and received his Ph.D. from the Indian Association for the Cultivation of Science (India). He did postdoctoral research with Professor James Espenson at Iowa State University (USA), and then moved to the polyurethane business R&D division of Dow Chemical Company (USA) where he led several breakthrough and implementation research projects. Currently, he is an Associate Research Scientist at Purdue University and is pursuing research on utilization of bio-renewable feedstocks for chemicals and fuels production via effective catalysis. Prior to joining Purdue University, he worked at Delhi University (India) as an Associate Professor. |
 Mahdi M. Abu-Omar | Mahdi is a native of Jerusalem, Palestine. He moved to the U.S. in 1988 to pursue higher education. He completed his Ph.D. in Inorganic Chemistry in 1996 from Iowa State University under the guidance of James Espenson. After a postdoc at Caltech in the group of Harry Gray, Mahdi started his independent academic career at UCLA and moved to Purdue University in 2003, where he is now professor of chemistry and chemical engineering. Mahdi's research interest is in the design and development of transition-metal catalysts for renewable energy and environmental applications. |
1. Introduction
The awareness of climate change and rapid depletion of nonrenewable fossil sources, such as petroleum, and the high volatility in crude oil price caused by the imbalance in demand and supply, have put enormous pressure on the global economy.1 Thus, the development of environmentally and economically viable synthetic routes and related technologies for producing chemicals and fuels from non-fossil carbon sources has been reenergized in recent years after the U.S. Department of Energy published a list of “top ten biobased chemicals”.1 In this context, lignocellulosic biomass-derived 5-hydroxymethylfurfural (HMF) and furfural have emerged as important platform chemicals to meet the chemical and fuel needs of the next generation.1,2 The driving factors for this initiative from biorenewable sources are not only limited to developing new energy platforms and CO2 minimization, but also creating opportunities to secure the local supply of energy and support agricultural economics.3,4The production of chemicals and fuels from lignocellulosic biomass requires effective pretreatment and hydrolysis of cellulose and hemicellulose polymers of the biomass to the corresponding pentose and hexose sugar units, followed by catalytic dehydration of sugars to the corresponding furfural and HMF products. The production of HMF from mono- and polysaccharides, including pretreated biomass substrates, using homogeneous mineral acid, Brønsted acidic ionic liquids (IL), Lewis acidic metal halides and recyclable heterogeneous catalysts in pure organic or aqueous solvents has been reported.5–7 The subsequent transformation of HMF into other value added chemicals, such as promising next generation polyester building block monomers (2,5-furandicarboxylic acid (FDCA),8,9 2,5-bis(hydroxymethyl)furan (BHMF),10 and 2,5-bis(hydroxymethyl)tetrahydrofuran (BHMTF)) and potential biofuel candidates (2,5 dimethylfuran (DMF),11,12 5-ethoxymethylfurfural (EMF),13 ethyl levulinate (EL) and γ-valerolactone (gVL))2 (Fig. 1) has also been explored by different researchers using HMF as a starting substrate or directly from biomass in a one-pot process.
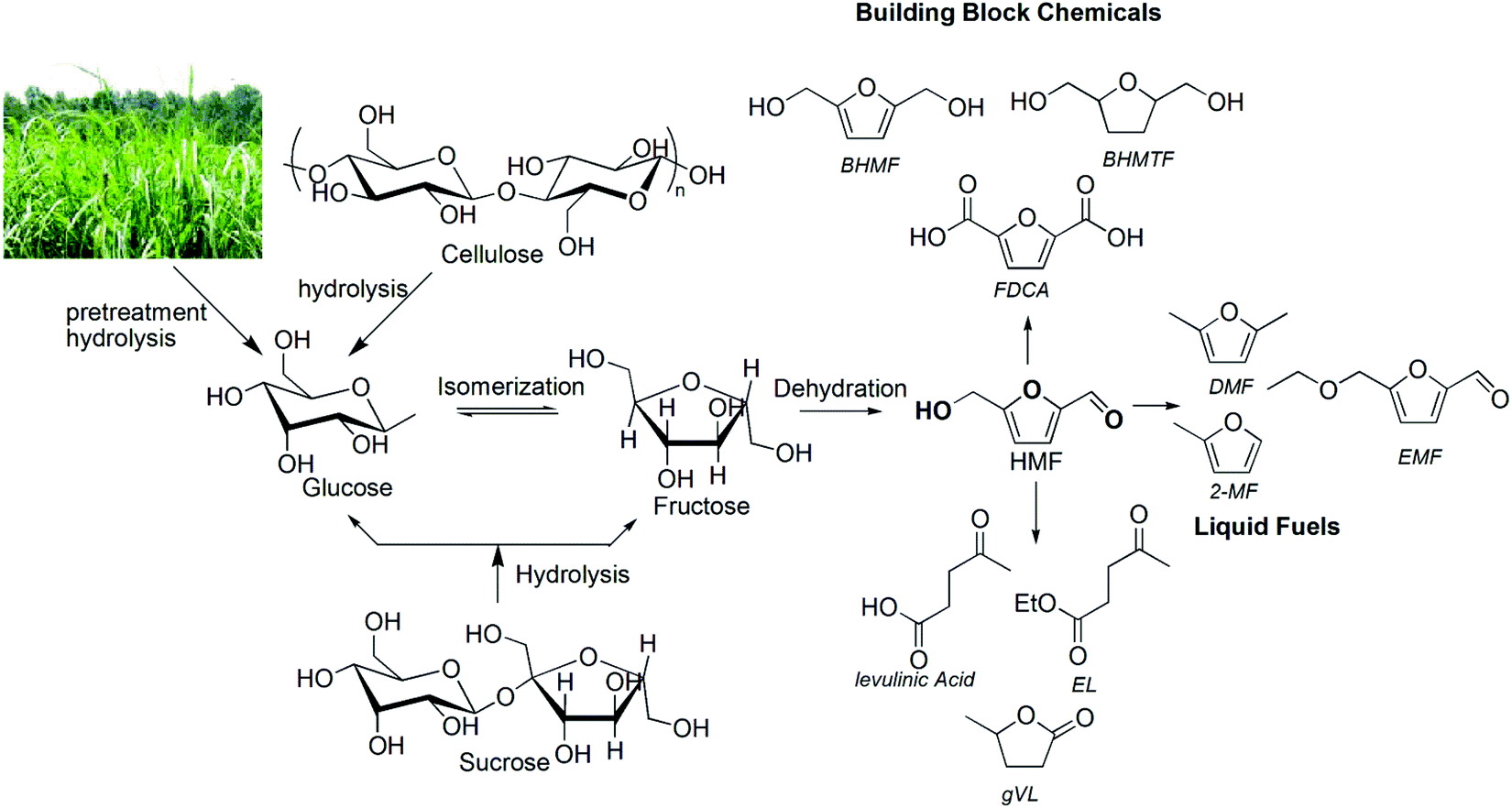 | ||
| Fig. 1 HMF production and its utilization routes for chemicals and liquid fuels. | ||
Additionally, HMF also functions as an anti-sickling agent for intermolecular sickle haemoglobin without inhibition by plasma and tissue proteins or other undesirable sequences.14 Experimental results show that HMF forms stable high-affinity Schiff base adducts with intramolecular hemoglobin and hence pre-treatment of transgenic sickle mice with HMF inhibited the formation of sickle cells. Despite the versatile application of HMF, the high production cost of HMF, owing to low product selectivity and high separation cost, is a major drawback to its sustainable utilization as a potential replacement for petroleum feedstock.
Besides the effectiveness of the catalyst for high yield and high selective in HMF production, solvents also play an important role in enhancing HMF yields; its purity and ease of separation make the process economically and environmentally competitive. Although the chemistry and engineering aspects of HMF production and its effective separation technology have advanced in recent years, most research efforts over the past decade were limited to achieving higher carbohydrate conversions in high boiling point organic solvents. These solvents include dimethylsulfoxide (DMSO), N,N-dimethylformide (DMF), N,N-dimethylacetamide (DMA), methyl isobutyl ketone (MIBK) and ILs. Several catalytic systems have been employed for the conversion of mono-, di-, polysaccharides and cellulose into HMF by utilizing the DMSO solvent system. Tong et al. reported fructose dehydration with Brønsted acidic IL catalysts, N-methyl-2 pyrrolidonium methyl sulfonate ([NMP]+[CH3SO3]−) and N-methyl-2-pyrrolidonium hydrogen sulfate ([NMP]+[HSO4]−) in DMSO, enabling maximum 72 mol% HMF yield with 87% selectivity from a reaction between 5.8 mmol fructose and 0.44 mmol [NMP]+[CH3SO3]− catalyst for 2 h at 90 °C.15 Other homogeneous and heterogeneous catalytic systems including SnCl4/tetra-butyl-ammonium bromide in DMSO,16 Amberlyst-70 in DMSO and N,N-demethylformamide,17 sulfonic acid (–HSO3) and IL functionalized mesoporous NPs in DMSO,18 1-hydroxyethyl-3-methylimidazolium tetrafluoroborate ([C2OHMIM]BF4) IL in DMSO,19 supported Sn-catalyst in DMSO,20 metal halides in DMSO and ILs,21 bifunctional SO42−/ZrO2–Al2O3 catalyst in DMSO–H2O,22 GeCl4 in DMSO–IL,23 CrCl2 in DMA–1-ethyl-3-imidazolium chloride ([EMIM]Cl),24 combined CuCl2–CrCl2 catalyst in ILs,25 and Brønsted–Lewis-surfactant-combined heteropolyacid (HPA), Cr[(DS)H2PW12O40]3, catalyst in ILs26 have shown moderate to high catalytic activity in HMF production. The most recent publication in this area of research showed the formation of 99 and 91 mol% HMF from fructose using Ti-containing and Ti-free sulfonated carbonaceous catalysts in DMSO.27,28 However, a potential drawback of the ILs solvent mediated process is that ILs are expensive and the separation of HMF from high boiling ILs is energy intensive. Besides the cost factor, ILs also deactivate by water, which is formed during the dehydration reaction. The high boiling points of DMSO and DMF also pose similar challenges for HMF separation, and therefore these processes are economically unfavorable on a commercial scale. Additionally, high concentrations of oligomeric species, humins, also form as by-products in organic solvent mediated dehydration reactions.29
To overcome these challenges, HMF production has been attempted in pure water. Zhu and co-workers have used a Lewis acidic ZnCl2 catalyst for the conversion of mono- and disaccharides into HMF in pure water.30 Mineral acid catalyzed hydrolysis and dehydration of carbohydrates in subcritical water was ineffective due to high reaction temperature (320 °C) and poor HMF selectivity.31 The aluminosilicate catalyzed dehydration of fructose in pure water suffered because of the low selectivity of the desired product, producing only 20–32 mol% HMF at 145 °C, perhaps due to rapid rehydration of HMF with Brønsted acidic aluminosilicate catalyst.32 In this context, Lewis and Brønsted acidic niobic acid, niobium or vanadium phosphate catalysts certainly claimed higher HMF yield (50 mol%) in pure water from fructose and inulin.33 This is perhaps the highest HMF yield reported to date in pure water without using any extracting solvent. The mesoporous TiO2 and H3PO4 treated Nb2O3 catalysts are reported to produce poor HMF (∼30%) in pure water.34,35 Thus, HMF production in monophasic solvent using pure water is not a viable option either, presumably due to the slow rate of HMF formation and rapid rehydration to furan ring opening products, levilininc acid (LA) and formic acid36,37 (Fig. 2).
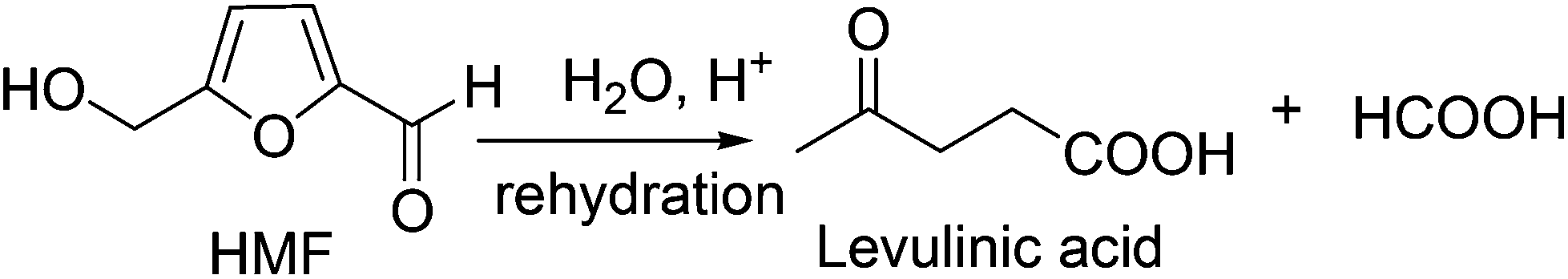 | ||
| Fig. 2 Rehydration of HMF with water. | ||
2. Advantages of biphasic solvents
Because of the aforementioned disadvantages of the monophasic solvent system using high boiling point organic solvents, ILs or pure water, current research efforts for HMF production have been directed towards the utilization of biphasic reaction systems in batch or continuous biphasic reactors. Aqueous or modified aqueous solution is used as the reactive phase for acid catalyzed conversion of starting substrates to HMF. The organic layer of the biphasic system acts as an extracting phase for continuous accumulation of HMF into the organic phase immediately after its formation in the reactive phase. Thus, lower concentration of HMF in the aqueous phase limits the rate of side reactions and thereby improves HMF yields.38 This method allows easy separation and reusability of the reactive aqueous phase containing spent homogeneous or heterogeneous catalysts. The partition coefficient (R), which is the ratio of HMF in the organic phase to that in the aqueous phase, is an important parameter in determining overall effectiveness of the biphasic media. Higher partitioning of HMF into the organic layer improves effective extraction and hence increases HMF selectivity. Besides the nature of the organic solvents in determining effective extraction, the presence of inorganic salt, e.g. NaCl, in the aqueous phase also plays an important role in improving the partition coefficient of HMF.Dumesic and co-workers measured the R values for HMF extraction using several organic solvents as the extracting phase in biphasic reaction systems in the presence and absence of NaCl in the reactive phase.39,40 A test reaction for fructose dehydration with a mineral acid catalyst at 150 °C and pH 0.6 shows that the R values for all organic solvents increased in the presence of NaCl in the aqueous phase and the relative increase of the R value depends on the solvent nature (Fig. 3). For example, 1-butanone showed a two-fold increase in the R-value from 1.6 to 3.2, whereas 2-butanone revealed a three-fold increase from 1.8 to 5.4. This significant improvement in the R value can be explained by the salting-out effect41,42 in which electrolytes alter the intermolecular bonding interactions between liquid components and decrease the mutual solubility of the aqueous and organic phases. The extent of the salting-out effect depends on the nature of ionic interactions between all components, nature of salts, concentrations, temperature and pressure.42
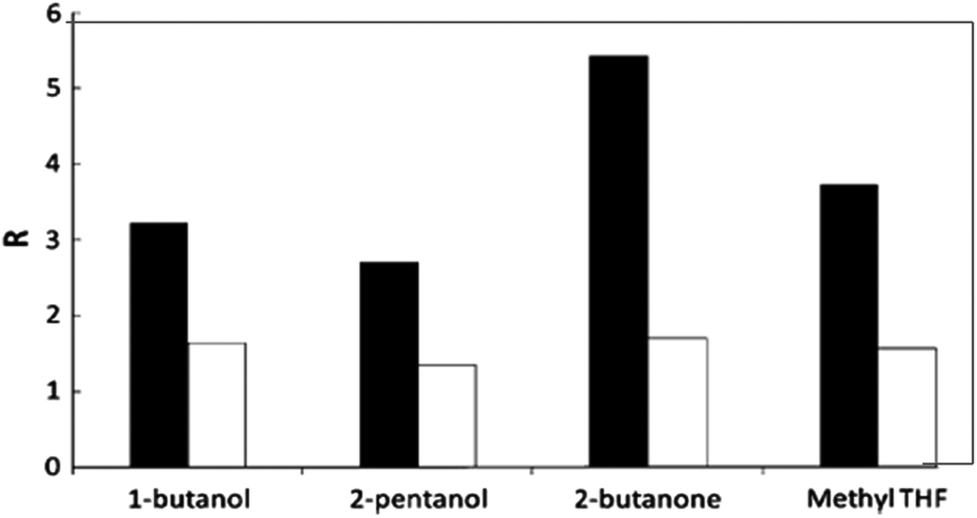 | ||
| Fig. 3 Effect of NaCl on the partition coefficient R of HMF extraction in representative biphasic systems. Black bars correspond to systems saturated with NaCl, while white bars represent systems without NaCl (modified from Fig. 2 of ref. 39). | ||
To further test the extent of the salting-out effect on the nature of salts, the authors further studied fructose dehydration in 1-butanol by varying the charge and size of cations and anions.39 Among several chloride salts, Na+ and K+ salts generated the highest partition coefficient and consequently produced HMF with maximum selectivity. This observation is attributed to the fact that smaller hydrated ions have the strongest influence on the salting out effect.43 Interestingly, Na2SO4 is found to be most effective at salting-out HMF in 1-butanol, giving a surprisingly high R-value of 8.1. Under comparable reaction conditions, a maximum of 68 wt% HMF yield is recorded in the 2-propanol and 2-butanone systems. The THF, 1-butanol and biorenewable Me–THF systems produced a maximum of 60, 59 and 66 wt% HMF, respectively. The presence of NaCl in the reactive aqueous phase also influenced higher partitioning of HMF into the organic phase in 0.25 M HCl catalyzed dehydration of fructose.11 In addition to a significant improvement of the R value from 1.5 to 3.5 with an increase of NaCl concentrations from 0 to 30 wt% in the water–2-butanol biphasic system, the selectivity of the HMF product also increased from 65 to 90% (Fig. 4), indicating a more efficient removal of HMF from the aqueous phase prevents undesired side products.
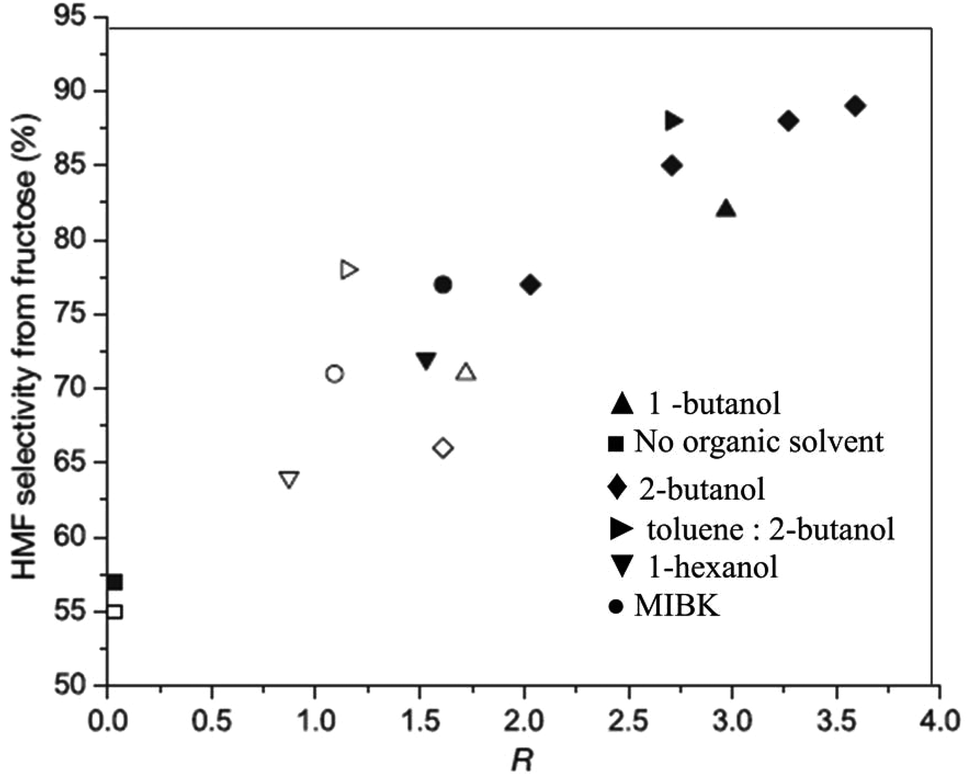 | ||
| Fig. 4 A correlation of partition coefficient of different organic solvents for HMF extraction from fructose dehydration versus HMF selectivity. Experiments without NaCl in the aqueous phase are represented by open symbols. Experiments with 30% NaCl in the aqueous phase are presented by solid symbols (modified from Fig. 3 of ref. 11). | ||
3. Scope
Because of the several benefits of the biphasic reaction systems, namely (i) high HMF yield and selectivity by preventing undesired side reactions, (ii) easy separation and reusability of the reactive phase containing spent catalysts and (iii) cost-effective separation of the desired product from lower boiling point extracting solvents, current research efforts in this area are directed towards the design and implementation of more effective biphasic solvents as well as reactor systems. As a result, dozens of journal and patent articles have been published over the past few years describing the beneficial effect of biphasic solvents on homogeneous and heterogeneous catalyzed carbohydrates and lignocellulosic biomass conversions, advancement of extracting organic phase from petroleum based solvents to environmentally friendly biorenewable solvents and the role of Lewis and Brønsted acidic sites of the catalyst on HMF yield and selectivity. The present review article is devoted exclusively to the latest advancement of HMF production technology in biphasic media with descriptive illustration of the results obtained by using different catalytic systems consisting of Brønsted acids, Lewis acids and a combination of both. Attention is paid to discussing the kinetic and mechanistic aspects of HMF formation, especially from glucose, which includes a very important step of isomerization of glucopyranose to fructofuranose.4. HMF production using homogeneous catalysts
To address the limitation of the HMF production technology suffered by poor product selectivity and separation issue, Dumesic et al.44 designed a modified biphasic reaction system (Fig. 5) using high boiling point polar aprotic solvents modified aqueous medium as a reactive phase for mineral acid (HCl) catalyzed dehydration of concentrated fructose solution (30–50 wt%). The modified reactive phase containing DMSO or poly(1-vinyl-2-pyrrolidinone) (PVP) or a combination of both DMSO–PVP as an additive with total mass up to 50%, based on mass of water, showed a noticeable increase in HMF yield in comparison to the reactive phase without a modifier. Among several organic solvents, such as methyl isobutyl ketone (MIBK), 1-butanol, 2-butanol, 1-hexane, MIBK–2 butanol mixture (7/3 w/w) and toluene–2 butanol mixture (5/5 w/w), that were tested as the organic phase, the modified system comprising water–DMSO–PVP (∼4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 weight ratio) as a reactive phase and MIBK–2 butanol (7:3 volume ratio) as an extracting phase offered the best HMF yield (76%) with 85% selectivity at 180 °C. The authors extended the application of the modified water–organic biphasic systems to HCl catalyzed conversions of mono-, di- and polysaccharides of C6/C5 sugar units to the corresponding furfural products.45 Under comparable reaction conditions, a biphasic system containing a water–DMSO mixture as the aqueous phase and MIBK–2 butanol (7:3 w/w) as the organic phase produced a maximum of 23, 50, 26, 27 and 75 wt% HMF from glucose, sucrose, starch, cellobiose and inulin, respectively at 170 °C (Table 1).
:1:2 weight ratio) as a reactive phase and MIBK–2 butanol (7:3 volume ratio) as an extracting phase offered the best HMF yield (76%) with 85% selectivity at 180 °C. The authors extended the application of the modified water–organic biphasic systems to HCl catalyzed conversions of mono-, di- and polysaccharides of C6/C5 sugar units to the corresponding furfural products.45 Under comparable reaction conditions, a biphasic system containing a water–DMSO mixture as the aqueous phase and MIBK–2 butanol (7:3 w/w) as the organic phase produced a maximum of 23, 50, 26, 27 and 75 wt% HMF from glucose, sucrose, starch, cellobiose and inulin, respectively at 170 °C (Table 1).
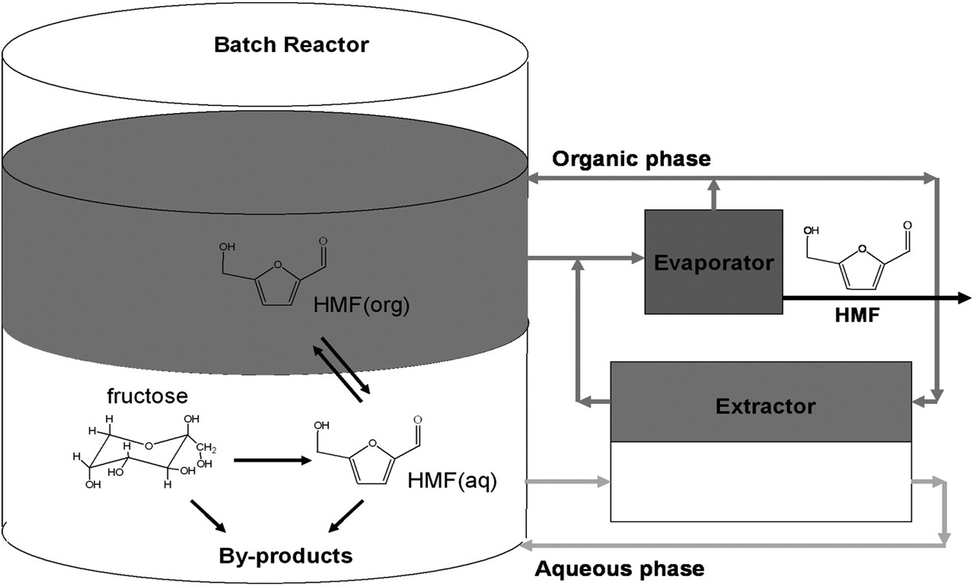 | ||
| Fig. 5 Schematic diagram of a biphasic reactor for HMF production via concurrent extraction and evaporation steps. The aqueous phase contains fructose, DMSO, PVP, and catalyst, and the organic phase contains MIBK and 2-butanol (modified from ref. 44). | ||
| Substrates | Reactive phase composition | pH | Time (min) | Conv. (%) | HMF selectivity (%) |
|---|---|---|---|---|---|
| Glucose | Water | 1.0 | 50 | 17 | 28 |
| Glucose | 4:6 H2O:DMSO |
1.0 | 10 | 43 | 53 |
| Glucose | 5:5 H2O:DMSO |
1.5 | 60 | 48 | 34 |
| Glucose | 5:5 H2O:DMSO |
1.0 | 17 | 50 | 47 |
| Xylose | 5:5 H2O:DMSO |
1.0 | 12 | 71 | 91 |
| Inulin | 5:5 H2O:DMSO |
1.5 | 5 | 98 | 77 |
| Sucrose | 4:6 H2O:DMSO |
1.0 | 5 | 65 | 77 |
| Cellobiose | 4:6 H2O:DMSO |
1.0 | 10 | 52 | 52 |
| Starch | 4:6 H2O:DMSO |
1.0 | 11 | 61 | 43 |
| Xylan | 5:5 H2O:DMSO |
1.0 | 25 | 100 | 66 |
Using this continuous reactor for subsequent hydrogenation of extracted HMF to the promising biofuel candidate, DMF46,47via hydrogenolysis of C–O bonds to HMF over a RuCu/C catalyst, the authors demonstrated that the formation of undesired side products was suppressed to a significant extent in the presence of a modifier in the reactive aqueous phase. The reason for suppression of by-products formation can be explained by more efficient HMF extraction facilitated by higher partition coefficient in the presence of a modifier. High extraction of HMF in the organic phase, induced by high concentration of the ZnCl2 catalyst (68 wt%) as well as the DMSO modifier in the aqueous phase, is also seen in the case of cotton cellulose conversion.48 Additionally, this process of HMF production does not require the separation of extracting organic solvents (2-butanol, 1-butanol, 1-hexanol, MIBK, and toluene–MIBK mixture) from HMF because of inherent fuel properties of the solvent themselves. The only disadvantage of this process is the complex and energy intensive separation of HMF from the high boiling point modifier as the modifying agent also expects to accumulate in the organic phase. Aspen model simulation for predicting the energy requirement for the separation of HMF from a low-boiling point solvent (pure MIBK) and from a high boiling point solvent (pure DMSO) shows that the vacuum evaporation method would evaporate 99.5% MIBK at 13 mbar and 70 °C with a 2.5% loss of HMF, whereas DMSO evaporation under similar conditions would cause 30% HMF loss.49 The simulation also predicts that HMF separation from DMSO with minimal loss would require a more expensive vacuum distillation process and 40% more energy than that from MIBK.
In a subsequent communication, Okano et al. used an acidic ionic liquid, 1-methyl-3-(butyl-4-chlorosulfonyl) imidazolium chlorosulfate ([MBCIm]SO3Cl) as a modifier for phase separation between water and acetonitrile in the water–acetonitrile biphasic system.50 In this process, a [MBCIm]SO3Cl modifier also acted as a dehydration catalyst for the conversion of fructose. The water–[MBCIm]SO3Cl–acetonitrile biphasic composition is reported to improve HMF yield and selectivity, enabling 81% HMF at 80 °C for 3 h. To claim the benefit of this biphasic system, control experiments were conducted in monophasic solvents using acetonitrile–[MBCIm]SO3Cl and water–[MBCIm]SO3Cl–HCl compositions. Significantly lower HMF yields from the monophasic systems (48 and 26 mol% HMF from acetonitrile–[MBCIm]SO3Cl and water–[MBCIm]SO3Cl–HCl compositions, respectively) suggest an involvement of secondary side reactions in the monophasic system. Among several varying compositions of water, acetonitrile and [MBCIm]SO3Cl in the biphasic system, the composition shown in the following phase diagram (Fig. 6) accounted for maximum HMF yields (89%). A larger scale reaction under optimized conditions produced 82% HMF, which is slightly lower than the test tube scale reaction due to mass transfer limitation of the liquid–liquid system. While [MBCIm]SO3Cl IL is an effective phase separator and an acid catalyst, its contamination with HMF in the organic phase is imminent and hence, possess the problem of expensive separation of this high boiling point IL from HMF.
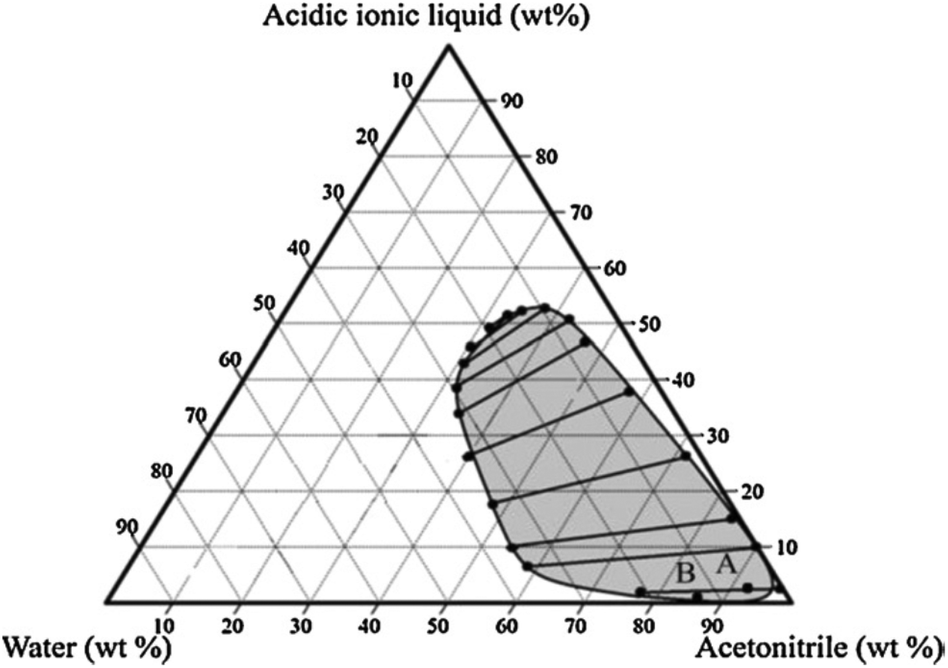 | ||
| Fig. 6 An experimental pseudo ternary phase diagram for the water–acetonitrile–[MBCIm]SO3Cl biphasic system with tie lines at room temperature (point A is the composition of a typical experiment and point B is the composition providing the highest yield of HMF in the acetonitrile phase). This figure is obtained from ref. 50. | ||
To avoid the disadvantages of expensive separation of a high boiling point modifier from HMF, Hu et al. developed a biphasic system based on deep eutectic solvent, such as chlorine chloride (ChoCl)–malonic acid, ChoCl–oxalic acid, ChoCl–citric acid, as the reactive phase and ethyl acetate as the organic phase.51 Deep eutectic solvents have characteristics similar to those of ILs, but are less expensive and less toxic than most ILs, and are sometimes biodegradable. Among several of these biorenewable ILs tested as the reactive phase as well as the catalyst for fructose conversion in a batch reactor, the ChoCl–citric acid reaction system enabled maximum fructose conversion (97%) and HMF selectivity (92%) in 1 h under reactive to organic phase mass ratio of 1.0 and mild reaction temperature (80 °C). A comparative study shows a 9% higher HMF yield in the biphasic system than the corresponding monophasic solvent (ChoCl–citric acid) without using ethyl acetate. Although ethyl acetate accounts for a slight increase in yield of the desired product, it facilitated easy separation of the reactive phase from the organic layer and hence allowed the recycling of the reactive phase up to 8 cycles with consistent HMF yields. In a subsequent communication, the authors extended the benefit of the ChoCl–citric acid–ethyl acetate and ChoCl–oxalic acid–ethyl acetate biphasic systems for the production of HMF from inulin.52 The kinetic profile of inulin conversion shows the formation of fructose as an intermediate, followed by its subsequent conversion to HMF. Under comparable reaction conditions, the ChoCl–citric acid–ethyl acetate and ChoCl–oxalic acid–ethyl acetate biphasic systems produced ∼55 and 64% HMF, respectively, which are 8–9% higher than the respective monophasic reaction systems without using ethyl acetate as an extracting solvent. In contrast to a previous report showing higher HMF yield from inulin than that from fructose in the water–DMSO–MIBK–2 butanol biphasic system,45 both ChoCl–citric acid–ethyl acetate and ChoCl–oxalic acid–ethyl acetate biphasic systems produced lower HMF from inulin than that from fructose. This difference may be due to lower partitioning of HMF into the organic phase in the present biphasic medium, thus causing rapid rehydration of HMF with water. Separate experiments to test the effect of water on HMF selectivity indeed show a rapid decrease in yield upon increasing the molar ratio of added water to fructose units in inulin above 20 (Fig. 7).
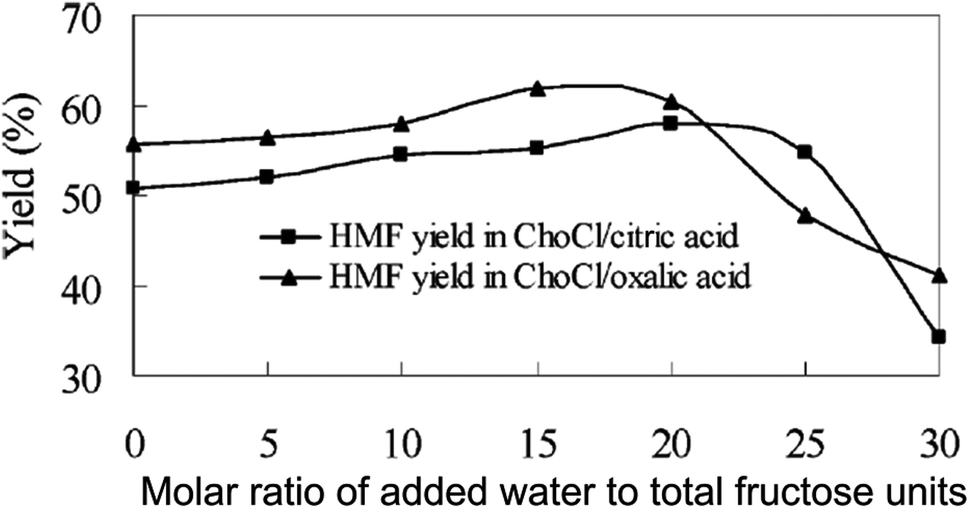 | ||
| Fig. 7 Effect of water on HMF yield from inulin in ChoCl–citric acid–ethyl acetate and ChoCl–oxalic acid–ethyl acetate biphasic systems (modified from Fig. 2 of ref. 52). | ||
As direct conversion of inexpensive and abundant raw biomass to HMF is the focus of many biorenewable research programs, Amiri and co-workers tested the effectiveness of biphasic reaction systems for the production of HMF from rice straw, a cheap, abundant and mainly unused agricultural waste.53 Rice straw contains 21% xylan, 37.5% glucan and 12% lignin. Because of the presence of xylan and glucan, simultaneous hydrolysis and dehydration of rice straw with 0.5 wt% H2SO4 produced HMF and furfural as major products in a biphasic medium consisting of 30 wt% NaCl saturated aqueous medium as the reactive phase and an organic solvent from the list of THF, 1-butanol, MIBK, 2-propanol and acetone as the extracting phase. The results showed a 2–3 fold increase in HMF yield in the biphasic system compared to the corresponding monophasic aqueous solvent. Depending on the nature of organic solvents and temperatures in the range of 120–180 °C, HMF yields varied in the range of 45–60 g per kg of rice straw. Among all organic solvents tested, THF and 1-BuOH were the most effective extracting solvents, giving 60 g HMF per kg of rice straw. These results are consistent with other literature data showing THF and 1-BuOH as good extracting solvents for HMF in HCl catalyzed dehydration of fructose.39
One aspect of process technology development is continuous operation rather than batch mode. To translate the benefit of the biphasic reaction system from a batch reactor to a continuous reactor, Brasholz et al. designed54 and implemented a continuous biphasic flow reactor for HCl catalyzed conversion of fructose to HMF using 0.25 M aqueous HCl as the reactive phase and MIBK as the organic phase (Fig. 8). This flow reactor was designed in such a way that a flow of 10 wt% fructose solution in aqueous HCl is mixed with a flow stream of the extracting phase (MIBK) in defined alternating segments in a heated PFA flow coil of 10 mL volume and 1 mm inner diameter. A back pressure regulator of 8 bar was connected to the reactor outlet to ensure continuous flow of the liquid stream. Among other reaction parameters, the flow rates of the reactive and extracting phases are found to have a significant influence on overall HMF yield and selectivity. The best results were obtained when the flow rate of MIBK stream is adjusted at 3 times higher than the flow rate of fructose stream and the resulting flow stream is heated at 140 °C for 15 min. A reaction under the optimized condition produced 74 wt% HMF with 88% selectivity. It is possible to further improve the selectivity of HMF by tuning the flow rates of both streams. Thus, this continuous biphasic flow reactor offers a simple, efficient and practical device for high HMF yield in reasonable reaction time. It should be noted that translating this flow reactor which uses homogeneous fructose solution to a reactor that employs biomass constitutes a significant challenge because of expected plugging of liquid flow streams by solid biomass residue. Perhaps, a modified flow reactor with a higher inner diameter may resolve the plugging issue.
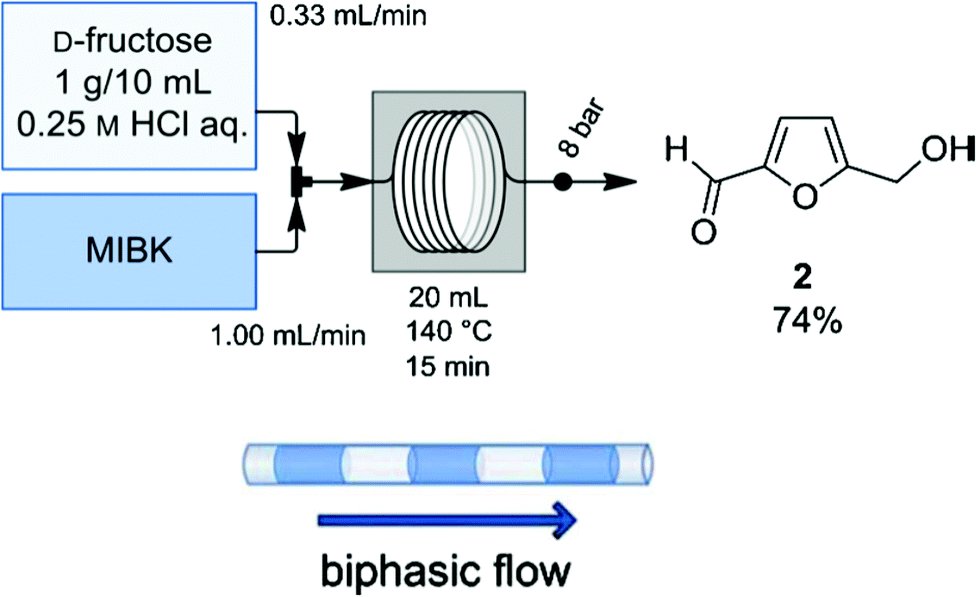 | ||
| Fig. 8 Biphasic continuous flow reactor for fructose dehydration.54 | ||
The water–MIBK biphasic solvent system is also used for anhydrous AlCl3 catalyzed dehydration of carbohydrates under microwave assisted heating at 120 °C.55 Among several experiments carried out for fructose conversion at varying reaction conditions in pure water and biphasic media, water–MIBK solvent combination accounted for 11% more HMF than that obtained from monophasic reactions in pure water. The respective HMF yields in water–MIBK and pure water were 62 and 51 mol% for 5 min. Consistent with previous studies, THF appears to be a better extracting solvent resulting in comparable yield (58 mol%) of the desired product at significantly lower temperature (100 °C). Because of the inherent benefits of the microwave assisted heating, this method allows reducing the reaction time from hours to minutes and also improves HMF yield and selectivity when compared with results for fructose dehydration under conventional oil-bath heating. The latter reaction under oil-bath heating produced only 33 mol% HMF in 1 h. A Lewis acidic AlCl3 catalyst also produced moderate HMF (24–43 mol%) from difficult carbohydrate substrates, such as glucose, starch and inulin, which require sequential hydrolysis, isomerization and dehydration steps. However, product selectivity is an issue in this process as 30% HMF exists in the form of chlorinated species, 5-chloromethylfurfural (CMF). 1H NMR spectrum of the isolated oily product confirms the presence of HMF and CMF in 2:1 molar ratio. CMF, a precursor to promising biofuel component 5-ethoxymethylfurfural (EMF), can easily be converted to EMF via a simple etherification reaction with ethanol.56
However, CMF formation was not an issue when Yang et al.57 investigated the catalytic activity of hydrated AlCl3 for the conversion of glucose by using the water–THF (1:3 v/v) biphasic system, where THF is a better extracting solvent than MIBK. Under microwave assisted heating at 160 °C and AlCl3 to glucose molar ratio of 2.5:1, the water–THF biphasic system produced 30 mol% more HMF than the monophasic system in pure water (Table 2). Additionally, high partition coefficient of the biphasic system led to a continuous separation of HMF into the THF phase. As a result, HMF recovery in the biphasic system reached as high as 94% with a significant decrease in by-products (lactic acid and LA) formation. As seen in Table 2, a further increase in HMF yield and selectivity is recorded in the presence of 35 wt% NaCl in the aqueous phase due to further increase of the R value because of the salting-out effect.41,42
| Solvent | Conv. (%) | HMF yield (%) | Lactic acid yield (%) | LA yield (%) |
|---|---|---|---|---|
| Single phase (water) | 98 | 22 | 17 | 10 |
| Biphasic (water–THF) | 99 | 52 | 13 | Trace |
| Single phase (water–NaCl) | 98 | 17 | — | 29 |
| Biphasic (water–NaCl/THF) | 99 | 61 | — | 1 |
| Biphasic (water–NaCl/THF) | 30 | 12 | — | 2 |
While the exact pathway for HMF formation has been debated over the years, the kinetic profile of AlCl3 catalyzed dehydration of glucose shows direct evidence for fructose formation as an intermediate species, followed by its rapid disappearance to HMF (Fig. 9). This result supports the fact that HMF formation with the Lewis acid catalyst occurs via isomerization of glucose to fructose. Analysis of relevant kinetic models for the observed intermediate and product formation revealed that the rate of HMF formation is approximately 4 times faster than the glucose isomerization to fructose. In a previous study,55 glucose dehydration with a AlCl3 catalyst has been monitored by 1H NMR spectroscopy to gain a better understanding of the mechanism of isomerization. 1H NMR spectra showed a line broadening of –OH proton signals of glucose in the beginning of the reaction. This line broadening of –OH protons indicated H-bonding interaction between the Cl atom of AlCl3 and –OH protons of glucose forming an AlCl3–glucose adduct. The line broadening of –OH protons corresponding to –H⋯Cl– interaction disappeared upon heating the reaction mixture at 100 °C for 40 min, with appearance of a new signal at 9.49 ppm corresponding to the –CHO proton signal of HMF. Similar line broadening of the –OH proton signals of glucose has been observed in the case of SnCl4,55,58 Yb(OTf)3 (OTf = trifluoromethanesulfonate)55 and Zr(O)Cl259 catalyzed dehydration of glucose. Based on these observations, the authors proposed a catalytic cycle for glucose isomerization to fructose in which Al[(OH)(H2O)5]2+ is formed by hydrolysis of AlCl3 with water (Fig. 10). This active catalytic species, Al[(OH)(H2O)5]2+ acts as a potential electrophile and reacts with α-glucopyranose to form intermediate A, as shown in Fig. 10. Intermediate A is possibly converted to fructofuranose via a hydride transfer, followed by cyclization.60 The transformation of fructofuranose to intermediate E occurs via a cyclic pathway assisted by a proton that is generated during AlCl3 hydrolysis. In agreement with earlier findings,24 the transformation of oxonium ion (intermediate C) to intermediate D is proposed to occur via two pathways in which Cl− acts as a base as well as a nucleophile for abstracting proton from the oxonium ion.
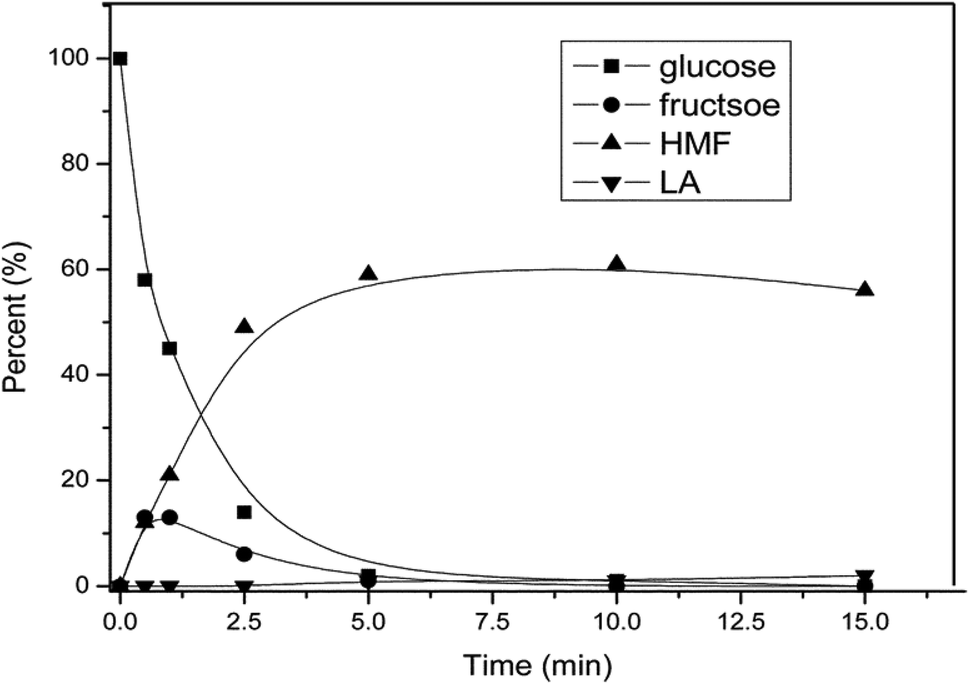 | ||
| Fig. 9 Conversion of glucose as a function of time using hydrated AlCl3 catalyst in water–NaCl/THF biphasic medium (obtained from ref. 57). | ||
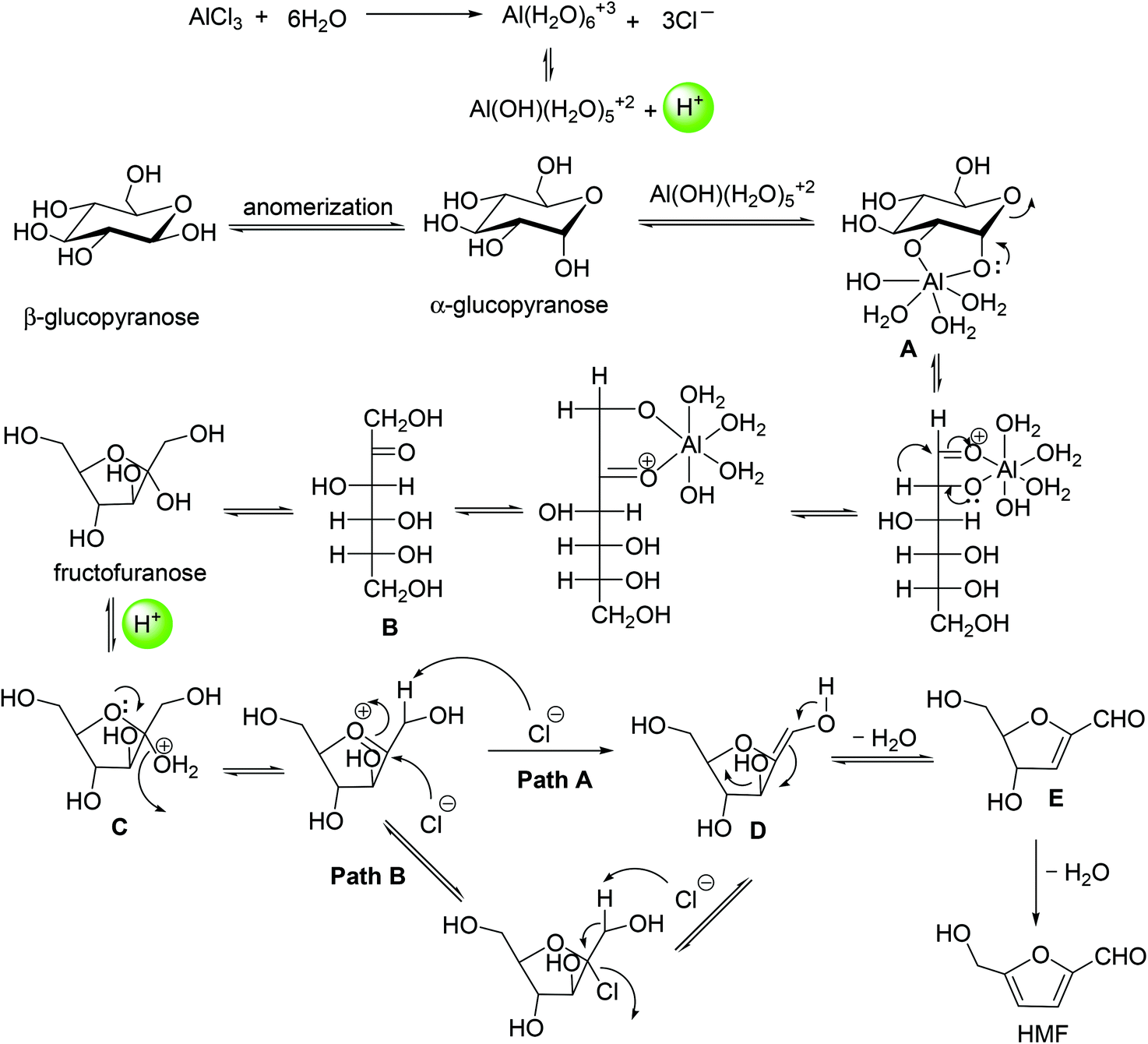 | ||
| Fig. 10 Proposed mechanism for glucose dehydration to HMF with AlCl3 catalyst. | ||
The potential of the combined AlCl3/water–NaCl–THF biphasic system has been further demonstrated for the conversion of intact lignocellulosic biomass (corn stover, pinewood, switchgrass, and poplar).61 Because of the presence of hemicellulose and cellulose biopolymers in the biomass, both HMF and furfural were produced concurrently upon catalysis (Table 3). However, the relative yield of HMF (19–26 mol%) was significantly lower than that of furfural (51–66 mol%) based on the literature values of total hexose and pentose units by weight.62,63 This difference in HMF and furfural yields may be due to the fact that cellulose is more recalcitrant than hemicellulose.64 The complex structure of lignocellulose, containing cross-linkers of hemicellulose with cellulose fibers into microfibrils and cellulose with lignin,65 makes it harder to convert intact biomass to the reactive sugar components of cellulose. In this context, the water–LiCl/dichloroethane biphasic system has yielded promising results for HCl catalyzed conversion of pure microcrystalline cellulose and lignocellulosic biomass, e.g., filter paper cotton, newsprint, wood, corn stover and straw.56 An integrated one-pot reaction between 10 g biomass substrates (filter paper, cotton and newspaper were cut into 0.25–1 cm2 pieces; wood, corn stover and straw were made into sawdust or smaller pieces and then ball-milled to powder) and 75 mL concentrated HCl in the presence of 10 g LiCl in a batch reactor at 65 °C for 30 h and continuous extraction of organic fragments into the extracting phase resulted in 72, 72, 60, 45, 43 and 36 wt% furanic product from filter paper, cotton, newsprint, wood, corn stover and straw, respectively. However, high concentration of chloride ions in the reaction medium is perhaps the reason for substitution of the hydroxyl group of HMF giving CMF as a major fraction along with small amount of HMF, furan monocarboxylic acid and LA. Furthermore, ball-milling renders the biomass more amenable to hydrolysis, but such pretreatment is not scalable to industrial scale production.
| Biomass | Temp (°C) | Furfural (%) | Xylose (%) | HMF (%) | Glucose (%) |
|---|---|---|---|---|---|
| Corn stover | 160 | 55 | <1 | 14 | 1 |
| Pinewood | 160 | 38 | <1 | 42 | 1 |
| Switchgrass | 160 | 56 | 1 | 13 | <1 |
| Poplar | 160 | 64 | <1 | 15 | <1 |
| Cellulose/xylan | 160 | 66 | 0 | 14 | 1 |
| Pinewood | 180 | 61 | <1 | 35 | 1 |
| Cellulose | 180 | — | — | 37 | <1 |
THF has also been reported as an effective extracting solvent in the THF–tetraethyl ammonium chloride (TEAC)/fructose/NaHSO4·H2O melt biphasic system for the production of HMF from fructose.66 This biphasic medium containing a mixture of 50 wt% fructose, TEAC and CrCl3 catalyst and 5 mol% of NaHSO4 (with respect to glucose) as the reactive phase produced 93.5 wt% HMF at 120 °C in 70 min. Low viscosity of the reactive melt at 120 °C favored mass transfer and easy separation of HMF from the reactive phase into the organic phase. Although this biphasic medium is beneficial in improving HMF yield by about 13 wt% as compared to the corresponding monophasic system without THF, the selectivity of HMF did not improve by this method.
Dumesic et al. also investigated the conversion of glucose to HMF using combined HCl/metal chloride catalysts, including AlCl3, to elucidate the mechanistic role of both Brønsted and Lewis acidic catalytic materials in the said conversion process by carrying out the reactions in water–NaCl/2-sec-butylphenol (SBP) and water–NaCl/THF solvent systems (organic to aqueous mass ratio = 2).67 In the absence of Lewis acidic metal chlorides, glucose conversion with Brønsted acidic HCl at pH 2.5 produced HMF with very low yield and selectivity. This result agrees well with Abu-Omar's findings showing very low HMF yield and selectivity from glucose conversion with the HCl catalyst alone.57 Consistent with AlCl3 catalyzed reaction, a significant improvement in HMF yield is noted in the presence of Lewis acidic metal chlorides. Depending on the nature of the metal chloride, HMF yields varied in the range of 60–38% (Table 4). Most importantly, the kinetic profile for progression of glucose conversion with a combined HCl/metal chloride catalyst showed the formation of fructose as an intermediate, the concentration of which varied depending on the metal chloride salt used. Al3+, being the hardest Lewis acid among all metal ions studied, strongly interacts with the oxygen atoms of the hydroxyl groups of glucose, and hence accounts for higher fructose concentration and consequently higher HMF yields. It should be noted here that glucose conversion with HCl alone did not show the formation of fructose intermediates, which suggested an involvement of a different mechanism for direct conversion of glucose to HMF with the Brønsted acid catalyst. It has been suggested that a carbocation is formed at the C-2 position of open-chain glucose, which reacts with the hydroxyl group of glucose at the C-5 position to form a tetrahydro-3,4-dihydroxy-5-(hydroxymethyl)-2-furanaldehyde intermediate.68 As shown in Fig. 11, HMF is formed via sequential removal of water from the latter intermediate.
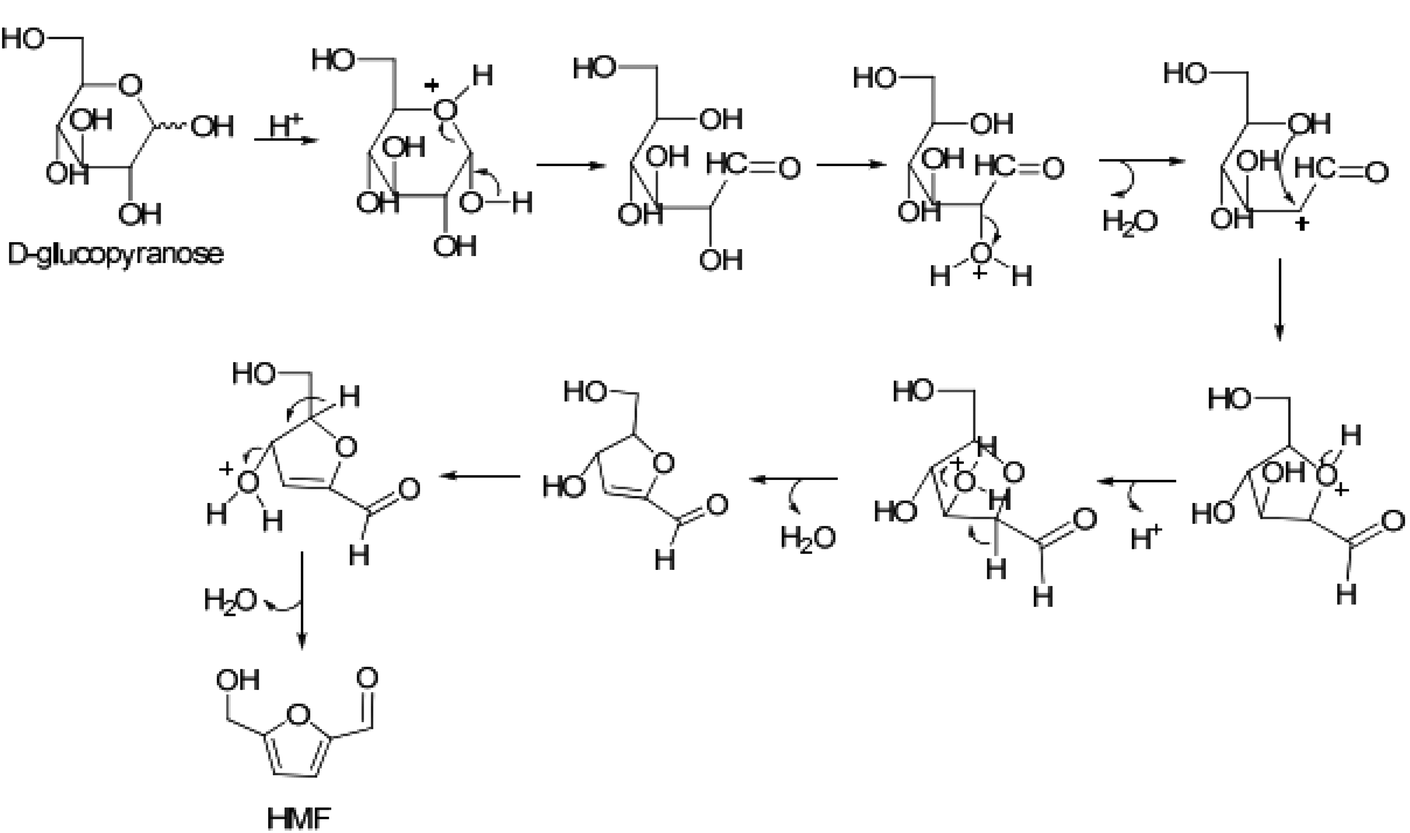 | ||
| Fig. 11 Reaction scheme for Brønsted acid catalyzed dehydration of glucose to HMF.67,68 | ||
| Catalyst | Time (min) | Conv. (%) | HMF selectivity (%) |
|---|---|---|---|
| HCl | 30 | 15 | 52 |
| HCl | 420 | 91 | 30 |
| HCl/AlCl3 | 40 | 91 | 68 |
| HCl/SnCl4 | 45 | 90 | 58 |
| HCl/VCl3 | 90 | 92 | 53 |
| HCl/GaCl3 | 120 | 90 | 50 |
| HCl/InCl3 | 150 | 86 | 52 |
| HCl/YbCl3 | 120 | 93 | 46 |
| HCl/DyCl3 | 160 | 93 | 41 |
| HCl/LaCl3 | 240 | 87 | 44 |
As mentioned above, bi-functional HCl/metal chloride catalyzed dehydration of glucose progresses via two routes, namely (i) isomerization of glucose to fructose and (ii) direct conversion of glucose with the formation of tetrahydro-3,4-dihydroxy-5-(hydroxymethyl)-2-furanaldehyde intermediate. To explore the role of Lewis acidic metal chloride catalyst alone, the conversion of glucose and other carbohydrate substrates with the lanthanide based metal chloride catalysts (YbCl3, DyCl3 and LaCl3) has been reinvestigated at pH 5.5 in the water–NaCl/SBP biphasic solvent system.69a Using SBP to aqueous phase mass ratio of 2, glucose dehydration at pH 5.5 without an isomerization catalyst showed a small conversion of the substrate (20 mol%) for 80 min. A similar reaction using a 25 mM YbCl3 catalyst improved both glucose conversion (90 mol%) and HMF yield (41 mol%) to the same extent as that observed with the bi-functional catalyst, HCl (pH 1.8)/YbCl3.67 This comparison suggests that the glucose to fructose isomerization step dominates in glucose dehydration with both catalytic systems. Interestingly, the bi-functional Brønsted/Lewis acid, HCl (pH 1.8)/YbCl3, catalysis reaction took twice the time (150 min) that of the YbCl3 catalysis reaction (80 min) to achieve a similar glucose conversion and HMF yield. While the reasons for the slower reaction with the LnCl3 catalyst at higher HCl concentrations are unclear, the above comparison reveals that the LnCl3 catalysis at pH 5.5 is more practical from both economical and environmental points of view. The kinetic isotope labeling and NMR spectroscopic data for the interaction of catalytic species and substrate reveals the involvement of an intra-molecular H-transfer from C-2 to C-1 in glucose/fructose isomerization,69b,70 and supports the proposed mechanism observed for AlCl3 catalyzed conversion of glucose.55
Biomass derived SBP solvent demonstrating higher HMF yield (68%) with better HMF extraction (97%) than the THF solvent (57% yield, 93% HMF extraction) under comparable reaction conditions is indeed attractive as environmentally friendly extracting solvents gain ground in biorefinery processes.67 The use of an SBP solvent having high R values (90 and 50 with and without NaCl saturation in the aqueous phase)71 is advantageous because of (i) fast HMF extraction, (ii) low amounts of mineral acids dissolved in SBP thereby eliminating energy intensive separation of mineral acid from the product layer, and (iii) SBP can be derived from biomass lignin. While the authors list SBP's higher boiling point than that of the HMF product as an advantage, this feature can be interpreted as a drawback because it requires intensive energy to remove the product via distillation.
One aspect of HMF process technology development is minimization of operational temperature (i) to reduce production cost by lowering energy requirement and (ii) to improve HMF selectivity by eliminating undesired secondary reactions. A biphasic system consisting of imidazolium based IL ([BMIM]Cl) as the reactive phase and THF as the organic phase has addressed this issue by demonstrating effective fructose dehydration with WCl6 catalyst at lower temperature ranging from room temperature to 50 °C.72 A test reaction between fructose and WCl6 catalyst in a biphasic reactor having capability of concurrent extraction of the desired product and evaporation of the organic phase results in 72% HMF at 50 °C in 1 h. One of the problems of biphasic systems containing high viscous IL is poor mixing between the reactive and organic phases, which hinders HMF extraction and hence can often give lower HMF yield. This is exactly observed in fructose dehydration with WCl6 catalyst in the [BMIM]Cl–toluene solvent system where HMF yield was suppressed by 25% in comparison to the [BMIM]Cl–THF system under comparable reaction conditions. A summary of HMF yields under best reaction conditions using different homogeneous catalysts in biphasic solvent systems is shown in Table 5.
| Entry (ref.) | Substrate | Reactive phase | Organic phase | Catalyst | Temp (°C) | Time (min) | HMF yield (%) |
|---|---|---|---|---|---|---|---|
| 139 | Fructose | H2O–Na2SO4 | 1-Butanol | HCl | 180 | 20 | 68 |
| 244 | Fructose | H2O–DMSO–PVP (4:1:2) |
MIBK–2-butanol (7:3) |
HCl | 180 | 3 | 77 |
| 345 | Inulin | H2O–DMSO (5:5) |
MIBK–2-butanol (7:3) |
HCl | 170 | 5 | 75 |
| 450 | Fructose | H2O–[MBCIm]SO3Cl | Acetonitrile | [MBCIm]SO3Cl | 80 | 180 | 89 |
| 552 | Fructose | ChoCl–citric acid | Ethyl acetate | Citric acid | 80 | 60 | 89 |
| 652 | Inulin | ChoCl–oxalic acid | Ethyl acetate | Oxalic acid | 80 | 60 | 64 |
| 753 | Rice straw | H2O | THF | H2SO4 | 180 | 180 | 16 |
| 854 | Fructose | H2O–NaCl | MIBK | HCl | 140 | 15 | 74 |
| 955 | Fructose | H2O | MIBK | AlCl3 | 120 | 5 | 62 |
| 1057 | Glucose | H2O–NaCl | THF | AlCl3 | 160 | 5 | 61 |
| 1161 | Cellulose | H2O | THF | AlCl3 | 140 | 60 | 37 |
| 1266 | Fructose | H2O–TEAC–NaHSO4 melt | THF | CrCl3 | 120 | 70 | 93.5 |
| 1367 | Glucose | H2O–NaCl | 2-sec-Butylphenol | HCl–AlCl3 | 170 | 40 | 61 |
| 1469a | Glucose | H2O–NaCl | 2-sec-Butylphenol | YbCl3 | 170 | 80 | 41 |
| 1572 | Fructose | [BMIM]Cl | THF | WCl6 | 50 | 60 | 72 |
5. HMF production using heterogeneous catalysts
Collective research efforts of HMF production technology over the past decade and better understanding of the mechanistic roles of the Brønsted and Lewis acids has contributed significantly to developing a more effective homogeneous catalysis strategy, process design and extraction methodologies for high selective HMF formation by minimizing undesired side reactions. However, energy intensive separation of HMF from the reaction media remains an issue. Although this issue has been addressed by developing biphasic reaction systems, partial dissolution of mineral acids and metal halides in the product phase and hence their contamination with the crude product still requires energy intensive separation of HMF from contaminants. To avoid this disadvantage, several heterogeneous catalytic materials having controlled acid sites and shape have been tested in recent years by utilizing biphasic media. Unlike the homogeneous catalyst, several factors of heterogeneous catalysts such as preparation condition of the catalyst, structural properties, accessibility of acid sites, surface area, pore volumes and nature of acid sites play important roles in determining the efficiency of solid catalyzed conversion of hexose rich carbohydrates to HMF. For example, a strong Brønsted acidic cation exchange resin, Dowex 50wx8–100 of 50–100 mesh beads, was prepared from sulfonated copolymer and divinyl benzene, and used for fructose conversion in the water–acetone biphasic system.73 Using the water to acetone weight ratio of 7:3, recyclable Dowex 50wx8–100 catalyst yielded 73 mol% HMF with 91% fructose conversion at 150 °C. To compare the partition efficiency of the water–acetone biphasic system, the authors performed another experiment in pure water which produced only 34 mol% HMF yield with 83% fructose conversion, suggesting that the investigated biphasic system has a high partition coefficient. Although continuous extraction of HMF into the extracting phase eliminated HMF rehydration to a significant extent, formation of humin oligomers catalyzed by strong Brønsted acidic sulfonate groups is the reason for the difference in observed HMF yield and fructose conversion.
In contrast to the disadvantage of Brønsted acidic Dowex 50wx8–100 catalyst in humins formation, Lewis acidic zeolite beta topology (Sn-beta) and Brønsted acidic HCl combination seems beneficial to achieve higher HMF selectivity.74 Using water–NaCl/THF biphasic media, Sn-beta catalyzed glucose dehydration shows 79 wt% conversion with 72% HMF selectivity under glucose to Sn molar ratio of 200. While the biphasic media itself accounted for 66% higher HMF selectivity and 34 wt% higher glucose conversion than the corresponding monophasic system without THF, the bi-functional HCl/Sn-beta catalyst resulted in a further increase in HMF selectivity compared to the Sn-beta catalyst alone. Similar to xylose dehydration with the Sn-beta catalyst in which the reaction progressed via isomerization of xylose to xylulose intermediate,75 glucose conversion with Lewis acidic Sn also takes place via the formation of a fructose intermediate. Besides dehydration via a isomerization pathway, a parallel pathway for direct conversion of glucose to HMF in the presence of HCl catalyst is perhaps the reason for higher HMF selectivity with the bi-functional Sn-beta/HCl catalyst.67
While the dehydration reaction requires an acid catalyst, acid density of the catalyst is not only the critical parameter in determining overall effectiveness of the catalyst as well as HMF selectivity. To justify this fact, Zhao et al. have used a series of recyclable solid acid catalysts for the conversion of concentrated fructose solution (30–50 wt%) to HMF in water–MIBK biphasic solvent.76 As seen in Table 6, heteropolyacid, Cs2.5H0.5PW12O40, catalyst having lower acid density (1.52 mmol g−1) than the Brønsted acidic Amberlyst-15 (acid density = 2.9 mmol g−1) or sulfated zirconia (acid density = 1.87) mmol g−1 is more effective, giving 95% HMF selectivity with 74% overall yield. However, a longer reaction time is found to have a negative effect on HMF selectivity due to its rehydration with water. Such rehydration predominantly occurrs with the Brønsted acidic catalytic materials (Amberlyst-15 and sulfated zirconia) resulting in a significantly lower selectivity of the desired product. Besides rapid rehydration, the high rate of oligomerization by the Amberlyst-15 and sulfated zirconia catalysts can also contribute to their low product selectivity. The higher catalytic activity of solid catalysts having lower acid density is also demonstrated by Yang et al.77 For example, modified hydrated niobium oxide (NA-p), prepared by treating niobium oxide (NA) with 1 M phosphoric acid, followed by calcination of the resultant sold at 300 °C for 3 h, is found to have a higher surface area (214.9 m2 g−1) and acid density (4.4 mmol g−1) than the parent NA material. A modified hydrated tantalum oxide (TA-p) catalyst, prepared by a similar phosphoric acid treatment and calcination method, has a lower surface area (141.5 6 m2 g−1) and acid density (1.5 mmol g−1) than the NA-p material.78 However, catalytic activities of both NA-p and TA-p materials were comparable for fructose conversion, giving 89 and 91 wt% HMF, respectively, in water–2-butanol biphasic media. Interestingly, NA-p catalyzed conversion of glucose and inulin produced significantly lower HMF than the corresponding reactions with the TA-p catalyst; respective HMF yields from glucose with NA-p and TA-p catalysts are 49 and 70 wt% and those from inulin are 54 and 95 wt%. A comparative analysis of these results reveals that the NA-p catalyst is less effective even though it has higher acid density and surface area than the TA-p catalyst. Although the authors neither elucidated the reasons for lower activity of the NA-p catalyst nor gave any data for by-products formation, it is presumed that the higher acid density of the NA-p catalyst could accelerate HMF rehydration36,79 and humin formation in the reactive aqueous phase. Differences in other material properties, e.g., pore diameter, could also be the reason for lower activity of the NA-p catalyst. In a recent publication,27 biorenewable glucose supported sulfonated carbonaceous material having total Brønsted acid density of 2.0 mmol g−1 is shown to be less effective in HMF selectivity than a similar carbonaceous catalysts containing Lewis acidic TiO2 mesoporous sites (0.071 mmol g−1) as well as significantly lower Brønsted acid density (0.027 mmol g−1).28 Although total acidity of the Ti-containing carbonaceous material is significantly lower than the Ti-free carbonaceous catalyst, the superior HMF selectivity in the product by the Ti-containing catalyst has been explained by the presence of its Lewis acidic sites as well as higher surface area (42.5 m2 g−1) than the Ti-free material (<1 m2 g−1).
| Catalysts | Acid density (mmol g−1) | Reaction time (h) | Conversion (%) | Selectivity (%) | Yield (%) | TOF (h−1) |
|---|---|---|---|---|---|---|
| Zeolite | 1.94 | 1 | 58 | 92 | 54 | 3.47 |
| Cs2.5H0.5PW12O40 | 1.52 | 1 | 78 | 95 | 74 | 3.66 |
| 2 | 95 | 82 | 77 | 2.25 | ||
| SO42−/ZrO2 | 1.87 | 1 | 79 | 62 | 49 | 4.67 |
| Amberlyst-15 | 2.9 | 1 | 100 | 48 | 48 | 6.00 |
Because of the benefit of biphasic media as well as ease of recyclability of a solid catalyst, several researchers have adopted this method for HMF production in recent years. In this direction, a self-assembled Lewis acidic mesoporous TiO2 nanoparticulate catalyst of particle size 10–20 nm has been prepared using sodium salicylate as a templating agent.80 This catalyst, having acid density of 0.45 mmol g−1 but high surface area of 326 m2 g−1, showed moderate activity for hydrolysis and dehydration of a number of carbohydrate substrates into HMF in water–MIBK biphasic solvent. Using the water to MIBK volume ratio of 1:2 and at mild reaction temperature of 130 °C, maximum reported HMF yields from fructose and glucose are 40 and 26 mol%, respectively, which are about 10 mol% higher than the corresponding yields in single phase using pure water as a solvent. A similar HMF yield (35 mol%) from fructose in water–MIBK biphasic solvent is also reported in a subsequent publication using the Lewis acidic titanium phosphate nanoparticle catalyst (MTiP-1).81 MTiP-1, prepared by using Pluronic P123 as a structure directing agent and having large pore diameter of 7 nm, was separated by a simple centrifugal method and recycled for several cycles without a significant loss in its catalytic activity. Recently biopolymer sodium alginate templated porous TiO2 nanocatalysts (Fig. 12), containing strong Lewis acidic sites as determined by pyridine-IR and NH3-TPD methods, have been prepared for the production of HMF from unutilized abundant sugar derivatives, such as D-mannose, D-galactose and lactose, in water–MIBK biphasic solvent.82 Total acid density of the TiO2 NPs material prepared under three different conditions (room temperature, 0 °C and hydrothermal at 60 °C), varied as a function of their synthetic conditions. Among these three varieties, the material prepared under hydrothermal condition (represented as TiO2–H) is most effective even though its total acid density (1.10 mmol g−1) is lower than the TiO2–R catalyst having a total acid density of 1.21 mmol g−1.
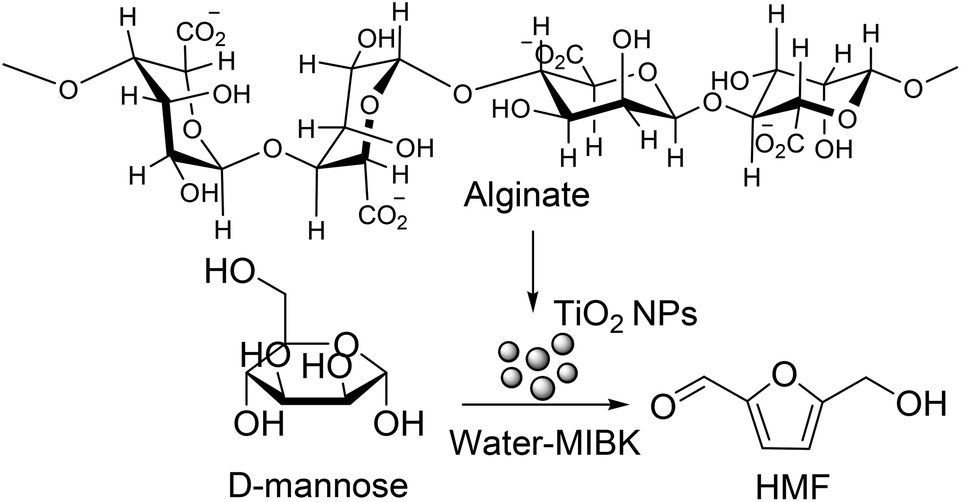 | ||
| Fig. 12 Conversion of mannose to HMF using alginate derived TiO2 NPs catalyst. | ||
The role of organic solvents of the biphasic media in improving the HMF yield and selectivity by minimizing the deactivation of the external pore volume of zeolites and amorphous aluminosilicate catalysts has been investigated by Ordomsky et al.83 In the absence of organic solvent (e.g., MIBK), fructose dehydration with zeolite and aluminosilicate catalysts having Si/Al ratio in the range of 11–16 and acid densities in the range of 0.2–1.1 mmol g−1, achieved 80–100% conversion in 5 h at 165 °C. However, poor HMF selectivity (maximum 50%) and very low LA/formic acid in the isolated product suggested the formation of a significant amount of humins by-product. The initial induction period in the kinetic profile of fructose conversion and decrease in HMF selectivity as a function of fructose conversion for all zeolite catalysts led to the hypothesis that fructose transformation takes place inside the pores of the catalyst and that HMF or its precursor cyclic intermediate84 forms humin inside the pore before HMF can desorb from the pore. One way to validate this hypothesis is to test fructose dehydration with these catalysts in a biphasic solvent system for concurrent extraction of HMF as it is formed in the aqueous phase. As expected, HMF selectivity improved to almost 98% in the beginning of the reaction at 20% conversion, but slowly decreased to ∼60% as the conversion increased to almost 100%. An initial increase in HMF selectivity is attributed to the fact that HMF was readily desorbed from the pore of the catalyst by MIBK, and thereby suppressed humins formation inside the pore. This argument supports the result of aluminosilicate catalysis reactions having lower acid density. While lower acid density of the aluminosilicate catalyst explains its lower catalytic activity, this catalyst without any micropores showed no improvement in HMF selectivity in the presence of MIBK solvent such as that observed for zeolite catalyzed reactions. This observation supports the fact that fructose dehydration occurs inside the pore of the catalyst.
To further elucidate the role of the pore diameter in HMF selectivity as well as its secondary transformation to unwanted side products, Ordomsky et al. prepared a series of heterogeneous materials with different pore diameters and different amount of Lewis and Brønsted acid density (Table 7) for fructose dehydration.85 While the catalytic activity of fructose conversion correlates with the strength of the Lewis acidity of the catalysts, the MOR zeolite containing highest acid density (1.1 mmol g−1) exhibits the lowest fructose conversion, which is due to the smallest pore diameter of this material. On the other hand, HMF selectivity in the product correlated with the Brønsted acid density of the catalyst. Thus, maximum selectivity in HMF yield is seen with the MOR and Amberlyst-15 catalysts having strong Brønsted acidity, whereas the aluminosilicate catalyst with weak Brønsted acidity shows lower HMF selectivity. This behavior of the Lewis and Brønsted acidic sites in the dehydration reaction also reflected their activity pattern towards by-products formation via secondary HMF transformations (Fig. 13). For example, Brønsted acid sites of the MOR and Amberlyst-15 catalyst strongly interacts with the carbonyl group of HMF, facilitating its desorption in the presence of organic solvents. As a result, HMF selectivity improved to about 75–80% with the Amberlyst-15 catalyst in the water–MIBK biphasic solvent (water–MIBK volume ratio = 5). While the authors explain that the Brønsted acidity of Amberlyst-15 is the reason for its high selective HMF formation, this observation can be interpreted by the high pore diameter of the Amberlyst-15 catalyst. On the other hand, Lewis acid sites of the alumina, aluminosilicate, niobic acid and zirconium phosphate catalysts tend to adsorb fructose on the surface of the catalyst by multiple surface interactions of carbonyl and hydroxyl groups with Lewis acid sites before HMF can be desorbed. Thus, fast cross-oligomerization between fructose and HMF takes place on the catalyst surface and forms humin by-products.
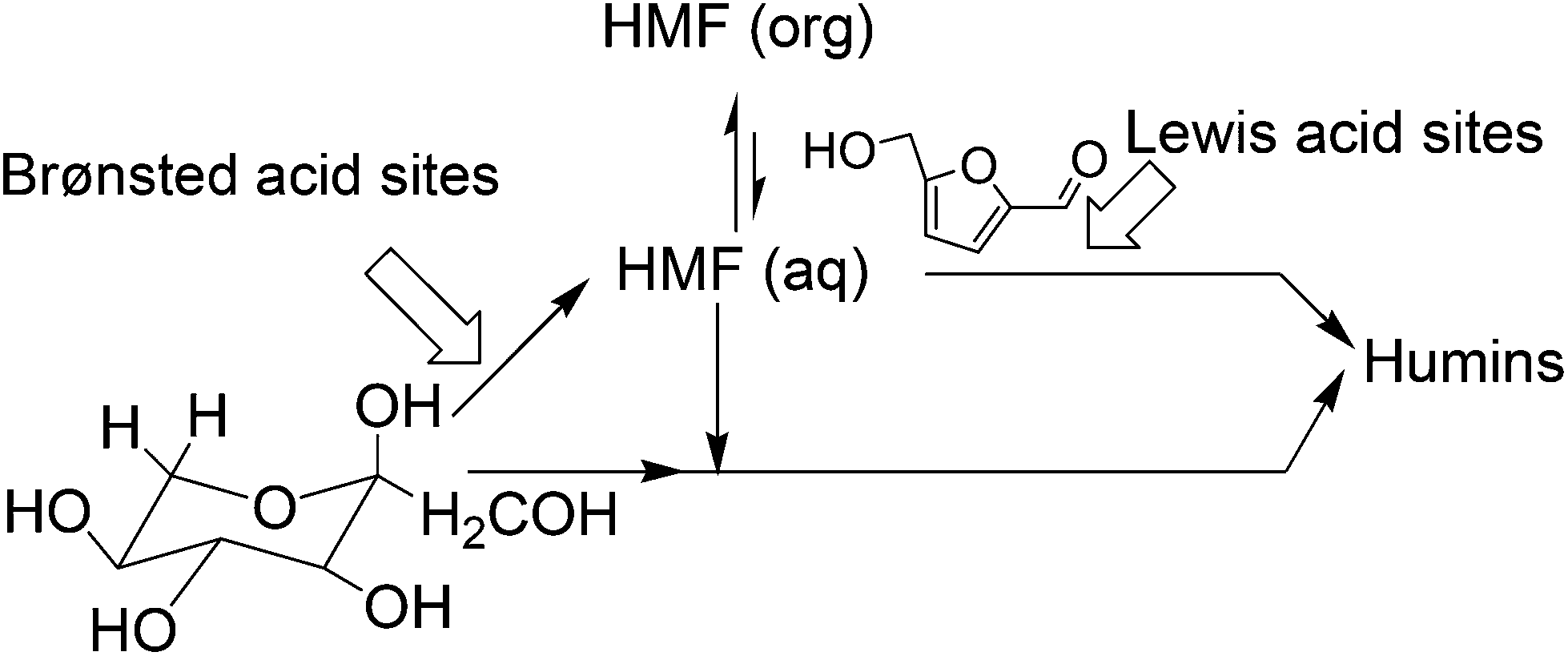 | ||
| Fig. 13 Fructose dehydration and HMF oligomerization routes. | ||
| Sample | Surface area (m2 g−1) | Pore size (nm) | TDP NH3 (mmol g−1) | L/B |
|---|---|---|---|---|
| a B = Brønsted acidity; L = Lewis acidity. | ||||
| SiO2–Al2O3 | 327 | 9.5 | 0.012 | 2 |
| Al2O3 | 262 | 9.8 | 0.072 | — |
| ZrPO4 | 93 | 8.5 | 0.11 | 2 |
| Nb2O3 | 108 | 8.0 | 0.24 | 2.4 |
| MOR | 420 | 0.5–0.8 | 1.1 | 0.2 |
| Amberlyst-15 | 53 | 30 | — | — |
6. Conclusions
HMF is a promising biomass derived platform chemical for the production of high value chemicals and liquid fuels that are currently obtained from petroleum feedstock. Development of effective catalysts and reactors designed to achieve high yield and selectivity of HMF from abundant biomass containing C6-sugars is a viable strategy for the modern biorefinery. HMF production technology over the past decade has primarily focused on the utilization of homogeneous mineral acids and metal halide catalysts in pure water or high boiling point solvents (DMSO, DMF, and ILs) alone or in modified high boiling point solvents mixed with aqueous media. These reactions suffered from low conversion and selectivity of the desired product due to the involvement of secondary reactions leading to the degradation of HMF to undesired side products via rehydration and oligomerization pathways. Besides low yield and selectivity, energy intensive separation and purification of HMF from high boiling point solvents and by-products are among the reasons for its high production cost, which creates a bottleneck for its sustainable utilization for making other value chemicals and fuels on commercial scale.Leveraging the learning from these drawbacks and better understanding of the mechanistic roles of both Brønsted and Lewis acidic sites for HMF and by-products formation has directed the current research efforts of HMF production towards development of biphasic reaction systems as well as heterogeneous catalysts containing balanced Brønsted and Lewis acid sites. As a result, several improvements have been noted on a laboratory scale for continuous extraction of HMF into the organic phase by using solvents having a high partition coefficient as well as by modifying the reactive phase with inorganic salts. Among several organic solvents tested, biomass derived 2-MeTHF and petroleum based THF, 2-butanol and 2-butanone solvents are found to have high partition coefficient values. While continuous improvement of biphasic media has enhanced HMF yield and selectivity by minimizing secondary reactions, there are fewer reports on the development of continuous reactors design or testing of the laboratory scale processes on a larger scale reaction to examine their production viability, particularly from intact biomass feedstock. Investigation of solid acid catalysts containing variable pore diameters and Lewis and Brønsted acid densities suggest that the mesoporous materials having large pore size, high surface area and balanced Lewis to Brønsted acid densities perform the best for high selective HMF production. A combination of both chemistry and engineering approaches to develop more effective catalytic systems and process chemistry, and cost-effective purification of the desired product are necessary to lower HMF production costs.
Acknowledgements
The authors acknowledge financial support from the Center for direct Catalytic Conversion of Biomass to Biofuels (C3Bio), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences under Award Number DE-SC0000997.References
- T. Werpy and G. Peterson, Top Value Added Chemicals from Biomass, Pacific Northwestern National Laboratory, 2004, vol. 1 Search PubMed.
- S. G. Wettstein, D. M. Alonso, E. I. Gürbüz and J. A. Dumesic, Chem. Eng., 2012, 1, 218 CAS.
- J. P. Lange, Catalysis for Renewables: From Feedstock to Energy Production, ed. G. Cento and R. A. van Santen, Wiley-VCH, Weinheim, 2007, pp. 21–51 Search PubMed.
- J. P. Lange, Biofuels, Bioprod. Biorefin., 2007, 1, 39 CrossRef CAS.
- S. Dutta, S. De and B. Saha, ChemPlusChem, 2012, 22, 259 CrossRef.
- S. Dutta, S. De, B. Saha and Md. I. Alam, Catal. Sci. Technol., 2012, 2, 2025 CAS.
- M. E. Zakrzewska, E. Bogel-Luasik and R. Bogel-Lukasik, Chem. Rev., 2011, 111, 397 CrossRef CAS PubMed.
- B. Saha, D. Gupta, M. M. Abu-Omar, A. Modak and A. Bhaumik, J. Catal., 2012, 299, 316 CrossRef.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138 CrossRef CAS PubMed.
- H. E. van Dam, A. P. G. Kieboom and H. van Bekkum, Starch/Stäerke, 1986, 38, 95 CrossRef CAS.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982 CrossRef CAS PubMed.
- S. De, S. Dutta and B. Saha, ChemSusChem, 2012, 5, 1826 CrossRef CAS PubMed.
- M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924 CrossRef CAS PubMed.
- O. Abdulmalik, M. K. Safo, Q. Chen, J. Yang, C. Brugnara, K. Ohene-Frempongm, D. J. Abraham and T. Asakura, Br. J. Haematol., 2005, 128, 522 CrossRef PubMed.
- X. Tong and Y. Li, ChemSusChem, 2010, 3, 350 CrossRef CAS PubMed.
- G. Tian, X. Tong, Y. Cheng and S. Xue, Carbohydr. Res., 2013, 370, 33 CrossRef CAS PubMed.
- X. Hu, L. Wu, Y. Wang, D. Mourant, C. Lievens, R. Gunawan and C. Z. Li, Green Chem., 2012, 14, 3087 RSC.
- Y. Y. Lee and K. C.-W. Kevin, Phys. Chem. Chem. Phys., 2012, 14, 13914 RSC.
- Y. Qu, C. Huang, Y. Song, J. Zhang and B. Chen, Bioresour. Technol., 2012, 121, 462 CrossRef CAS PubMed.
- J. Wang, J. Ren, X. Liu, X. J. Xi, Q. Xia, Y. Zu, G. Lu and Y. Wang, Green Chem., 2012, 14, 2505 Search PubMed.
- C. B. Rasrendra, J. N. M. Soetedio, I. G. B. N. Makertihartha, S. Adisasmito and H. J. Heeres, Top. Catal., 2012, 55, 543 CrossRef CAS.
- Y. Tang, X. Xiang, D. Tong, C. Hu and M. M. Abu-Omar, Bioresour. Technol., 2012, 116, 302 CrossRef PubMed.
- Z. Zhang, B. Liu and Z. Zhao, Carbohydr. Polym., 2012, 88, 891 CrossRef CAS.
- (a) J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS PubMed; (b) C. Li, Z. Zhang and Z. B. K. Zhao, Tetrahedron Lett., 2009, 50, 5403 CrossRef CAS.
- Y. Su, H. M. Brown, X. Huang, X.-D. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117 CrossRef CAS.
- S. Zhao, M. Cheng, J. Li, J. Tian and X. Wang, Chem. Commun., 2011, 47, 2176 RSC.
- J. Wang, W. Xu, J. Ren, X. Liu, G. Lu and Y. Wang, Green Chem., 2011, 13, 2678 RSC.
- M. Mazzotte, D. Gupta, B. Saha, A. K. Patra, A. Bhaumik and M. M. Abu-Omar, unpublished results.
- S. J. Dee and A. T. Bell, ChemSusChem, 2012, 4, 1166 CrossRef PubMed.
- T. Deng, X. Cui, Y. Qi, Y. Wang, X. Hou and Y. Zhu, Chem. Commun., 2012, 48, 5494 RSC.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2006, 45, 2163 CrossRef CAS.
- R. L. De Souze, H. Tu, F. Rataboul and N. Essayem, Challenges, 2012, 3, 212 CrossRef , and references therein.
- P. Carniti, A. Gervasini, S. Biella and A. Auroux, Catal. Today, 2006, 118, 373 CrossRef CAS.
- K. Nakajima, Y. Baba, R. Noma, M. Kitano, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2011, 133, 4224 CrossRef CAS PubMed.
- S. Dutta, S. De, A. K. Patra, M. Sasidharan, A. Bhaumik and B. Saha, Appl. Catal., A, 2011, 409–410, 133 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Green Chem., 2009, 11, 1327 RSC.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2007, 46, 7703 CrossRef CAS.
- E. I. Gürbüz, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383 CrossRef PubMed.
- Y. Roman-Leshkov and J. A. Dumesic, Top. Catal., 2009, 52, 297 CrossRef CAS.
- J. A. Dumesic, Y. Roman-Leshkov and J. N. Chheda, 2009, U.S. Patent, 7572925 B2 Search PubMed.
- E. O. Eisen and J. Joffe, J. Chem. Eng. Data, 1966, 11, 480 CrossRef CAS.
- T. C. Tan and S. Aravinth, Fluid Phase Equilib., 1999, 163, 243 CrossRef CAS.
- M. Gorgenyi, J. Dewulf, H. Van Langenhove and K. Heberger, Chemosphere, 2006, 65, 802 CrossRef PubMed.
- J. A. Dumesic, Y. Roman-Leshkov and J. N. Chheda, 2007, International Patent, WO 2007/146636 A1 Search PubMed.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342 RSC.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446 CrossRef CAS PubMed.
- Y. Roman-Leshkon, C. J. Barrett, Z. Y. Liu and J. A. Demesic, Nature, 2007, 227, 982 CrossRef PubMed.
- X. N. Lv, F. Yang, P. Gao, Z. H. Liu, L. Meng and X. Q. Yu, Ind. Eng. Chem. Res., 2013, 52, 197 CrossRef.
- Y. Roman-Leshkov, J. N. Cheda and J. A. Dumesic, Science, 2006, 312, 1933 CrossRef CAS PubMed.
- T. Okano, K. Qiao, Q. Bao, D. Tomida, H. Hagiwara and C. Yokoyama, Appl. Catal., A, 2013, 451, 1 CrossRef CAS.
- S. Hu, Z. Zhang, Y. Zhou, B. Han, H. Fan, W. Li, J. Song and Y. Xie, Green Chem., 2008, 10, 1280–1283 RSC.
- S. Hu, Z. Zhang, Y. Zhou, J. Song, H. Fan and B. Han, Green Chem., 2009, 11, 873 RSC.
- H. A. Amiri, K. Karimi and S. Roodpeyma, Carbohydr. Res., 2010, 345, 2133 CrossRef CAS PubMed.
- M. Brasholz, K. V. Kanel, C. H. Hornung, S. Saubern and J. Tsanaktsidis, Green Chem., 2011, 13, 1114 RSC.
- S. De, S. Dutta and B. Saha, Green Chem., 2011, 13, 2859 RSC.
- M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 423 CrossRef CAS PubMed.
- Y. Yang, C. W. Hu and M. M. Abu-Omar, Green Chem., 2012, 14, 509 RSC.
- S. Hu, Z. Zhang, J. Song, Y. Zhou and B. Han, Green Chem., 2009, 11, 1746 RSC.
- S. Dutta, S. De, I. Alam, M. M. Abu-Omar and B. Saha, J. Catal., 2012, 288, 8 CrossRef CAS.
- Y. Roman-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954 CrossRef CAS PubMed.
- Y. Yang, C. W. Hu and M. M. Abu-Omar, ChemSusChem, 2012, 5, 405 CrossRef CAS PubMed.
- Y. L. Lu and N. S. Mosier, Biotechnol. Prog., 2007, 23, 116 CrossRef CAS PubMed.
- Y. L. Lu and N. S. Mosier, Biotechnol. Bioeng., 2008, 101, 1170 CrossRef CAS PubMed.
- S. E. Jacobson and C. E. Wyman, Appl. Biochem. Biotechnol., 2000, 84–86, 81 CrossRef.
- E. M. Rubin, Nature, 2008, 454, 841 CrossRef CAS PubMed.
- Q. Cao, X. Guo, J. Guan, X. Mu and D. Zhang, Appl. Catal., A, 2011, 403, 98 CrossRef CAS.
- Y. J. Pag′an-Torres, T. Wang, J. M. R. Gallo, B. H. Shanks and J. A. Dumesic, ACS Catal., 2012, 2, 930 CrossRef.
- J. F. Robyt, in Essentials of Catbohydrates Chemsitry, Springer, New York, 1998, pp. 48–74 Search PubMed.
- (a) T. Wang, Y. J. Pagan-Torres, E. J. Combs, J. A. Dumesic and B. H. Shanks, Top. Catal., 2012, 55, 657Y CrossRef; (b) Roman-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954 CrossRef CAS PubMed.
- R. W. Nagorski and J. P. Richard, J. Am. Chem. Soc., 2001, 123, 894 Search PubMed.
- E. I. Gürbüz, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383 CrossRef PubMed.
- J. Young, G. Chan and Y. Zhang, ChemSusChem, 2009, 2, 731 CrossRef PubMed.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith Jr., Green Chem., 2008, 10, 799 RSC.
- E. Nikolla, Y. Roman-Leshkov, M. Molinder and M. E. Davis, ACS Catal., 2011, 1, 408 CrossRef CAS.
- V. Choudhary, A. B. Pinar, S. I. Sandler, D. G. Vlachos and R. F. Lobo, ACS Catal., 2011, 1, 1724 CrossRef CAS.
- Q. Zhao, L. Wang, S. Zhao, X. Wang and S. Wang, Fuel, 2011, 90, 2289 CrossRef CAS.
- F. Yang, Q. Liu, X. Bai and Y. Du, Bioresour. Technol., 2011, 102, 3424 CrossRef CAS PubMed.
- F. Yang, Q. Liu, M. Yue, X. Bai and Y. Du, Chem. Commun., 2011, 47, 4471 Search PubMed.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2007, 46, 7703 CrossRef CAS.
- S. Dutta, S. De, A. K. Patra, M. Sasidharan, A. Bahumik and B. Saha, Appl. Catal., A, 2011, 409–410, 133 CrossRef CAS.
- A. Dutta, A. K. Patra, S. Dutta, B. Saha and A. Bhaumik, J. Mater. Chem., 2012, 22, 14094 RSC.
- S. De, S. Dutta, A. K. Patra, B. S. Rana, A. K. Sinha, B. Saha and A. Bhaumik, Appl. Catal., A, 2012, 435–436, 197 CrossRef CAS.
- V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijihuis, J. Catal., 2012, 287, 68 CrossRef CAS.
- M. J. Antal, W. S. L. Mok and G. N. Richards, Carbohydr. Res., 1990, 199, 91 CrossRef CAS PubMed.
- V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, ChemSusChem, 2012, 5, 1812 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |