Oxidation of phenolic compounds catalyzed by immobilized multi-enzyme systems with integrated hydrogen peroxide production†
Javier
Rocha-Martin
a,
Susana
Velasco-Lozano
b,
José M.
Guisán
*a and
Fernando
López-Gallego
*a
aDepartamento de Biocatálisis, Instituto de Catálisis y Petroleoquímica, CSIC, Campus UAM, Cantoblanco, 28049 Madrid, Spain. E-mail: jmguisan@icp.csic.es; flg802@gmail.com; Fax: +34 915854760; Tel: +34 915854809/+34 915855477
bDepartamento de Biotecnología, Universidad Autónoma Metropolitana Iztapalapa, Av. San Rafael Atlixco 186, Col. Vicentina 09340, D.F. México
First published on 15th October 2013
Abstract
Suicide inactivation of peroxidases by hydrogen peroxide is the major deterrent to using such biocatalysts in oxidative processes. This has been successfully addressed by the in situ generation of H2O2. In this study, we have developed a novel multi-enzyme biocatalyst that has been immobilized on agarose-type carriers to oxidize phenols using oxygen and formic acid as indirect oxidants. This original system couples the in situ production of H2O2 to the phenol oxidation via an enzymatic cascade that involved three different enzymes (formate dehydrogenase, NADH-oxidase and peroxidase) and two different redox cofactors: nicotinamide and flavin derivatives. The cascade reaction only works when enzymes are immobilized on the solid support since soluble enzymes are dramatically inactivated under the reaction conditions. The highest oxidation efficiency was achieved by combining two different solid biocatalysts: (1) formate dehydrogenase and NADH-oxidase co-immobilized onto agarose beads activated with glyoxyl groups and (2) peroxidase immobilized onto agarose beads as well but activated with boronate groups. Unlike conventional peroxidase-mediated oxidations with exogenous H2O2, this novel system enables the quantitative oxidation of phenol without the addition of H2O2. Furthermore, this novel system can use a broad range of redox cofactors with similar oxidative effectiveness. Therefore, this novel immobilized tri-enzyme system removes important pollutants such as hydroxylated aromatic derivatives (phenol, 4-aminophenol, 2,4-dichloro-phenol or α-naphthol) using formic acid and molecular oxygen as substrates. In addition, this system generates CO2 as waste beyond the oxidized phenols that can be easily separated from the aqueous solution. The sustainability of this system is supported by an E-factor of 1.3 and an atom economy of 43%.
Introduction
Peroxidases (E.C. 1.11.1.7) are versatile oxidative biocatalysts that need hydrogen peroxide as a co-substrate. They have been used in many biotechnological applications such as biosensors, bioremediation and wastewater treatment or organic synthesis.1 Peroxidases, and mainly peroxidase from horseradish (HRP), have been exploited to remove phenolic compounds from synthetic model effluents as well as from real industrial water.2–5 The oxidation of these pollutants catalyzed by HRP facilitates the remediation of wastewater.6 However, HRP requires large amounts of H2O2 to treat large volumes of wastewater which has two drawbacks: (1) chemical synthesis of H2O2 is not environmentally friendly, so its sustainability as a “green oxidant” is overestimated,7 and (2) high concentrations of H2O2 trigger the suicide inactivation of peroxidases.8 Therefore, the addition of large volumes of H2O2 presents process sustainability limitations.Several solutions to overcome the limitation of using high H2O2 concentrations as oxidants have been proposed. Among those solutions, in situ production of H2O2 from molecular oxygen is revealed as one of the most successful ones9–14 through both physicochemical and enzymatic methodologies.10,13,14 The physicochemical systems using electrochemical or optical generation of H2O2 require complex reactor designs that decrease the cost efficiency. In contrast, a fully enzymatic system can require only simple equipment and can work under mild conditions, enabling enhanced sustainability. As far as we know, there is only one enzymatic method for the in situ production of H2O2 for use in peroxidase-mediated oxidation reactions. This method involves flavin-dependent oxidases which transfer hydrogen from reduced substrates to molecular oxygen yielding H2O2 as a by-product.11,15 Glucose oxidase is the most commonly used in situ producer of H2O2 in these processes.9,11,15 However this system presents several limitations: (1) the glucose as a co-substrate might diminish the cost-efficiency of the oxidative process, (2) the in situ H2O2 production yields gluconic acid as a by-product that may need to be removed at the end of the process and (3) operational enzyme inactivation that hampers their industrial use.
Enzyme immobilization can be used to address these inactivation issues. A large number of enzymes have been immobilized on a pre-existing ‘carrier’, resulting in high stabilization factors that pave their way to industrial applications. Previously, our group developed a series of methodologies to stabilize several flavin-dependent oxidases based on enzyme immobilization.16–18 Immobilized preparations allow continuous operations in flow-type fixed bed reactors. Moreover, the enzyme immobilization protocol determines the final properties of the biocatalyst. Therefore, selection of immobilization chemistries is the key to achieving an optimal heterogeneous biocatalyst. This is even more relevant for co-immobilized multi-enzyme systems where optimal immobilization chemistries for each enzyme must be coordinated in order to obtain an active and stable heterogeneous biocatalyst.
Herein, we propose an immobilized multi-enzyme system for the in situ production of H2O2 without yielding undesired by-products. This system is based on the NADH oxidase from Thermus thermophilus HB27 (NOX) to produce H2O2 from O2. This oxidation is mediated by flavins and uses catalytic amounts of NADH because it is regenerated in situ by formate dehydrogenase from Pseudomonas sp. (FDH), which utilizes NAD+ to oxidize formic acid to carbon dioxide. In this work, preparation of co-immobilized NOX and FDH has been further coupled to immobilized preparation of HRP which can utilize the produced H2O2 to oxidize pollutants in aqueous solutions under mild conditions. We have applied this heterogeneous multi-enzyme system to phenol oxidation that yields solid products which can be further separated by simple decantation (Fig. 1). Using this methodology, we have demonstrated the oxidation of phenols from aqueous solutions, with obvious applications towards wastewater treatment systems.
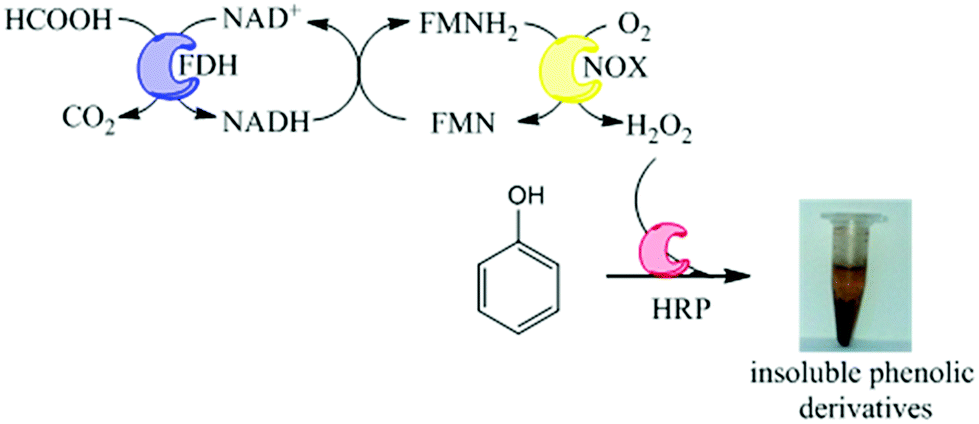 | ||
| Fig. 1 Reaction scheme of phenol removal with in situ and enzyme-based H2O2 production. | ||
Results and discussion
Novel tri-enzymatic system catalyzes phenol degradation through in situ H2O2 production
We have developed a novel two-enzyme system for the in situ production of H2O2 that can be further used by HRP to perform oxidation reactions. This system can be carried out in one-pot as a cascade reaction. Firstly, FDH (1st enzyme) oxidizes formic acid concomitantly reducing NAD+ to NADH and yielding CO2 which easily diffuses to the air phase, shifting the equilibrium to the oxidation reaction. NADH is further used by NOX (2nd enzyme) – which is a flavin-protein – to reduce molecular oxygen, yielding H2O2. Finally, HRP uses such in situ produced H2O2 to oxidize phenols. The coordination of FDH and NOX activities enables the in situ recycling of the redox cofactor pair (NADH/NAD+) (Fig. 1). Therefore, in this reaction scheme, 1 mol of formic acid is needed to oxidize 1 mol of phenolic compound using catalytic amounts of redox cofactors.Unexpectedly, the soluble tri-enzyme system did not significantly oxidize phenol from aqueous solutions. This result might be a consequence of the thermophilic origin of NOX that would not work optimally under mild temperatures. However, as we reported that NOX functioned under mesophilic temperatures (25–37 °C),16 this should not be the reason why the system did not work. A more plausible explanation might be related to the enzyme inactivation promoted either by the radicals created from the phenol oxidation or by the in situ generated H2O2. For this reason we immobilized the three-enzymes onto agarose-type carriers aiming to stabilize and make them suitable for this biotechnological application.
Immobilization and co-immobilization protocols for FDH, NOX and HRP
FDH, NOX and HRP were individually immobilized on different carriers. NOX (Table 1, entry 1) and FDH (Table 1, entry 2) were immobilized on agarose beads activated with glyoxyl groups (Ag-G),16,19 while we used agarose beads activated with boronate groups (Ag-B) to immobilize HRP (Table 1, entry 3).20 Since the co-immobilization of multi-enzyme systems has been unveiled as an efficient methodology to increase the catalytic efficiency of given reaction cascades, we have also designed different co-immobilization patterns: (i) bi-enzyme biocatalysts containing both FDH and NOX immobilized on Ag-G (Table 1, entry 4), and (ii) tri-enzyme biocatalysts where NOX, FDH and HRP were co-immobilized on an agarose-type carrier activated with both glyoxyl and boronate groups (Ag-BG) (Table 1, entry 5). This hetero-functional surface is necessary to co-immobilize HRP with NOX and FDH onto the same solid surface, since glyoxyl chemistry does not work for HRP and boronate chemistry does not for either NOX or FDH.| Entry | NOX | FDH | HRP | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Ψ a (%) | Ar![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b [U gcarrier−1] b [U gcarrier−1] |
RArc (%) |
Ψ a (%) | Arb [U gcarrier−1] |
RArc (%) |
Ψ a (%) | Arb [U gcarrier−1] |
RArc (%) |
|
| a Immobilization yield, (Ψ) = 100 − [(supernatant activity/blank activity) × 100]. b Residual activity (Ar) means the enzyme activity after the immobilization protocol and is defined as enzyme units (see the Experimental section) per gram of carrier measured after immobilization protocol was completed. c Relative residual activity, (RAr) = immobilized activity (U gcarrier−1)/Ar (U gcarrier−1). The immobilized activity expresses the immobilized enzyme according to the immobilization yield but it does not consider the enzyme inactivation promoted through the immobilization protocol. Entries correspond to (1) NOX immobilized on Ag-G. (2) FDH immobilized on Ag-G. (3) HRP immobilized on Ag-B. (4) NOX and FDH co-immobilized on Ag-G. (5) HRP, NOX and FDH co-immobilized on Ag-BG. | |||||||||
| 1 | 100 | 3 ± 0.15 | 34 | — | — | — | — | ||
| 2 | — | — | 97 | 2.6 + 0.13 | 18 | — | — | ||
| 3 | — | — | — | — | 60 | 63 + 3 | 42 | ||
| 4 | 100 | 1.7 + 0.17 | 18 | 60 | 2.5 + 0.08 | 30 | — | — | |
| 5 | 100 | 1.7 + 0.15 | 18 | 50 | 2.5 + 0.13 | 37 | 60 | 5.8 + 0.3 | 2 |
Table 1 shows the immobilization parameters of NOX, FDH and HRP for their single immobilization (entries 1–3) and co-immobilization protocols (entries 4 and 5). On the one hand, individual immobilization of NOX and FDH on Ag-G was quantitative, whereas the HRP immobilization yield on Ag-B was 60% of offered enzyme activity. On the other hand, the single immobilization protocols (entries 1–3) caused a reduction of the residual activity for the three enzymes. We found similar immobilization yields for NOX when it was co-immobilized with FDH on Ag-G (entry 4). However, such a co-immobilization protocol decreased the FDH immobilization yield (entry 4). When the three enzymes were co-immobilized on the same porous surface (Ag-BG), we observed a dramatic reduction on the HRP residual activity.
As our group previously reported, the immobilization of NOX and FDH notably reduced their residual activities in both separately immobilized and co-immobilized preparations using the glyoxyl chemistry.16,19 However, residual activities were sufficient to produce hydrogen peroxide and, most important, enzyme stabilization promoted by glyoxyl immobilization allowed the further use of both co-immobilized and separately immobilized preparation for the phenol removal (Fig. S1†).16,19 In contrast, the co-immobilization of the three enzymes resulted in an extremely low HRP activity. Likely, the glyoxyl groups on the surface of Ag-BG dramatically inactivated HRP, hampering its optimal co-immobilization along with NOX and FDH on the same solid surface. This inactivation may rely on the features of glyoxyl chemistry.21,22 HRP is immobilized on Ag-BG in a three-step mechanism;23 the enzyme is firstly immobilized through its glycosylation sites, and then the vicinal lysine residues react with the highly reactive glyoxyl groups under alkaline conditions. This latter reaction yields reversible Schiff's base bonds that must be finally reduced to convert them into irreversible ones. This process was carried out using NaBH4 as a reducing agent. We observed how the post-immobilization reduction resulted in the 98% loss of HRP activity (Table 1, entry 5). Nevertheless, incubation of the soluble HRP with NaBH4 did not promote enzyme inactivation. Therefore, we propose that HRP inactivation is provoked by turning the flexible and reversible Schiff's base bonds between the enzyme and the carrier into irreversible and rigid secondary amine bonds. The spacer arms of glyoxyl groups on the support surface are very short, which greatly reduces the distance between both the protein and the carrier surface. Therefore, HRP will see reduced conformation flexibility, and such tight links between the protein and the carrier can even trigger structural distortions as has been described for other proteins.21,22 Pramparo et al. found dramatic reduction of enzyme activity when HRP was immobilized on solid carriers through aldehyde chemistry.24 Therefore, HRP could not be used in its irreversibly co-immobilized form, but it could be used as reversibly immobilized on agarose activated with boronate groups (Ag-B).25 The residual activity of such a preparation was 42% of the immobilized enzyme, although HRP was attached to the carrier in a reversible manner. Therefore, the higher residual activity of the HRP immobilized preparation is more important than the irreversible covalent attachment between the enzyme and the carrier surface.
Finally, we evaluated the stability of the immobilized preparations of NOX, FDH and HRP (Fig. S1†). These immobilized preparations were thermally inactivated under different temperatures and compared to their soluble counterparts. Immobilized enzymes were more stable than soluble ones except HRP, which was almost as stable as its soluble form (Fig. S1†). In general terms, the multi-enzyme system is much more robust when the three enzymes are immobilized. Therefore, although immobilization efficiency was not very high for all the enzymes involved on the multi-enzyme system the stability gained by mainly NOX and FDH drove us to using these insoluble preparations as biocatalysts to oxidize phenols with integrated H2O2 production.
Novel immobilized tri-enzymatic system catalyzes phenol degradation with in situ H2O2 production
Different preparations of immobilized FDH, NOX and HRP were used to evaluate 4 different enzymatic systems to oxidize and thus remove phenol from aqueous solutions using different H2O2 sources. System A was the conventional method to enzymatically oxidize phenols using HRP (immobilized on Ag-B carriers) and adding H2O2 as a co-substrate.24,26 System B employed NOX, FDH and HRP separately immobilized on different carriers (entries 1–3, Table 1). System C employed FDH and NOX co-immobilized on Ag-G (entry 4, Table 1) and HRP immobilized on Ag-B (entry 3, Table 1). And finally system D employed NOX, FDH and HRP co-immobilized on Ag-BG (entry 5, Table 1). The four resulting enzymatic systems were tested in phenol oxidative reactions. In the case of system A, we exogenously added 4 mM H2O2 as a co-substrate to ensure the HRP maximum oxidation rate,24,26–28 while system B–D endogenously produced H2O2.Fig. 2 shows the phenol oxidation rates of different immobilized preparations. The best results were observed with system C. The resulting tri-enzymatic system immobilized on two different carriers catalyzed the quantitative phenol oxidation with in situ H2O2 generation. However when H2O2 was exogenously added as an oxidation co-substrate, system A – without in situ H2O2 production system – only removed 50% of phenol (in 4 hours), afterwards the enzyme was completely inactive. Hence, system C likely accumulates very low H2O2 concentration in the reaction medium which avoids HRP suicide inactivation, explaining the good results in terms of phenol oxidation yield.
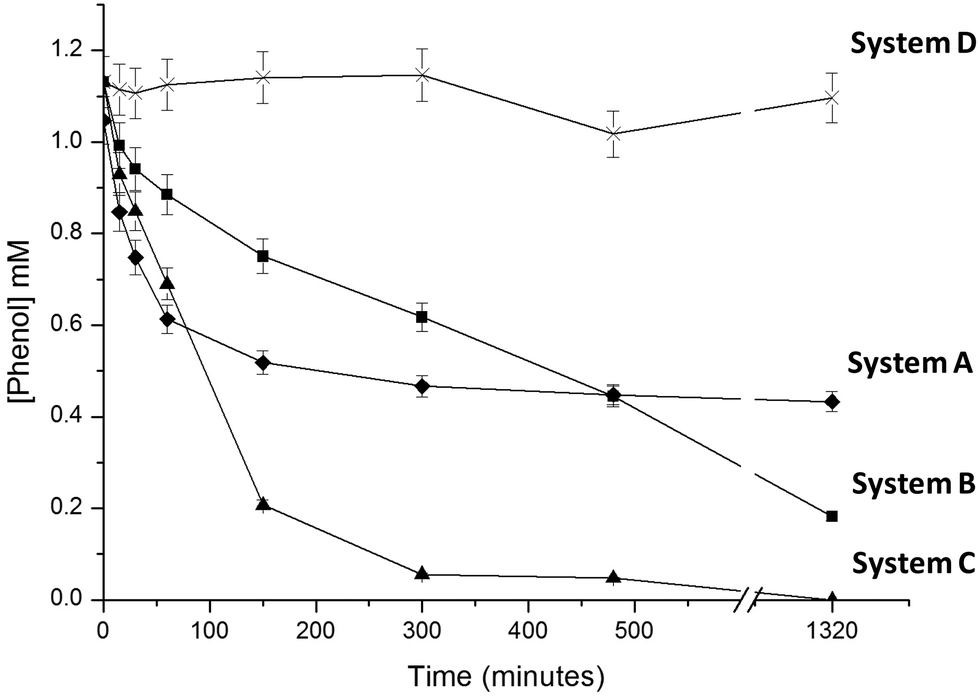 | ||
| Fig. 2 Phenol removal catalyzed by different immobilized enzymatic systems. Reactions were catalyzed by three different immobilized enzyme systems: system A (◆) is catalyzed by HRP immobilized on Ag-B. System B (■) is catalyzed by HRP immobilized on Ag-B and NOX and FDH separately immobilized on Ag-G. System C (▲) is catalyzed by NOX and FDH co-immobilized on Ag-G and HRP immobilized on Ag-B. Finally, system D (X) is formed by HRP, NOX and FDH co-immobilized on Ag-BG. The reaction medium contained 1 mM phenol and 100 mM formic acid dissolved in 100 mM sodium phosphate buffer at pH 6 and 25 °C. In system A, 4 mM H2O2 was used as a co-substrate, while in systems B–D, H2O2 is replaced by adding 0.2 mM NADH and 0.15 mM FADH2. These latter cofactors are required by NOX to in situ produce H2O2. The formic acid is needed to in situ regenerate the NADH. | ||
Using the same HRP amount per reaction, we observed how the HRP initial rate (V0(HRP)) for phenol oxidation varied in relation to the source of H2O2. V0(HRP) with exogenous addition of H2O2 was 0.22 μmol h−1 mL−1 while system C and system B with in situ H2O2 generation showed V0(HRP) of 0.21 and 0.14 μmol h−1 mL−1 respectively. These data suggest that in situ H2O2 production is more rapid when both NOX and FDH were co-localizing across the same porous surface. In this scenario, the cofactor recycling needed to reduce molecular oxygen and form hydrogen peroxide will take place within the same porous microenvironment as has been recently reported by our group.29 Consequently the cofactors (NADH/NAD+) would not have to diffuse in and out between two different porous surfaces to be recycled. Moreover, the V0 (HRP) of system C was close to that observed by exogenously adding H2O2 (system A). It suggests that the co-immobilized system supplies enough H2O2 to saturate HRP to work at a maximum rate – similar to that found with exogenous H2O2 – but avoiding the HRP suicide inactivation that hampers maximum yields. In comparison, system D demonstrated a very slow V0 (HRP) of 0.018 μmol h−1 mL−1 resulting in a poor phenol oxidation yield of 5% in 24 hours.
In principle the co-immobilization of the three enzymes should be positive for system performance; however, the immobilization chemistry for both NOX and FDH was incompatible for HRP, which resulted in a low residual peroxidase-activity. Such poor HRP activity made impossible the activity coordination between the three enzymes. In system C, the activity ratio HRP:NOX was 37:1 while in system D it was only 3.3:1 (Table 1). Likely, the low peroxidase-activity excess observed in system D led to higher H2O2 accumulation that might promote the enzyme inactivation of the system, making this preparation a poor choice for phenol oxidation.
Summing up, we have demonstrated that an optimally immobilized tri-enzyme system in situ oxidizes phenol without exogenous addition of H2O2 as a co-substrate without inactivating HRP along the investigated reaction time. Since the system depends on many different parameters, we next evaluated several parameters that may affect system performance.
Effect of hydrogen peroxide source on the HRP inactivation and total turnover number
As we have mentioned above, in situ generation of H2O2 circumvents HRP inactivation. However, such inactivation might be also avoided by the addition of higher HRP concentration in the reaction mixture because the inactivation degree lies on the molar ratio H2O2:HRP.30 In this context, the more hydrogen peroxide molecules per HRP molecule, the higher and faster the HRP inactivation. Thus, HRP concentration will affect not only the reaction rate, but also the final yield in systems with exogenous H2O2. Fig. 3 shows how phenol removal yield decreased along with the HRP concentration using system A (exogenous H2O2). In contrast, the reaction yield remained constant regardless of HRP concentration when H2O2 was generated in situ. This fact dramatically impacted on the HRP total turnover number (TTN) under low enzyme concentration conditions (5 μg mL−1); TTN with exogenous H2O2 was 1428 while TTN with enzymatically in situ produced H2O2 was 6785 (almost 5-fold higher). Hence we hypothesize that the in situ and dynamic production of H2O2 would be limiting the oxidant concentration in the reaction mixture, and consequently H2O2 would be utilized to oxidize phenol rather than to trigger the HRP suicide inactivation.
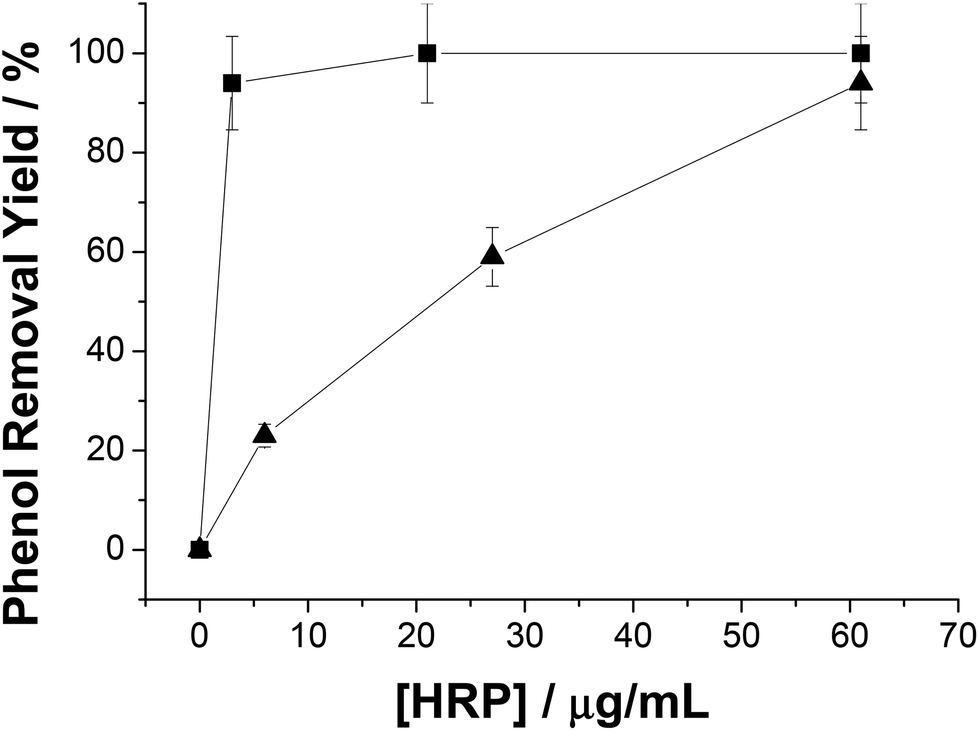 | ||
| Fig. 3 Effect of HRP concentration on the phenol removal yield (PRY). Phenol was removed using two different enzyme systems with different concentrations of HRP. In system A (▲) HRP immobilized on Ag-B catalyzed the phenol removal using the pre-existing H2O2 as a co-substrate. In system C (■) HRP immobilized on Ag-B catalyzed the phenol removal by using the H2O2 produced in situ by NOX and FDH co-immobilized on Ag-G using NAD+, FADH2 (as cofactors); and formic acid (as a substrate). | ||
Role of the redox cofactor in the phenol oxidation
The three-enzymatic system requires two different redox cofactors: the nicotinamide cofactor (utilized by NOX and FDH) and the flavin cofactor (utilized by NOX) to synthesize H2O2 from formic acid and molecular oxygen. The nicotinamide cofactor transfers the electron from the formic to the flavin cofactor that reduces the molecular oxygen to yield hydrogen peroxide. To improve the efficiency of system C, we investigated the effect of varying the specific cofactors utilized by NOX (Table 2).| Reaction | Cofactor concentration (mM) |
V
0a (mM h−1) |
PRYb (%) | ||||
|---|---|---|---|---|---|---|---|
| NADH | NAD+ | FAD+ | FMN | Riboflavin | |||
| a V 0 This values means the amount of phenol (μmol) removed in one hour per mL of reaction mixture containing 1 mM phenol. b Phenol removing yield is calculated after 27 hours of reaction. | |||||||
| 1 | — | 0.2 | — | 0.15 | — | 0.2 ± 0.02 | 87 ± 9 |
| 2 | — | 0.2 | 0.15 | — | — | 0.22 ± 0.01 | 100 ± 3 |
| 3 | — | 0.2 | — | — | 0.15 | 0.2 ± 0.02 | 88 ± 10 |
| 4 | 0.2 | — | — | 0.15 | — | 0.06 ± 0.01 | 63 ± 5 |
| 5 | 0.2 | — | 0.15 | — | — | 0.13 ± 0.01 | 95 ± 3 |
| 6 | 0.2 | — | 0 | — | 0.15 | 0.04 ± 0.01 | 40.6 ± 8 |
Triggering the reaction with the oxidized nicotinamide cofactor (NAD+) enabled the most rapid phenol degradation regardless of the flavin cofactor utilized, yielding >85% of phenol removal for all cases. However, using the reduced nicotinamide cofactor (NADH), we only observed high phenol removal yields when the flavin cofactor was FAD+, and the phenol degradation rate was slower than the rate achieved with NAD+.
The optimal concentration of both nicotinamide and flavin cofactors is displayed in Fig. 4. In the case of the nicotinamide cofactor, we found 0.2 mM as the optimal nicotinamide concentration to remove phenol (Fig. 4A). It is noteworthy that higher NADH concentrations decreased the phenol removal efficiency in terms of both rate and yield. This fact may be pointing out an interference of the nicotinamide cofactor in the oxidation reaction catalyzed by HRP. We suggest a potential competition between H2O2 and NADH that would decrease the oxidative activity of HRP (Fig. S2†).
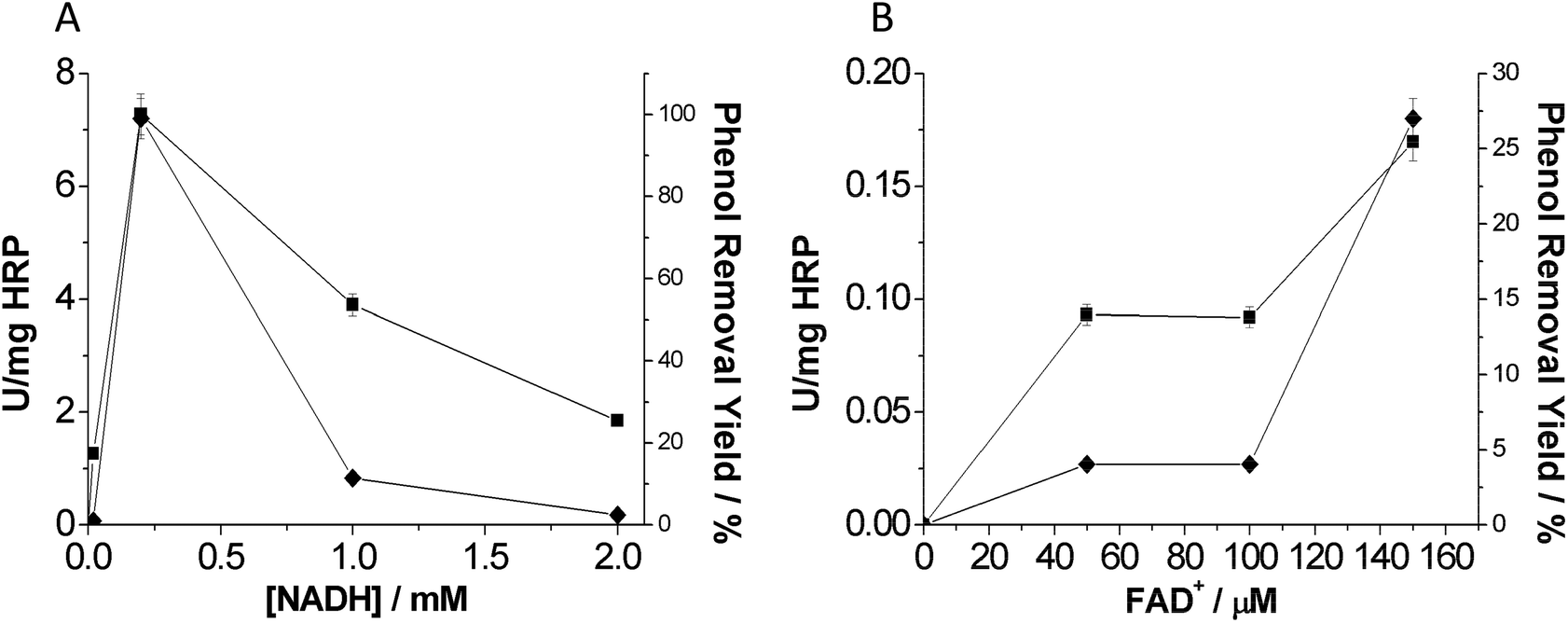 | ||
| Fig. 4 Effect of cofactor concentration on final HRP activity (◆) and phenol removal yield (■). (A) NADH; reaction were carried out with system C and 0.15 mM FADH2. (B) FADH2; reactions were carried out with system C and 2 mM NADH. 1 activity unit (U) is defined as the amount of enzyme able to oxidize 1 mmol of phenol per minute and per mg of HRP. For further methodological details see the Experimental section. | ||
For HRP-mediated oxidation, activity increased with the flavin nucleotide concentration. The highest activity was observed with 0.15 mM flavin nucleotide in the reaction mixture (Fig. 4B). This result agreed with the maximum flavin concentration to provide the highest NOX activity.16 Therefore, in the presence of the optimal amount of flavin cofactor, NOX produces H2O2 at the highest rate. Unlike the nicotinamide cofactor, the flavin cofactor did not seem to interfere with the heme-mediated catalysis. Furthermore, this tri-enzyme system presents a high flexibility since it is able to use different types of flavin cofactors: FAD, FMN or even riboflavin. A similar fact was also observed by Perez et al.10 by using a photo-chemical system to produce H2O2 together with a flavin cofactor electron mediator. Interestingly, having such versatile systems allows the use of the most economically viable flavin cofactors such as riboflavin.
Removal of different recalcitrant compounds using the immobilized tri-enzymatic system
HRP enables the oxidation of a broad range of substrates that are mainly related to aromatic acids and alcohols.1 Some of these compounds are extremely recalcitrant and their removal is of much interest for the wastewater treatment industry. Table 3 shows the removal yield of several phenols using system C. Oxidation was observed for all recalcitrant compounds although reaction yields varied depending on the aromatic compound (Table 3); system C oxidized phenol itself much more efficiently than the other aromatic compounds. In fact, 100% oxidation of α-naphthol required 100 times more immobilized HRP than quantitative phenol oxidation, and partial 2,4-dichlorophenol and 4-aminophenol oxidation was only possible with 200 times more immobilized HRP. These results show that HRP oxidizes less efficiently those aromatic compounds that contain heteroatoms in addition to oxygen (Table 3). Hence, it seems that radicals formed during the oxidation of those latter phenolic derivatives may be more toxic for the multi-enzyme systems than radicals derived from phenol. In spite of potential negative effects of different radicals on the performance of this multi-enzyme system, the final oxidation yields in 7 hours (using only 0.3% (w/v) of immobilized HRP catalyst at the maximum) were comparable to other productivity data previously reported.24 The efficiency of this biotransformation using this multi-enzyme system may be improved by using higher amounts of catalyst or higher loads of HRP in the immobilized preparations.| Compound |
|
|
|
|
|---|---|---|---|---|
| Remove yield (%) | 95 ± 91 | 70 ± 32 | 50 ± 32 | 91 ± 73 |
Sustainability of in situ H2O2 production catalyzed by a novel immobilized tri-enzyme system
The use of enzymatic process within the chemical technology field is justified by their better fitness to the green chemistry principles.31 In this context, the FDH/NOX system for the in situ generation of H2O2 has shown better green chemistry values such as E-factor and atom economy than other in situ H2O2 production systems (Table 4). The reason behind these better values lies in the production of fewer wastes because this system mainly produces carbon dioxide as a by-product with the additional advantage that it is easily dissipated to the air phase. In contrast the non-enzymatic in situ H2O2 production using light, flavin cofactor and EDTA generates 6-fold more wastes than the NOX/FDH system proposed herein. In the same way, H2O2-production mediated by glucose oxidase (GOX) generates 1 mol of gluconic acids as by-products to produce 1 mol of H2O2 which results in a 4 times higher E-factor and 3 times lower atom economy compared to the multi-enzyme system reported in this work. Hence, this novel system matches the green chemistry principles because almost half of the substrate atoms (formic acid and molecular oxygen) are contained in the final product (H2O2), waste production is very low, the wastes (H2O and CO2) are completely innocuous, and finally the system uses biocatalysis to accelerate the reaction. In fact, while the glucose and EDTA systems produce other organic acids that must be removed, the novel system we present here only produces CO2 as wastes that can be easily dissipated and even re-usable in further CO2-transformation technologies.| System | Raw material (g mol−1) | Wastesa (g mol−1) | H2O2 (g mol−1) | E-factorb | Atom economyc (%) | Ref. |
|---|---|---|---|---|---|---|
| a H2O was not considered as waste. b E-factor = ((raw material (g mol−1) − product − product (g))/product (g)). c Atom economy (%) = (mass of atom in the desired product/mass of atom in the raw material) × 100. The cofactors and the solid biocatalysts were not included in the calculation of the green chemistry parameters since they were used in catalytic amounts meaning less than 2% (w/v) of the reaction mixture. | ||||||
| Glucose/GOX | 212 | 178 | 34 | 5.2 | 16 | 10 |
| EDTA/light/FMN | 374 | 264 | 34 | 7.8 | 9.1 | 9 |
| HCOOH/NADH/Flavin | 78 | 44 | 34 | 1.3 | 43 | This work |
Conclusions
Formate dehydrogenase, NADH oxidase and peroxidase have been immobilized through different chemistries on agarose beads. This immobilized tri-enzyme system significantly avoids the peroxidase suicide inactivation. The best scheme to efficiently oxidize phenols without exogenously added H2O2 involves two carriers: (1) both NADH oxidase and formate dehydrogenase co-immobilized on agarose beads activated with glyoxyl groups and (2) peroxidase immobilized on agarose beads activated with boronate groups. Hence, these two carriers were combined in the same vessel in order to quantitatively oxidize phenols with in situ production of H2O2 from oxygen and formic acid. Furthermore, the system requires both nicotinamide and flavin redox cofactor in catalytic amounts to in situ produce H2O2. Nevertheless the system accepted different types of flavin-type derivatives, even riboflavin, while its performance was negatively affected by NADH. This latter effect was enhanced at very low H2O2 concentrations in the reaction vessel. In addition, this system was proved to work with different substituted phenols although with different oxidation efficiencies. Finally, a simple sustainability study demonstrated that using formic acid and molecular oxygen as substrates to in situ generate H2O2 improves “green chemistry” parameters such as E-factor and atom economy compared to other systems for in situ H2O2 production. Herein, we have applied this enzymatic system of dynamic H2O2 generation to phenol oxidation, but it could potentially be applied to other reactions, both biological and chemical, where H2O2 is the oxidant co-substrate.Experimental section
Chemicals
Nicotinamide adenine dinucleotides (NAD+ and NADH) and flavin nucleotides (FAD, FMN and riboflavin) were purchased from GERBU Biotechnik GmbH (Wieblingen, Germany). Horseradish peroxidase (HRP), 2-amino-phenyl boronic acid and phenol were supplied by Sigma-Aldrich Co (St. Louis, IL). Formate dehydrogenase from Pseudomonas sp. (FDH) was purchased from Jülich Fine Chemicals (Codexis, Redwood city, CA). Crosslinked agarose beads (4%) were from Agarose Beads Technology (Madrid, Spain). Agarose beads activated with boronate groups (Ag-B) were prepared as previously described.23 Glyoxyl agarose beads (Ag-G) were prepared as previously described.32 Heterofunctional agarose carrier activated with both boronate and glyoxyl groups (Ag-BG) was prepared as previously described.23 NADH oxidase (NOX) from T. thermophilus overexpressed in Escherichia coli was produced and purified as published elsewhere.16 Protein concentration was determined using Bradford's method.33 All other used reagents were of analytical grade.Immobilization of multi-enzyme systems
For each system, enzymes were mixed with different agarose-type supports under gentle stirring. Samples of the supernatants and the support-enzyme suspensions were withdrawn at different times, and different enzymatic activities were assayed to monitor the immobilization progress. Blanks containing enzyme solutions under the same conditions, but without solid carriers, were monitored in parallel to the immobilization experiment.Immobilization of HRP on Ag-B
1 g of Ag-B was incubated with 10 mL of 25 U mL−1 of HRP dissolved in 10 mM sodium phosphate at pH 7. The suspension was kept under mild stirring at 25 °C. Once the maximum immobilization yield was reached, the immobilization suspension was vacuum filtered to remove the non-bound protein.Immobilization of NOX on Ag-G
1 g of Ag-G was incubated with 10 mL of 0.8 U mL−1 of NOX dissolved in 100 mM sodium carbonate at pH 10. The suspension was kept under mild stirring at 25 °C. Once the maximum immobilization yield was reached, sodium borohydride up to 1 mg mL−1 final concentration was added to the immobilization suspension. Finally the reduced immobilized preparation was vacuum filtered to remove the non-bound protein.Immobilization of FDH on Ag-G
1 g of Ag-G was incubated with 10 mL of 1.4 U mL−1 of FDH dissolved in 100 mM sodium carbonate at pH 10. The suspension was kept under mild stirring at 25 °C. Once the maximum immobilization yield was reached, sodium borohydride up to 1 mg mL−1 final concentration was added to the immobilization suspension. Finally the reduced immobilized preparation was vacuum filtered to remove the non-bound protein.Co-immobilization of NOX and FDH on Ag-G
Firstly, 1 g of Ag-G was incubated with 10 mL of 0.8 U mL−1 of NOX dissolved in 100 mM sodium carbonate at pH 10 for 22 h and later 1 mL of 14 U mL−1 of FDH solution was added to the suspension and it was allowed to react for 1.5 hours. The suspension was always kept under mild stirring at 25 °C. Once the maximum immobilization yields were reached for both enzymes, sodium borohydride up to 1 mg mL−1 final concentration was added to the co-immobilization suspension. Finally the reduced immobilized preparation was vacuum filtered to remove the non-bound protein.Co-immobilization of HRP, NOX and FDH on Ag-BG
Firstly, 1 g of Ag-BG was incubated with 10 mL of 25 U mL−1 HRP dissolved in 10 mM sodium phosphate at pH 7. The suspension was kept under mild stirring at 25 °C. Once the maximum immobilization yield was reached, the immobilization suspension was vacuum filtered to remove the non-bound protein. Then, 10 mL of 0.8 U mL−1 NOX solution in 100 mM sodium carbonate at pH 10 was incubated for 22 h with 1 g of solid carrier where HRP had been immobilized. Once NOX was immobilized, 1 mL of 14 U mL−1 FDH solution was added to the immobilization suspension and it was allowed to react for 1.5 hours. Sodium borohydride up to 1 mg mL−1 final concentration was added to the co-immobilization suspension. Finally the reduced immobilized preparation was vacuum filtered to remove the non-bound protein.Enzyme assays
The redox activities of NOX and FDH were spectrophotometrically measured by monitoring the absorbance at 340 nm that varies based on the concomitant production or consumption of the corresponding nicotinamide cofactor according to the specific methods elsewhere described.16,29 HRP activity was measured in 0.1 M sodium phosphate at pH 6 using 42 mM pyrogallol and 0.026% (v/v) H2O2 as substrates. The reaction mixture was incubated at 25 °C with magnetic stirring and the reaction was triggered with different amounts of HRP in both soluble and immobilized forms. The oxidative reaction was spectrophotometrically monitored at 420 nm in order to determine the enzymatic activity based on ε = 12000 M−1 cm−1. This activity was expressed in international units (U), which means the amount of enzyme needed to oxidize 1 mol of pyrogallol per minute in the reaction conditions described above.Enzymatic oxidation of phenolic compounds
The reaction mixture contained 1 mM of the aromatic compound (phenol, 2,4-dichlorophenol, 4-aminophenol or α-naphthol), 0.1 M formic acid and different concentrations of nicotinamide (NAD+ or NADH) or flavin (FAD, FMN or riboflavin) dissolved in 0.1 M sodium phosphate at pH 6. The reaction was triggered with different amounts of enzymes in either their soluble or immobilized forms. Using soluble enzymes, samples of reaction mixture were withdrawn at different times and then ultra-filtered using Amicon Ultra centrifugal tubes with 10000 Da cut-off (Milipore, MA, USA). In the case of the reaction catalyzed with the immobilized enzymes, the supernatant was directly withdrawn at different times. The reaction courses were followed via reverse-phase HPLC (Spectra Physic SP 100 pump coupled with an UV detector Spectra Physic SP 8450) using a Kromasil C18 column (15 cm × 0.46 cm) supplied by Análisis Vínicos (Spain). All the analyses were performed at a flow rate of 1 mL min−1. For phenol oxidation, the mobile phase was a mixture of methanol–Milli-Q water (30/70, v/v) and the analyses were performed by recording the absorbance at 270 nm. For 2,4-dichlorophenol oxidation, the mobile phase was a mixture of methanol–Milli-Q water (60/40, v/v) and the analyses were performed by recording the absorbance at 280 nm. For 4-aminophenol the mobile phase was a mixture of methanol–Milli-Q water (20/80, v/v) and the analyses were performed by recording the absorbance at 270 nm. In the case of α-naphthol, the mobile phase was a mixture of methanol–Milli-Q water (45/55, v/v). The retention times for phenol, 2,4-dichlorophenol, 4-aminophenol and α-naphthol were 5.1, 14.5, 2.5 and 26 min respectively.
Acknowledgements
This work was sponsored by the Spanish Ministry of Economy and Innovation (projects BIO-2012-36861 and CTQ2009-07568). In addition, we would like to thank the Spanish Government for the Juan de la Cierva fellowship funding to PhD Fernando López Gallego. We would also like to acknowledge Dr Jacob Vick who helped us to edit the manuscript.References
- A. M. Azevedo, V. C. Martins, D. M. F. Prazeres, V. Vojinović, J. M. S. Cabral and L. P. Fonseca, Biotechnol. Annu. Rev., 2003, 9, 199–247 CrossRef CAS.
- Q. Husain, Rev. Environ. Sci., Biotechnol., 2010, 117–140 CrossRef CAS.
- J. A. Nicell, J. K. Bewtra, N. Biswas and E. Taylor, Water Res., 1993, 27, 1629–1639 CrossRef CAS.
- A. M. Klibanov, T. M. Tu and K. P. Scott, Science, 1983, 221, 259–261 CAS.
- S. Dalal and M. N. Gupta, Chemosphere, 2007, 67, 741–747 CrossRef CAS PubMed.
- Q. Husain and R. Ulber, Crit. Rev. Environ. Sci. Technol., 2011, 41, 770–804 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- B. Valderrama, M. Ayala and R. Vazquez-Duhalt, Chem. Biol., 2002, 9, 555–565 CrossRef CAS.
- D. Jung, C. Streb and M. Hartmann, Microporous Mesoporous Mater., 2008, 113, 523–529 CrossRef CAS PubMed.
- D. I. Perez, M. M. Grau, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2009, 6848–6850 RSC.
- H. Uyama, H. Kurioka and S. Kobayashi, Polym. J., 1997, 29, 190–192 CrossRef CAS.
- F. Van Rantwijk and R. A. Sheldon, Curr. Opin. Biotechnol., 2000, 11, 554–564 CrossRef CAS.
- S.-H. Cho, J. Shim, S.-H. Yun and S.-H. Moon, Appl. Catal., A-Gen., 2008, 337, 66–72 CrossRef CAS PubMed.
- J. Xu, T. Tang, K. Zhang, S. Ai and H. Du, Process Biochem., 2011, 46, 1160–1165 CrossRef CAS PubMed.
- R. Schoevaart, F. Van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2000, 70, 353–357 CrossRef.
- J. Rocha-Martin, D. Vega, J. M. Bolivar, C. A. Godoy, A. Hidalgo, J. Berenguer, J. M. Guisan and F. Lopez-Gallego, BMC Biotechnol., 2011, 11, 101 CrossRef CAS PubMed.
- L. Betancor, F. Lopez-Gallego, A. Hidalgo, N. Alonso-Morales, G. Dellamora-Ortiz, J. M. Guisan and R. Fernández-Lafuente, J. Biotechnol., 2006, 121, 284–289 CrossRef CAS PubMed.
- F. Lopez-Gallego, L. Betancor, A. Hidalgo, G. Dellamora-Ortiz, C. Mateo, R. Fernandez-Lafuente and J. M. Guisan, Enzyme Microb. Technol., 2007, 40, 278–284 CrossRef CAS PubMed.
- J. M. Bolivar, L. Wilson, S. A. Ferrarotti, R. Fernandez-Lafuente, J. M. Guisan and C. Mateo, Biomacromolecules, 2006, 7, 669–673 CrossRef CAS PubMed.
- J. M. Abad, M. Velez, C. Santamaría, J. M. Guisan, P. R. Matheus, L. Vázquez, I. Gazaryan, L. Gorton, T. Gibson and V. M. Fernández, J. Am. Chem. Soc., 2002, 124, 12845–12853 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CAS.
- C. Mateo, J. M. Palomo, M. Fuentes, L. Betancor, V. Grazu, F. Lopez-Gallego, B. C. C. Pessela, A. Hidalgo, G. Fernandez-Lorente, R. Fernandez-Lafuente and J. M. Guisan, Enzyme Microb. Technol., 2006, 39, 274–280 CrossRef CAS PubMed.
- C. Mateo, J. M. Bolivar, C. A. Godoy, J. Rocha-Martin, B. C. Pessela, J. A. Curiel, R. Muñoz, J. M. Guisan and G. Fernandez-Lorente, Biomacromolecules, 2010, 11, 3112–3117 CrossRef CAS PubMed.
- L. Pramparo, F. Stüber, J. Font, A. Fortuny, A. Fabregat and C. Bengoa, J. Hazard. Mater., 2010, 177, 990–1000 CrossRef CAS PubMed.
- R. Torres, B. Pessela, M. Fuentes, R. Munilla, C. Mateo, R. Fernandez-Lafuente and J. M. Guisan, J. Biotechnol., 2005, 120, 396–401 CrossRef CAS PubMed.
- Z. Tong, Z. Qingxiang, H. Hui, L. Qin, Z. Yi and Q. Min, Chemosphere, 1998, 37, 1571–1577 CrossRef CAS.
- J. L. Gomez, A. Bodalo, E. Gómez, J. Bastida, A. M. Hidalgo and M. Gomez, Enzyme Microb. Technol., 2006, 39, 1016–1022 CrossRef CAS PubMed.
- J.-Z. Liu, H.-Y. Song, L.-P. Weng and L.-N. Ji, J. Mol. Catal. B: Enzym., 2002, 18, 225–232 CrossRef CAS.
- J. Rocha-Martin, B. d. l. Rivas, R. Muñoz, J. M. Guisan and F. Lopez-Gallego, ChemCatChem, 2012, 4, 1279–1288 CrossRef CAS.
- V. A. Cooper and J. A. Nicell, Water Res., 1996, 30, 954–964 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- J. Guisan, Enzyme Microb. Technol., 1988, 10, 375–382 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3gc41456f |
| This journal is © The Royal Society of Chemistry 2014 |