 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Renewable production of phthalic anhydride from biomass-derived furan and maleic anhydride†
Eyas
Mahmoud
a,
Donald A.
Watson
b and
Raul F.
Lobo
*a
aCatalysis Center for Energy Innovation, Department of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE 19716, USA. E-mail: lobo@udel.edu; Fax: +302-831-1048; Tel: +302-831-1261
bDepartment of Chemistry and Biochemistry, University of Delaware, Newark, Delaware 19716, USA
First published on 14th October 2013
Abstract
A route to renewable phthalic anhydride (2-benzofuran-1,3-dione) from biomass-derived furan and maleic anhydride (furan-2,5-dione) is investigated. Furan and maleic anhydride were converted to phthalic anhydride in two reaction steps: Diels–Alder cycloaddition followed by dehydration. Excellent yields for the Diels–Alder reaction between furan and maleic-anhydride were obtained at room temperature and solvent-free conditions (SFC) yielding 96% exo-4,10-dioxa-tricyclo[5.2.1.0]dec-8-ene-3,5-dione (oxanorbornene dicarboxylic anhydride) after 4 h of reaction. It is shown that this reaction is resistant to thermal runaway because of its reversibility and exothermicity. The dehydration of the oxanorbornene was investigated using mixed-sulfonic carboxylic anhydrides in methanesulfonic acid (MSA). An 80% selectivity to phthalic anhydride (87% selectivity to phthalic anhydride and phthalic acid) was obtained after running the reaction for 2 h at 298 K to form a stable intermediate followed by 4 h at 353 K to drive the reaction to completion. The structure of the intermediate was determined. This result is much better than the 11% selectivity obtained in neat MSA using similar reaction conditions.
1. Introduction
The consumption of fossil fuels for the production of energy and materials has led to increasing levels of CO2 in the atmosphere1 and, in response, there has been an intense research effort towards the conversion of renewable biomass feedstocks into fuels and chemicals.2–6 Lignocellulosic biomass, which comprises up to 70 wt% of cellulose and hemicellulose, is one promising renewable biomass feedstock.7One important chemical intermediate that could be produced from biomass is phthalic anhydride. Phthalic anhydride is used for the manufacture of plasticizers, unsaturated polyesters, and alkyd resins.8 In 2000, the worldwide production of phthalic anhydride was estimated to be 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 232000 tonnes per year.9,10 Phthalic anhydride is currently produced from oil feedstocks by the vapor phase oxidation of o-xylene and naphthalene via the Gibbs phthalic anhydride process.11 Myriant has suggested the use of renewable bio-succinic acid as an alternative to phthalic anhydride.10 Although this route may be promising, it would be preferable to produce phthalic anhydride renewably.
232000 tonnes per year.9,10 Phthalic anhydride is currently produced from oil feedstocks by the vapor phase oxidation of o-xylene and naphthalene via the Gibbs phthalic anhydride process.11 Myriant has suggested the use of renewable bio-succinic acid as an alternative to phthalic anhydride.10 Although this route may be promising, it would be preferable to produce phthalic anhydride renewably.
Phthalic anhydride can be produced renewably from furan (1) and maleic anhydride (2) as shown in Scheme 1. Industrially, furan is produced by the decarbonylation of furfural in high yields12–14 and maleic anhydride can be obtained renewably by the oxidation of furfural using a VOx/Al2O3 catalyst15 or by the oxidation of 5-hydroxymethylfurfural in the liquid phase.16 Furan and maleic anhydride can be used to produce phthalic anhydride (4) in a two-step process: Diels–Alder cycloaddition to produce 3 followed by dehydration. A related product that can be prepared following this approach is the synthesis of p-xylene from dimethylfuran and ethylene.17 In the latter system, selectivity decreases rapidly with decreasing substitution on the furan ring: with dimethylfuran an 80% selectivity to p-xylene has been achieved while in reactions run using furan only an 18% selectivity to benzene is achieved.18 This reduction in yield is due to furan polymerizing in the presence of a Bronsted acid.19,20 To minimize these decomposition pathways, the Diels–Alder and dehydration reactions were carried out in series for higher yields of phthalic anhydride.
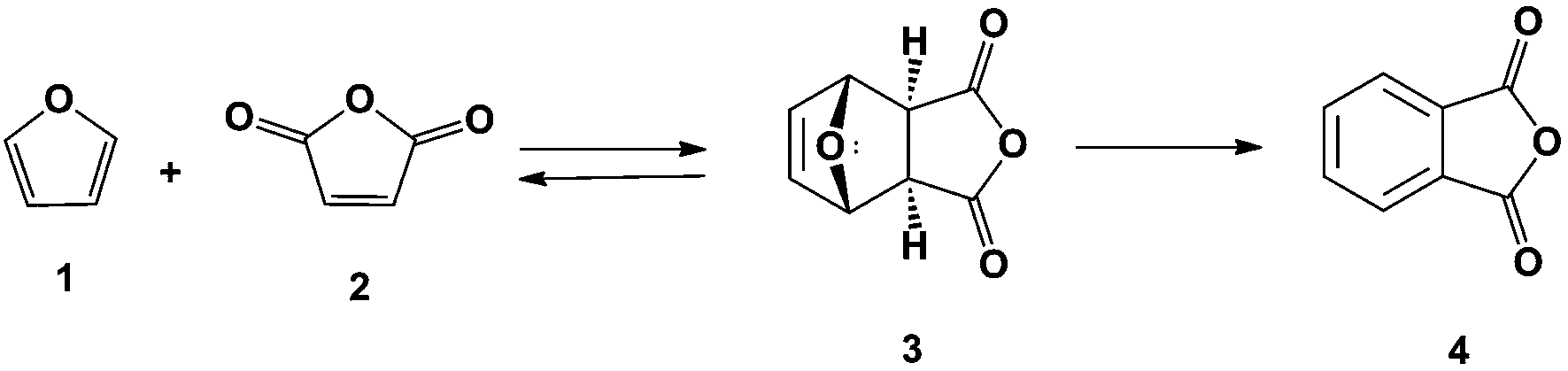 | ||
| Scheme 1 Route to renewable phthalic anhydride from biomass-derived furan and maleic anhydride. | ||
The Diels–Alder cycloaddition of furan and maleic anhydride has been investigated by several groups.21–25 The reactions were usually run in diethyl ether,21,22,26,27 acetonitrile,23,24 or tetrahydrofuran28 with yields of only 35% after 24 h of reaction at room temperature.26 To control the temperature of solvent free Diels–Alder reactions and to accelerate their rate, Windmon and Dragojlovic29 added minimal amounts of water as a variation of the “on water” reaction procedure originally reported by Sharpless and coworkers for a series of Diels–Alder reactions.30 They found that it is possible to control the temperature of multi-gram scale Diels–Alder reactions by adding a minimal amount of water to neat reactants. Furthermore, for some reactions the presence of water resulted in an increased reaction rate and formation of a higher purity product.
The second step (Scheme 1) is the dehydration of the oxanorbornene intermediate (3) which is inherently difficult because oxanorbornene intermediates are sensitive to heat and can readily undergo the retro-Diels–Alder reaction.31 Moreover, depending on the degree and the substitution on the furan ring, the boiling point of the furan may be as low as 304 K and may be susceptible to polymerization.19 The dehydration of 3, the adduct of 1 and 2, has been conducted using HBr in glacial acetic acid, but few details were reported.32 Similarly, the dehydration of the adduct of 2-methylfuran and maleic anhydride was conducted in 85% sulfuric acid at 273 K to produce 3-methylphthalic acid in 25% selectivity.33 It was shown that the retro-Diels–Alder reaction competes with the acid catalyzed dehydration. A maximum of 66% yield of 3-methylphthalic anhydride was obtained at 218 K (too low for industrial practice) when binary mixtures of sulfolane and sulfuric acid were used as a solvent. The dehydration of the adduct of 2-acetoxyfuran and maleic anhydride was carried out in acetic anhydride with catalytic amounts of sulfuric acid and only a 57% yield of the anhydride was achieved.34 A more systematic, high yield, and environmentally friendly system for the dehydration of oxanorbornenes still needs to be developed.35 Furthermore, side-reaction pathways have to be investigated (Scheme 2).33
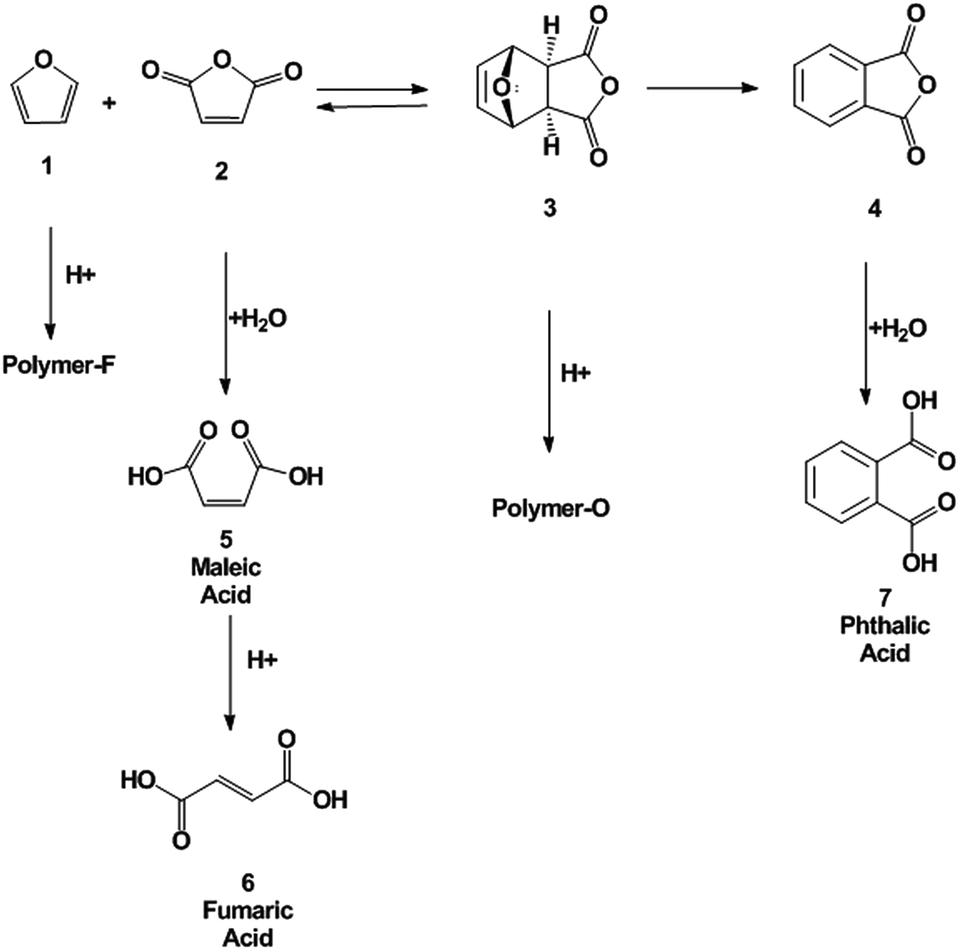 | ||
| Scheme 2 Reaction channels for the formation of byproducts. | ||
In this report we first investigate the solvent-free Diels–Alder reaction of furan and maleic anhydride and we assess whether the “on water” effect applies for this reaction based on the solubility and reactivity of maleic anhydride in water. Secondly, we report an alternative method for the dehydration of 3 using binary mixtures of methanesulfonic acid (MSA) and acetic anhydride. Acetic anhydride reacts with sulfonic acids to generate mixed sulfonic carboxylic anhydrides.36 With these strong acylating agents,37 the oxanorbornene dicarboxylic anhydride ring may readily be cleaved at room temperature in the presence of acid and the intermediate may be simply converted to phthalic anhydride by heating. We provide mechanistic insights into the progression of this reaction and provide a scalable strategy for low temperature and high-yield dehydration of oxanorbornenes for the production of aromatics from furans.
2. Experimental section
Diels–Alder reactions
Furan (99+% Sigma-Aldrich), 2,5-dimethylfuran (99% Sigma-Aldrich), 2-methylfuran (99%, contained 0.024% BHT as a stabilizer, Sigma Aldrich), and maleic anhydride (99.0+%, Sigma-Aldrich) were stored under an inert atmosphere and used as received. Pilot plant scale reactions were performed in a 600 mL Parr reactor (Parr Instrument Company – model 452HC3) equipped with a pressure and temperature sensor. For these reactions, maleic anhydride was first placed into the 600 mL Parr reactor under a nitrogen atmosphere. If the reaction was run “on-water”, water was added at this point. After this step, an equimolar amount of furan was added to the reactor. The reactor was then sealed, flushed 3 times with nitrogen, and pressurized to 1.70 bar. During set-up, light exposure was avoided during loading. The mixture was allowed to react for 240 min, initially stirring at a rate of 650 rpm while monitoring the temperature and pressure of the reactor. Immediately following reaction completion, the solid was ground with a mortar and pestle, and a small portion was taken for NMR. The yield was calculated based on the ratio of 3 to the sum of the peak area of maleic anhydride [1H NMR (400 MHz, DMSO) δ 7.48 (2H, s, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) )], maleic acid [1H NMR (400 MHz, DMSO) δ 6.23 (2H, s, –CH)] and oxanorbornene dicarboxylic anhydride (3) evaluated using quantitative nuclear magnetic resonance spectroscopy (qNMR):38
)], maleic acid [1H NMR (400 MHz, DMSO) δ 6.23 (2H, s, –CH)] and oxanorbornene dicarboxylic anhydride (3) evaluated using quantitative nuclear magnetic resonance spectroscopy (qNMR):38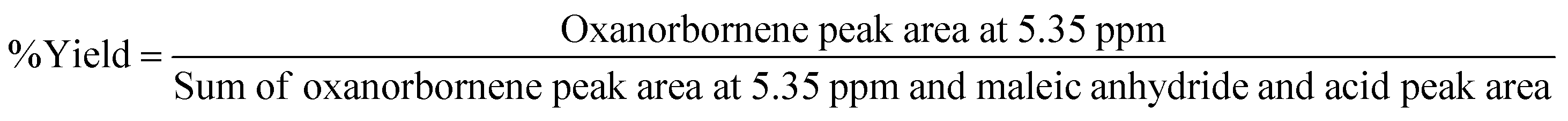
The yield calculated based on the weight of the isolated anhydride (3) after washing with diethyl ether (cooled to 253 K), filtering and drying was consistent with results found by qNMR. The product was analyzed by 1H NMR and found to be exo-4,10-dioxa-tricyclo[5.2.1.0]dec-8-ene-3,5-dione. For reactions run “on-water”, excess water was first filtered off and the reported yield was evaluated by the weight of the isolated oxanorbornene after washing with diethyl ether (cooled to 253 K) rather than by qNMR.
1H NMR (400 MHz, DMSO) δ 6.58 (2H, s, –CH), 5.35 (2H, s, –CH–O–), 3.33 (2H, s, –CH–) ppm; IR (neat) ν 575, 634, 675, 690, 733, 849, 877, 883, 920, 922, 1020, 1080, 1150, 1210, 1220, 1280, 1310, 1780 cm−1.
Equilibrium experiments were conducted in a 600 mL Parr reactor (Parr Instrument Company – model 452HC3) equipped with a pressure and temperature sensor. 0.10 moles of oxanorbornene dicarboxylic anhydride (3) were loaded under a nitrogen atmosphere (10 psig of N2). The temperature of the reactor was increased stepwise as the pressure was monitored. For equilibrium experiments conducted “on-water”, 3.09 mL of water were first loaded into the reactor before the addition of 3.
exo-cis-1-Methyl-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic anhydride, the adduct of 2-methylfuran and maleic anhydride, was prepared by reacting 0.0666 moles (6.53 g) of maleic anhydride and 0.0666 moles (5.47 g) of 2-methylfuran in a 45 mL closed Parr reactor (series 4703–4714, General Purpose Pressure Vessels) for 24 h at room temperature under a nitrogen atmosphere. Following the reaction, the solid was washed with diethyl ether (253 K) to give 10.068 g of a white product (84% yield).
1H NMR (400 MHz, DMSO-d6) δ 6.58 (dd, J = 5.5, 1.8 Hz, 7H), 6.41 (d, J = 5.6 Hz, 7H), 5.22 (d, J = 1.9 Hz, 7H), 3.44 (d, J = 6.6 Hz, 8H), 3.20 (d, J = 6.6 Hz, 7H), 1.62 (s, 22H); IR (neat) ν 579, 615, 661, 673, 735, 845, 876, 895, 922, 955, 982, 1070, 1090, 1140, 1230, 1320, 1390, 1770, 1840 cm−1.
The adduct of 2,5-dimethylfuran and maleic anhydride, 1,7-dimethyl-4,10-dioxa-tricyclo[5.2.1.02,6]dec-8-ene-3,5-dione, was similarly prepared by reacting 0.0618 moles (6.06 g) of maleic anhydride and 0.0618 moles (5.94 g) of 2,5-dimethylfuran in a 45 mL closed Parr reactor for 24 h. The solid was washed with diethyl ether (cooled to 253 K) to give 8.981 g of a white solid (75% yield).
1H NMR (400 MHz, DMSO-d6) δ 6.41 (s, 5H), 3.32 (s, 6H), 1.58 (s, 16H); IR (neat) ν 579, 625, 669, 744, 831, 862, 918, 976, 1040, 1080, 1210, 1240, 1320, 1390, 1440, 1760, 1850, 2990 cm−1.
Dehydration reaction
Methanesulfonic acid (99.5%+, Sigma Aldrich), acetic anhydride (99.4%, Fisher Scientific), glacial acetic acid (99.9%, Fisher Scientific), methanesulfonic anhydride (97%, Sigma Aldrich) maleic anhydride (99.0+%, Sigma-Aldrich) and sulfolane (99%+, Sigma Aldrich) were used as received and stored under a dry inert atmosphere. In a 20 mL disposable scintillation vial the desired amount of MSA was loaded in addition to 3.01 mmol (0.362 grams) of sulfolane (99%+, Sigma Aldrich) which was used as an internal standard. Sulfolane was selected as internal standard because of its high stability against strong acids as well as thermal stability.39 (For reactions performed using binary mixtures, the second chemical was added to this mixture at this point.) The mixture was then stirred for 5 minutes at room temperature. In the case of acetic anhydride, MSA and acetic anhydride reacted to form acetyl methanesulfonate [1H NMR (400 MHz, CDCl3) δ 2.28 (3H, s, COCH3), 3.33 (3H, s, SO2CH3)]36 which was accompanied by an exotherm. The reaction was then heated to the desired temperature and 6.02 mmoles of oxanorbornene were slowly added while monitoring the temperature. The rate of addition was maintained such that the reaction temperature did not exceed the desired temperature. The vial was then flushed with nitrogen. Conversion and selectivity were calculated using the internal standard by qNMR. All the reactions were conducted such that the total solution volume was 5 mL to avoid the confounding effects associated with the side-reactions.Following reaction, the reaction product was cooled to room temperature and extracted with anhydrous toluene (99.8%, Sigma Aldrich) (10 mL, 3 times). The toluene fractions were combined and the solvent was removed in a rotary evaporator. The isolated solid was then recrystallized from acetic anhydride and washed with cold hexane to give phthalic anhydride (white crystal). Yields calculated based on the weight of phthalic anhydride were consistent with those determined by qNMR.
1H NMR (400 MHz, DMSO) δ 8.09 (2H, dd, –CH), 8.01 (2H, dd, –CH); IR (neat) ν 534, 642, 677, 710, 798, 839, 903, 1000, 1110, 1160, 1260, 1360, 1470, 1600, 1760, 1790, 1840 cm−1; m/z 148.
(rac) (3aS,4S,5S,7aS)-1,3-Dioxo-1,3,3a,4,5,7a-hexahydroisobenzofuran-4,5-diyl diacetate was isolated by running the reaction at conditions such that only this reaction intermediate formed (0.039 moles of MSA and 0.0264 moles of acetic anhydride, 24 h, 298 K). The intermediate was separated using a 2:1 mixture of hexanes (99.9%, Fisher Scientific) and ethyl acetate (99.9%, Fisher Scientific) by flash chromatography using Supelco® spherical flash silica (45–75 μm). The intermediate was analyzed by 2D NMR correlation spectroscopy (COSY), heteronuclear multiple bond correlation (HMBC), and heteronuclear multiple quantum coherence (HMQC)40 (see Fig. S1 in the ESI† section).
1H NMR (400 MHz, Chloroform-d) δ 6.30 (dd, J = 10.0, 3.8 Hz, 1H), 6.22–6.12 (m, 1H), 5.47–5.40 (m, 1H), 5.23 (dd, J = 5.4, 3.8 Hz, 1H), 3.82 (ddd, J = 9.3, 3.8, 2.6 Hz, 1H), 3.73 (dd, J = 9.3, 4.2 Hz, 1H), 2.10 (s, 3H), 2.05 (s, 3H). HRMS (EI) m/z, calcd for [C12H12O7]: 269.0661; found: 269.0661.
The yields of 3,6-dimethyl phthalic anhydride and 3-methyl phthalic anhydride were calculated by qNMR. For the dehydration of the cycloadduct 2,5-dimethylfuran and maleic anhydride the peaks at 7.68 (2H, s, –CH) and 7.62 (2H, s, –CH) corresponding to 3,6-dimethyl phthalic anhydride and 3,6 dimethyl phthalic acid were integrated. For the dehydration of the cycloadduct of 2-methylfuran and maleic anhydride the yield was calculated by integrating the peaks found between 7.9 to 7.3 ppm corresponding to the three protons of 3-methyl phthalic anhydride and 3-methyl phthalic acid in the aromatics region.
Polymer separation
As a control, a furan polymer was prepared by slowly adding 6.02 mmoles (0.44 mL) of furan to methanesulfonic acid. The mixture was allowed to react for 3 h. The filtered solid was washed three times with water and dried for 48 h at 353 K. The oxanorbornene polymer was isolated by slowly pouring the reaction mixture onto ice. The solid was the washed with ice-cold water three times and dried for 48 h at 353 K before analysis.Analytical nuclear magnetic resonance experiments (NMR)
Attenuated total reflectance (ATR) infrared spectroscopy
A Nicolet 8700 FTIR spectrometer equipped with a DTG detector and a Golden Gate single-reflection diamond ATR was used for all IR spectroscopy measurements. The instrument was equipped with a purge gas of de-humidified air to remove water vapor. The resolution of the instrument was set at 4 cm−1 and the number of scans was set to 16.Elemental analysis
All elemental analysis was conducted at Galbraith Laboratories (Tennessee). The carbon and hydrogen contents of the isolated polymers were analyzed using a PerkinElmer 2400 Series II CHNS/O analyzer at Galbraith Laboratories (Tennessee). The oxygen content was determined using a Thermo Finnigan FlashTM Elemental Analyzer and the sulfur content was determined using a LECO SC-432DR.Karl-Fischer titration
Karl-Fischer (KF) titration was performed to determine the water content in MSA using a Mettler Toledo V20 Volumetric KF titrator. For optimal execution of the KF titration, the sample was buffered with imidazole (Sigma 99%) so that the pH of the solution was in the range between 4 and 8 in dry methanol (Hydranal®). MSA was found to have a water content of 1.80 weight% water.3. Results and discussion
Diels–Alder reaction
The Diels–Alder reaction between an equimolar amount of furan and maleic anhydride was performed at multi-gram scales at room temperature under SFCs. NMR of samples taken after 4 h of reaction revealed the presence of the oxanorbornene dicarboxylic anhydride (3), 1, and 2 (Fig. S2†). A 96% yield of oxanorbornene dicarboxylic anhydride (3) was obtained for reactions run using a 1.4 mol basis of furan and maleic anhydride. No other side-products were detected by 1H NMR if care was taken to avoid both light and air exposure. For reactions run using 1.4 mole basis of furan and maleic anhydride, the temperature of the reactor increased to 357 K after 40 minutes of reaction (Fig. S3†) and the pressure of the reactor increased as well due to the evaporation of furan into the head space of the reactor (Fig. S2†).Running the Diels–Alder reaction at three different scales (0.69 mol, 1.4 mol, 2.1 mol of each reactant) revealed that the reaction is resistant to thermal runaway (Fig. S3†) due to the reduction of the equilibrium constant at high temperatures (Fig. S4†). This observation is contrary to the solvent-free normal Diels–Alder reaction which can thermally runaway and may form explosive mixtures.29 Here, doubling and tripling the loading of both reactants did not lead to an increase in the peak reaction temperature (365 K). In fact, as the reaction temperature approached 423 K, the oxanorbornene dicarboxylic anhydride (3) can be converted into the starting materials by the retro-Diels–Alder reaction.41 Separate experiments in which isolated 3 was heated to different temperatures confirmed this observation with the oxanorbornene converting to the starting reactants at higher temperatures by the retro-Diels–Alder reaction before melting. Finally, the reaction was run under pseudo-adiabatic conditions (Fig. S5†): the reaction temperature plateaued at ∼365 K suggesting again that the reversibility controls run-away.
Specific guidelines have been set to determine whether a reaction occurs “in-water” versus “on-water” and whether substantial rate accelerations by Marcus trans-phase H-bonding are possible based on solubility.42 The Diels–Alder reaction of furan and maleic anhydride was conducted “on-water”. It has been shown that running this reaction on-water results in rate increases and serves to absorb some of the heat of the reaction.29 As shown in Table 1, however, adding 21 mL of water to the reaction mixture resulted in a selectivity reduction from 81% to 66% after 1 h of reaction. This composition and reaction time is identical to the one used by Windmon.29 Adding 35 mL of water resulted in a yield of only 32%. Adding only 10 mL of water to the Diels–Alder reaction resulted in the hydrolysis of the anhydride functionality with increasing temperature. Furthermore, equilibrium experiments reveal that the conversion of the oxanorbornene dicarboxylic anhydride (3) to the starting materials increases in the presence of water at similar temperatures (Fig. S6†). Maleic anhydride is hydrolyzed to maleic acid at room temperature in water.43 No rate enhancements for the Diels–Alder reaction between furan and maleic anhydride were observed when using water. Water solubility places maleic anhydride in the transition boundary between ‘on water’ and ‘in-water’ where opposing normal H-bonding effects on both reactants in water can also inhibit the ‘on-water’ effect, despite the physical appearance of the two-phase system.42
| Solvent and amount | Selectivity to oxanorbornene (3) (%) |
|---|---|
| 20 mL, heptane | 74 |
| SFC | 81 |
| 10 mL, water | 68 |
| 21 mL, water | 62 |
| 35 mL, water | 32 |
Dehydration of exo-4,10-dioxa-tricyclo[5.2.1.0]dec-8-ene-3,5-dione (3)
The dehydration of oxanorbornene dicarboxylic anhydride (3) was investigated in neat MSA at 298, 353, and 393 K. MSA was selected as the acid because it is a non-oxidizing, biodegradable and thermally stable acid alternative to sulfuric acid.44,45 Within 5 minutes of reaction a brownish, black tar-like residue appeared at the three temperatures investigated. After reaction completion, phthalic anhydride (4), phthalic acid (7), maleic anhydride (2), maleic acid (5), and fumaric acid (6) were all detected by proton NMR (Scheme 3). No furan was detected in the NMR spectra.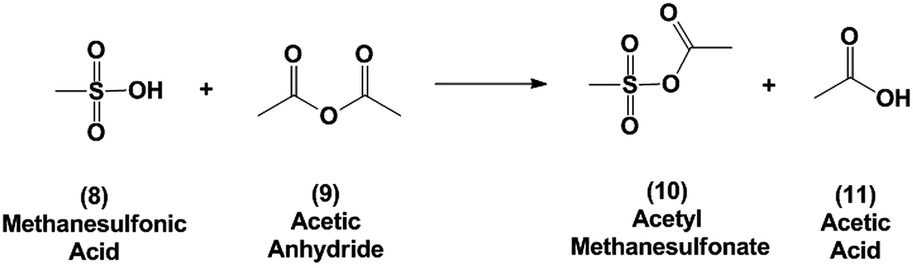 | ||
| Scheme 3 Formation of mixed sulfonic-carboxylic anhydride from MSA and acetic anhydride. | ||
The formation of byproducts was assigned to two major pathways: decomposition of oxanorbornene dicarboxylic anhydride (3) by the retro-Diels–Alder reaction to maleic anhydride and furan (1) which readily polymerized in acid19 and the polymerization of the oxanorbornene dicarboxylic anhydride. To confirm that the latter polymerization pathway exists, the dehydration of the Diels–Alder adduct of 2,5-dimethylfuran and maleic anhydride, 1,7-dimethyl-4,10-dioxa-tricyclo[5.2.1.02,6]dec-8-ene-3,5-dione, and the Diels–Alder adduct of 2-methylfuran and maleic anhydride, 1 exo-cis-1-methyl-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic anhydride were conducted in neat MSA at 298 K (Table 2). The yield drastically increased with increasing substitution of the oxanorbornene, suggesting that the methyl groups act as protecting groups for polymerization. Minimal amounts of maleic anhydride were detected for these experiments. Separate experiments confirm that maleic anhydride, maleic acid, and fumaric acid are stable at 298 K in MSA. Therefore, the presence of maleic anhydride was used as a measure of the extent of the retro-Diels–Alder reaction. Finally, to confirm that there are two byproduct side reactions, the reaction was run in neat MSA at 298 K for 3 h and the resulting solid product was isolated and analyzed by ATR-IR spectroscopy and by elemental analysis. The ATR-IR spectrum of the isolated polymer (polymer O) and a polymer formed by the polymerization of neat furan in MSA (polymer F) at the same conditions have distinctly different absorption bands (Fig. S7†). In the region characteristic of carbonyl stretching frequencies (1670–1820 cm−1), the oxanorbornene polymer has several absorption bands while the furan polymer does not. The IR spectrum of furan was consistent with characteristic vibrations of the furyl group in polyfuran previously found.19 The elemental composition of the two polymers are different (Polymer F – 64.08, 5.66, 29.31% C, H, O and Polymer O – 55.92, 4.74, 37.10%, C, H, O), supporting the idea that there are two decomposition pathways. 1H NMR samples taken of the isolated polymers revealed that the samples are free of residual phthalic acid and furan.
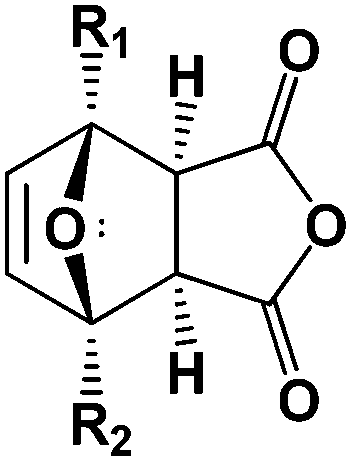
As observed by others for similar oxanorbornenes,33 we found that for the oxanorbornene dicarboxylic anhydride dehydration, the retro-Diels–Alder reaction competes with the acid promoted dehydration (Table 3). Running at lower reaction temperatures decreased the losses associated with the retro-Diels–Alder reaction to 24%. The optimal results in neat MSA were obtained at 298 K where a 14% yield of phthalic anhydride was achieved after 3 days of reaction (23% yield of phthalic anhydride plus phthalic acid). This yield is similar to the results reported for other oxanorbornene dicarboxylic anhydride systems in neat acids.33,35 However, 62% of the oxanorbornene dicarboxylic anhydride was converted directly to unidentified byproducts.
| Temperature (K) | Maleic anhydride, maleic acid, and fumaric Acid | Phthalic anhydride and phthalic acid | Oxanorbornene polymerization/char formation |
|---|---|---|---|
| 298 | 24 | 14 | 62 |
| 353 | 31 | 11 | 58 |
| 393 | 36 | 7 | 57 |
Mixed-sulfonic carboxylic anhydrides
Because of the poor yields obtained using neat MSA, dehydration of the oxanorbornene dicarboxylic anhydride was also conducted using mixed sulfonic carboxylic anhydrides. Mixed-sulfonic carboxylic anhydrides are strong acylating agents that have been shown to rapidly cleave the ether rings with high selectivity to their ring-opened products.37 Mixtures of acetic anhydride and methanesulfonic acid formed acetyl methanesulfonate (10) at room temperature after 5 minutes of reaction (Scheme 3). The formation of acetyl methanesulfonate (10) was tracked with varying ratios of MSA and acetic anhydride in the mixture (Fig. S8†).The dehydration of 3 was further studied using the mixtures of MSA and acetic anhydride at 298 K to cleave the ether ring (Fig. 1). In this reaction the mixed anhydride is consumed, generating MSA and acetic acid and the oxanorbornene is dehydrated to phthalic anhydride. Unlike the reaction run in pure MSA, the reaction medium was yellow until 2 h of reaction. The reaction products and byproducts were monitored by qNMR and it was found that the oxanorbornene dicarboxylic anhydride signal disappeared after two hours of reaction with the appearance of phthalic anhydride (4) as well as another unidentified compound (this compound is the diacetate (12), see below). The reaction temperature was then increased to 353 K for 4 h and the disappearance of the unidentified compound was correlated to the appearance of phthalic anhydride, indicating that this compound is an intermediate. After 4 hours, phthalic anhydride, maleic anhydride, and acetic acid peaks due to the formation of the mixed anhydride were detected by proton NMR (Fig. S9†). No maleic acid or fumaric acid was detected indicating that the reaction medium was nearly water-free, and no furan was detected by NMR as well. In addition, only 5% of the oxanorbornene dicarboxylic anhydride was converted to maleic anhydride by the retro-Diels–Alder reaction. Running the reaction to completion, an 80% selectivity to phthalic anhydride and an 87% selectivity to aromatics (phthalic anhydride and acid) was obtained.
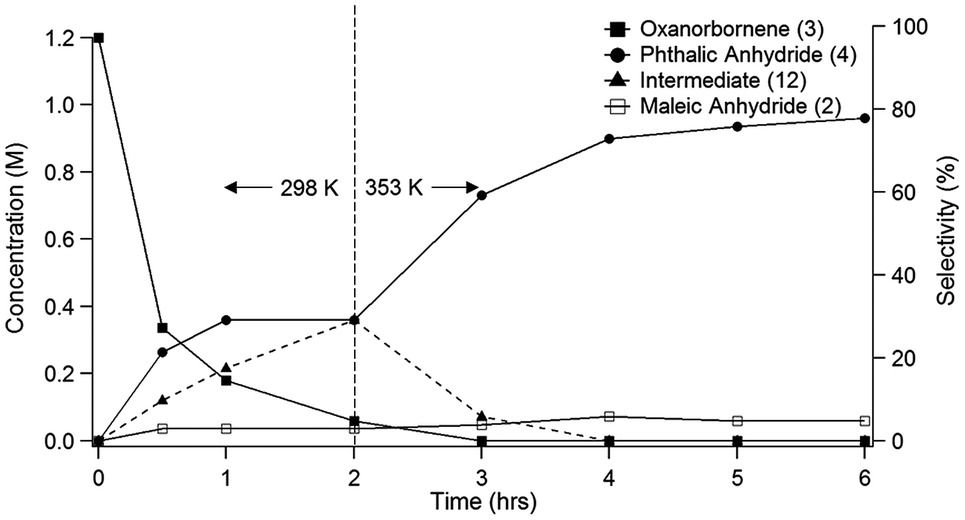 | ||
| Fig. 1 Time evolution of product (3) and intermediate (12) selectivity. The reaction was run at 298 K for 2 h followed by a 4 h period at 353 K in 62 mmol of MSA and 11 mmol of acetic anhydride. | ||
As a control experiment, the dehydration was run using the same composition of MSA and oxanorbornene dicarboxylic anhydride, but replacing acetic anhydride with maleic anhydride, acetic acid, or methanesulfonic anhydride (Table 4). As in the neat MSA experiments, using maleic anhydride the reaction mixture turned blackish brown after a few minutes of reaction. Maleic anhydride did not react with MSA to form a mixed anhydride. A 14% yield of phthalic anhydride was obtained at 298 K for 2 h and then at 353 K for 4 h. This shows that despite maleic anhydride's ability to uptake water to form maleic acid in the presence of MSA, the increase in yield of the reaction is not due to the absence of water. A separate experiment was conducted using the same composition of MSA and oxanorbornene dicarboxylic anhydride, but using an equivalent molar ratio of acetic acid as maleic anhydride and acetic anhydride. The mixture also turned blackish-brown after a few minutes of reaction and a 4% yield of phthalic anhydride was obtained following 2 h of reaction at 298 K and then 4 h at 353 K. Finally, an experiment was conducted using the same composition of MSA and oxanorbornene dicarboxylic anhydride, but using an equivalent molar ratio of methanesulfonic anhydride as maleic anhydride and acetic anhydride. The mixture turned blackish-brown after a few minutes of reaction and a 23% yield of phthalic anhydride was obtained at 298 K for 2 h and then at 353 K for 4 h. Although methanesulfonic anhydride has a similar structure as acetic anhydride, its effect on the reaction is drastically different, indicating the favorable properties of acetic anhydride.
| Solvent composition | Selectivity to phthalic anhydride (%) |
|---|---|
| 78 mmol MSA | 11 |
| 62 mmol MSA, 11 mmol acetic acid | 6 |
| 62 mmol MSA, 11 mmol maleic anhydride | 15 |
| 62 mmol MSA, 11 mmol acetic anhydride | 80 |
| 62 mmol MSA, 11 mmol methanesulfonic anhydride | 23 |
The observation of the diacetate (12) by NMR suggests a plausible reaction mechanism (Scheme 4) for this dehydration. The mixed sulfonic-carboxylic anhydride (10) is attacked by the bridging oxygen of the oxanorbornene leading to a ring-opened product. The added acetal group then leans over the resonance stabilized product and is subsequently attacked by acetic acid to form the diacetate observed (12). In the presence of acid and heat the diacetate is converted to phthalic anhydride (4).
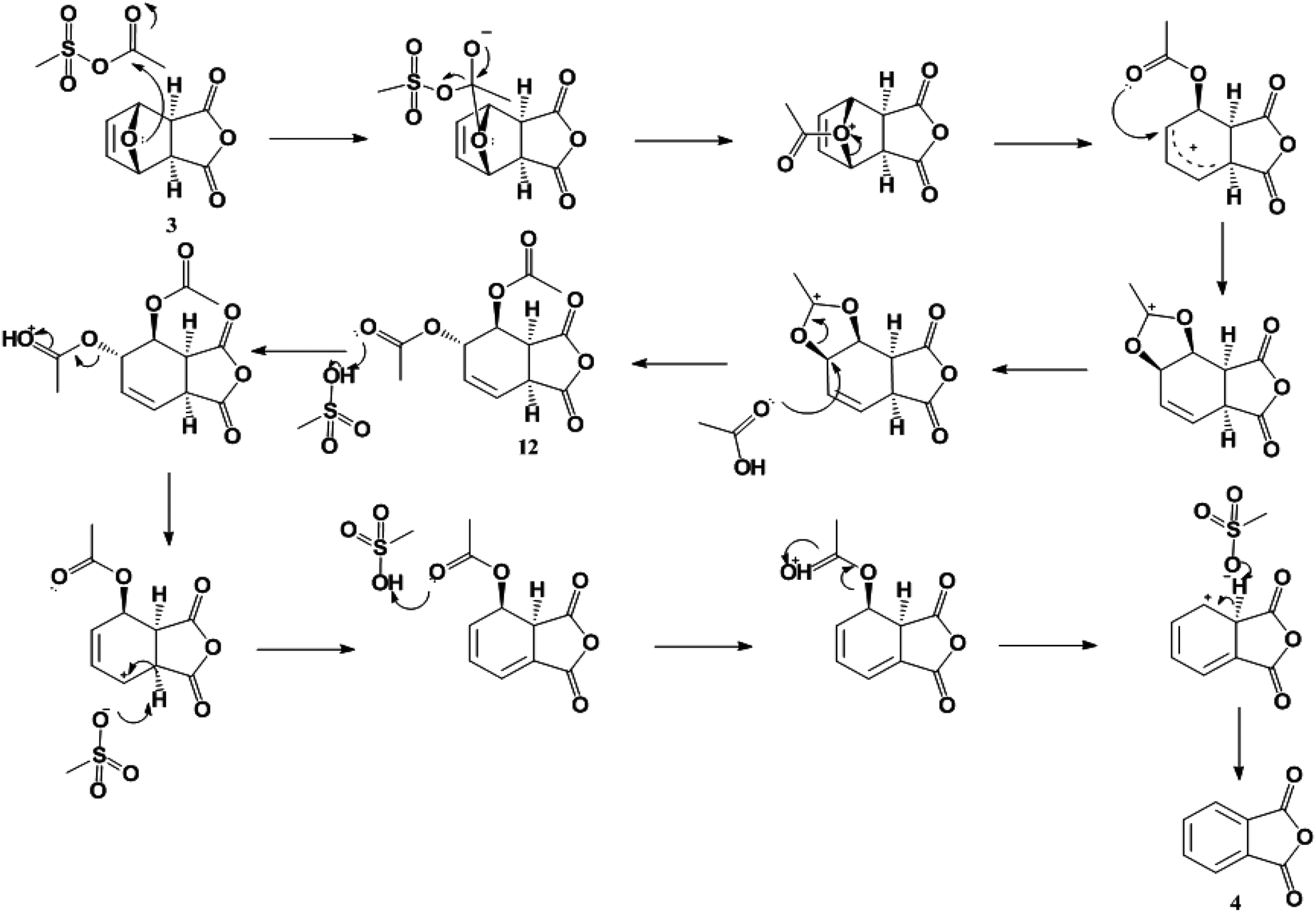 | ||
| Scheme 4 Proposed reaction mechanism for the dehydration of 3. | ||
Running the reaction at different molar ratios of MSA (8) to acetic anhydride (9) provides further insights into the mechanism for the conversion of the oxanorbornene dicarboxylic anhydride to phthalic anhydride. For these experiments, the total volume of the reactor was held constant and the reactor temperature was maintained at 298 K for 1 day. Conversion of the oxanorbornene increased with increasing ratios of MSA (8) to acetic anhydride (9) (Fig. 2). This is because some acidity is required to open the oxanorbornene ring. The selectivity of phthalic anhydride is maximized in the presence of excess acid supporting the proposed reaction mechanism. At intermediate molar ratios, selectivity to the diacetate intermediate increased, going through a maximum. At larger molar ratios of MSA (8) to acetic anhydride (9) the carbon balance did not completely close, suggesting some selectivity losses associated with the polymerization of the oxanorbornene dicarboxylic anhydride. Thus, the acid promoted dehydration competes with ring-opening using mixed-sulfonic carboxylic anhydrides. With decreasing ratios of MSA to acetic anhydride, more maleic anhydride appeared suggesting that the rate of ring-opening decreases with decreasing acidity as indicated by the drop in conversion.
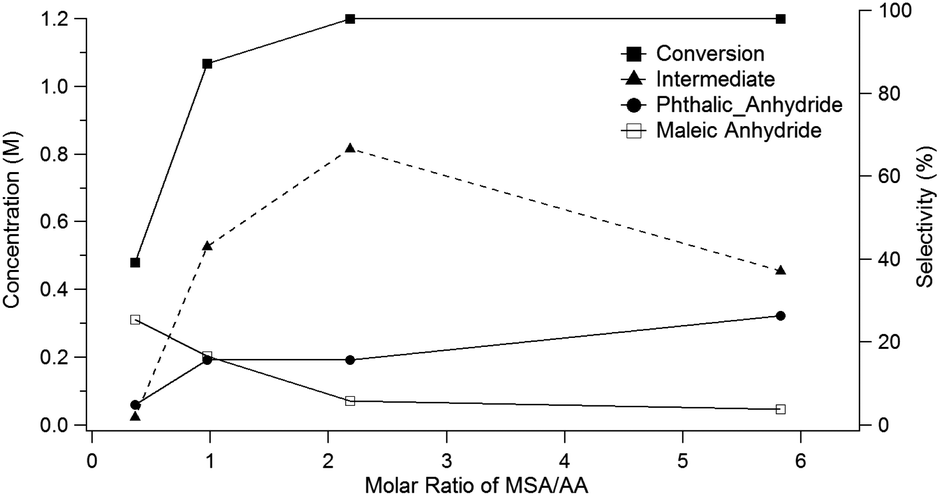 | ||
| Fig. 2 Yield of phthalic anhydride as a function of the molar ratio acetic anhydride to MSA (24 h at 298 K using a 1.2 M concentration of 3). | ||
Selective dehydration of oxanorbornene compounds is inherently difficult. Oxanorbornene dicarboxylic anhydride (3) is heat sensitive and undergoes the retro-Diels–Alder reaction leading to the formation of furan and maleic anhydride. In the acidic environment required by dehydration reactions, furan readily polymerizes, leading to low selectivity. These limitations can be avoided by running the reaction in binary mixtures of MSA (8) and acetic anhydride (9) in two stages: in the first stage, the oxanorbornene ring is cleaved at 298 K leading to the formation of a stable intermediate (12), and in the second stage, the reaction temperature is increased to 353 K to drive the reaction to completion leading to a selectivity as high as 80% to phthalic anhydride. At industrial scales, phthalic anhydride may be extracted or separated from the reaction mixture by chromatography or by precipitation. Acetic acid and residual anhydride may be distilled from the reaction mixture and the acetic anhydride regenerated via ketene.46 MSA may also be reused recycling all of the key reactants.
4. Conclusions
A highly selective and scalable route for the production of phthalic anhydride at high yields has been demonstrated. The process consists of two steps: Diels–Alder cycloaddition of furan and maleic anhydride followed by dehydration of the oxanorbornene dicarboxylic anhydride to produce phthalic anhydride. High yields (96%) for the Diels–Alder cycloaddition of furan and maleic anhydride were obtained under SFC. We demonstrate that the Diels–Alder reaction of furan and maleic anhydride is kinetically self-limiting for thermodynamic reasons. By using binary mixtures of MSA and acetic anhydride to ring-open the oxanorbornene, the selectivity of phthalic anhydride can be increased from 11% in neat MSA to 80% using mixed sulfonic-carboxylic anhydrides under the same conditions. This ring-opening strategy may find use for the dehydration of other oxanorbornene dicarboxylic anhydrides and for the conversion of furans to aromatics.Acknowledgements
This work was supported as part of the Catalysis Center for Energy Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award no. DE-SC0001004. This material is also based upon the work supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1247394. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the National Science Foundation. Mass spectral data was acquired at UD on instruments obtained with the assistance of NSF (MIR CHE1229234).References
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520–540 RSC.
- C. H. Christensen, J. Rass-Hansen, C. C. Marsden, E. Taarning and K. Egeblad, ChemSusChem, 2008, 1, 283–289 CrossRef CAS PubMed.
- P. N. R. Vennestrom, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- B. H. Shanks, Ind. Eng. Chem. Res., 2010, 49, 10212–10217 CrossRef CAS.
- T. Pinnarat and P. E. Savage, Ind. Eng. Chem. Res., 2008, 47, 6801–6808 CrossRef CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- C. M. Park and R. J. Sheehan, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley and Son Inc., 2000, vol. 10, pp. 1–45 Search PubMed.
- S. Bizzari, SRI International, Chemical Economics Handbook, Syracuse, NY, 2001 Search PubMed.
- Bio-based Replacement Products, http://www.myriant.com/products/replacement-products.cfm, Accessed July 26, 2013, 2013.
- C. E. Andrews, US Pat., 1,336,182, 1920 Search PubMed.
- H. Singh, M. Prasad and R. D. Srivastava, J. Chem. Technol. Biotechnol., 1980, 30, 293–296 CrossRef CAS.
- W. Zhang, Y. L. Zhu, S. Niu and Y. W. Li, J. Mol. Catal. A: Chem., 2011, 335, 71–81 CrossRef CAS PubMed.
- W. M. V. R. H. E. Hoydonckx, W. Van Rhijn, D. E. De Vos and P. A. Jacobs, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co., 2007 Search PubMed.
- N. Alonso-Fagundez, M. L. Granados, R. Mariscal and M. Ojeda, ChemSusChem, 2012, 5, 1984–1990 CrossRef CAS PubMed.
- Z. T. Du, J. P. Ma, F. Wang, J. X. Liu and J. Xu, Green Chem., 2011, 13, 554–557 RSC.
- C. L. Williams, C. C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- D. Wang, C. M. Osmundsen, E. Taarning and J. A. Dumesic, ChemCatChem, 2013, 5, 2044–2050 CrossRef CAS.
- J. Kresta and H. K. Livingst, J. Polym. Sci., Part B: Polym. Lett., 1970, 8, 795–803 CrossRef CAS.
- G. Piancatelli, M. Dauria and F. Donofrio, Synthesis, 1994, 867–889 CrossRef CAS PubMed.
- O. Diels and K. Alder, Ber. Dtsch. Chem. Ges., 1929, 62, 554–562 Search PubMed.
- R. B. Woodward and H. Baer, J. Am. Chem. Soc., 1948, 70, 1161–1166 CAS.
- F. A. L. Anet, Tetrahedron Lett., 1962, 1219–1222 CrossRef CAS.
- M. W. Lee and W. C. Herndon, J. Org. Chem., 1978, 43, 518–518 CrossRef CAS.
- M. J. S. Dewar and A. B. Pierini, J. Am. Chem. Soc., 1984, 106, 203–208 CrossRef CAS.
- M. R. Johnson, J. F. Gauuan, C. Guo, P. R. Guzzo, V. D. Le, R. A. Shenoy, J. Hamby, H. Roark, M. Stier and J. E. Mangette, Synth. Commun., 2011, 41, 2769–2793 CrossRef CAS.
- A. McCluskey, S. P. Ackland, M. C. Bowyer, M. L. Baldwin, J. Garner, C. C. Walkom and J. A. Sakoff, Bioinorg. Chem. Appl., 2003, 31, 68–79 CAS.
- C. L. Zhang, T. H. Li, S. H. Niu, R. F. Wang, Z. L. Fu, F. Q. Guo and M. Yang, Bioinorg. Chem. Appl., 2009, 8 Search PubMed.
- N. Windmon and V. Dragojlovic, Green Chem. Lett. Rev., 2008, 1, 155–163 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CAS.
- J. A. Norton, Chem. Rev., 1942, 31, 319–523 CrossRef CAS.
- A. P. Dunlop and F. N. Peters, The Furans, Reinhold Publishing Corporation, Baltimore, 1953 Search PubMed.
- M. S. Newman and V. Lee, J. Org. Chem., 1977, 42, 1478–1479 CrossRef CAS.
- M. P. Cava, C. L. Wilson and C. J. Williams, J. Am. Chem. Soc., 1956, 78, 2303–2304 CrossRef CAS.
- H. N. C. Wong, T. K. Ng, T. Y. Wong and Y. Dexing, Heterocycles, 1984, 22, 875–890 CrossRef CAS.
- M. H. Karger and Y. Mazur, J. Org. Chem., 1971, 36, 528–531 Search PubMed.
- M. H. Karger and Y. Mazur, J. Org. Chem., 1971, 36, 532–540 CrossRef.
- F. Malz and H. Jancke, J. Pharm. Biomed. Anal., 2005, 38, 813–823 CrossRef CAS PubMed.
- U. Tilstam, Org. Process Res. Dev., 2012, 16, 1273–1278 CrossRef CAS.
- N. E. Jacobsen, NMR Spectroscopy Explained, John Wiley and Sons, Inc., Hoboken, 2007 Search PubMed.
- J. T. Manka, A. G. Douglass, P. Kaszynski and A. C. Friedli, J. Org. Chem., 2000, 65, 5202–5206 CrossRef CAS.
- R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302–6337 CrossRef CAS PubMed.
- J. M. Rosenfel and C. B. Murphy, Talanta, 1967, 14, 91–96 CrossRef.
- S. C. Baker, D. P. Kelly and J. C. Murrell, Nature, 1991, 350, 627–628 CrossRef CAS.
- A. S. Thompson, N. J. P. Owens and J. C. Murrell, Appl. Environ. Microbiol., 1995, 61, 2388–2393 CAS.
- D. Arnold, J. Bartels, H. Lenzmann, G. Jacobsen, H. Wendt and M. Stoltenberg, Germany Pat., 4,455,439, 1984 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3gc41655k |
| This journal is © The Royal Society of Chemistry 2014 |