Visible light enhanced oxidant free dehydrogenation of aromatic alcohols using Au–Pd alloy nanoparticle catalysts†
Sarina
Sarina
a,
Sagala
Bai
b,
Yiming
Huang
a,
Chao
Chen
a,
Jianfeng
Jia
c,
Esa
Jaatinen
a,
Godwin A.
Ayoko
a,
Zhaorigetu
Bao
*b and
Huaiyong
Zhu
*a
aSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology, Brisbane, QLD4001, Australia. E-mail: hy.zhu@qut.edu.au; Fax: +61 7 3138 1804; Tel: +61 7 3138 1581
bSchool of Chemistry, Inner Mongolia Normal University, Hohhot, China. E-mail: zrgt@imnu.edu.cn
cSchool of Chemical and Material Science, Shanxi Normal University, Linfen 041004, China
First published on 17th October 2013
Abstract
We find that visible light irradiation of gold–palladium alloy nanoparticles supported on photocatalytically inert ZrO2 significantly enhances their catalytic activity for oxidant-free dehydrogenation of aromatic alcohols to the corresponding aldehydes at ambient temperatures. Dehydrogenation is also the dominant process in the selective oxidation of the alcohols to the corresponding aldehydes with molecular oxygen. The alloy nanoparticles strongly absorb light and exhibit superior catalytic and photocatalytic activity when compared to either pure palladium or gold nanoparticles. Analysis with a free electron gas model for the bulk alloy structure reveals that the alloying increases the surface charge heterogeneity on the alloy particle surface, which enhances the interaction between the alcohol molecules and the metal NPs. The increased surface charge heterogeneity of the alloy particles is confirmed with density function theory applied to small alloy clusters. Optimal catalytic activity was observed with a Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd molar ratio of 1:186, which is in good agreement with the theoretical analysis. The rate-determining step of the dehydrogenation is hydrogen abstraction. The conduction electrons of the nanoparticles are photo-excited by the incident light giving them the necessary energy to be injected into the adsorbed alcohol molecules, promoting the hydrogen abstraction. The strong chemical adsorption of alcohol molecules facilitates this electron transfer. The results show that the alloy nanoparticles efficiently couple thermal and photonic energy sources to drive the dehydrogenation. These findings provide useful insight into the design of catalysts that utilize light for various organic syntheses at ambient temperatures.
:Pd molar ratio of 1:186, which is in good agreement with the theoretical analysis. The rate-determining step of the dehydrogenation is hydrogen abstraction. The conduction electrons of the nanoparticles are photo-excited by the incident light giving them the necessary energy to be injected into the adsorbed alcohol molecules, promoting the hydrogen abstraction. The strong chemical adsorption of alcohol molecules facilitates this electron transfer. The results show that the alloy nanoparticles efficiently couple thermal and photonic energy sources to drive the dehydrogenation. These findings provide useful insight into the design of catalysts that utilize light for various organic syntheses at ambient temperatures.
Introduction
Photocatalytic processes are generally able to drive reactions at ambient temperature and pressure. As a result, product selectivity is improved and unstable intermediates of thermal reactions may be produced as products of photocatalytic reactions. However, the bulk of all reported photocatalytic studies focus on semiconductor photocatalysts such as TiO2, ZnO and CdS and their application in decomposing organic pollutants,1 the development of new solar cells and the production of hydrogen and oxygen from water.2 To date, only limited progress has been reported in applying TiO2 and Nb2O5 semiconductor photocatalysts to synthesize organic chemicals.3–5 This is largely because of the limited light absorption and low efficiency of these photocatalysts. Due to their band structure, the well-known TiO2 photocatalysts only show significant light absorption of ultraviolet (UV) light.1,2,6,7 Consequently, the efficiency of the semiconductor photocatalysts, expressed as photon yield, is typically low.7 To overcome this, various methods have been developed to produce efficient visible light photocatalysts from semiconducting materials.8 In recent years, photocatalysis with nanoparticles (NPs) of plasmonic metals, such as gold (Au), silver (Ag) and copper (Cu), has attracted significant interest because of their exceptional absorption of visible light and thus their potential as visible light photocatalysts.9–17 Plasmonic metal nanoparticles exhibit strong visible-light absorption due to the localised surface plasmon resonance (LSPR).18–20 In addition they also strongly absorb ultraviolet (UV) light due to inter-band electron transitions (e.g. for Au, between the 5d and 6sp bands).21–23 The NP conduction electrons gain the irradiation energy, resulting in high energy electrons at the surface. These energetic electrons, which remain in an excited ‘hot’ state for up to 0.5–1 ps,24 can promote chemical reactions of molecules on the surface of the NPs.25,26The underpinning photocatalytic mechanisms of the Au and Ag NPs supported by insulating solids with very wide band gaps are quite distinct from those of semiconducting photocatalysts.9–12,25,26 These highly efficient NP photocatalysts have several important advantages over their extensively studied semiconductor counterparts. (1) The NP conduction electrons gain light energy, resulting in high energy electrons at the NP surface which is advantageous for activating molecules adsorbed on the NPs for chemical reactions. (2) Since both light harvesting and the catalysing reaction take place on the NPs, the photon efficiency is not significantly affected by photo-excited charge migration. (3) The density of the conduction electrons at the NP surface is much higher than that at the surface of any semiconductor, so once photo-energized more electrons are available to drive reactions. (4) The metal NPs have a strong affinity for some reactants, such as CO and organic compounds, making the NPs superior photocatalysts for organic synthesis reactions compared to semiconductor photocatalysts.
However, the total number of chemical reactions that can be catalysed by the three plasmonic metals is relatively few when compared to those thermally catalysed by non-plasmonic transition metals. To develop catalysts for light driven synthesis of a broad range of organic chemicals, we presented a unique but viable approach: alloying gold and a transition metal such as Pd, which is thermally catalytically active for many reactions.13 Thus, the light energy absorbed by the gold can enhance the intrinsic catalytic activity of Pd at moderate temperatures. A typical example is the selective (or partial) oxidation of aromatic alcohols to the corresponding aldehydes, which can be catalysed by Pd catalysts but not Au catalysts. Here we show that Au–Pd alloy NPs exhibit superior catalytic activity when exposed to visible light irradiation at ambient temperatures than that displayed by NPs made from either pure component metals.
Selective oxidation of alcohols is widely considered one of the most fundamental transformations in both laboratory and industrial synthetic chemistry because the product carbonyl compounds can serve as important and versatile intermediates for fine chemical synthesis.27–30 Permanganate and dichromate have been traditionally employed31,32 but they are expensive and/or toxic. For environmental and economic reasons, metal-catalysed reactions using molecular oxygen as an oxidant are particularly attractive. Many of the reactions are conducted under heating and/or high pressure to achieve a better reaction efficiency. Since the first successful example of palladium-catalysed aerobic oxidation of alcohols in 1977 by Blackburn and Schwartz,33 subsequent efforts have extended the substrate scope and efficiency of palladium catalysts. However, the reported PdII salt based homogeneous catalysts34–37 require high catalyst concentration and an excess of ligands or bases. On an industrial scale the problems related to corrosion and plating out on the reactor wall, handling, recovery, and reuse of the catalyst represent serious limitations of these processes.38 Solid catalysts active in the liquid phase under mild conditions have a much broader application range. Relatively few heterogeneous Pd (supported Pd nanoparticles and PdII) are available. For example, Pd on hydrotalcite,39,40 carbon,39,41 Al2O3,39 SiO2,39 pumice,42 SiO2–Al2O3 mixed oxide,43 TiO244 and polymer supported Pd.45 Those catalysts, both in the metallic NPs or immobilized ionic state, can catalyse the benzyl alcohol oxidation at elevated temperatures and/or high pressure. Recently it was reported that activation of molecular oxygen is the key step in selective oxidation of aromatic alcohols using TiO2 as photocatalysts under UV irradiation.
The use of Au and Pd alloy NPs as visible light photocatalysts for the selective oxidation of aromatic alcohols will have an underlying mechanism different from those aforementioned processes. It is known that during selective oxidation, the aromatic alcohols may undergo abstraction of a hydrogen atom bonded to the α-carbon atom (the carbon atom of the methylene group bonded to the hydroxyl group), which is denoted as α-H, followed by abstraction of the hydrogen atom from the hydroxyl group.46,47 If the first hydrogen abstraction is the rate-determining step and can take place via a photocatalytic process, high reaction temperature and high oxygen pressure are not necessary for selective oxidation. In the present study, we find that light irradiation can drive the α-H abstraction of aromatic alcohols with Au–Pd alloy NPs and thus the transformation from alcohol into the corresponding aldehyde can be achieved in an oxidant free environment at ambient temperatures. The α-H abstraction is the rate-determining step of the selective oxidation. Theoretical calculations by two independent methods show that the alloying of gold and palladium enhances the interaction between the alcohol molecules and the alloy NPs. The strong interaction facilitates the transfer of light excited electrons of the alloy NPs to the alcohol molecules adsorbed on the NPs, and such an electron transfer enables the hydrogen abstraction under moderate conditions. Understanding this mechanism is useful for developing photocatalytic processes for other important syntheses.
Results and discussion
Performance of the photocatalysts
In the present study, Au and Pd alloy NPs with varying relative ratios were prepared on a ZrO2 support (abbreviated as Au–Pd@ZrO2, and details are given in ESI†). The metal nanoparticles on the support were well dispersed avoiding aggregation of the particle. ZrO2 has a wide band gap (5 eV), and exhibits no visible light absorption. Hence, the support does not contribute to photocatalytic activity. Fig. 1 shows the photocatalytic performance of the Au–Pd@ZrO2 photocatalysts with various Au:Pd mass ratios in catalysing the dehydrogenation of aromatic alcohols. All experiments were conducted under an Ar gas atmosphere after a strict freeze–pump–thaw degassing process to remove O2. The conversion rates achieved under light irradiation (in blue) are compared with those obtained in the dark (in black).
 | ||
| Fig. 1 Dehydrogenation of aromatic alcohols with the Au–Pd@ZrO2 catalysts of various Au:Pd mass ratios under visible light irradiation (blue bar) and in the dark (black bar). All of the data represent the averages of triplicate runs with a mean variation of less than ±3%. The quantum yield (Q.Y., %) and its calculation method are given in ESI.† A(1) Benzyl alcohol dehydrogenation using the catalyst of 3% Au–Pd@ZrO2; the reaction proceeded for 5 h. A(2) 4-Methyloxy benzyl alcohol dehydrogenation, reaction for 2 h. A(3) 1-Phenylethanol dehydrogenation, reaction for 22 h. Reaction conditions: 2 mmol of the reactant, 50 mg of catalyst in trifluorotoluene solvent at 45 °C and under 1 atm of Ar after O2 removal using freeze–pump–thaw degassing. B(1) Benzyl alcohol dehydrogenation, 2% Au–Pd@ZrO2, reaction for 16 h. B(2) 2-Phenylethanol dehydrogenation, 2% Au–Pd@ZrO2, reaction for 16 h. B(3) 3-Phenylpropanol dehydrogenation, 2% Au–Pd@ZrO2, reaction for 16 h. Reaction conditions: 1 mmol of the reactant, 50 mg of catalyst in trifluorotoluene solvent at 45 °C and under 1 atm of Ar after O2 removal using freeze–pump–thaw degassing. | ||
The results in Fig. 1 demonstrate that visible light irradiation increased the product yield of the dehydrogenation product of aromatic alcohols – corresponding aldehydes. For example, the conversion rate of benzyl alcohol with the Au–Pd@ZrO2 catalyst with an Au:Pd mass ratio of 1:1 is 100%. In contrast the rate is 43% when the reaction was conducted in the dark. Similar trends are observed in other reactants, for example, the conversion rate of 4-methoxybenzyl alcohol is 99% under light irradiation but 54% in the dark at the same reaction temperature. Blank experiments without metal NPs were also conducted with just the photocatalytically inactive ZrO2 supports dispersed in a toluene solution of benzyl alcohol. Under otherwise identical conditions, no alcohol conversion was observed under visible light irradiation or in the dark. We found that at identical reaction temperatures and identical reaction periods, the conversion rates of the oxygen-free dehydrogenation of aromatic alcohols (under an argon atmosphere) are comparable to those of the selective oxidation conducted under an oxygen atmosphere. This demonstrates that the rate-determining step of the transformation from alcohols into the corresponding aldehydes with Au–Pd@ZrO2 photocatalysts is dehydrogenation, and light irradiation significantly enhances the dehydrogenation.
Compared to the Au–Pd@ZrO2 catalyst containing 3% of alloy NPs, the Au–Pd@ZrO2 catalyst with lower metal loading (2%), the reaction time required to achieve the same conversion rate with identical reaction conditions was longer, as shown in Fig. 1B. The impact on the photocatalytic activity of varying mass ratios of the two metals with 2% metal loading has the same trend as that observed with the catalysts with 3% metal loading.
As can be seen in Fig. 1, the alloy NP catalysts exhibited significantly higher activity than pure Au NPs (Au:Pd ratio of 1:0) or pure Pd NPs (Au:Pd ratio of 0:1) for the dehydrogenation of alcohols under visible light irradiation (>420 nm). This fact implies that Au–Pd@ZrO2 catalysts are not collections of Au NPs and Pd NPs; instead, Au and Pd exist as binary alloy particles in these samples.
Transmission electron microscopy (TEM) analysis of the NPs (Fig. 2) shows that the mean diameters of the Au–Pd alloy NPs are less than 10 nm. Fig. 2B is a line profile of the energy dispersion X-ray (EDX) spectrum of a typical Au–Pd alloy NP in Fig. 2A, which shows the elemental distribution along the radial direction of the metal NP. The line profile indicates that the NP consists of both Au and Pd distributed spherically around a common centre. This means that the two metals exist as binary alloy NPs in this sample. No diffraction peaks corresponding to either metallic Au or metallic Pd were observed from X-ray diffraction (XRD) patterns of the Au, Pd and Au–Pd samples (not shown) because of the low metal content of the samples.
 | ||
| Fig. 2 (A) TEM image of 1.5% Au–1.5% Pd@ZrO2 catalyst. The arrows indicate Au–Pd NPs. (B) High resolution TEM images of an alloy particle in the catalyst. (C) EDX spectrum line profile analysis of a typical Au–Pd NP indicated along the blue arrow in Fig. 2A, providing information of the elemental composition and distribution of the NP. | ||
The formation of Au–Pd alloy NPs is also supported by the significant change of light absorption properties of the sample as shown in Fig. 3.48–50 Therefore, the catalytic properties of this sample resulted from Au–Pd alloy NPs rather than a collection of discrete Au NPs and Pd NPs.
 | ||
| Fig. 3 Diffuse reflectance UV-visible (DR-UV-vis) spectra of the photocatalysts and ZrO2 support. | ||
ZrO2 has a band gap of about 5 eV,51 and exhibits weak visible light absorption with no charge carriers generated under irradiation of light with wavelengths above 400 nm (Fig. 3). Therefore, the ZrO2 support by itself does not contribute to photocatalytic activity. The absorption peak at 520 nm in the spectrum of the pure Au (3 wt%) sample is due to the LSPR absorption of the Au NPs.21,25,26,48–50,52–55 The presence of the support and its interaction with the Au NP can shift the resonance to longer wavelengths and broaden the LSPR absorption peak. The LSPR absorption band of Pd NPs is in the UV range at a wavelength of 330 nm.18 However, this absorption is not observed in the extinction spectrum of the pure Pd NPs on ZrO2.
Interestingly, we observed a clear light absorption near the Pd absorption band (330 nm) in the spectrum of Au–Pd alloy particles on ZrO2 (trace b, Fig. 4B), which is believed to be associated with Au–Pd alloy NPs.18 In the spectrum of the alloy sample, the characteristic LSPR absorption peak of Au NPs at 520 nm is much weaker when compared to the spectrum of the pure Au sample, but larger than the absorption observed for the pure Pd sample.
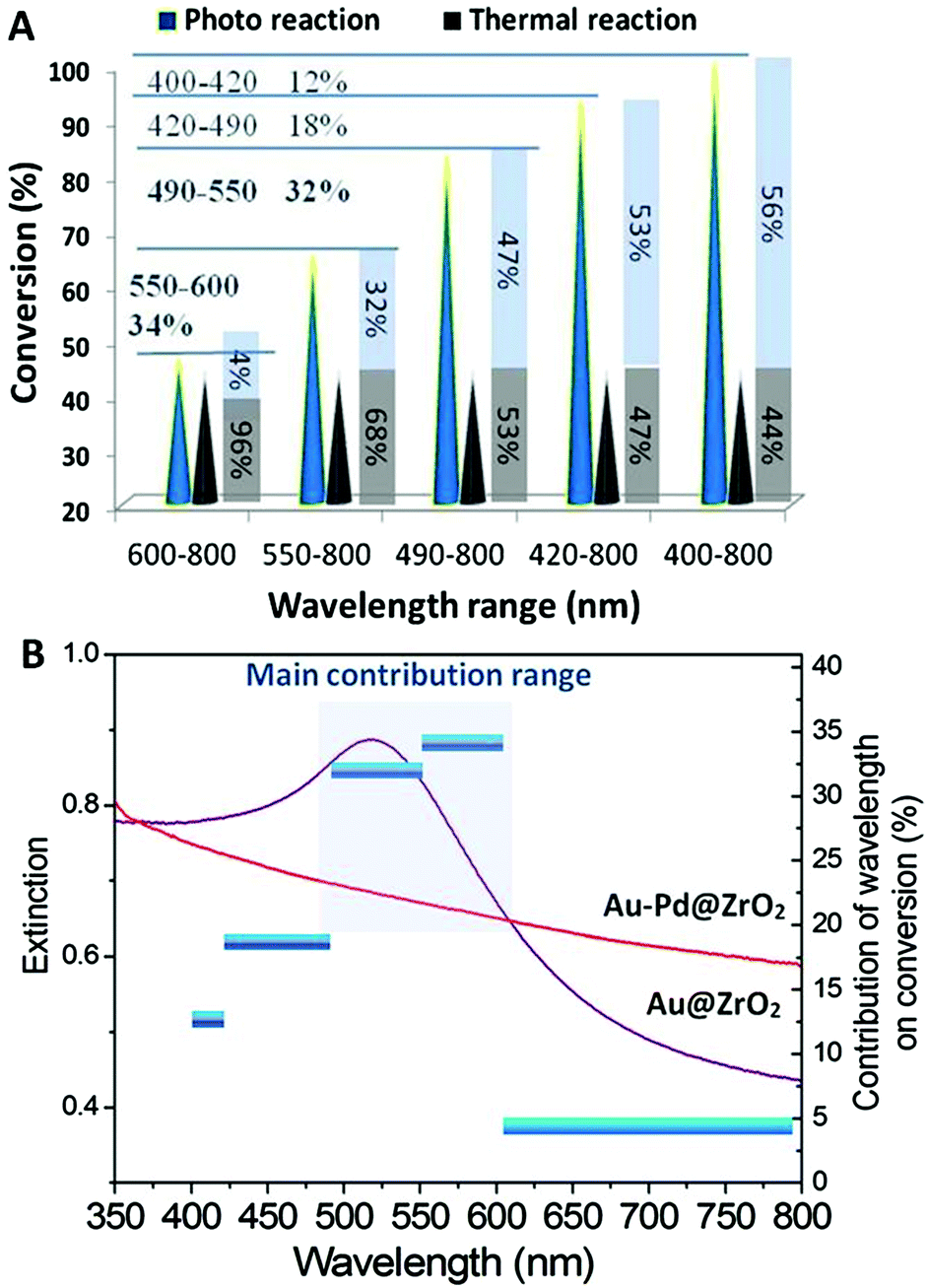 | ||
| Fig. 4 (A) The dependence of the catalytic activity of the Au–Pd@ZrO2 catalyst for the benzyl alcohol dehydrogenation on the wavelength of light irradiation. Both the light-driven reaction and the reaction in the dark were conducted at 45 °C ± 1 °C. (B) Action spectrum based on the data in (A), and the light absorption spectra of Au–Pd@ZrO2 and Au@ZrO2 are given for comparison. | ||
The influence of light irradiation
The wavelength and intensity are important irradiation energy parameters and, hence, they are the critical factors influencing the performance of photocatalysts in reactions. The dependence of the photocatalytic dehydrogenation of benzyl alcohol on the wavelength and intensity was investigated. The results are illustrated in Fig. 4.Optical filter glasses were applied to block irradiation below specific cut-off wavelengths. Therefore, we were able to tune the wavelength of the light to clarify the influence of the wavelength range on the catalytic activity of the Au–Pd@ZrO2 catalyst for benzyl alcohol oxidation. When light with wavelengths in the full 400 nm to 800 nm range irradiated the reaction system a 100% reaction conversion was observed. Applying a filter that blocked wavelengths below 490 nm resulted in a decrease in the conversion of the reaction to 83%. Increasing the cut-off wavelength to 550 nm and then 600 nm resulted in the conversion decreasing to 65% and 46%, respectively. Given that the thermal conversion rate at this temperature is 44%, the contribution of light irradiation in the wavelength range of 400–800 nm to the overall catalytic activity is 56%.
From the results acquired from the irradiation of tuned wavelength we can estimate the contribution of the light in a narrow wavelength range. For example, the yield of the reaction is 83% when the light with wavelengths below 490 nm was cut off. Since the reaction temperature was held constant, the contribution of the thermal reaction remains constant regardless of the filter used. Therefore, the observed decrease in the yield of 17% (= 100%–83%) can be attributed to light irradiation by wavelengths in the 400 nm–490 nm range. The enhancement in the yield due to the light irradiation by wavelengths in the 490 nm–800 nm range is 39% (= 83%–44%), which accounts for 47% of the overall yield achieved (83%). When the system was irradiated with light with wavelengths in the 600 nm–800 nm range, the enhancement in the yield decreased to 4%. As shown by the results in Fig. 4A, light irradiation in the wavelength range of 490 nm–600 nm results in the largest enhancement in yield, accounting for 66% of the total light irradiation enhancement (conversion rate difference between photocatalytic reaction and that observed in the dark), 32% results from the light irradiation with wavelengths between 490 nm and 550 nm, 34% from light between 550 nm and 600 nm, while the light with wavelengths between 400–490 nm and 600–800 nm account for the remaining 34%. The total light energy absorbed by the NPs was estimated from the overlap of the absorption spectrum of the Au–Pd@ZrO2 catalyst, and the spectral distribution of the light irradiated. It was found that 36.2% of the total light energy absorption by the NPs was in the 490 nm–600 nm wavelength range. Plotting the enhancement caused by light irradiation from the different wavelength ranges (the vertical axis on the right hand side) against the light wavelength reveals the action spectrum (Fig. 4B). It shows which light wavelengths are most effectively used in specific chemical reactions. Given that the LSPR peak of Au NPs is in the wavelength range between 500 nm and 600 nm, these results suggest that for Au–Pd@ZrO2 photocatalysts, it is the gold that harvests visible light and that the gold nanostructure's LSPR plays an important role in enhancing the reaction yield in alloy NP catalysed reactions. Furthermore, from the spectrum, it is seen that the rate-determining step of the alcohol dehydrogenation should occur with Au–Pd alloy NPs.
A different wavelength range is found to produce the most significant enhancement in performance of the pure Au NP photocatalyst from that observed for Au–Pd@ZrO2. In our recent study on selective reactions catalysed by the Au NPs under visible light, acetophenone hydrogenation to 1-phenyl ethyl alcohol and styrene oxide reduction to styrene,14,15 light irradiation in the wavelength range between 490 nm and 550 nm made the most important contribution to driving the reaction. The contributions from light in this wavelength range for the two reactions are 41% and 65%, respectively. The contribution of light in the wavelength range of 550–600 nm is much less (24% and 0% for the two reactions, respectively), while for the Au–Pd@ZrO2 catalyst in the present study, the contribution of light within the wavelength range of 550–600 nm is even more significant than that of light in the range of 490–550 nm. In other words, the effective wavelength range of Au–Pd alloy NPs is broader (490–600 nm) than that of Au NPs. This phenomenon can be attributed to the formation of the alloy NPs, and it also reveals that Au–Pd alloy NPs have the potential to utilize light energy from a wider portion of the visible light spectrum for enhancing reactions than pure Au NPs.
The impact of the light intensity on the catalyst performance was investigated while keeping other experimental conditions unchanged. Fig. 5 shows the rate of benzyl alcohol dehydrogenation over the Au–Pd@ZrO2 catalyst with a Au:Pd molar ratio of 1:1 as a function of light intensity at 45 °C ± 1 °C and 60 °C ± 1 °C, respectively. When the light intensity increased (the reaction temperature of the reaction mixture was controlled at 45 °C; the only parameter changed is light intensity), the conversion of benzyl alcohol oxidation increased linearly up to a light intensity of 0.8 W cm−2. Further increase in light intensity results in much greater rate increases, and the relation between light intensity and reaction rate becomes nonlinear. This is a feature of the chemical processes driven by the light excited electrons of metals.16 It is also possible that when the light intensity is very high, multi-photon absorption occurs, increasing the number of excited metal electrons with sufficient energy to drive the reactions.
 | ||
| Fig. 5 Light intensity dependent photocatalytic activity of Au–Pd alloy NPs for the transformation from benzyl alcohol into benzaldehyde at 45 °C (A) and 60 °C (B). | ||
The light induced enhancement on the conversion was calculated by subtracting the observed conversion of a reaction performed in the dark from the conversion observed under light irradiation performed at the same temperature. This allows the photo-induced and thermal contributions to the conversions to be determined and expressed as a percentage for each process, as shown in Fig. 6. It shows clearly that higher light intensities result in a larger light enhanced contribution to the total conversion rate.
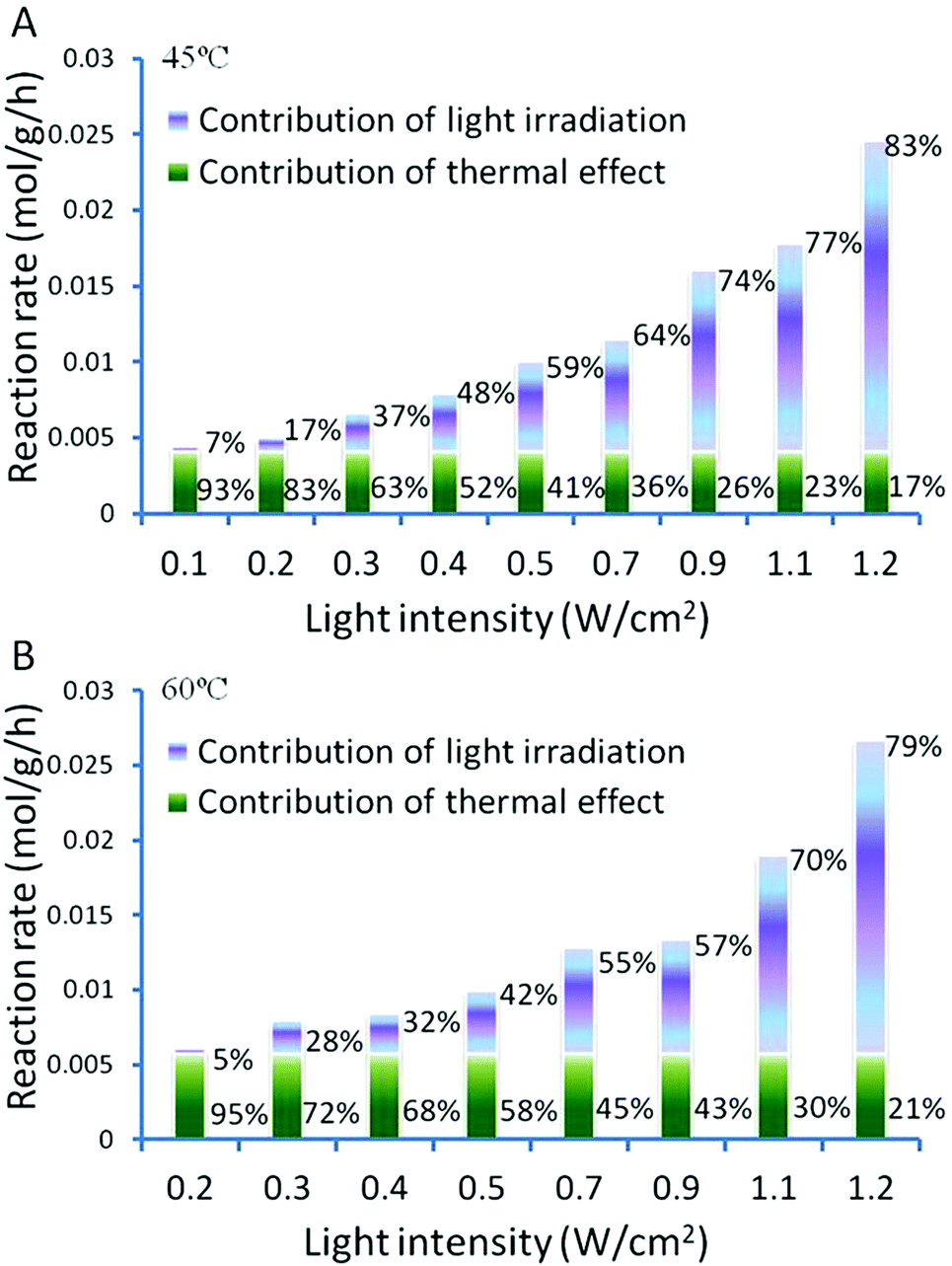 | ||
| Fig. 6 Light intensity dependent activity of Au–Pd@ZrO2 photocatalysts on oxidant free dehydrogenation of benzyl alcohol. | ||
The influence of reaction temperature
Increasing operating temperature was observed to increase the photocatalytic rate of benzyl alcohol oxidation. Fig. 7 shows that at a constant light intensity (we applied 0.4 W cm−2 as an example), increasing operating temperature results in a significant increase of the photocatalytic reaction rate. There are two critical aspects of the temperature effect on the photocatalytic rate. First, the relative population of the adsorbed reactant molecule with excited states increases according to the Bose–Einstein distribution at higher temperatures, which means that the reactant molecule (benzyl alcohol or oxygen molecule) will require less energy to overcome the activation barrier, and this “less energy” could be provided by light irradiation.16,56 Second, at higher temperature more electrons of the alloy NPs are in higher energy levels which can be excited by light irradiation yielding more electrons with sufficient energy to induce reactions in the alcohol molecules adsorbed on the alloy NPs. | ||
| Fig. 7 Dependence of the photocatalytic rate of benzyl alcohol oxidation on reaction temperature at a constant light intensity. | ||
The influence of the Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Pd component ratio
:Pd component ratio
As shown by the results in Fig. 1, the Au:Pd mass ratio of the alloy particles is a key influencing factor on the catalytic performance of the oxidant free dehydrogenation of the aromatic alcohols both under light illumination and in the dark. All reactions achieved the highest yield of target products when the Au:Pd mass ratio of the alloy NPs was 1:1 (molar ratio of 1:1.86). Alloy NPs with other Au:Pd mass ratios proved to be much less active. When the reactions were performed in the dark, the same dependence on the Au:Pd mass ratio was observed but with much lower conversion efficiencies. Catalytic processes driven by heating Au–Pd alloy catalysts have been reported in the literature. It was found that Au–Pd alloy NPs could catalyse the hydrogenation of cinnamaldehyde to cinnamyl alcohol.57 Toshima et al. reported that Au–Pd alloy NPs were more active in catalyzing the hydrogenation of 1,3-cyclooctadiene than either gold or palladium alone.58,59
A possible explanation for the superior catalytic activities of Au–Pd alloy NPs to NPs consisting of either a pure component is that Au can isolate active Pd sites within bimetallic systems.60 In the present study, to confirm the significant role that the Pd sites at the alloy interface play in the catalytic processes, a sample of NPs with a Pd core and an Au shell was prepared by reducing HAuCl4 on the Pd-ZrO2 support with H2. This sample has almost no Pd sites at the NP surface and exhibited low activity for selective oxidation of benzyl alcohol (10% conversion compared to 100% achieved by the Au–Pd alloy NPs with a similar Au:Pd ratio). However, the existence of Pd sites at the alloy surface does not explain why the optimal catalytic activity was observed when the Au:Pd molar ratio is 1:1.86. The underlying cause of this dependence on alloy composition is believed to be related to the higher surface charge heterogeneity of the alloy NPs, compared with those of either pure component NPs.13,61 Pure palladium has a slightly larger work function (ΦPd ∼ 5.6 eV) than pure gold (ΦAu ∼ 5.3 eV), so once the two metals are in contact, electrons will redistribute between gold and palladium until equilibrium is reached and the chemical potentials are equal everywhere in the NPs (see Fig. 8). This electron redistribution between Au and Pd enhances surface charge heterogeneity of the NPs (the surface charge of an NP of pure metal is not homogeneous). The greater surface charge heterogeneity results in an enhanced interaction between the alcohol and the NP. The enhanced interaction may lower the activation energy of the oxidation and thus enhance the catalytic activity. Furthermore, the Fermi level in alloy NPs, Φalloy, is higher than that in pure Pd NPs, ΦPd, so that the transfer of electrons at the Fermi level of the alloy NPs to the benzyl alcohol molecule adsorbed on the NPs is easier compared with that from the Fermi level of pure Pd NPs to the adsorbed molecule. The electron transfer causes the transformation of benzyl alcohol molecules as discussed later. The light absorption of gold results in energetic conduction electrons, which are in an even higher energy level Φalloy* and have a better ability to migrate to the Pd sites on the surface. This further increases the possibility of electron transfer from the alloy NPs to the reactant molecules.
 | ||
| Fig. 8 Schematic profiles showing the impact of alloying and visible light irradiation of an Au–Pd alloy NP. (A) The surface electronic properties of the alloy NPs are different from those of pure gold NPs as there are Pd islets on the alloy NP surface. The Pd sites are electron-rich because Pd has a slightly larger work function than gold and electrons will flow from gold to palladium until equilibrium is reached (the chemical potentials of the electrons are equal in the two metals, being Φalloy). (B) The light absorption by gold results in energetic conduction electrons, which migrate to the Pd sites on the surface. The Fermi level of the alloy NPs under light irradiation is higher (Φalloy*) than that without irradiation (Φalloy), which increases the charge transfer possibility from the NP to the reactant molecule. Thus the surface Pd sites with energetic electrons could exhibit significantly enhanced catalytic activity even at ambient temperatures. Since in such an alloy NP structure the energy of incident light is very efficiently utilised to enhance the intrinsic catalytic activity of palladium, efficient photocatalysts may be developed from it for the synthesis of organic chemicals. (C) Electron transfer from gold to palladium in the alloy NPs, expressed as ΔN, varies with the composition of the alloy NPs (the curve). ΔN reaches a maximum when the atomic ratio of Au and Pd in alloy NPs is 1:1.86 (mass ratio 1:1). The information on gold content of the alloy NPs (horizontal axis) and photocatalytic conversion (vertical axis on the right hand side) of the reactions in the present study is also given in the figure (the symbols). The photocatalytic efficiency of the alloy NP photocatalysts is strongly correlated to the electron transfer ΔN. | ||
Since the increased surface charge heterogeneity of the alloy NPs is due to the electron redistribution between Au and Pd, we estimated the increase (details are provided in ESI†) by using a free electron gas model.51 The analysis reveals that the number of electrons transferred between the two metals, (ΔN), is maximum when the molar ratio of the two metals in the alloy NPs is 1:1.86. A plot of the electron transfer (ΔN; refer to the axis on the left hand side) predicted by the model as a function of the gold mole fraction (%) in the Au–Pd alloy NPs is shown in Fig. 8C. The catalytic conversion rates (refer to the axis on the right hand side) of the photocatalysts for oxidant free dehydrogenation of benzyl alcohol are also given (the symbols; refer to the axis on the right hand side). The results in Fig. 8C demonstrate a strong correlation between ΔN and the catalytic conversion efficiency of reactants over the alloy NP catalysts. Note that this electron transfer ΔN analysis uses parameters of bulk particles and is independent of the size of alloy particles.
We also carried out simulations using the density function theory (DFT) for electron states with and without light irradiation; the irradiation wavelength range between 532 and 535 nm is chosen, which is around the LSPR absorption of Au. Calculation capacity limitations of our DFT simulation necessitated the examination of a Pd32, Au32, and Au12Pd20 cluster. The Au:Pd ratio of the Au12Pd20 cluster is 1.67 close to the ratio of 1:1.86 for the optimal Au–Pd@ZrO2 photocatalyst. The detailed calculation method and the calculated Mulliken charge distributions are given in ESI.† The DFT simulation results confirm that charge heterogeneity exists even in the monometallic Pd clusters and monometallic Au clusters (Fig. 9), and the alloy structure of Au and Pd increases the charge heterogeneity of the NP surface. This result is consistent with that of the free electron gas model analysis and previous reports.62,63 Furthermore, the comparison of simulation results without irradiation (blue lines) with the results under irradiation (red lines) suggests that light irradiation also promotes the charge heterogeneity in Au–Pd alloy NPs.
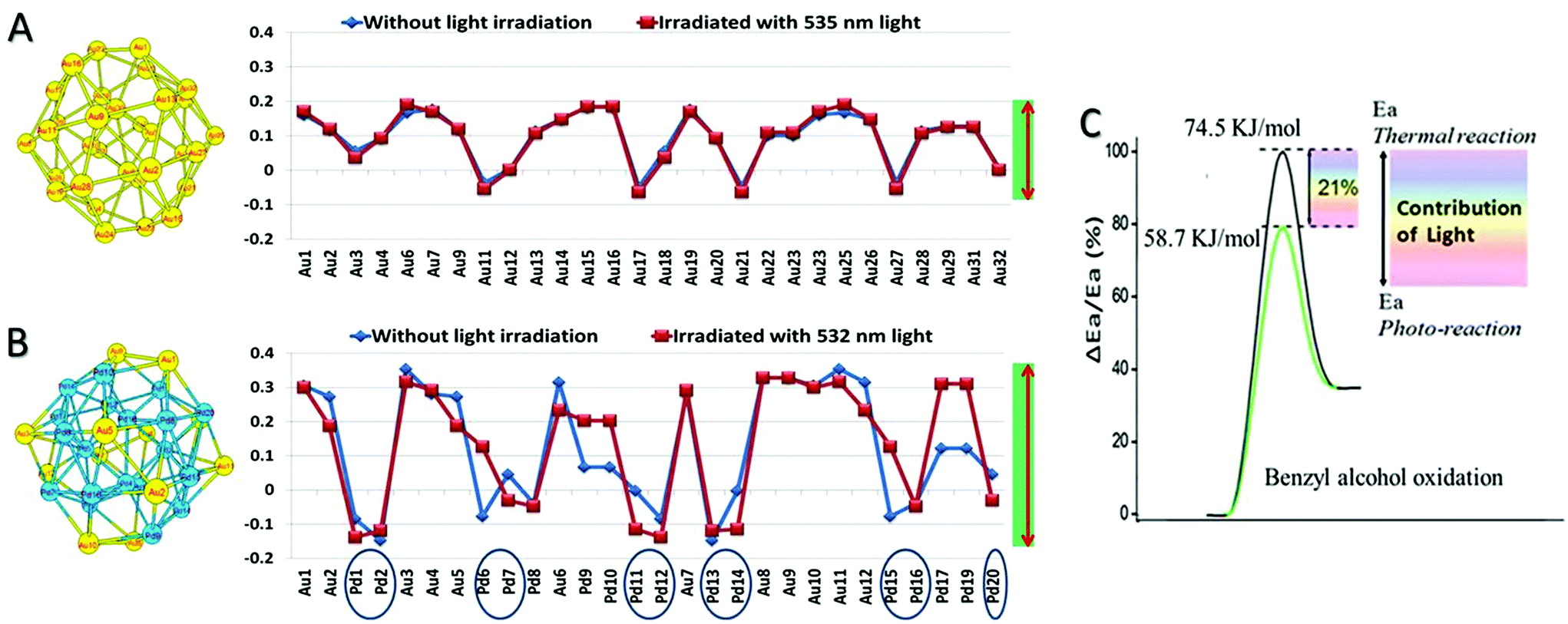 | ||
| Fig. 9 The optimized geometry and the natural charge distributions of the Au32 cluster (A) and Au12Pd20 clusters (B) in the ground state and the considered excited state. (C) Apparent activation energy reduction of benzyl alcohol dehydrogenation caused by the light irradiation on the Au–Pd alloy NP photocatalyst. | ||
The apparent activation energies of the oxidant free dehydrogenation of benzyl alcohol under light irradiation and in the dark were derived from the kinetic data of the reaction at different temperatures (details are provided in ESI†) using the Arrhenius equation. The difference between the activation energies under light irradiation and in the dark (ΔEa) indicates the contribution of the light irradiation to reducing the apparent activation energy.14,15 For example, as illustrated in Fig. 9C, the apparent activation energy for the dehydrogenation in the dark is ∼74.5 kJ mol−1 and it is ∼58.7 kJ mol−1 for the reaction under visible light illumination. The apparent activation energy in the dark is in agreement with those reported by Bavykin et al.64 (79 kJ mol−1 for the ruthenium-catalyzed oxidation of benzyl alcohol), and by Ilyas et al.65 (77.8 kJ mol−1 for a Pt/ZrO2 catalyst system). Visible light irradiation reduces the activation energy of the partial oxidation by 15.8 kJ mol−1, which represents 21% of the activation energy.
It was reported that the first step of alcohol oxidation with a Pd catalyst was alcohol dehydrogenation, which resulted in adsorbed hydrogen on the Pd surface and the formation of aldehyde.66 The rate-determining step of the alcohol oxidation on gold catalysts is believed to be the hydrogen abstraction from alcohol and the formation of Au–H species; the role of oxygen in this reaction is to remove hydrogen from the gold surface, leading to the catalytic cycle.67,68 In order to clarify the reaction mechanism we investigated the hydrogen abstraction from alcohol to the surface of Au–Pd NPs. 2,2′,6,6′-Tetramethylpiperidine N-oxyl (TEMPO) is a good hydrogen abstractor, and can abstract hydrogen from the surface of metals to form hydroxylamine but not from alcohol molecules.69 If the addition of TEMPO can result in alcohol dehydrogenation in the absence of oxygen, then it proves that hydrogen transfer from the alcohol to Au–Pd NPs occurs in the reaction because the TEMPO can only abstract the hydrogen on the NP surface. Here TEMPO plays a role similar to oxygen: removing hydrogen from the NP surface to complete the catalytic cycle so that the catalytic conversion of alcohol to the corresponding aldehyde could proceed.
Benzyl alcohol dehydrogenation with Au–Pd@ZrO2 (Au:Pd ratio of 1:1.86) catalyst at 45 °C under light irradiation was conducted under a N2 atmosphere with 300 mg of TEMPO added to the reaction system. After visible light irradiation, 21% of the benzyl alcohol was converted to benzaldehyde. No conversion was observed in the dark at 45 °C. When the reaction was conducted at 80 °C in the dark, 30% of the benzyl alcohol was converted by the TEMPO. Evidently in the absence of oxygen, TEMPO could abstract hydrogen atoms and drive alcohol oxidation either under visible light irradiation or in the dark at a higher temperature. Blank experiments of TEMPO addition without metal NPs (ZrO2 support only) were also conducted at both 45 °C and 80 °C. No conversion was observed even under visible light, indicating that TEMPO could not abstract hydrogen atoms directly from the alcohol but could capture them from the surface of Au–Pd NPs.
It is known that light excited electrons of plasmonic metal NP can populate unoccupied orbitals of the molecules adsorbed on the NPs yielding a transient anion species.16,25,70 Results of a DFT simulation (the detailed calculation method is given in ESI†) show that in a benzyl alcohol molecule, the distances between the α-C and the two H atoms are 1.098 and 1.100 Å, respectively, while in the transient benzyl alcohol anion species one C–H distance remains at 1.100 Å while the other elongates to 1.104 Å. The energy required to break one of the C–H bonds at the α-C in the molecule is 371 kJ mol−1, but only 242 kJ mol−1 is required to break the longer C–H bond of the α-C in the transient anion species. Hence the light irradiation can facilitate the hydrogen abstraction from the α-C through the excitation of NP electrons to the benzyl alcohol molecules adsorbed on them. Therefore, the irradiation reduces the apparent activation energy of the reaction.
On the basis of these facts, a tentative mechanism for selective benzyl alcohol oxidation is proposed as depicted in Scheme 1. The results of the influence of light intensity and the action spectrum indicate that the rate determining step of the partial oxidation takes place on the surface of the supported alloy NPs. The first step should be the abstraction of α-H atoms from the alcohol molecules. Once this abstraction is completed the subsequent abstraction of the hydrogen atom from the hydroxyl group of the transient anion species proceeds readily producing aldehyde as the final product while the negative charge of the transient anions returns to the alloy NPs. The charge injection to the reactant on the plasmonic metal NPs and the charge return to the NPs after reaction are known processes.25,26 Oxygen or TEMPO takes away the hydrogen on the NP surface yielding water or hydroxylamine, respectively, and thus, the NPs’ ability for dehydrogenation of alcohol is restored.
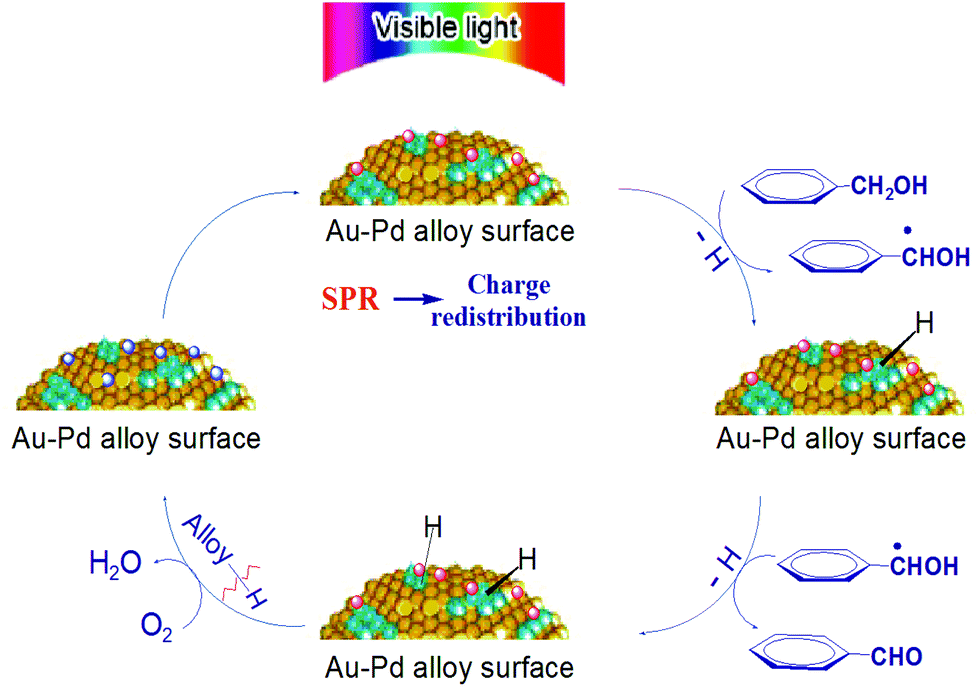 | ||
| Scheme 1 Proposed mechanism of aromatic alcohol oxidation over Au–Pd alloy NPs under visible light irradiation. | ||
The conduction electrons of the alloy NPs can gain the energy of the incident visible light via the LSPR effect and inter-band transition. These energetic electrons are available to Pd sites at the NP surface because of electron–electron collisions and electron redistribution between Au and Pd. The Pd sites have good affinity to the aromatic alcohol molecules and the interaction between the alcohol molecules and the NP surface is enhanced by the surface charge heterogeneity of the alloy NPs. The strong interaction facilitates the transfer of the light excited electrons of the NPs to the adsorbed alcohol molecules. As a result, the catalytic activity of alloy NPs is significantly enhanced at ambient temperatures, which allows the alloy NPs to efficiently catalyse the selective oxidation of aromatic alcohols to the corresponding aldehydes and ketones. This explains our observations that the conversion of a reaction under light irradiation is always higher than that of the corresponding reaction in the dark, and that the selective oxidation of benzyl alcohol could proceed at moderate temperatures (e.g. <45 °C) under irradiation but not in the dark.
Conclusions
In summary, it was found for the first time that visible light could significantly enhance the performance of Au–Pd alloy NPs supported on ZrO2 for the dehydrogenation of aromatic alcohols to yield the corresponding aldehydes in the absence of oxygen at ambient temperature. The rate determining step of the dehydrogenation is the abstraction of α-H atoms from the alcohol molecules. The dehydrogenation is also the rate determining step of selective oxidation of the alcohols to the corresponding aldehydes with molecular oxygen. The results of both the free electron gas model analysis and DFT simulation indicate that the alloy structure of Au and Pd increases the charge heterogeneity of the NP surface, which enhanced interaction between the alloy NPs and the alcohol molecules adsorbed on the NPs. The combination of light absorption of alloy NPs, the enhanced interaction and the intrinsic catalytic activity of the transition metal leads to a unique structure where the absorption of visible and UV radiation can yield energetic electrons available at catalytically active transition metal sites on the NP surface promoting the reaction of the molecules adsorbed on the NPs. The optimal activity for the alloy NPs was observed with an Au:Pd molar ratio of 1:1.86, being in good agreement with the simulation results. The Au–Pd@ZrO2 is an example of the alloy NPs formed by gold and a catalytically active transition metal, which can be used as a new superior catalyst for fine organic chemical synthesis under light irradiation. Since little input energy is consumed by other components of the reaction system, such as the solvent, support of the NPs, the atmosphere or container, this catalyst structure is highly efficient for driving various chemical reactions with sunlight. The knowledge acquired in this study is useful for designing suitable photocatalysts made from gold alloyed with other transition metals and may inspire further studies on new efficient photocatalysts of gold and other transition metals for a wide range of organic synthesis driven by sunlight.
Notes and references
- C. C. Cheng, W. H. Ma and J. C. Zhao, Chem. Soc. Rev., 2010, 39, 4206 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- M. Zhang, Q. Wang, C. C. Chen, L. Zang, W. H. Ma and J. C. Zhao, Angew. Chem., Int. Ed., 2009, 48, 6081 CrossRef CAS PubMed.
- X. J. Lang, H. W. Ji, C. C. Chen, W. H. Ma and J. C. Zhao, Angew. Chem., Int. Ed., 2011, 50, 3934 CrossRef CAS PubMed.
- T. Shishido, T. Miyatake, K. Teramura, Y. Hitomi, H. Yamashita and T. Tanaka, J. Phys. Chem. C, 2009, 113, 18713 CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr., Chem. Rev., 1995, 95, 735 CrossRef CAS.
- N. Serpone, J. Photochem. Photobiol., A, 1997, 104, 1 CrossRef CAS.
- K. Maeda, K. Teramura, D. L. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- X. Chen, H. Y. Zhu, J. C. Zhao, Z. F. Zheng and X. P. Gao, Angew. Chem., Int. Ed., 2008, 47, 5353 CrossRef CAS PubMed.
- H. Y. Zhu, X. B. Ke, X. Z. Yang, S. Sarina and H. W. Liu, Angew. Chem., Int. Ed., 2010, 49, 9657 CrossRef CAS PubMed.
- H. Y. Zhu, X. Chen, Z. F. Zheng, X. B. Ke, E. Jaatinen, J. C. Zhao, C. Guo, T. F. Xie and D. J. Wang, Chem. Commun., 2009, 7524 RSC.
- S. Sarina, E. R. Waclawik and H. Y. Zhu, Green Chem., 2013, 15, 1814 RSC.
- S. Sarina, H. Y. Zhu, E. Jaatinen, Q. Xiao, H. W. Liu, J. F. Jia, C. Chen and J. Zhao, J. Am. Chem. Soc., 2013, 135, 5793 CrossRef CAS PubMed.
- X. B. Ke, S. Sarina, J. Zhao, X. G. Zhang, J. Chang and H. Y. Zhu, Chem. Commun., 2012, 48, 3509 RSC.
- X. B. Ke, X. G. Zhang, J. Zhao, S. Sarina, J. Barry and H. Y. Zhu, Green Chem., 2013, 15, 236 RSC.
- P. Christopher, H. L. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044 CAS.
- A. Marimuthu, J. W. Zhang and S. Linic, Science, 2013, 339, 1590 CrossRef CAS PubMed.
- P. Mulvaney, Langmuir, 1996, 12, 788 CrossRef CAS.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209 RSC.
- L. M. Liz-Marzán, Langmuir, 2006, 22, 32 CrossRef PubMed.
- K. Yamada, K. Miyajima and F. Mafun, J. Phys. Chem. C, 2007, 111, 11246 CAS.
- S. Link, C. Burda, Z. L. Wang and M. A. El-Sayed, J. Chem. Phys., 1999, 111, 1255 CrossRef CAS.
- C. Voisin, N. Del Fatti, D. Christofilos and F. Vallee, J. Phys. Chem. B, 2001, 105, 2264 CrossRef CAS.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409 CrossRef CAS.
- L. Brus, Acc. Chem. Res., 2008, 41, 1742 CrossRef CAS PubMed.
- C. D. Lindstrom and X. Y. Zhu, Chem. Rev., 2006, 106, 4281 CrossRef CAS PubMed.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- C. L. Hill, in Advances in Oxygenated Processes, ed. A. L. Baumstark, JAI Press, London, 1988, vol. 1, p. 1 Search PubMed.
- M. Hudlucky, Oxidations in Organic Chemistry; ACS Monograph Series, American Chemical Society, Washington, DC, 1990 Search PubMed.
- Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, U.K., 1991 Search PubMed.
- G. Cainelli and G. Cardillo, Chromium Oxidants in Organic Chemistry, Springer, Berlin, 1984 Search PubMed.
- D. G. Lee and U. A. Spitzer, J. Org. Chem., 1970, 35, 3589 CrossRef CAS.
- T. F. Blackburn and J. Schwartz, J. Chem. Soc., Chem. Commun., 1977, 157 RSC.
- G. J. Brink, I. W. C. E. Arends and R. A. Sheldon, Science, 2000, 287, 1636 CrossRef PubMed.
- S. S. Stahl, J. L. Thorman, R. C. Nelson and M. A. Kozee, J. Am. Chem. Soc., 2001, 123, 7188 CrossRef CAS PubMed.
- M. A. Steinhoff, S. R. Fix and S. S. Stahl, J. Am. Chem. Soc., 2002, 124, 766 CrossRef PubMed.
- D. R. Jensen, M. J. Schultz, J. A. Mueller and M. S. Sigman, Angew. Chem., Int. Ed., 2003, 42, 3810 CrossRef CAS PubMed.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS PubMed.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657 CrossRef CAS PubMed.
- T. Nishimura, N. Kakiuchi, M. Inoue and S. Uemura, Chem. Commun., 2000, 1245 RSC.
- N. Dimitratos, A. Villa, D. Wang, F. Porta, D. Su and L. Prati, J. Catal., 2006, 244, 113 CrossRef CAS.
- L. F. Liotta, A. M. Venezia, G. Deganello, A. Longo, A. Martorana, Z. Schay and L. Guczi, Catal. Today, 2001, 66, 271 CrossRef CAS.
- J. Chen, Q. Zhang, Y. Wang and H. Wan, Adv. Synth. Catal., 2008, 350, 453 CrossRef CAS.
- K. M. Choi, T. Akita, T. Mizugaki, K. Ebitani and K. Kaneda, New J. Chem., 2003, 27, 324 RSC.
- Y. Uozumi and R. Nakao, Angew. Chem., Int. Ed., 2003, 42, 194 CrossRef CAS PubMed.
- M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, J. Am. Chem. Soc., 2009, 131, 7189 CrossRef CAS PubMed.
- A. Maldotti, A. Molinari, R. Juarez and H. Garćia, Chem. Sci., 2011, 2, 1831 RSC; K. Patel, S. Kapoor, D. P. Dave and T. Mukherjee, Res. Chem. Intermed., 2006, 32, 103 CrossRef CAS.
- Y. W. Lee, N. H. Kim, K. Y. Lee, K. Kwon, M. Kim and S. W. Han, J. Phys. Chem. C, 2008, 112, 6717 CAS.
- Y. H. Chen, Y. H. Tseng and C. S. Yeh, J. Mater. Chem., 2002, 12, 1419 RSC.
- A. Emeline, G. V. Kataeva, A. S. Rudakova, V. K. Ryabchuk and N. Serpone, Langmuir, 1998, 14, 5011 CrossRef CAS.
- K. Yamada, K. Miyajima and F. J. Mafun, Phys. Chem. C, 2007, 111, 11246 CrossRef CAS.
- C. Voisin, N. Del Fatti, D. Christofilos and F. Vallee, J. Phys. Chem. B, 2001, 105, 2264 CrossRef CAS.
- M. R. Hartings, Nat. Chem., 2012, 4, 764 CrossRef CAS PubMed.
- C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef PubMed.
- S. S. Stahl, Science, 2005, 309, 1824 CrossRef CAS PubMed.
- J. Gavnholt, T. Olsen, M. Engelund and J. Schiøtz, Phys. Rev. B: Condens. Matter, 2008, 78, 075441 CrossRef.
- V. I. Pârvulescu, V. Pârvulescu, U. Endruschat, G. Filoti, F. E. Wagner, C. Kubël and R. Richards, Chem.–Eur. J., 2006, 12, 2343 CrossRef PubMed.
- N. Toshima, M. Harada, Y. Yamazaki and K. Asakura, J. Phys. Chem., 1992, 96, 9927 CrossRef CAS.
- M. Harada, K. Asakura and N. Toshima, J. Phys. Chem., 1993, 97, 5103 CrossRef CAS.
- M. S. Chen and D. W. Goodman, Science, 2004, 306, 252 CrossRef CAS PubMed.
- D. Yuan, X. Gong and R. Wu, Phys. Rev. B: Condens. Matter, 2008, 78, 035441 CrossRef.
- A. Kotsifa, T. I. Halkides, D. I. Kondarides and X. E. Verykios, Catal. Lett., 2002, 79, 113 CrossRef CAS.
- P. P. Fang, J. F. Li, X. D. Lin, J. R. Anema, D. Y. Wu, B. Ren and Z. Q. Tian, J. Electroanal. Chem., 2012, 665, 70 CrossRef CAS.
- D. V. Bavykin, A. A. Lapkin, S. T. Kolaczkowski and P. K. Plucinski, Appl. Catal., A, 2005, 288, 175 CrossRef CAS.
- M. Ilyas and M. Sadiq, Chem. Eng. Technol., 2007, 30, 1391 CrossRef CAS.
- C. Keresszegi, D. Ferri, T. Mallat and A. Baiker, J. Phys. Chem. B, 2005, 109, 958 CrossRef CAS PubMed.
- M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, J. Am. Chem. Soc., 2009, 131, 7189 CrossRef CAS PubMed.
- A. Maldotti, A. Molinari, R. Juarez and H. Garcia, Chem. Sci., 2011, 2, 1831 RSC.
- J. P. Roth, J. C. Yoder, T. J. Won and J. M. Mayer, Science, 2001, 294, 2524 CrossRef CAS PubMed.
- M. Turner, V. B. Golovko, O. P. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. Johnson and R. M. Lambert, Nature, 2008, 454, 981 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, estimation of Au–Pd alloy NPs' ionic property by free gas model, proposed mechanism of oxidative coupling of benzylamine (A) and aromatic alcohol oxidation (B) over Au–Pd alloy NPs under visible light irradiation, calculation of apparent activation energy for the reactions. See DOI: 10.1039/c3gc41866a |
| This journal is © The Royal Society of Chemistry 2014 |