 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-responsive control of bacterial gene expression: precise triggering of the lac promoter activity using photocaged IPTG†
Dennis
Binder‡
a,
Alexander
Grünberger‡
b,
Anita
Loeschcke
a,
Christopher
Probst
b,
Claus
Bier
c,
Jörg
Pietruszka
bc,
Wolfgang
Wiechert
b,
Dietrich
Kohlheyer
b,
Karl-Erich
Jaeger
*ab and
Thomas
Drepper
*a
aInstitute of Molecular Enzyme Technology, Heinrich-Heine-University Düsseldorf, Research Center Jülich, Germany. E-mail: k.-e.jaeger@fz-juelich.de; t.drepper@fz-juelich.de
bInstitute of Bio- and Geosciences, IBG-1: Biotechnology, Systems Biotechnology, Research Center Jülich, Germany
cInstitute for Bioorganic Chemistry, Heinrich-Heine-University Düsseldorf, Research Center Jülich, Germany
First published on 16th May 2014
Abstract
Light can be used to control numerous cellular processes including protein function and interaction as well as gene expression in a non-invasive fashion and with unprecedented spatiotemporal resolution. However, for chemical phototriggers tight, gradual, and homogeneous light response has never been attained in living cells. Here, we report on a light-responsive bacterial T7 RNA polymerase expression system based on a photocaged derivative of the inducer molecule isopropyl-β-D-thiogalactopyranoside (IPTG). We have comparatively analyzed different Escherichia coli lac promoter-regulated expression systems in batch and microfluidic single-cell cultivation. The lacY-deficient E. coli strain Tuner(DE3) harboring additional plasmid-born copies of the lacI gene exhibited a sensitive and defined response to increasing IPTG concentrations. Photocaged IPTG served as a synthetic photo-switch to convert the E. coli system into an optogenetic expression module allowing for precise and gradual light-triggering of gene expression as demonstrated at the single cell level.
Insight, innovation, integrationOptogenetic approaches aim to trigger biological processes by light. For the establishment of a light-responsive E. coli expression system, we validate different lac promoter-controlled, T7 RNA polymerase-dependent expression modules. Using microfluidic techniques we were able to pin down and abolish bottlenecks of inducer-dependent regulatory response. By implementing a derivative of the synthetic inducer IPTG, which is coupled to the light-sensitive photocaging group 6-nitropiperonal, we assembled a precise photoswitch that can be controlled by UV-A light. |
Introduction
Synthetic biology requires the development of regulatory switches that facilitate dynamic regulation of target gene expression.1–4 In this context, optogenetic approaches demonstrated precise control over cellular functions by light.5–7 The unique variability of the stimulus light, including its color and intensity, allows for a specific triggering of cellular events in a non-invasive and highly resolving spatiotemporal fashion.5 Light-mediated control over gene expression basically relies on two principles which use either genetically encoded biological photoreceptors or chemically photocaged biomolecules.6–9 Recombinant photoreceptors, for example, have been successfully employed for light-mediated in vivo signal transduction in synthetic biological applications.10–12 The principle of photocaging poses an alternative approach to achieve light-mediated control over gene expression. Photocaged molecules are rendered biologically inactive through the addition of a photo-removable protection group, the so-called photocaging group or photocage. Functionality can be restored, both in vitro and in vivo, by the light-mediated release (uncaging) of the bioactive molecule.13 Plenty of biomolecules were subjected to photocaging, including proteins or small inducer molecules,7,14e.g. isopropyl β-D-thiogalactopyranoside (IPTG)15 and a doxycycline analog,16 which were able to activate lac and tet promoter-controlled microbial expression systems upon UV-A light exposure.Induction of lac promoter-dependent gene expression by sugar analogs with light-responsive photocaging was first described using 6-nitropiperonal (NP) photocaged IPTG.15 Here, the NP-photocaged IPTG was unable to bind the repressor LacI, while its biological activity was restored upon UV-A light exposure, leading to LacI binding and therefore to derepression of gene expression.15 However, currently available light-controlled systems, which operate with photocaged molecules (caged T7RP; caged IPTG; caged doxycycline),15–17 have not yet been employed for precise and homogeneous in vivo regulation of microbial gene expression.
The Escherichia coli T7-RNA polymerase (T7RP)-dependent expression system is regarded as the most widely used system for high-level gene expression.18–20 It consists of a lambda DE3 lysogenic E. coli strain carrying a chromosomally integrated copy of the T7RP gene whose expression is tightly controlled by the lac promoter20 and an appropriate expression plasmid allowing target gene expression from a T7 promoter. The highly processive phage polymerase exclusively targets its own promoter and therefore operates decoupled from other cellular processes.21 For this reason, the E. coli T7 system is recommended as a ‘what to try first’ system for the expression of pro- and eukaryotic proteins.20,22
One of the most prominent T7RP expression strains is E. coli BL21(DE3).19 However, this common system harbors the wild-type E. coli lac operon including the lactose permease-encoding lacY gene, whose expression is also lactose-dependent and causes a positive feedback loop by actively translocating inducer molecules into the cell.23 Thereby, it generates a non-gradual and also inhomogeneous induction behavior over a bacterial population, especially for low amounts of inducer molecules.24 Therefore, the precise fine regulation of gene expression using common T7RP- and lac-based expression modules appears to be difficult.
The expression of a target gene is usually analyzed within an entire bacterial population; however, to gain insights into exact regulation processes, the expression needs to be studied at the single cell level. Currently, batch cultivation is combined with reporter-imaging technologies such as single cell photography25 or flow cytometry analysis.26 A profound drawback of these methods is the system-inherent discontinuous environment.27 For instance, nutrient depletion and accumulation of metabolic products result in discontinuity over time within the cultivation vessel.28 Therefore, those methods are unable to distinguish between environmental or biological heterogeneity or even determine the origin of depicted heterogeneities.27,29 Furthermore, the above mentioned single cell analyses such as fluorescence-activated cell sorting (FACS) only provide a snapshot of cellular states rather than full information about ongoing behavior and could additionally produce artificial results as cells are analyzed “off-line” outside the cultivation device.
The challenge of understanding cellular heterogeneity resulted in an increasing amount of different devices and protocols for investigating single cells “on-line”. Systems range from basic agar-pads30 to advanced microfluidic single cell set-ups.31–33 The latter include single cell traps,34 single cell channels35,36 and monolayer growth chambers.37,38 Recently, a picoliter bioreactor for the investigation of single cell processes over many generations under constant conditions was developed.39 Since cells are cultured in a monolayer, the genealogical analysis of clonal colonies can be performed,40,41 allowing for the reconstruction of lineage trees and thus for accurately assessing population heterogeneity under constant environmental conditions in contrast to common agar-pad-based technologies.42
With this advanced microfluidic technology at hand, we characterized different E. coli T7RP expression systems in order to establish an efficient and light-responsive expression system in E. coli.
Materials and methods
Bacterial strains and plasmids
Escherichia coli strains DH5α,43 BL21(DE3)18 and Tuner(DE3) (Novagen) were grown in Luria–Bertani (LB) medium44 supplemented with kanamycin (50 μg ml−1) or chloramphenicol (50 μg ml−1) at 37 °C under constant agitation.The construction of expression vectors and recombinant DNA techniques were carried out in E. coli DH5α as described by Sambrook et al.44
The derivative of the pRhotHi-245 expression vector pRhotHi-2-LacI was constructed by excising the aphII gene from pBSL1546 with restriction enzyme BamHI. The resulting fragment was subsequently cloned into the BglII-site of pBBR22b,47 harboring a copy of the lacI gene. As the final expression vector, the variant was chosen, where aphII and T7 promoters were oriented in opposite directions. The EYFP-encoding reporter gene,48 which was isolated by hydrolyzing pRhotHi-2-EYFP with NdeI and XhoI, was cloned into pRhotHi-2-LacI resulting in pRhotHi-2-LacI-EYFP.
All bacterial strains and plasmids used in this study are listed in Table S1 (ESI†).
NP-photocaged IPTG synthesis modified according to Young & Deiters 200715
IPTG (100 mg, 0.42 mmol) and 6-nitropiperonal (245 mg, 1.26 mmol, for synthesis see ESI,† Methods) were dissolved in 1 ml of dimethylsulfoxide (DMSO). At 0 °C concentrated sulfuric acid (0.15 ml) was carefully added and the reaction was allowed to warm up to room temperature. After 24 h the reaction mixture was quenched with water and extracted with ethyl acetate. The combined organic layer was dried over magnesium sulfate and concentrated under reduced pressure. The residue was purified by flash-column chromatography on SiO2 (EtOAc/pentane 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) to receive (122.5 mg, 0.29 mmol, 72%) of a light yellow solid. After an additional cleaning step via MPLC we had (31.4 mg, 0.08 mmol) of a colorless pure product, with a yield of 18% in our hands. Analytical data are shown in the ESI,† Methods.
:3) to receive (122.5 mg, 0.29 mmol, 72%) of a light yellow solid. After an additional cleaning step via MPLC we had (31.4 mg, 0.08 mmol) of a colorless pure product, with a yield of 18% in our hands. Analytical data are shown in the ESI,† Methods.
Deep well plate cultivation and off-line measurement of in vivo fluorescence
Expression cultures were grown in 96 deep well plates (Master Block, Greiner Bio One) by shaking at 600 rpm. After inoculation with a cell density corresponding to an OD580 of 0.1 in a volume of 950 μl, expression cultures were incubated for 2 h until cells reached an OD580 of 0.4–0.6. The target gene expression was induced with 50 μl of inducer solution leading to final inducer concentrations in a range from 0 to 100 μM IPTG in a final volume of 1 ml.Cultures (1 ml) pre-supplemented with 40 μM NP-photocaged IPTG were grown in the dark for 1, 1.5 or 2 h and subsequently exposed to UV-A light for 0.25 to 10 minutes (hand lamp VL-315.BL, Vilber Lourmat, France; placed at a distance of 2.5 cm over the deep well plate). After 6 and 20 h of cultivation in the dark, respectively, in vivo fluorescence and cell densities (OD580) were measured in 96 flat bottom transparent polystyrene microplates (Thermo scientific-Nunclon) using a fluorescence microplate-reader (Tecan Infinite M1000 Pro). Prior to measurements samples were diluted 5-fold in 0.1 M Tris-HCl buffer (pH 8.0) resulting in a final volume of 100 μl. Emission of YFP was determined at 527 nm after excitation with blue light (λmax = 488 nm). Fluorescence units of diluted samples were normalized to a cell density of OD580 = 1.0.
Microfluidic cultivation
A single-use polydimethylsiloxane (PDMS) microfluidic chip fabricated as previously described39,49 was utilized to cultivate single cells and isogenic microcolonies (Fig. 1). A single chip used in this study (Fig. 1A) contained several hundred monolayer growth chambers (Fig. 1B and C) (dimensions: 1 μm × 40 μm × 40 μm) facilitating high-throughput single cell analysis. Each growth chamber was interconnecting two parallel 10-fold deeper supply channels, as illustrated in Fig. 1D. Throughout the operation both supply channels were infused with identical volume flow rates. This resulted in solely diffusion-based mass transport across the shallow cultivation chambers, permitting reliable single cell tracking for genealogical studies inside growing microcolonies.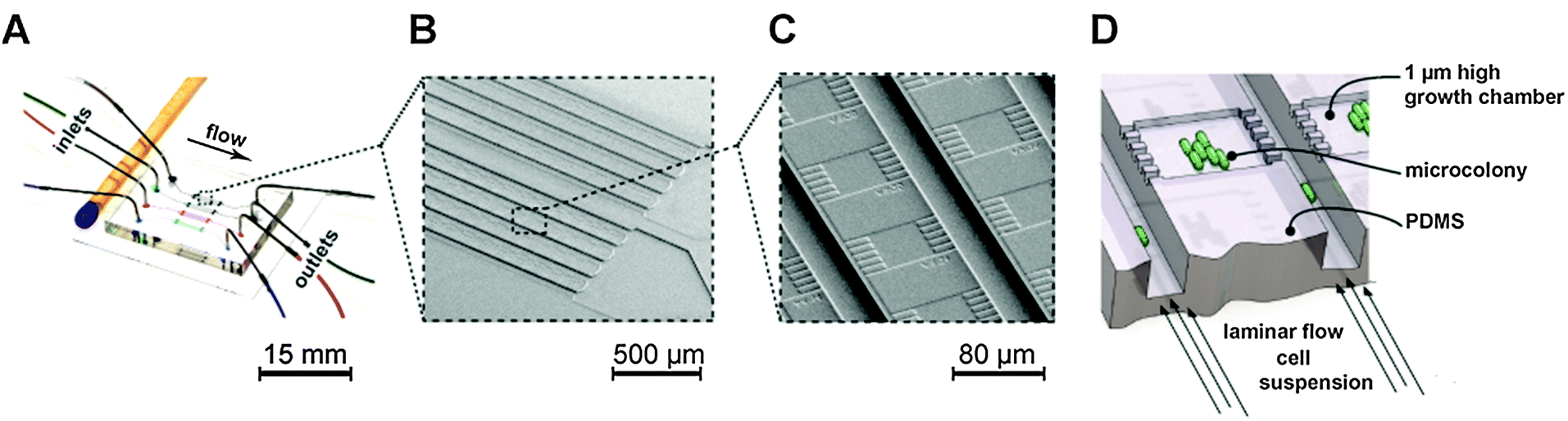 | ||
| Fig. 1 Microfluidic PDMS single cell cultivation devices. (A) Photograph of a PDMS cultivation chip next to a match. (B) SEM of monolayer cultivation sections containing several hundreds of single cultivation chambers (C). (D) Schematic illustration of microscale growth chamber that is perfused with cell suspensions and media for cultivation of trapped cells. | ||
Cell suspensions for chip perfusion were prepared by inoculating fresh cultures from a preculture to an OD580 of 0.05 and cultivated until the mid-logarithmic growth phase was reached.
This cell suspension was infused at 200 nl min−1 using 1 ml disposable syringes and high precision syringe pumps (neMESYS, Cetoni, Germany) to randomly inoculate single mother cells into the growth chambers. After sufficient single cells were trapped, the cell suspension was replaced with fresh LB medium infused at 100 nl min−1. After 1 h cultivation, cells were induced by IPTG supplemented LB cultivation media.
During cultivation at 37 °C the chip was continuously perfused with fresh medium to maintain constant environmental culture conditions. If desired, the perfusion of fresh medium was manually stopped to induce batch equivalent conditions inside the chambers (with nutrition depletion and byproduct accumulation). Media supplemented with NP-photocaged IPTG were exposed to UV-A light prior to use.
Time-lapse microscopy and image analysis
The microfluidic chip was mounted onto a motorized microscope (Nikon Eclipse Ti) equipped with an in-house developed incubator and a heated Nikon Apo TIRF 100× Oil DIC N objective (ALA OBJ-Heater, Ala Scientific Instruments, USA) for temperature control. Furthermore, the microscope was equipped with a Nikon perfect focus system compensating for thermal drift, an ANDOR LUCA R DL604 EMCCD camera (Andor Technology plc., Belfast, UK), a 300 W Xenon light source for fluorescence excitation (Lambda DG4, Sutter Instruments, USA), and YFP fluorescence filters (AHF Analysentechnik, Germany) (excitation: 500 nm/20, dichroic: 500 nm and emission: 535 nm/30). If not stated different, the fluorescence and camera exposure was 200 ms for EYFP, at zero camera gain and 100% lamp intensity. Fluorescence exposure times were minimized to avoid the impact on cellular growth or viability.Phase contrast and fluorescence microscopy images of multiple colonies were captured in a sequence every 10 min by automated time-lapse microscopy thereby facilitating image-based single cell analysis with spatiotemporal resolution. Final image sequences were analyzed using the Nikon NIS Elements AR software package to determine cell length and fluorescence intensity. The mean fluorescence intensity of each cell was determined by measuring the fluorescence values of each cell and subtracting the background fluorescence value obtained from an empty position of the cultivation chamber. The visualization of the lineage tree was realized using in-house developed Python-based software.
Results and discussion
We aimed to establish an optogenetic expression system in bacteria that provides minimal background activity as well as a gradual and homogenous light response within the entire cell population (Fig. 2A). Hence, we constructed a lac promoter-based E. coli T7RP expression system that provides exact controllability via photocaged inducer molecules (Fig. 2B). The characteristics and functions of all regulatory and metabolic elements involved in the control of lac promoter/operator activity in E. coli are well described.19,50,51 In this context, the repressor LacI, the lactose permease LacY and the diffusible artificial inducer IPTG play key roles in triggering lac gene expression. Firstly, the impact of LacY and LacI on the stringency and regulatory dynamics of lac promoter/operator-based gene expression was analyzed by characterization of different E. coli T7RP-based expression systems (Table 1) at both population and single cell level. The applied expression systems constitute combinations of E. coli strains and plasmids differing in their lacY and lacI configurations, whereas the expression of the genes encoding the T7 polymerase and the in vivo fluorescence reporter YFP is always controlled by the same lac promoter and operator, respectively.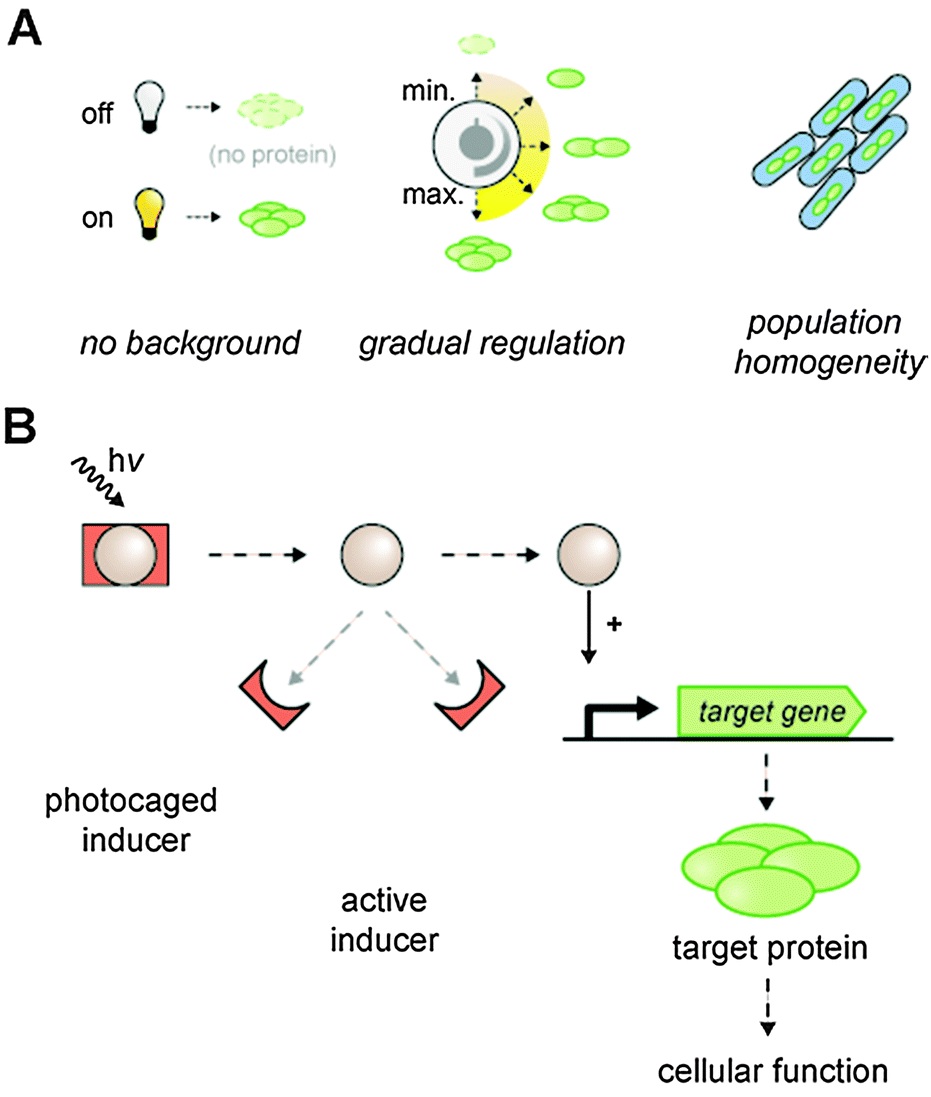 | ||
| Fig. 2 Principal characteristics of the aimed lac promoter-based optogenetic expression system. (A) Low background activity will ensure a defined switch from a clear OFF state to an ON state with high gene expression levels. A gradual induction response will allow a direct correlation between defined irradiation times and protein accumulation levels. Simultaneous and identical induction behavior of all cells will produce a homogeneous population. (B) Concept of exerting light-dependent control over gene expression using the photocaged inducer IPTG. NP-photocaged IPTG is released upon UV-A light irradiation. The decaged inducer activates the lac promoter and induces gene expression allowing for a light-responsive control of cellular behavior. | ||
| E. coli strain/plasmid | lacY | lacI |
|---|---|---|
| a chr: chromosome, pl: plasmid. | ||
| BL21(DE3)/pRhotHi-2-EYFP | chr | chr |
| Tuner(DE3)/pRhotHi-2-EYFP | — | chr |
| Tuner(DE3)/pRhotHi2-LacI-EYFP | — | chr/pl |
The role of lactose permease was studied by comparing IPTG-dependent responsiveness of YFP expression in the commonly used E. coli T7RP expression strain BL21(DE3)19,20,22 (lacY+, lacI+) to the so far rarely used permease-deficient E. coli T7RP expression strain Tuner(DE3) (lacY−, lacI+). E. coli Tuner(DE3) is suitable for inducer-dependent adjustment of gene expression levels52 and assumed to show homogeneous induction behavior that, however, has not been verified in a scientific study so far. The mid-copy T7RP expression plasmids pRhotHi-2 (lacI−)45 and pRhotHi-2-LacI (lacI+) additionally allowed us to adjust the intracellular levels of the repressor LacI.
Strict regulation of lac operator-controlled gene expression is impeded in E. coli standard expression host BL21(DE3)
Properties of lac regulation were first investigated in E. coli BL21(DE3) carrying the expression vector pRhotHi-2-EYFP. Cells were initially grown in a common batch cultivation set-up (Fig. 3A) using IPTG concentrations ranging from 0 to 100 μM. YFP in vivo fluorescence was quantified 6 and 20 h (representing the late logarithmic and stationary phase, respectively) after induction of gene expression (Fig. 3B). The results clearly demonstrated a high background expression level in non-induced cultures (0 μM IPTG). In all cases, the addition of IPTG led to a moderate increase of YFP-mediated in vivo fluorescence (a two-fold increase after 6 h and a three-fold increase after 20 h), irrespective of the applied inducer concentration. The results thus show that the first strain/vector system, where LacY is present and the amount of LacI is low, neither exhibits low background activity nor allows gradual induction of gene expression. Subsequently, homogeneity of the induction behavior was tested for the chosen expression system in a microfluidic perfusion set-up (Fig. 3C), which allows us to keep E. coli in the logarithmic growth phase under persistent cultivation conditions until the growth chamber is completely filled with cells. To this end, cells were trapped in microscale growth chambers and incubated for 1 h before YFP expression was induced applying media supplemented with different IPTG concentrations. To analyze the mean fluorescence during the development of microcolonies, single cell fluorescence values were monitored for a cultivation time of up to 500 min (Fig. 3D).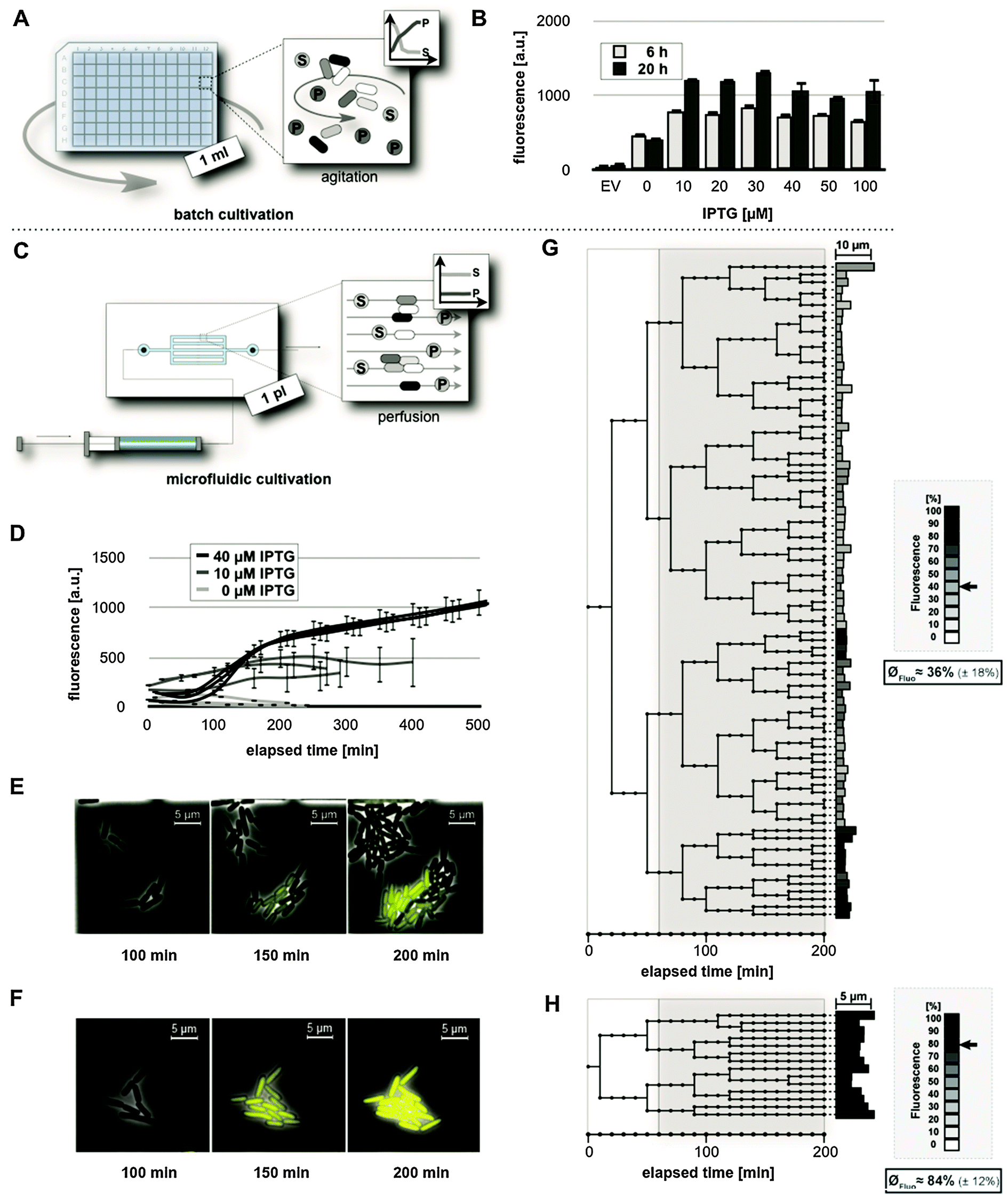 | ||
Fig. 3 Analysis of IPTG-induced expression in E. coli BL21(DE3)/pRhotHi-2-EYFP. (A) Schematic diagram of E. coli batch cultivation in deep well plates, where cells were agitated in a defined volume of cultivation medium. Metabolizable media components ( ) are consumed while metabolic products ( ) are consumed while metabolic products ( ) accumulate over time. (B) In vivo fluorescence of batch cultures at different IPTG concentrations. EV: empty vector control. Values are means of triplicate measurements. Error bars indicate the respective standard deviations. a.u.: arbitrary units. (C) Schematic diagram of microfluidic perfusion cultivation, where medium is constantly flushed through growth chambers. Here, both media components ( ) accumulate over time. (B) In vivo fluorescence of batch cultures at different IPTG concentrations. EV: empty vector control. Values are means of triplicate measurements. Error bars indicate the respective standard deviations. a.u.: arbitrary units. (C) Schematic diagram of microfluidic perfusion cultivation, where medium is constantly flushed through growth chambers. Here, both media components ( ) and metabolic products ( ) and metabolic products ( ) are maintained at constant levels. (D) Fluorescence of developing microcolonies during differently supplemented microfluidic cultivation. For each IPTG concentration, single cell fluorescence values were monitored for three independent microcolonies until the bacteria fully colonized the growth chambers. (E, F) Selected photographs from time lapse microscopy during microfluidic cultivation with 10 μM (E) and 40 μM IPTG (F). (G, H) Lineage trees of single cells, where YFP expression (grey highlighted) was induced with 10 μM (G) and 40 μM IPTG (H) after 1 h of precultivation. Lineage trees were generated from data of representative microcolonies over a period of 200 minutes. End point fluorescence (grayscale) and individual cell size (bar length) are plotted. In the box, the fluorescence mean value and standard deviation normalized to the highest value achieved in all microfluidic experiments is depicted. This fluorescence value is also marked by an arrow. ) are maintained at constant levels. (D) Fluorescence of developing microcolonies during differently supplemented microfluidic cultivation. For each IPTG concentration, single cell fluorescence values were monitored for three independent microcolonies until the bacteria fully colonized the growth chambers. (E, F) Selected photographs from time lapse microscopy during microfluidic cultivation with 10 μM (E) and 40 μM IPTG (F). (G, H) Lineage trees of single cells, where YFP expression (grey highlighted) was induced with 10 μM (G) and 40 μM IPTG (H) after 1 h of precultivation. Lineage trees were generated from data of representative microcolonies over a period of 200 minutes. End point fluorescence (grayscale) and individual cell size (bar length) are plotted. In the box, the fluorescence mean value and standard deviation normalized to the highest value achieved in all microfluidic experiments is depicted. This fluorescence value is also marked by an arrow. | ||
Low inducer concentrations (10 μM IPTG) resulted in a highly heterogeneous expression response of individual cells, as reflected by large error bars. With 40 μM IPTG, cells exhibited a homogeneously strong fluorescence. Apparently, the expression response was saturated at this concentration, as supplementation with 100 μM IPTG produced the same results (see Fig. S1, ESI†). Interestingly, evaluable time periods (i.e. the cultivation time needed by a microcolony to fully occupy the micro-incubation chamber) indicated an inverse correlation between lac induction and cellular growth (Fig. 3D). This was likewise observed for single cell traces of differently induced E. coli BL21(DE3)/pRhotHi-2-EYFP cells (see Fig. S3, ESI†).
Remarkably, in contrast to the observations made in batch cultures (Fig. 3B), cells in the microfluidic set-up (Fig. 3D) showed that very low YFP fluorescence intensities could be observed where YFP expression was induced with 40 instead of 10 μM IPTG. These observations might result from elementary differences in applied cultivation technologies, as in batch cultivation, media components such as glucose that are involved in carbon catabolite repression of the lac promoter are consumed over time, whereas in the microfluidic perfusion system cells are continuously supplied with fresh media. Therefore, media components relevant for catabolite repression may be maintained at ‘repressing’ concentrations without additional IPTG and also might impair full induction of YFP expression at intermediate inducer concentrations (i.e. 10 μM) during microfluidic but not batch cultivation.
To further analyze the fluorescence development at the single cell level during microfluidic cultivation, lineage trees were generated from data of representative microcolonies, each of which developed from a single cell, supplemented with 10 or 40 μM IPTG over a time-period of 200 minutes (Fig. 3G and H).
In a microcolony supplemented with 10 μM IPTG (Fig. 3G) variably fluorescing (the mean value of normalized YFP fluorescence: 36% ± 18%) and differentially growing sub-populations developed from the initial cell (see also Video S1, ESI†). At the time of induction, four cells gave rise to explicitly different branches. In the three upper branches where cells showed only low fluorescence, the bacteria divided 23 times on average. In contrast, in the lower branch, where cells showed relatively high fluorescence, bacteria divided only 11 times. As expected from the results shown in Fig. 3D and F, the tree ended with uniformly strong fluorescing cells (84% in average with a standard deviation of ±12%) when the medium was supplemented with 40 μM IPTG (Fig. 3H). Here, the correlation of lac induction with cellular growth becomes evident in an altogether drastically smaller tree. On average, cells divided only 2.5 times after induction. The observed inconsistency of growth rates at lower inducer concentrations may promote overgrowth of cells with lower expression levels and displacement of cells with higher expression levels during cultivation, yielding a rather unfavorable overall expression and unpredictable regulatory response.
In summary, applying microfluidic cultivation with time lapse microscopy allowed for the first time to demonstrate differences in the induction response in cells of the standard expression host E. coli BL21(DE3). In combination with pRhotHi-2-EYFP this strain does not allow precise control of gene expression as required for systems biology and optogenetic approaches. It exhibits a high expression background in the absence of IPTG, a non-gradual expression response to increasing inducer concentrations as well as an inhomogeneous and unpredictable behavior of individual cells at intermediate inducer concentrations. In another study, these characteristics have also been observed for expression of a reporter gene which was under direct control of the lac promoter.53 We show here that these same observations also hold for the T7RP expression system.
LacY-deficiency and elevated amounts of the LacI repressor enable precise control of T7RP-dependent gene expression
Deletion of lacY eliminates permease-mediated IPTG import and thus prevents the positive feedback loop.24 As a consequence, IPTG can only enter the cells via diffusion processes which thereby enables strict dependency of lac gene expression on supplemented inducer concentrations.23Hence, we next examined IPTG-responsiveness of the lactose permease-deficient strain E. coli Tuner(DE3) (lacY−, lacI+) in the absence or presence of an additional copy of the plasmid-born lacI gene. First, expression of the YFP reporter gene was analyzed in E. coli Tuner(DE3) carrying pRhotHi-2-EYFP (lacI−) at increasing inducer concentrations (0–100 μM) in a batch cultivation set-up (Fig. 4A). As expected, this strain showed a gradual expression response for low amounts of inducer up to 20 μM. Moreover, a lower background expression of YFP was observed compared to the expression in E. coli BL21(DE3), leading to a 5-fold (6 h) to 8-fold (20 h) increase of in vivo fluorescence intensities. These results confirm the characteristics of the T7RP expression strain E. coli Tuner(DE3) described before52 and corroborate that lacY-deficiency allows a gradual induction of lac promoter-dependent gene expression in response to increasing inducer concentrations.24 Notably, in our system the lac promoter-controlled expression is conveyed via T7RP to the fluorescence output. However, basal expression of this system was still too high for aspired optogenetic applications. In order to overcome this leaky basal expression observed in E. coli Tuner(DE3) with pRhotHi-2-EYFP, an additional copy of the lac repressor gene lacI was introduced. To this end, the new expression vector pRhotHi-2-LacI was constructed, harboring a copy of the lacI gene under the control of its natural constitutive promoter. Subsequently, E. coli Tuner(DE3)/pRhotHi-2-LacI-EYFP was subjected to expression studies applying inducer concentrations from 0 to 100 μM (Fig. 4B). Compared to both afore conducted expression studies (Fig. 3B and 4A), a clearly reduced background expression under non-induced conditions was observed. Furthermore, expression response strictly depended on inducer concentrations enabling a gradual response for IPTG concentrations up to 30 μM and 40 μM after 6 and 20 hours, respectively. Maximal induction of reporter gene expression finally resulted in a 15-fold (6 h) and 23-fold (20 h) increase of YFP-mediated fluorescence (Fig. 4B). Moreover, with an exception for induction with 100 μM IPTG, the ratio of the fluorescence signal detected after 6 and 20 hours of cell cultivation remained remarkably constant and is thus largely independent of the growth phase. The homogeneity of expression response within a cell population of E. coli Tuner(DE3) harboring expression plasmid pRhotHi-2-LacI-EYFP was tested by monitoring the fluorescence development of single cells using microfluidic techniques (Fig. 4C–E). Traces of representative microcolonies displayed a gradual (Fig. 4C) and homogeneous (Fig. 4D) fluorescence increase. However, final fluorescence values were much weaker than those previously observed in E. coli BL21(DE3) with pRhotHi-2-EYFP (Fig. 3D and 4C). Therefore, fluorescence development of cultures that were supplemented with 10 μM IPTG showed no significant increase in YFP in vivo fluorescence as also observed for uninduced cells. The generally lower fluorescence values of Tuner(DE3) might be explained by the faster growth that restricted the evaluable time period to 225 minutes. This assumption was corroborated by long-term microfluidic cultivation that revealed comparable final in vivo fluorescence values for both investigated strains (Fig. S4, ESI†). To analyze the fluorescence development of an initial single cell during repeated cell division, a lineage tree was generated over a time period of 200 minutes from a representative microcolony where target gene expression was induced by adding 40 μM IPTG (Fig. 4E). In contrast to E. coli BL21(DE3)/pRhotHi-2-EYFP, this tree branches to cells with equal end point fluorescence values of 7% ± 2% (see also additional histograms in Fig. S2B, ESI†). Moreover, no distinctive growth impairment occurred (also elucidated by additional single cell traces shown in Fig. S5, ESI†), as indicated by a high cell division rate after induction of gene expression (26 times on average). The detailed characterization of the E. coli T7RP expression strain Tuner(DE3) demonstrates that lac permease deficiency and elevated lacI copy numbers enable the precise control of gene expression levels. The respective strain showed low background expression and a gradual induction response to different inducer concentrations. Furthermore, this expression system exhibits a superior homogeneity in both expression behavior and cellular growth, which is independent of the applied cultivation conditions.
 | ||
| Fig. 4 Analysis of IPTG-induced expression in E. coli Tuner (DE3)/pRhotHi-2-EYFP (A) and Tuner(DE3)/pRhotHi-2-LacI-EYFP (B–E). (A, B) Development of in vivo fluorescence during batch cultivations of E. coli Tuner(DE3) with pRhotHi-2-eYFP (A) and pRhotHi-2-LacI-EYFP (B) after addition of increasing concentrations of IPTG. EV: empty vector control. Values are means of triplicate measurements. Error bars indicate the respective standard deviations. a.u.: arbitrary units. (C) Fluorescence development of microcolonies during microfluidic cultivation. Single cell fluorescence values were monitored for three independent microcolonies. For each IPTG concentration, fluorescence development of three microcolonies is plotted. Data points represent mean fluorescence values of all cells with the standard deviation as error bars. (D) Selected photographs from time lapse microscopy during microfluidic cultivation with 40 μM IPTG. (E) Lineage tree of a single cell, where YFP expression (grey highlighted) was induced with 40 μM IPTG after 1 h of precultivation. Lineage trees were generated from data of representative microcolonies over a period of 200 minutes. End point fluorescence (grayscale) and individual cell size (bar length) are plotted. Furthermore, the fluorescence mean value and standard deviation normalized to the highest value obtained in all microfluidic experiments are depicted. This fluorescence value is also marked by an arrow. | ||
Precise triggering of T7RP-dependent gene expression by light
The expression system composed of E. coli Tuner(DE3) and pRhotHi-2-LacI-EYFP was used to implement light-responsive gene expression. To this end, we synthesized an NP-photocaging group which was subsequently coupled to IPTG as described before15 (see Methods, ESI†), yielding NP-photocaged IPTG (Fig. 5A). After UV-A exposure, the resulting regioisomeric NP-nitrosocarbonyl esters are hydrolyzed in E. coli releasing IPTG (Fig. 5A).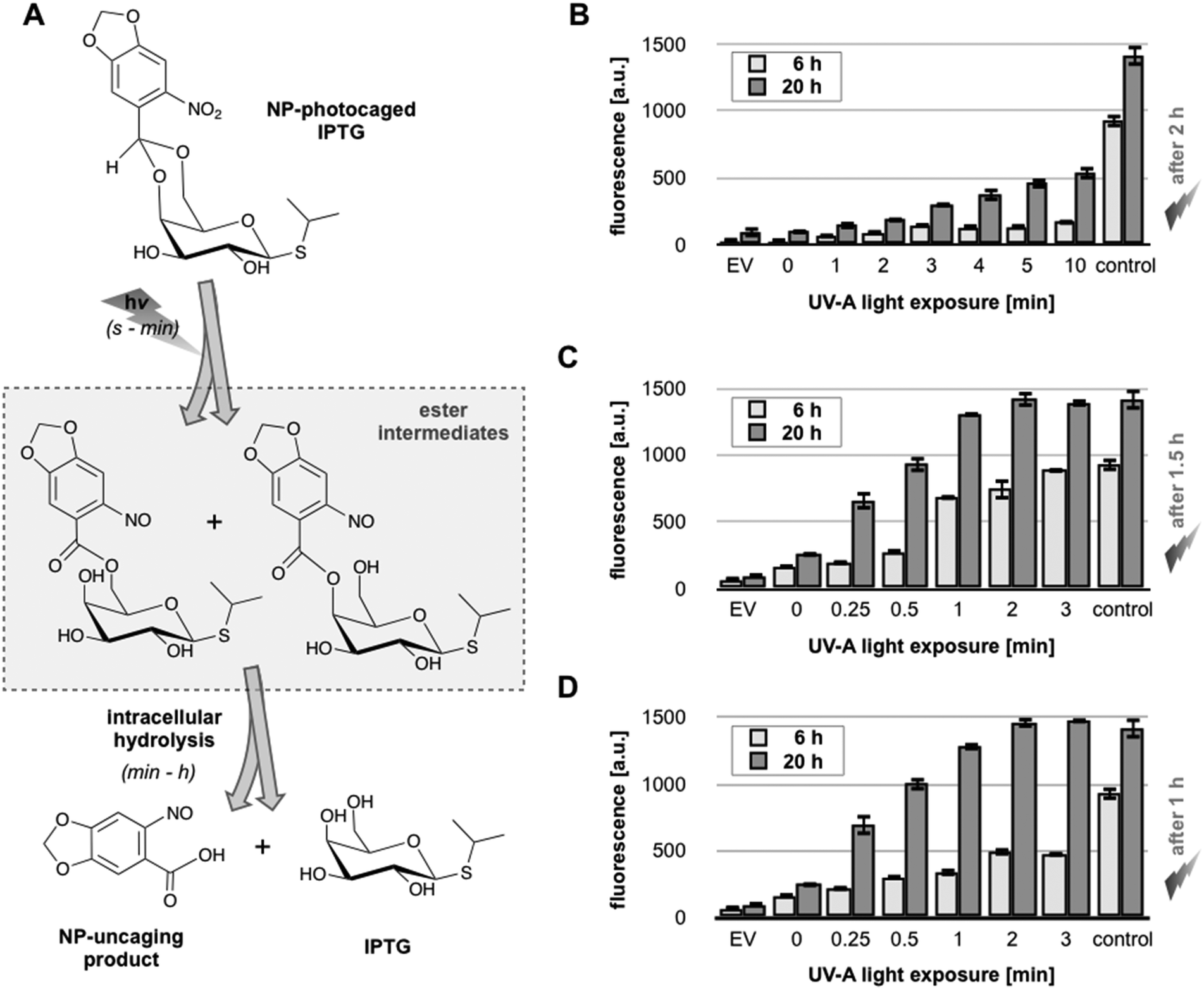 | ||
| Fig. 5 UV-A light-controlled regulation of gene expression in E. coli Tuner(DE3)/pRhotHi-2-LacI-EYFP using NP-photocaged IPTG. (A) Two-step release of NP-photocaged IPTG by UV-A light exposure and intracellular hydrolysis as described by Young & Deiters (2007).15 The reaction times ranging from seconds (s) over minutes (min) to hours (h) are given in brackets. (B–D) In vivo fluorescence of E. coli cultures supplemented with 40 μM NP-photocaged IPTG. Gene expression was specifically induced by increasing periods of UV-A light exposure. Cultures were induced after 2 h (B), 1.5 h (C) or 1 h (D) of pre-cultivation where cells were kept in darkness. Corresponding control cultures were supplemented with 40 μM uncaged IPTG. EV: empty vector control. Values are means of triplicate measurements. Error bars indicate the respective standard deviations. a.u.: arbitrary units. | ||
The light-responsiveness of this expression system was tested with E. coli Tuner(DE3) cells carrying plasmid pRhotHi-2-LacI-EYFP that were batch cultivated in LB medium supplemented with 40 μM NP-photocaged IPTG for two hours in the dark. T7RP-dependent YFP expression was induced by exposure to UV-A light (λmax = 365 nm) with increasing times ranging from 0 to 10 minutes (Fig. 5B). As shown in Fig. S6 (ESI†), UV-A illumination did not lead to phototoxic effects since exposure times of up to 30 minutes are not affecting cellular fitness. Subsequently, YFP in vivo fluorescence was recorded 6 and 20 hours after UV-A illumination and compared to results obtained with conventionally induced cultures (Fig. 5B). These first results clearly demonstrated that the increase of light exposure time provoked a gradual expression response with NP-photocaged IPTG. However, neither 6 nor 20 hours of YFP expression after light induction were sufficient to achieve in vivo fluorescence values comparable to IPTG-induced cultures (Fig. 5B, control). This observation can either be explained by a decreased stability of NP-photocaged IPTG molecules in comparison to IPTG or it could be speculated that a deferred intracellular hydrolysis of photo-cleaved ester intermediates (Fig. 5A) might result in a delayed release of IPTG and thus prevented fully efficient induction of gene expression. However, since the comparison of different time-points of NP-IPTG supplementation did not lead to enhanced YFP expression levels (Fig. S7, ESI†), in vivo instability of NP-photocaged IPTG can be neglected. Next, the time of E. coli precultivation was shortened in order to increase the efficiency of E. coli-mediated hydrolysis of its ester intermediate. As shown in Fig. 5C and D, earlier UV-A light exposure indeed yielded higher YFP expression levels: under these conditions, the in vivo fluorescence gradually increased in response to prolonged duration of light exposure and, finally, levels of conventionally induced cells were reached after 2 minutes of UV-A light excitation. The light-response of the novel expression system was also analyzed at the single cell level. Therefore, E. coli Tuner(DE3)/pRhotHi-2-LacI-EYFP was subjected to microfluidic cultivation in LB medium containing 40 μM NP-photocaged IPTG, which was pre-exposed to UV-A light for 1 minute (data not shown). Surprisingly, no light-induced YFP expression could be detected in the microfluidic set-up over the entire cultivation time, even when the concentration of caged inducer molecules was increased to 100 μM (Fig. 6A).
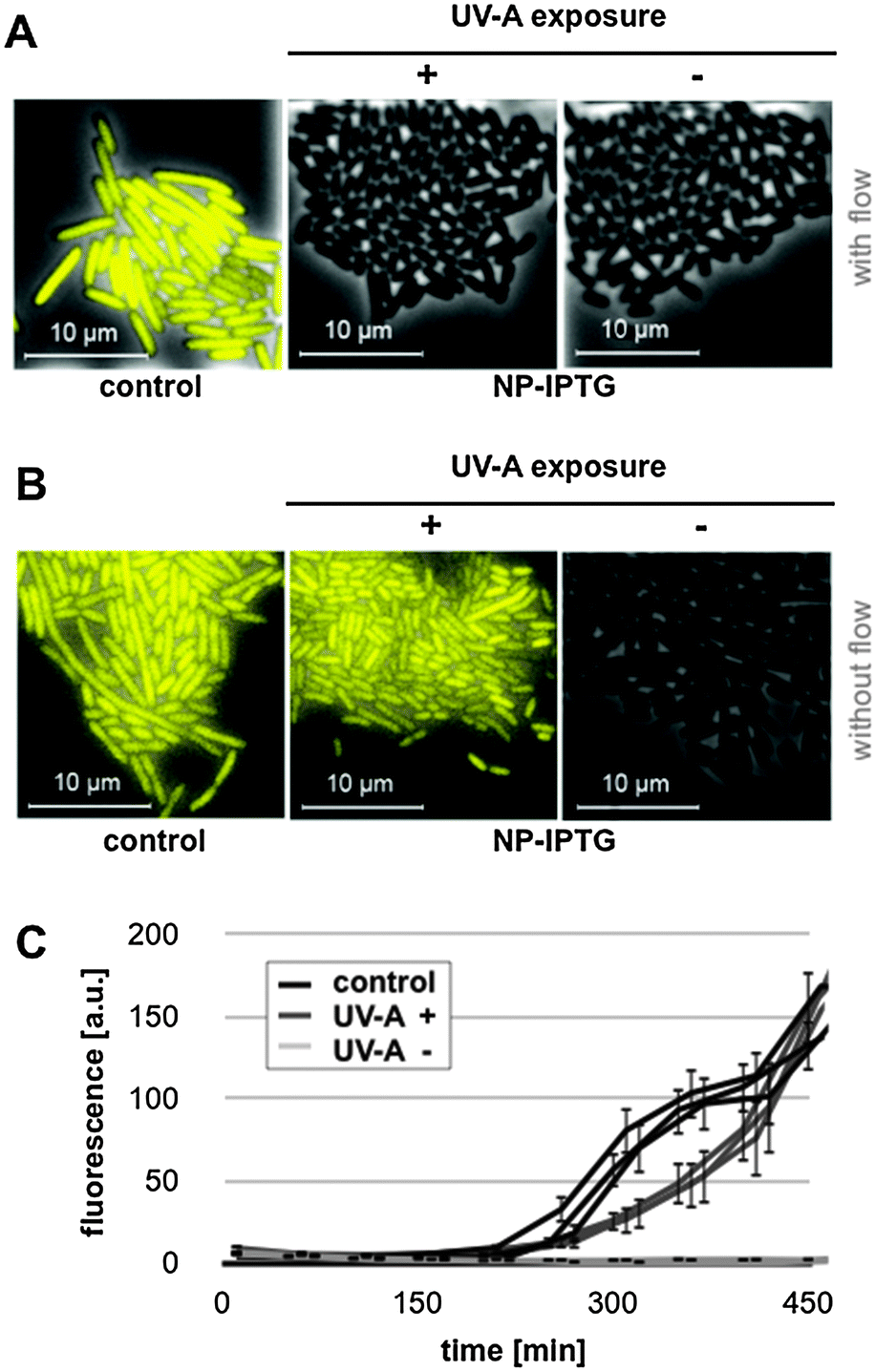 | ||
| Fig. 6 Light-controlled YFP expression in microcolonies of E. coli Tuner(DE3) cells carrying expression vector pRhotHi-2-LacI-EYFP. Photographs were taken from time lapse microscopy during microfluidic perfusion cultivation (A) and microscale batch cultivation (B) using LB medium supplemented with 100 μM (A) or 40 μM (B) of NP-photocaged IPTG and applied with (+) or without (−) pre-exposure to UV-A light. Control: LB medium supplemented with equivalent concentrations of conventional IPTG. (C) Fluorescence development of microcolonies during microscale batch cultivation. Single cell fluorescence values were monitored for three representative microcolonies. Control: medium supplemented with 40 μM conventional IPTG; UV-A+: NP-photocaged IPTG supplemented medium that was UV-A exposed for 1 min prior to cultivation. UV-A−: unexposed NP-photocaged IPTG supplemented medium. | ||
Two aspects might be considered to explain this observation: (1) IPTG molecules are intracellularly hydrolysed and immediately washed out of the cells due to free bidirectional diffusion over the cell membrane. (2) The results shown in Fig. 5D suggested that hydrolase levels in the early logarithmic growth phase were too low to promptly release IPTG in the cytoplasm. In the microfluidic cultivation set-up, exactly this growth phase seems to be mimicked due to persistent nutrient supply.54 To overcome these specific limitations during microfluidic cultivation, the same experimental set-up was chosen as before with the subtle difference that media flow was turned off after rinsing trapped cells with light-exposed medium. Fig. 6B clearly shows that microscale batch cultivation indeed resulted in a light-induced expression response. Furthermore, at the end of the experiment (i.e. after 450 min), UV-A light-induced YFP expression was comparable to that of conventionally induced cells (Fig. 6C). Online monitoring of the fluorescence development of microcolonies further revealed that the expression response upon UV-A light exposure was decelerated in comparison to conventionally induced cultures (Fig. 6C). Moreover, these data clearly document the remarkable homogeneity of light-dependent expression response. Thus, the combination of tightly controlled lac promoter-based T7RP-dependent gene expression with the use of photocaged IPTG molecules allowed a non-invasive and precise light control over gene expression in E. coli.
To finally elucidate the role of LacY in NP-photocaged IPTG-dependent triggering of lac-based gene expression, light responsiveness was monitored in the E. coli lacY+ strain BL21(DE3)/pRhotHi-2-EYFP (Fig. S8, ESI†). Surprisingly, a gradual light response during batch cultivation was observed, suggesting a diffusion-based instead of a LacY-mediated uptake of the caged inducer molecule before photo-cleavage (Fig. S8A, ESI†). However, microscale batch cultivation clearly revealed a distinctive heterogeneity of expression at the single cell level (Fig. S8B, ESI†) demonstrating that LacY indeed conveyed a positive feedback loop due to the specific uptake of decaged IPTG after light-mediated cleavage.
To the best of our knowledge the here presented optogenetic set-up, consisting of the lacY-deficient E. coli strain Tuner(DE3) and NP-photocaged IPTG, represents the first gradually light-regulated T7RP-dependent expression system in bacteria. The NP-photocaged IPTG-based system exhibits several outstanding features including precise and gradual regulation, high population homogeneity and low background expression. Moreover, the implementation of T7RP allows the expression of large and complex gene clusters55 and enables broad applicability to various alternative expression hosts,56 that is, however, clearly dependent on the actual growth phase (compare Fig. 5C and D and Fig. 6A and B), the corresponding expression host and the applied cultivation approach.
In contrast, photoreceptor-based light control is often hampered by their high basal activities10 and their extremely sharp transitions from inactive to active signaling states.57 Furthermore, the use of photoreceptors as light switches is usually restricted to certain hosts, as they specifically interact with corresponding signal transduction proteins and/or promoters.
Nevertheless, novel caged inducer molecules that are directly activated in a one-step photocleavage reaction are required to ensure high temporal resolution of light-regulated gene expression that is mostly independent of growth conditions. Furthermore, the establishment of advanced single cell batch cultivation systems seems to be vitally important,58 as it was shown within this study that environmental discontinuity is a crucial limitation for some synthetic biology approaches.
Conclusions
Exact control of gene expression by light allows the regulation of simple to complex cellular functions in living microorganisms with high spatial and temporal resolution. The results presented here clearly demonstrate that well characterized expression modules can be easily converted into a versatile photo-switch by implementing photo-caged effector molecules, such as NP-photocaged IPTG. The here described light switch is a valuable optogenetic tool applicable for biomedicine, systems biology, functional genomics, and biotechnology. Moreover, this optogenetic module can be implemented as a “photo-biobrick” into light-controlled higher-order artificial networks useful for a variety of synthetic biology approaches.Acknowledgements
Special thanks go to Vera Ophoven and Thomas Classen for valuable assistance during NP-photocaged IPTG synthesis. Further, we thank Stefan Helfrich and Katharina Nöh for Phyton-based lineage tree visualization. This work was partly performed at the Helmholtz Nanoelectronic Facility (HNF) of Research Center Jülich and supported by grants from Federal Ministry of Education and Research (OptoSys, FKZ 031A16).References
- S. Mukherji and A. van Oudenaarden, Nat. Rev. Genet., 2009, 10, 859–871 CAS.
- K. D. Bhalerao, Trends Biotechnol., 2009, 27, 368–374 CrossRef CAS PubMed.
- M. H. Medema, R. Breitling, R. Bovenberg and E. Takano, Nat. Rev. Microbiol., 2011, 9, 131–137 CrossRef CAS PubMed.
- G. Stephanopoulos, ACS Synth. Biol., 2012, 1, 514–525 CrossRef CAS PubMed.
- J. M. Christie, J. Gawthorne, G. Young, N. J. Fraser and A. J. Roe, Mol. Plant, 2012, 5, 533–544 CrossRef PubMed.
- T. Drepper, U. Krauss, S. Meyer zu Berstenhorst, J. Pietruszka and K.-E. Jaeger, Appl. Microbiol. Biotechnol., 2011, 90, 23–40 CrossRef CAS PubMed.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed.
- U. Krauss, T. Drepper and K.-E. Jaeger, Chem. – Eur. J., 2011, 17, 2552–2560 CrossRef CAS PubMed.
- C. W. Riggsbee and A. Deiters, Trends Biotechnol., 2010, 28, 468–475 CrossRef CAS PubMed.
- J. J. Tabor, A. Levskaya and C. A. Voigt, J. Mol. Biol., 2011, 405, 315–324 CrossRef CAS PubMed.
- R. Ohlendorf, R. R. Vidavski, A. Eldar, K. Moffat and A. Möglich, J. Mol. Biol., 2012, 416, 534–542 CrossRef CAS PubMed.
- J. Melendez, M. Patel, B. L. Oakes, P. Xu, P. Morton and M. N. McClean, Integr. Biol., 2014, 6, 366–372 RSC.
- A. Deiters, ChemBioChem, 2010, 11, 47–53 CrossRef CAS PubMed.
- D. D. Young and A. Deiters, Org. Biomol. Chem., 2007, 5, 999–1005 CAS.
- D. D. Young and A. Deiters, Angew. Chem., Int. Ed., 2007, 46, 4290–4292 CrossRef CAS PubMed.
- S. B. Cambridge, D. Geissler, S. Keller and B. Cürten, Angew. Chem., Int. Ed., 2006, 45, 2229–2231 CrossRef CAS PubMed.
- C. Chou, D. D. Young and A. Deiters, ChemBioChem, 2010, 11, 972–977 CrossRef CAS PubMed.
- F. W. Studier and B. A. Moffatt, J. Mol. Biol., 1986, 189, 113–130 CrossRef CAS.
- K. Terpe, Appl. Microbiol. Biotechnol., 2006, 72, 211–222 CrossRef CAS PubMed.
- J. C. Samuelson, in Methods in molecular biology, ed. T. C. Evans and M.-Q. Xu, Humana Press, Totowa, NJ, 1st edn, 2011, vol. 705, pp. 195–209 Search PubMed.
- I. Iost, J. Guillerez and M. Dreyfus, J. Bacteriol., 1992, 174, 619–622 CAS.
- S. Gräslund, P. Nordlund, J. Weigelt, B. M. Hallberg, J. Bray, O. Gileadi, S. Knapp, U. Oppermann, C. Arrowsmith, R. Hui, J. Ming, S. Dhe-Paganon, H. Park, A. Savchenko, A. Yee, A. Edwards, R. Vincentelli, C. Cambillau, R. Kim, S.-H. Kim, Z. Rao, Y. Shi, T. C. Terwilliger, C.-Y. Kim, L.-W. Hung, G. S. Waldo, Y. Peleg, S. Albeck, T. Unger, O. Dym, J. Prilusky, J. L. Sussman, R. C. Stevens, S. A. Lesley, I. A. Wilson, A. Joachimiak, F. Collart, I. Dementieva, M. I. Donnelly, W. H. Eschenfeldt, Y. Kim, L. Stols, R. Wu, M. Zhou, S. K. Burley, J. S. Emtage, J. M. Sauder, D. Thompson, K. Bain, J. Luz, T. Gheyi, F. Zhang, S. Atwell, S. C. Almo, J. B. Bonanno, A. Fiser, S. Swaminathan, F. W. Studier, M. R. Chance, A. Sali, T. B. Acton, R. Xiao, L. Zhao, L. C. Ma, J. F. Hunt, L. Tong, K. Cunningham, M. Inouye, S. Anderson, H. Janjua, R. Shastry, C. K. Ho, D. Wang, H. Wang, M. Jiang, G. T. Montelione, D. I. Stuart, R. J. Owens, S. Daenke, A. Schütz, U. Heinemann, S. Yokoyama, K. Büssow and K. C. Gunsalus, Nat. Methods, 2008, 5, 135–146 CrossRef PubMed.
- A. Fernández-Castané, C. E. Vine, G. Caminal and J. López-Santín, J. Biotechnol., 2012, 157, 391–398 CrossRef PubMed.
- A. Marbach and K. Bettenbrock, J. Biotechnol., 2012, 157, 82–88 CrossRef CAS PubMed.
- I. G. de Jong, J.-W. Veening and O. P. Kuipers, Environ. Microbiol., 2012, 14, 3110–3121 CrossRef CAS PubMed.
- S. Müller and G. Nebe-von-Caron, FEMS Microbiol. Rev., 2010, 34, 554–587 Search PubMed.
- A. R. Lara, E. Galindo, O. T. Ramírez and L. A. Palomares, Mol. Biotechnol., 2006, 34, 355–381 CrossRef CAS.
- S. Unthan, A. Grünberger, J. van Ooyen, J. Gätgens, J. Heinrich, N. Paczia, W. Wiechert, D. Kohlheyer and S. Noack, Biotechnol. Bioeng., 2013, 111, 359–371 CrossRef PubMed.
- S. Huang, Development, 2009, 136, 3853–3862 CrossRef CAS PubMed.
- J. W. Young, J. C. W. Locke, A. Altinok, N. Rosenfeld, T. Bacarian, P. S. Swain, E. Mjolsness and M. B. Elowitz, Nat. Protoc., 2012, 7, 80–88 CrossRef CAS PubMed.
- B. Lin and A. Levchenko, Curr. Opin. Chem. Biol., 2012, 16, 307–317 CrossRef CAS PubMed.
- A. Grünberger, W. Wiechert and D. Kohlheyer, Curr. Opin. Biotechnol., 2014, 29, 15–23 CrossRef PubMed.
- F. S. O. Fritzsch, K. Rosenthal, A. Kampert, S. Howitz, C. Dusny, L. M. Blank and A. Schmid, Lab Chip, 2013, 13, 397–408 RSC.
- C. Probst, A. Grünberger, W. Wiechert and D. Kohlheyer, J. Microbiol. Methods, 2013, 95, 470–476 CrossRef PubMed.
- P. Wang, L. Robert, J. Pelletier, W. L. Dang, F. Taddei, A. Wright and S. Jun, Curr. Biol., 2010, 20, 1099–1103 CrossRef CAS PubMed.
- G. Schendzielorz, M. Dippong, A. Grünberger, D. Kohlheyer, A. Yoshida, S. Binder, C. Nishiyama, M. Nishiyama, M. Bott and L. Eggeling, ACS Synth. Biol., 2014, 3, 21–29 CrossRef CAS PubMed.
- W. Mather, O. Mondragón-Palomino, T. Danino, J. Hasty and L. S. Tsimring, Phys. Rev. Lett., 2010, 104, 208101 CrossRef.
- G. Ullman, M. Wallden, E. G. Marklund, A. Mahmutovic, I. Razinkov and J. Elf, Philos. Trans. R. Soc. London, Ser. B, 2013, 368, 20120025 CrossRef CAS PubMed.
- A. Grünberger, N. Paczia, C. Probst, G. Schendzielorz, L. Eggeling, S. Noack, W. Wiechert and D. Kohlheyer, Lab Chip, 2012, 12, 2060–2068 RSC.
- N. Mustafi, A. Grünberger, D. Kohlheyer, M. Bott and J. Frunzke, Metab. Eng., 2012, 14, 449–457 CrossRef CAS PubMed.
- N. Mustafi, A. Grünberger, R. Mahr, S. Helfrich, K. Nöh, B. Blombach, D. Kohlheyer and J. Frunzke, PLoS One, 2014, 9, e85731 Search PubMed.
- L. Robert, G. Paul, Y. Chen, F. Taddei, D. Baigl and A. B. Lindner, Mol. Syst. Biol., 2010, 6, 1–12 Search PubMed.
- D. Hanahan, J. Mol. Biol., 1983, 166, 557–580 CrossRef CAS.
- J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory Press, New York City, USA, 2nd edn, 1989 Search PubMed.
- N. Katzke, S. Arvani, R. Bergmann, F. Circolone, A. Markert, V. Svensson, K.-E. Jaeger, A. Heck and T. Drepper, Protein Expression Purif., 2010, 69, 137–146 CrossRef CAS PubMed.
- M. F. Alexeyev, Biotechniques, 1995, 18, 52–56 CAS.
- F. Rosenau and K. Jaeger, in Enzyme functionality: design, engineering and screening, ed. A. Svendson, Dekker, Marcel Inc, New York, 1st edn, 2004, pp. 617–630 Search PubMed.
- J. Potzkei, M. Kunze, T. Drepper, T. Gensch, K.-E. Jaeger and J. Buechs, BMC Biol., 2012, 10, 28 CrossRef CAS PubMed.
- A. Gruenberger, C. Probst, A. Heyer, W. Wiechert, J. Frunzke and D. Kohlheyer, J. Visualized Exp., 2013,(82), e50560 Search PubMed.
- C. J. Wilson, H. Zhan, L. Swint-Kruse and K. S. Matthews, Cell. Mol. Life Sci., 2007, 64, 3–16 CrossRef CAS PubMed.
- M. Santillán and M. C. Mackey, J. R. Soc., Interface, 2008, 5, 29–39 CrossRef PubMed.
- D. Hartinger, S. Heinl, H. E. Schwartz, R. Grabherr, G. Schatzmayr, D. Haltrich and W.-D. Moll, Microb. Cell Fact., 2010, 9, 62 CrossRef PubMed.
- E. M. Ozbudak, M. Thattai, H. N. Lim, B. I. Shraiman and A. Van Oudenaarden, Nature, 2004, 427, 737–740 CrossRef CAS PubMed.
- A. Grünberger, J. van Ooyen, N. Paczia, P. Rohe, G. Schiendzielorz, L. Eggeling, W. Wiechert, D. Kohlheyer and S. Noack, Biotechnol. Bioeng., 2013, 110, 220–228 CrossRef PubMed.
- A. Loeschcke, A. Markert, S. Wilhelm, A. Wirtz, F. Rosenau, K.-E. Jaeger and T. Drepper, ACS Synth. Biol., 2013, 2, 22–33 CrossRef CAS PubMed.
- S. C. Troeschel, S. Thies, O. Link, C. I. Real, K. Knops, S. Wilhelm, F. Rosenau and K.-E. Jaeger, J. Biotechnol., 2012, 161, 71–79 CrossRef CAS PubMed.
- A. Levskaya, A. A. Chevalier, J. J. Tabor, Z. B. Simpson, L. A. Lavery, M. Levy, E. A. Davidson, A. Scouras, A. D. Ellington, E. M. Marcotte and C. A. Voigt, Nature, 2005, 438, 441–442 CrossRef CAS PubMed.
- J. Dai, S. H. Yoon, H. Y. Sim, Y. S. Yang, T. K. Oh, J. F. Kim and J. W. Hong, Anal. Chem., 2013, 85, 5892–5899 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ib00027g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |