 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nano-particulate pressed powder tablets for LA-ICP-MS
Dieter
Garbe-Schönberg
* and
Samuel
Müller
CAU Kiel University, Institute of Geosciences, Ludewig-Meyn-Strasse 10, D-24118 Kiel, Germany. E-mail: dgs@gpi.uni-kiel.de
First published on 16th April 2014
Abstract
We developed an advanced method for producing undiluted nano-particulate pressed powder tablets without addition of any binder applying wet-milling protocols in aqueous suspension using a high power planetary ball mill and agate tools. The method was tested and optimized for the milling time, ball size, ball-to-powder ratio and water-to-powder ratio using pulverized diorite DR-N containing abundant zircon crystals that are especially difficult to break down by both milling and acid-digestion methods. The new method produces nano-particulate powders with typical grain size d50 < 1.5 μm allowing us to form pressed powder tablets with excellent cohesion and homogeneity suitable for laser ablation micro-analysis. The optimized milling protocol was applied to a selection of other natural rock CRMs (BHVO-2, JGb-1, UB-N, AC-E, GA) covering a wide range of rock types, and formed powder tablets were analyzed by LA-ICPMS. Precision was found to be in the same range of <2–5%RSD as it is obtained from laser ablation analysis of glasses (e.g., volcanic glasses, vitrified rock powders), and preferred values could be reproduced within uncertainty for most of the 38 trace and ultra-trace elements analyzed. Our new method accomplishes the preparation of refractory sample materials or materials containing refractory mineral phases (e.g., plutonic rocks and sediments, ultramafic rocks, oxides as in banded iron formation, ceramics) for subsequent analysis by LA-ICPMS. Even more importantly, this method opens up new prospects for the manufacturing (and possibly isotopic spiking) of homogenized reference materials from natural minerals that are urgently needed for true matrix-matched calibration and validation of LA-ICP-MS geochemical and isotope data.
1 Introduction
Over the last decade significant progress has been made improving accuracy, precision, and detection limits of laser ablation inductively-coupled plasma mass spectrometry (LA-ICP-MS) leading to an ever accelerating increase of applications of this technique in many research fields.1 This is partly due to improving instrumentation of both laser ablation and ICP-MS: on the laser side shorter wavelengths and pulse lengths to generate fine aerosols and improved instrument stability in combination with dedicated ablation cells for efficient aerosol transport and rapid washout of laser-generated aerosols into the ICP; on the MS side faster scanning and data acquisition, improved interface designs and ion optics for increased ion transmission, extended dynamic range of detectors, and interference reduction by collision/reaction cell and/or multiple quadrupoles.2–4 Furthermore, elaborate operating protocols using additional gases (nitrogen,5 helium,6 hydrogen,7 and oxygen8) further reduce effects from non-stoichiometric sampling and aerosol transport, and ionisation in the ICP. However, before matrix-independent calibration or even standard-free data generation can be achieved, lots of fundamental processes remain to be studied and understood. Adequate matrix matched reference materials (RM) are still rare and non-matrix matched instrument calibration with silicate glasses NIST SRM610-614 is commonly used in many labs for the analysis of e.g., volcanic glasses, minerals and other materials. Non-matrix matched calibration may lead to systematic bias of the results for a number of elements since ablated mass, aerosol size distribution, vaporization, ionization regions of the particles within the ICP etc. might be significantly different.9,10 Recently, Korh11 demonstrated that non-matrix matched standardization of allanite with a zircon standard led to considerable matrix-induced errors in age determination.Either way, to accomplish both matrix-matched calibration and validation of generated analytical data a large variety of adequate certified reference materials (CRMs) are urgently needed. Today, we are confronted with a tremendous deficiency of available CRMs that are both homogeneous enough for certification of trace and ultra-trace elements on the micro-scale (e.g., 10–30 μm), and that match with the matrix of our samples. Furthermore, data for many newly generated CRMs are derived from analyses using the NIST glasses for calibration, which will perpetuate systematic bias in the data for real samples, and not help to improve the knowledge about capabilities of LA-ICPMS.
Increasingly, laser ablation ICP-MS is also used for bulk analysis of heterogeneous mineral assemblages (e.g., rocks, sediments, soils, ores, biominerals) circumventing tedious wet-lab sample preparation and problems associated with the dissolution of refractory mineral phases. The accurate determination of ultra-trace elements in geological samples containing refractory minerals – e.g., zircon in plutonic rocks and sediments, spinel in ultramafic rocks and eclogites – is still challenging. Standard hot plate or microwave-assisted digestion protocols fail in completely dissolving these minerals, and alternative procedures including vitrification with and without addition of a flux also have serious shortcomings, e.g. volatilisation of elements, inhomogeneity, contamination, and strong memory from the flux material. Pressurized bomb digestion yields mostly accurate results but is time-consuming and tends to form fluorides of e.g., Al and Mg that are difficult to dissolve. Hence, the direct analysis of pulverized and homogenized samples by laser ablation is a desirable approach.
In geochemistry and environmental sciences a broad assortment of reasonably well-characterized CRMs for silicate and carbonate rocks, soils, sediments, ores, bio-/minerals etc. is available from many suppliers (cf., GeoReM online database) but these are mainly in the form of coarse/fine grain powders and, rarely in the form of monomineralic crystals (e.g., CNRS glauconite GL-O, and some sheet silicates). Only very few crystallized mineral standards have been developed so far from either natural (e.g., zircon12,13) or synthesized (e.g., sulphides14) materials that are partially suitable for trace element and isotope studies. Homogeneous natural minerals in quantities sufficient to serve as CRMs are extremely rare and difficult to find, and the synthesis of such materials with a homogeneity of trace elements on a scale of 10–50 μm is more than challenging. There are homogeneous natural volcanic glasses but very few have been characterized as CRMs so far (e.g., obsidian15). Glassy CRMs manufactured from vitrified rock powder with satisfying homogeneity have also been produced by a few specialized labs (e.g., USGS basalts BHVO-2G, BIR-1G, BCR-2G;16 Bayreuth MPI-DING-glasses17). However, the ablation behaviour of these materials is dissimilar to that of crystalline rocks, rock powders, or natural mineral phases.
Mainly four strategies for generating RMs with micrometer-scale homogeneity suitable for LA micro-analysis are currently followed: (i) synthesis of minerals, (ii) selection of homogeneous natural minerals, (iii) vitrification of pulverized samples, and (iv) manufacturing of pressed powder pellets or tablets from natural or synthetic materials. Producing homogeneous synthetic minerals (e.g., oxides, sulphides, silicates, carbonates) and materials is difficult with respect to trace element composition under given p,T-conditions and diffusion during crystallization, and only very few standards are reported in the literature (e.g.ref. 14). Recently, a rapid technique for vitrification of pulverized (geological) samples has been re-invented and improved18–20 using a temperature-controlled heated iridium or tungsten strip for shock-melting under inert (argon atmosphere) conditions. Homogeneous glass beads, however, are only obtained if the liquid is thoroughly stirred during melting. To some extent, volatile elements are lost from the liquid under atmospheric pressure conditions,21 and contamination from the strip material (Ir, W, and impurities) is significant. Some sample types (e.g., ultramafic rocks like harzburgite, highly differentiated rocks like granite, granodiorite) cannot be entirely melted without matrix modification.18 Matrix modification, in turn, and addition of fluxes (e.g., lithiumborate) inherently bear the risk of introducing contaminant elements, influence the LODs due to dilution, and may cause tremendous memory effects (e.g., Li and B).
Pressed powder tablets (PT) from pulverized samples using a binder for achieving sufficient cohesion to be pressed by means of a tablet press have been repeatedly reported as a strategy for the direct bulk analysis of solids by laser ablation. Gray22 was one of the first who described laser ablation for sample introduction into ICP-MS using powder tablets that were manufactured similar to the procedure used for XRF-analysis of powders. More detailed studies compared results from pressed powder tablets with data obtained from fused glass discs and concluded that fused glasses delivered better precision and accuracy.23–25 Nevertheless, further studies aiming at in situ analysis of natural solid samples using LA-ICP-MS demonstrated that the analysis of PTs can deliver results of considerable analytical quality.26–28 A number of studies investigated in detail the binders that were used for producing cohesive powder tablets. While PVA and PVC are commonly used as binders in XRF analysis, cellulose or cellulose mixture spiked with internal standards,23,29,30 vanillic acid, pyrazinoic acid and nicotinic acid,31 synthetic resin,32,33 and silver powder30 were evaluated as binders for laser ablation. In a recent study, sample particles were bound with a zinc-oxide–eugenol-complex after homogenization and milling.34 Homogenization of the sample powder and binder was achieved by fine-grinding.35,36 Internal standards were added in the form of pulverized CRM,24,37,38 as solutions,39,40 or as solids for isotopic dilution.41
Far less attention has been paid to the manufacturing of undiluted PTs without addition of any binder (e.g., Imai42). Procedures for manufacturing undiluted PTs directly from synthetic compounds are described by Motelica et al.43 Only recently, the aspect of the grain size has been recognized as being important for producing homogeneous and cohesive undiluted PTs and were further investigated in more detail. Arroyo et al.44 dry-ground soil and sediment samples for 20 minutes by means of a high power mill and obtained particles with an average grain size of d50 < 10 μm. Ito and coworkers45 picked up the concept of dry fine-grinding and studied matrix effects on 238U and 232Th related to the grain size and non-matrix matched calibration. There is a consensus from these studies that small grain sizes <10 μm are a prerequisite for homogeneity and acceptable data quality obtainable with ablation spot sizes of 80–200 μm.
In situ microanalysis of minerals typically requires smaller LA spot sizes of <20–50 μm in order to obtain sufficient spatial resolution that is needed for analyzing discrete growth zones or for avoiding micro-inclusions of other mineral phases, melts, or fluids. Consequently, utilizing the pulverized CRM in the form of PT for matrix matched calibration and validation requires even smaller, nano-scale particles in order to obtain representative (and homogeneous) sample aliquots during laser sampling. Minimum grain sizes achievable with dry-grinding lie typically somewhere in the 5–10 μm range (W. Hopfe, pers. commun.). However, hematite (Fe2O3) nanoparticles with a minimum particle size of 76.6 nm have been obtained with a dedicated protocol after dry milling for 10 hours.46 Wet mechanical milling in e.g., hydrous suspension is a more efficient “top-down” approach for producing the required nano-scale particles.47 This approach, however, has not been studied in the context of laser ablation analysis so far. A different “bottom-up” approach is the production of nanoparticles by flame synthesis that were mixed and pressed into pellets suitable as standards for laser ablation sampling.48
In this study we optimized procedural parameters like the milling ball size, ball-to-powder ratio, sample-to-water ratio, total milling time etc. and show that undiluted pressed powder tablets can be successfully used for ultra-trace element analysis of rocks (serpentinite to granite) after pulverisation into nano-particles of grain size d50 < 1.5 μm. A variety of wet milling-produced materials were analysed for 38 elements by laser ablation ICP-MS and figures of merit, such as precision and accuracy as well as the limits of detection were determined and are reported.
2 Experimental
2.1 Milling, freeze-drying, and tablet pressing
A high power planetary ball mill (Fritsch Pulverisette 7 classic line, centrifugal acceleration 50 g) with two 45 mL agate vials and variable number and size of agate milling balls (cf., Table 2) was used at a nominal speed of 700 to 800 rpm. The starting material for all experiments was the original rock powder <75 μm (200 mesh) as distributed from the certifying institutions (e.g., USGS Denver, CRPG-CNRS Nancy). After loading each vial with milling balls approx. 2 g sample powder were added. Depending on the experimental protocol up to 8 mL ultrapure water (ELGA, >18.5 MΩ cm−1) were pipetted into the vials. Grinding was carried out at consecutive time intervals of 3 min with 20 s pause for cooling. After completion, the grinding vials were opened and balls together with liquid sample suspension were transferred into a plastic sieve (polyethylene) on-top of a sample beaker (polycarbonate). Vials and balls were carefully rinsed with ultrapure water from a pipette until the rinsing fluid collected in the beaker was clear. The beaker with sample suspension was then closed and stored in a freezer overnight. After 24 hours beakers were covered with filter paper and freeze-dried (Alpha 1-2 LD, Martin Christ GmbH). The freeze-dried powder was re-homogenized for 5 min by hand using an agate mortar and then transferred into a sample vial (polycarbonate). Approximately 300 mg of this dry homogenized, nano-particulate sample powder were loaded into a standard tablet press (Specac Atlas 15 t manual) with a 13 mm die set and pressed into a tablet applying 10 t (0.74 × 106 kPa) for 1 min. All agate vessels were thoroughly cleaned using a portion of the next sample that was then discarded. Washed analytical grade quartz sand turned out to have abundant heavy minerals making it inappropriate for cleaning the agate tools in this application. Synthetic quartz (sand) as obtainable from crystal glass factories might be an alternative.49| ICP-MS | ||
| Forward power | 1500 | W |
| Carrier gas flow (He) | 1.0 | L min−1 |
| Optional gas (H2) | 14 | mL min−1 |
| Oxide formation rate (ThO/Th) | <0.2 | % |
| Doubly charged formation rate (Ca2+) | <0.3 | % |
| Plasma condition (U/Th) | 1.05 | |
| Dwell time per m/z | 10 | ms |
| Total dwell time per sweep | 0.484 | ms |
| Total integration time | 80 | s |
| Laser ablation | ||
| Pulse length | 17–20 | ns |
| Energy density on sample | 13.5 | J cm−2 |
| Pulse frequency | 10–15 | Hz |
| No. pulses | 400–600 | |
| Ablation spot size | 80 | μm |
| Milling balls | BPR | |||
|---|---|---|---|---|
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
20:1 |
|||
| Diameter [mm] | Quantity | Weight [g] | Weight powder [g] | |
| 15 | 7 | 32.5 | 3.25 | 1.63 |
| 10 | 25 | 35 | 3.5 | 1.75 |
| 7 | 70 | 32.5 | 3.25 | 1.63 |
2.2 Grain size analysis by laser diffraction
For particle size analysis 65–170 mg of the sample were weighed and mixed with 20 mL ultrapure water. The efficiency of laser diffraction analysis and, in consequence, the data quality is highly influenced by the opacity of the analyzed suspension. For that reason the sample aliquot weight was adapted according to the powder's color index (more sample if color is brighter). For improved dispersion 1 mL of sodium pyrophosphate was added to each suspension before shaking it for two days using a circular tube rotator. Measurements were carried out using a laser counter (Beckman Coulter LS 13320) applying twenty reading cycles with a cycle duration of 1 min. During particle counting the suspension was sonicated. For the particle size analysis the Fraunhofer diffraction model was used instead of the Mie scattering model. While it is known that the Fraunhofer model underestimates particles <10 μm,50,51 the Mie scattering algorithm needs the refraction index of the liquid medium as well as that of the sample to be known exactly to create an optical model. Since the refraction index of natural rock powders with complex mineralogical composition is highly variable and, therefore, unique for each sample, the Fraunhofer diffraction model was applied here.2.3 LA-ICP-MS analysis
A 193 nm ArF excimer laser ablation system (GeoLasPro Plus, Coherent) coupled to an Agilent 7500s ICP-MS was used for all measurements. Samples were loaded into a Zurich-type low dispersion high capacity laser ablation cell (LDHCLAC, Fig. 1)52 flushed with 1.0 L min−1 He as the carrier gas. Addition of 14 mL min−1 H2 into the He carrier gas stream before entering the ablation cell led to an increase in sensitivity and reduction of oxide formation. Operating conditions are summarized in Table 1. Each measurement comprised data acquisition intervals of 20 s background, 40–60 s sample ablation, and 20 s washout monitoring. To reduce the potential error from surface contamination, the first 5–10 s of each sample acquisition were discarded from data integration. The GLITTER software package53 was used for data reduction using the graphical visualization tool for setting integration intervals for each analyzed spot. In GLITTER external calibration was done with silicate glass NIST SRM612 using 48Ca for internal standardization.54 For these data obtained with NIST-glass calibration typically display a systematic bias of the results as a consequence of different sample matrices a second matrix-matched calibration step was applied using external spreadsheet software. Results for at least 2–3 pressed powder tablet CRMs with a composition matching with that of unknown samples as closely as possible are used for calculating average “bias factors” for each element. These factors were then applied to NIST-glass calibrated sample data. All data were background corrected and represent averages of 4–10 replicates. | ||
| Fig. 1 Nano-particulate powder tablets (13 mm) loaded into the Zurich-type large-volume ablation cell. In the centre is a 1′′ mount with polished NIST SRM610-614 glass standards in epoxy. | ||
3 Results and discussion
Typically, pure pulverized rocks and minerals with “analytical-grade” grain sizes of <200–100 mesh or <75 μm, respectively, as produced in many labs cannot be pressed into cohesive pellets or tablets suitable for laser ablation analysis without addition of a binder or further grinding to smaller grain sizes. The motivation for our study was to find a method to produce homogeneous pulverized minerals and rocks for direct analysis by laser ablation. In addition, emphasis was put on “difficult” rock types containing abundant refractory minerals such as zircon, spinel and other oxides that are problematic to digest. These minerals represent “containers” of geochemically important trace metals but are difficult to pulverize or digest during grinding and acid digestion, respectively. Residual mineral micro-fragments may survive acid digestion procedures leading to low recoveries for trace metals like Zr, Hf, Nb, Ta, Th, REE, and Cr. Larger mineral fragments in otherwise homogeneous fine-grained powder pellets and tablets, respectively, hinder the representative sampling with a micro-analytical technique like LA-ICP-MS. For our optimization study we used pulverized granodiorite DR-N (CRPG-CNRS) with abundant and highly refractory zircon, and serpentinite UB-N (CRPG-CNRS) containing spinel which is characterized by very low concentrations of a number of trace elements. Basalt BHVO-2 (USGS) being a very well characterized CRM was analysed along with these samples.3.1 Materials for the grinding bowl and balls
The crucial point for pulverization of e.g., silicate and oxide particles with a planetary ball mill (in this study: centrifugal acceleration of 50 g) is the use of collision energy for the disintegration of sample particles. Consequently, mills delivering high acceleration forces and grinding materials with high densities (and hardness) are favorable for fast and efficient crushing of particles. However, high-density materials commonly used for this purpose have high concentrations of elements that are of geochemical interest: zirconium oxide (ZrO2, ∼5.9 g cm−3) contains considerable amounts of Hf and is stabilized with Mg and/or Ce, Y; silicon nitride (Si3N4, 3.25 g cm−3) contains significant Al and Y and many other trace elements. Sintered corundum (Al2O3, 3.8 g cm−3) is relatively coarse-grained with a rough surface and not suitable for grinding into very small particle sizes. Materials with highest densities and reasonable to very good abrasion resistance are steel (7.8 g cm−3) and tungsten carbide (14.95 g cm−3) but are contaminant sources of numerous transition metals and other trace elements. The only remaining material appropriate for trace element analytical applications but with low density is agate (2.65 g cm−3). Therefore, we used agate milling balls of 15, 10 and 7 mm diameter and found no significant contamination of our samples except for elevated SiO2 from abrasion of the agate. The recommended procedure for cleaning agate tools is grinding with pure quartz sand. This procedure had to be abandoned for natural quartz sand that also contains refractory heavy minerals (e.g., zircon). In some experiments, micro-fragments from these minerals were found being transferred into the next sample despite thorough cleaning.3.2 Dry milling versus wet milling in suspension
Our initial attempt was dry re-grinding of the original sample powder (grain size <75 μm, d90 = 35 μm) with small milling balls (7 mm diameter) avoiding any addition of liquid or other milling aids. After milling for 30 min, the grain size could be reduced (d90 = 29 μm; Fig. 2). The handling of the fine-grained powder, however, was very difficult due to its stickiness to surfaces and re-agglomeration of particles. Moreover, the minimum grain size achievable with dry-milling protocols is limited to 5–10 μm (pers. commun. W. Hopfe). Hence, no further evaluation of dry milling protocols was done. The significant progress towards small particles was made by addition of a liquid to the sample powder and milling in suspension as shown in Fig. 2 (see also the discussion below). This change from dry to wet milling reduced the grain size of DR-N to d90 = 8 μm (Table 4). Adding a liquid (here: ultrapure DIW) inherently bears an additional risk of contamination during handling of the sample suspension. Hence, utmost care was taken to keep the workplace clean and liquid handling in a clean bench environment was found to be favorable. Milling in suspension with other fluids e.g., ethanol, isopropanol, or acetone might be an alternative making drying of the sample easier but this aspect was not investigated in this study. Our data eventually showed some indication for contamination from Zn, Pb (lab environment, handling) and Mo, W (tablet press dies).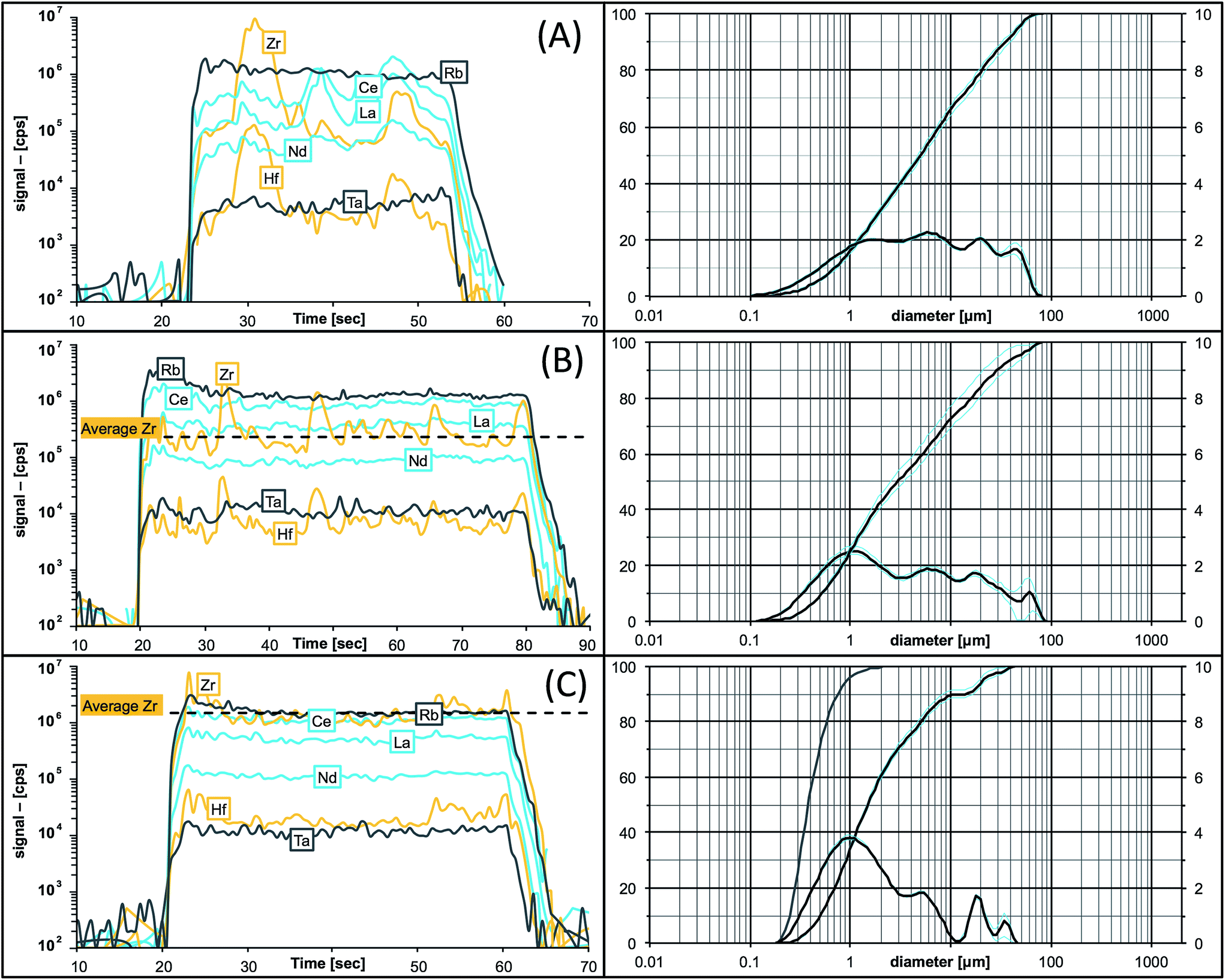 | ||
| Fig. 2 Traces of signal intensities (in [cps], left) for selected elements in granodiorite DR-N during time-resolved laser ablation analysis compared to the grain size distribution in the pressed powder tablet (right). (A) Original sample powder (KBr binder); (B) after dry-milling for 30 min; (C) after 45 min wet-milling. Note the secular intensity maxima for Zr and Hf during ablation of large zircon fragments in (A) and improvements towards both homogeneous and smooth ablation patterns, and the increased average ablation yield for Zr and Hf after re-milling in (B) and (C). The grain size improved from d50 = 5 μm in (A) to d50 = 1.5 μm in (C) with more than 95% of all particles being <1 μm (see the additional cumulative curve for the number of particles in (C)). | ||
| Step 1 | Step 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ball size [mm] | Duration [min] | Water [mL] | Ball size [mm] | Duration [min] | Water [mL] | Total time [min] | WPR | ||
| a WPR = water-to-powder ratio. | |||||||||
| 1 | 15 | 10 | 5 | → | 7 | 30 | +7 | 40 | 6 |
| 2 | 15 | 10 | 8 | → | 7 | 30 | +8 | 40 | 8 |
| 3 | 15 | 15 | 5 | → | 7 | 45 | +7 | 60 | 6 |
| 4 | 10 | 15 | 5 | → | 7 | 45 | +7 | 60 | 6 |
| 5 | 15 | 30 | 5 | 30 | 2.5 | ||||
| 6 | 10 | 30 | 5 | 30 | 2.5 | ||||
| 7 | 7 | 30 | 5 | 30 | 2.5 | ||||
| Grain size of pulverized DR-N | |||
|---|---|---|---|
| Initial [μm] | Protocol | d 90 [μm] | d 50 [μm] |
| <75 | Original powder | 35.3 | 5.0 |
| Dry, 30 min, 7 mm balls | 29.2 | 2.9 | |
| Wet, 30 min, 7 mm balls | 7.9 | 1.4 | |
3.3 Milling ball size, number of balls, and ball-to-powder ratio (BPR)
The selection of the milling ball size depends first hand on the sample grain size, larger particles requiring stronger forces from larger (heavier) balls for disintegration. Secondly, smaller particles can be obtained with smaller milling balls and, commonly, smallest nanoparticles <10–100 nm are produced using sub-millimeter-sized (glass) beads. Consequently, rapid progress in particle size reduction can be achieved if balls are changed to increasingly smaller sizes in subsequent milling steps. The total mass of milling balls or the ball-to-powder ratio (BPR) must be kept constant. We combined subsequent milling steps with 15 mm and 7 mm as well as 10 mm and 7 mm milling balls, respectively. However, the results in terms of precision obtained by LA-ICPMS did not show significant improvements (Table 3). Moreover, the exchange of milling balls bears a high risk of additional contamination during handling of the milling balls, and adds excess rinsing water to the suspension lowering the milling efficiency. Therefore, we decided to avoid changing milling balls in this application on the expense of slightly (∼15 min) longer milling times. The favorable number of milling balls depends on the ball size and the volume of the bowl. With a 45 mL milling bowl the manufacturer recommends 7, 18, or 180 balls with 15, 10, or 5 mm diameter, respectively. In order to maintain a constant mass, the milling balls were weighed to 32.5–35 g resulting in 7, 25, and 70 agate balls with nominal diameters of 15, 10, and 7 mm, respectively (Table 2). Following recommendations of the manufacturer the ball-to-powder mass ratio should be in the range of 10–20. In our experiments the sample weight was kept constant at ∼2 g resulting in a BPR of ∼17.3.4 Water-to-powder ratio (WPR)
The addition of a liquid maintains all particles in a suspension and their stochastic distribution within the milling bowl during milling. Adding too much liquid, however, will reduce the collision energy for the liquid and reduces the speed of the milling balls (protocol #2, Table 3). On the other hand, adding too little water leads to enhanced stickiness of the suspension making further handling extremely difficult. During our experiments a water-to-powder (WPR) ratio between 2.5 and 6 yielded similar promising results and all subsequent experiments were conducted with a WPR of 2.5 (i.e., ∼2 g sample powder suspended in 5 mL ultrapure DIW). The optimum WPR depends on the sample material and was also investigated. For example, hydrated minerals like sheet silicates and clays need more liquid for continued suspension during milling. So far, we have only used water as the solvent for the suspension. Future experiments will also investigate non-polar solvents (e.g., isopropanol, ethanol).3.5 Milling time and intervals
The total milling time was varied from 30 to 60 minutes at intervals of 3 min milling and 20 s cooling to avoid overheating of the sample. The best results in terms of precision <3%RSD for most elements were obtained with protocols #4, #6 and #7 (Table 3). The good results obtained with protocol #4 must be ascribed to the changing of milling balls rather than the total milling time, but were not better than with protocols #6 and #7. The milling efficiency decreases over time as a consequence of the relationship between the ball size and the powder grain size since incident forces from rotating milling balls can no longer be transferred to the small particles. However, it was not unambiguously clear from these initital “proof of concept” results how much the milling process continues after 30 minutes and how much that improves the analytical figures of merit. Furthermore, it needs to be mentioned that the exchange of milling balls had to be avoided, and that the handling of 25 balls is easier than handling 70 balls. Based on these observations, all subsequent experiments were done with protocol #6 but with an extended milling time of 45 min.3.6 Laser ablation-generated transient signals in dependence on the particle grain size
Particle size distributions for all produced powders were measured using a laser particle counter. Table 4 gives an overview of the grain size distribution for pulverized DR-N in its original analytical grade form, and after dry and wet re-grinding with 7 mm grinding balls. It can be seen that the major improvements towards smaller grain sizes were achieved by wet grinding in aqueous suspension. However, the final grain size obtainable with wet-milling using a single protocol is still dependent on the material: similar rock types like basalts (BIR-1, BHVO-2) and gabbro (JGb-1) show similar grain size distributions with smaller particles at d50 (∼1.3 μm) and d90 (∼5 μm) percentiles when compared to e.g., granites (AC-E, GA: d50 = 2.3 and 1.9 μm, d90 = 22.8 and 17.6 μm), diorite (DR-N: d50 = 1.5 μm, d90 = 15.5 μm) and serpentinite (UB-N: d50 = 1.5 μm, d90 = 10.2 μm) (Table 5). Our wet-milling procedure shifts the particle size distribution towards a larger fraction of sub-micron particles, and more than 95% of all particles in DR-N are <1 μm (cf., Fig. 2c, the cumulative curve for the number of particles). It should be emphasized here that the challenge of milling e.g., plutonic rocks lies in their heterogeneity: the mineral composition and mineral grain size with contrasting physical properties like e.g., hardness and cleavage may vary over a wide range. Consequently, it will be much more difficult to grind hard crystals (e.g., zircons) in a soft matrix (e.g., feldspars or clay minerals) than grinding mineral assemblages with a similar hardness (e.g., mono-mineralic quartz sand) where all particles contribute to particle–particle collision.| CRM | Initial [μm] | d 90 [μm] | d 50 [μm] |
|---|---|---|---|
| BIR-1 | <75 | 5.3 | 1.3 |
| BHVO-2 | <75 | 5.2 | 1.3 |
| UB-N | <75 | 10.2 | 1.5 |
| JGb-1 | <149 | 4.8 | 1.3 |
| DR-N | <75 | 15.5 | 1.5 |
| AC-E | <75 | 22.8 | 2.3 |
| GA | — | 17.6 | 1.9 |
As soon as particles are smaller than 100 nm the effects of the Coulomb electrostatic force and the van der Waals force will re-aggregate these particles explaining the high cohesion of nano-particulate powders.47 The benefit of improved cohesion of nano-particulate pressed powders for laser ablation is illustrated in Fig. 3 showing smooth surfaces of the tablet, and of crater walls and bottom that are not very different from ablation pits in glass. The quality of controlled ablation in nano-particulate sample powders, particularly of basalts, resembles that of glass and produces very stable ion signals in the ICP-MS (Fig. 4) leading to a precision better than 2%RSD for most of the analyzed elements in BHVO-2 (cf., Table 6). It should be mentioned here that the Zurich-type ablation cell has a very rapid washout for most elements (<2 s for signal decay by 2–3 magnitudes after the last laser pulse), and no aerosol-homogenization device was used so that any instability during ablation of the powder becomes more recognizable. A closer look at the time resolved signals for a variety of rocks allows some qualitative conclusions on the grain size distribution of the ablated powder. Fig. 2 compares the grain size distribution in pulverized granodiorite DR-N with the signal intensity stability of trace elements during laser ablation, and progress towards smaller grain sizes of even the refractory zircon crystals is reflected in the increasing signal stability of e.g., La, Ce and Nd, and Zr, Hf, Nb and Ta. The time-resolved ablation pattern during analysis of the original (Fig. 2A) and dry-milled sample powder (Fig. 2B) of DR-N shows a smooth and constant signal for Rb (<2%RSD) but a very irregular ablation pattern for all other elements shown here. Especially for the original sample powder high peaks for Zr and Hf can be interpreted as a consequence of large zircon crystal fragments. Individual peaks for other elements like Ce, La, and Nd are most likely a result of ablating individual monazite (La, Ce, Nd)PO4 crystal fragments. The corresponding cumulative grain size distribution curve shows that the fraction >1 μm has a volume of about 80%. After dry re-milling of DR-N powder the monazite induced REE peaks are eliminated (Fig. 2B) suggesting that monazite crystals are pulverized and statistically distributed in the pressed powder tablet. However, smaller but still discernible peaks for Zr and Hf show that zircon crystals “survived” this milling step. The grain size distribution curve for the dry milled powder shows, on the one hand, an increase of fine particles with a maximum at about 1 μm when compared to the original sample but, on the other hand, a broader particle size distribution. This is possibly caused by re-aggregation of the dry-milled powder. The grain size distribution curve is still polymodal, the fraction of large particles >10 μm was only slightly reduced, and the slope of the cumulative curve showed not much improvement. The accuracy of the measured total Zr is poor. Fig. 2C shows the ablation pattern and particle size distribution for powder obtained with the final protocol. Whereas the time resolved signal for Zr and Hf is still not as stable as that for other elements (e.g., Rb and Nd), the average signal for Zr (and Hf) shows a significant increase compared to the average Zr (Hf) of graph 2B and yields good accuracy for Zr and Hf. A coarse-grained fraction with a diameter >10 μm can still be seen in the particle size distribution graph but after wet milling it represents only a volume of about 10% (compared to >30% shown in Fig. 2A and B). The particle size distribution below 10 μm is mostly homogeneous with the major peak at 1 μm and a small peak at 4 μm.
 | ||
| Fig. 3 SEM image of an 80 μm ablation crater in nano-particulate pressed powder of basalt BHVO-2. Note the smooth surfaces of the tablet and of crater walls and bottom (ablation with 400 pulses @ 10 Hz, 13.5 J cm−2). | ||
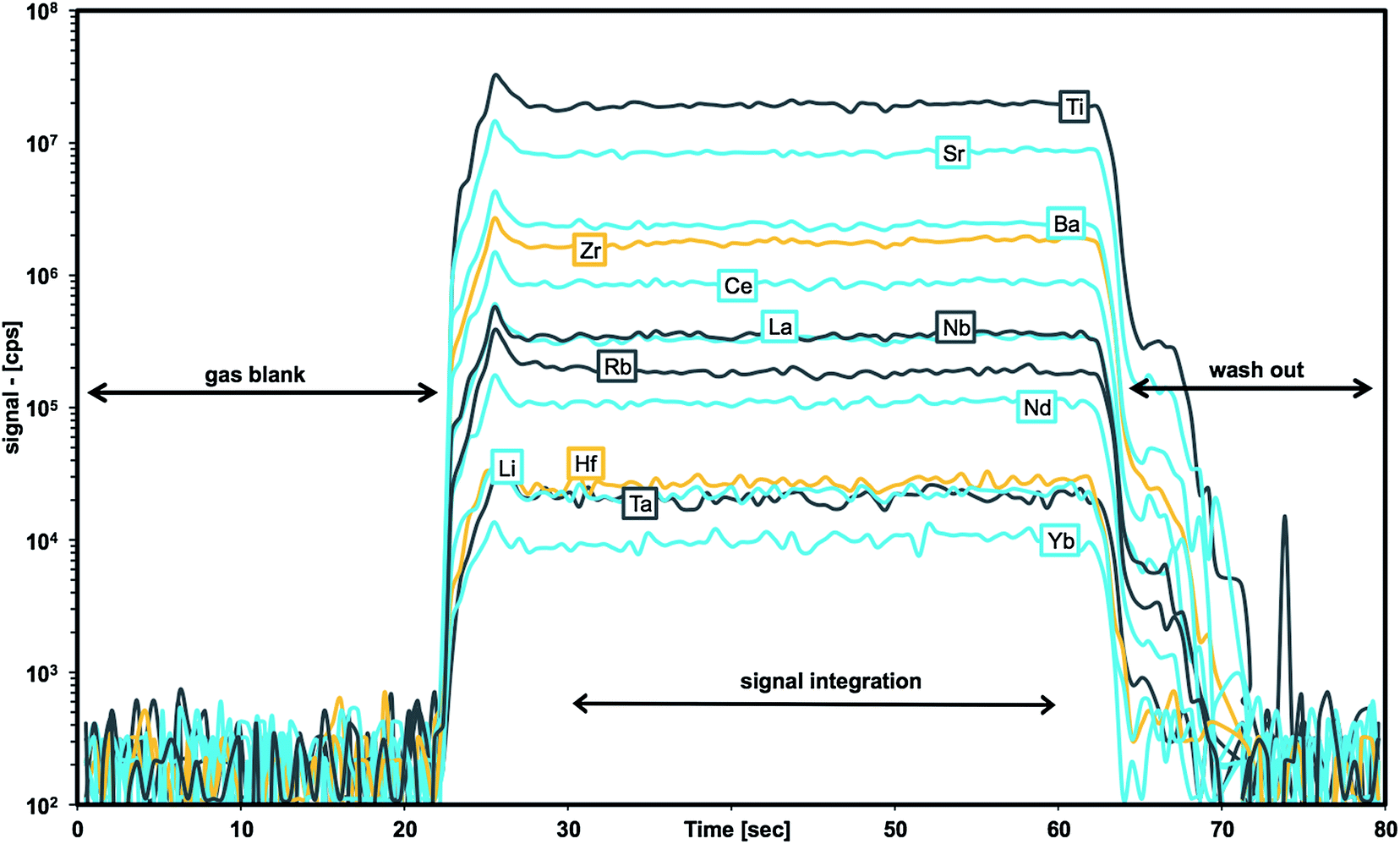 | ||
| Fig. 4 Traces of signal intensities (in [cps]) for selected m/z during laser ablation time-resolved analysis of BHVO-2 nano-particulate pressed powder. | ||
| BHVO-2 | JGb-1 | UB-B | DR-N | AC-E | GA | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. [ppm] | This Study [ppm] | RSD [%] | Ref. [ppm] | This Study [ppm] | RSD [%] | Ref. [ppm] | This Study [ppm] | RSD [%] | Ref. [ppm] | This Study [ppm] | RSD [%] | Ref. [ppm] | This Study [ppm] | RSD [%] | Ref. [ppm] | This Study [ppm] | RSD [%] | |
| Li | 4.8 | 4.9 | 3.2 | 4.59 | 4.42 | 0.7 | 27 | 28.73 | 0.3 | 40 | 40 | 3.9 | 93 | 88 | 3.5 | 90 | 83.6 | 0.1 |
| SiO2 (%) | 49.4 | 49.4 | 0.8 | 43.6 | 43.4 | 0.7 | 39.4 | 37.8 | 2.2 | 52.8 | 49.8 | 0.5 | 70.4 | 62.2 | 4.9 | 70 | 61.6 | 0.1 |
| Sc | 32 | 32.5 | 0.4 | 35.8 | 35.8 | 0.6 | 13 | 13.3 | 2.3 | 28 | 28 | 1.1 | 0.11 | 1.76 | 5.0 | 7 | 8.2 | 2.3 |
| TiO2 (%) | 2.7 | 2.7 | 0.4 | 1.6 | 1.6 | 0.2 | 0.1 | 0.09 | 0.8 | 1.1 | 1.1 | 2.3 | 0.1 | 0.1 | 4.3 | 0.4 | 0.4 | 3.2 |
| V | 317 | 317 | 0.4 | 640 | 657 | 0.6 | 75 | 62.3 | 0.5 | 220 | 220 | 2.8 | 3 | 0.176 | 3.1 | 38 | 37.5 | 1.1 |
| Cr | 280 | 290 | 1.0 | 57.8 | 61 | 1.6 | 2300 | 2340 | 1.5 | 40 | 35 | 2.4 | 3.4 | 1.6 | 10 | 12 | 8.8 | 10 |
| Mn | 1450 | 1340 | 0.9 | 1508 | 1540 | 0.7 | 929 | 973 | 1.1 | 1704 | 1790 | 0.8 | 449 | 407 | 4.8 | 697 | 592 | 1.5 |
| Co | 45 | 44 | 0.9 | 61.6 | 58 | 1.9 | 100 | 99 | 2.9 | 35 | 38 | 6.5 | 0.2 | 0.088 | 7.0 | 5 | 4.24 | 0.6 |
| Ni | 119 | 119 | 0.7 | 25.4 | 23 | 9.2 | 2000 | 1973 | 2.6 | 15 | 16 | 0.7 | 1.5 | 0.75 | 4.5 | 7 | 2.78 | 0.9 |
| Cu | 127 | 127. | 1.0 | 86.8 | 84 | 7.3 | 28 | 26 | 9.3 | 50 | 49.2 | 1.4 | 4 | 7.8 | 4.0 | 16 | 4.6 | 8.7 |
| Zn | 103 | 115 | 1.2 | 109 | 103 | 1.1 | 85 | 111 | 6.3 | 145 | 146 | 2.5 | 224 | 204 | 2.8 | 80 | 55.0 | 1.9 |
| Ga | 22 | 20.6 | 1.4 | 18.9 | 18.1 | 1.4 | 3 | 3.02 | 3.2 | 22 | 21.4 | 2.6 | 39 | 37 | 5.3 | 16 | 14.8 | 1.5 |
| Rb | 9.11 | 9.379 | 0.1 | 5.9 | 5.6 | 5.1 | 4 | 3.91 | 0.9 | 73 | 71 | 3.9 | 152 | 133 | 5.5 | 175 | 155.7 | 0.4 |
| Sr | 396 | 387 | 0.6 | 321 | 328 | 0.6 | 9 | 7.49 | 1.1 | 400 | 403 | 1.6 | 3 | 2.7 | 6.0 | 310 | 273 | 0.8 |
| Y | 26 | 25.76 | 0.3 | 9 | 9.7 | 1.1 | 2.5 | 2.7 | 3.7 | 26 | 26.1 | 1.1 | 184 | 167 | 5.0 | 21 | 18.6 | 3.1 |
| Zr | 172 | 170 | 0.3 | 28.7 | 29 | 3.2 | 4 | 3.7 | 5.8 | 125 | 129 | 14 | 780 | 804 | 5.8 | 150 | 130 | 4.3 |
| Nb | 18.1 | 18.6 | 0.7 | 2.4 | 2.38 | 1.2 | 0.05 | 0.055 | 8.5 | 7 | 7.57 | 1.2 | 110 | 108 | 6.0 | 12 | 12.0 | 6.5 |
| Sn | 1.7 | 2.04 | 0.5 | 0.48 | 0.48 | 2.3 | 0.37 | 0.39 | 6.2 | 2 | 4.3 | 1.0 | 13 | 17 | 6.7 | 2.7 | 3.0 | 5.5 |
| Sb | 0.13 | 0.156 | 6.1 | 0.08 | 0.07 | 17 | 0.3 | 0.121 | 5.8 | 0.4 | 0.48 | 6.2 | 0.4 | 0.387 | 1.6 | 0.2 | 0.14 | 7.7 |
| Cs | 0.1 | 0.1 | 0.2 | 0.21 | 0.199 | 0.7 | 10 | 13.57 | 0.7 | 6.3 | 6.5 | 4.9 | 3 | 2.6 | 7.1 | 6 | 5.7 | 2.8 |
| Ba | 131 | 129.6 | 0.6 | 63 | 63.0 | 1.3 | 27 | 21.6 | 1.1 | 385 | 363 | 1.5 | 55 | 51 | 5.1 | 840 | 750.0 | 0.1 |
| La | 15.2 | 15.6 | 0.9 | 3.6 | 3.61 | 1.0 | 0.35 | 0.3 | 7.3 | 21.5 | 20.0 | 5.6 | 59 | 51 | 6.4 | 40 | 34 | 10 |
| Ce | 37.5 | 36.3 | 0.6 | 8.08 | 8.1 | 1.9 | 0.8 | 0.74 | 5.1 | 46 | 45.0 | 3.6 | 154 | 131 | 6.0 | 76 | 59.0 | 3.4 |
| Pr | 5.35 | 5.25 | 0.7 | 1.14 | 1.14 | 3.0 | 0.12 | 0.113 | 5.7 | 5.7 | 5.6 | 2.6 | 22.2 | 18.0 | 5.4 | 8.3 | 6.9 | 9.3 |
| Nd | 24.5 | 24.4 | 0.8 | 5 | 5.3 | 1.0 | 0.6 | 0.59 | 5.1 | 23.5 | 23.5 | 1.7 | 92 | 80 | 5.2 | 27 | 24 | 7.6 |
| Sm | 6.07 | 6.15 | 0.7 | 1.49 | 1.45 | 3.0 | 0.2 | 0.21 | 7.5 | 5.4 | 5.32 | 0.9 | 24.2 | 22 | 5.4 | 5 | 4.4 | 4.1 |
| Eu | 2.07 | 2.07 | 1.9 | 0.62 | 0.617 | 1.4 | 0.08 | 0.08 | 4.8 | 1.45 | 1.46 | 2.0 | 2 | 1.74 | 5.5 | 1.08 | 0.93 | 2.4 |
| Gd | 6.24 | 6.1 | 1.7 | 1.63 | 1.64 | 1.4 | 0.3 | 0.32 | 4.6 | 4.7 | 5.24 | 1.0 | 26 | 24 | 5.0 | 3.8 | 3.4 | 3.4 |
| Tb | 0.92 | 0.943 | 0.6 | 0.26 | 0.28 | 4.7 | 0.06 | 0.064 | 4.6 | 0.77 | 0.77 | 1.9 | 4.8 | 4.5 | 4.6 | 0.6 | 0.52 | 3.1 |
| Dy | 5.31 | 5.28 | 1.3 | 1.56 | 1.78 | 3.4 | 0.38 | 0.42 | 3.9 | 4.6 | 4.84 | 0.8 | 29 | 28 | 4.8 | 3.3 | 3.0 | 4.1 |
| Ho | 0.98 | 1.004 | 0.6 | 0.33 | 0.37 | 3.1 | 0.09 | 0.099 | 3.2 | 1 | 0.96 | 1.9 | 6.5 | 6.1 | 5.0 | 0.7 | 0.61 | 5.8 |
| Er | 2.54 | 2.54 | 1.8 | 1 | 1.06 | 1.8 | 0.28 | 0.3 | 5.0 | 2.5 | 2.78 | 1.8 | 17.7 | 17.6 | 5.2 | 1.9 | 1.7 | 6.5 |
| Tm | 0.33 | 0.34 | 3.7 | 0.14 | 0.147 | 3.8 | 0.045 | 0.045 | 4.5 | 0.39 | 0.39 | 2.5 | 2.6 | 2.6 | 5.1 | 0.3 | 0.26 | 7.7 |
| Yb | 2 | 2 | 1.1 | 1.06 | 0.96 | 2.9 | 0.28 | 0.309 | 2.5 | 2.5 | 2.57 | 1.9 | 17.4 | 16.8 | 5.3 | 2 | 1.8 | 6.1 |
| Lu | 0.274 | 0.279 | 2.6 | 0.129 | 0.141 | 4.6 | 0.045 | 0.047 | 3.6 | 0.4 | 0.389 | 1.2 | 2.45 | 2.3 | 5.8 | 0.3 | 0.26 | 8.7 |
| Hf | 4.36 | 4.43 | 0.9 | 0.84 | 0.83 | 4.0 | 0.1 | 0.13 | 6.5 | 3.5 | 3.1 | 11 | 27.9 | 27 | 6.5 | 4 | 3.8 | 17 |
| Ta | 1.14 | 1.141 | 0.6 | 0.13 | 0.138 | 2.3 | 0.02 | 0.009 | 24 | 0.6 | 0.56 | 2.2 | 6.4 | 6.2 | 5.8 | 1.3 | 1.13 | 5.7 |
| Pb | 1.6 | 1.77 | 3.8 | 1.64 | 1.64 | 2.7 | 13 | 15 | 6.7 | 55 | 60 | 4.2 | 39 | 34 | 7.2 | 30 | 25.3 | 1.5 |
| Th | 1.22 | 1.24 | 1.0 | 0.45 | 0.35 | 6.4 | 0.07 | 0.048 | 7.6 | 5 | 5.0 | 33 | 18.5 | 18 | 11 | 17 | 15 | 16 |
| U | 0.403 | 0.45 | 1.6 | 0.1 | 0.1 | 3.7 | 0.07 | 0.053 | 9.0 | 1.5 | 1.65 | 4.8 | 4.6 | 4.4 | 7.6 | 5 | 4.33 | 1.4 |
3.7 Accuracy of analytical results for some geochemical rock CRMs
After optimization of our method for generating precise data from undiluted nano-particulate pressed powder tablets of diorite DR-N and basalt BHVO-2, a more comprehensive selection of certified reference materials was used for testing the accuracy of this advanced sample preparation technique. The geochemical rock CRMs UB-N (serpentinite), JGb-1 (gabbro), and AC-E, GA (granites) were re-milled and analyzed by LA-ICPMS.A large number of reports on trace element determinations using LA-ICPMS conclude that after an initial non-matrix matched calibration with NIST glasses a second calibration step with matrix-matched standards is obligatory to obtain accurate results. Non-matrix matched calibration with NIST SRM610-614 glasses fails for some elements.45 Here, matrix-matched calibration was achieved by calculating average “bias factors” for each element that were derived from the analysis of pressed powder tablets of basalts BCR-2 and BIR-1. These factors were applied to rock types with broadly “similar” chemical matrix composition, for example basalt BHVO-2 and diorite DR-N. Fig. 5 compares our results for DR-N and BHVO-2 with preferred values.55 Concentrations of 38 trace elements in BHVO-2 can be reproduced within ±5%rel. for most elements, only Sn, Sb, W, and U show a larger deviation of ∼10%rel. Larger errors of the results for these elements are also known from compiled data forming the database for preferred values in GeoReM. Typically, these data were generated from the LA multi-element analysis of glasses with no dedicated analytical protocol for these elements. This underlines the need for more robust data for such elements that are more volatile and occur typically in concentrations close to detection limits of current LA-ICP-MS systems. Elevated Zn and Pb in our data might be due to contamination from sample handling during the wet-milling procedure. The results for DR-N are similar but with higher deviation ≦10%rel. Additional data for CRMs are summarized in Table 6 and demonstrate the improved capabilities of this sample preparation technique with respect to precision and accuracy for a large number of elements (preferred values for BHVO-2,56 JGb-1,57 UB-N, DR-N, AC-E, and GA55).
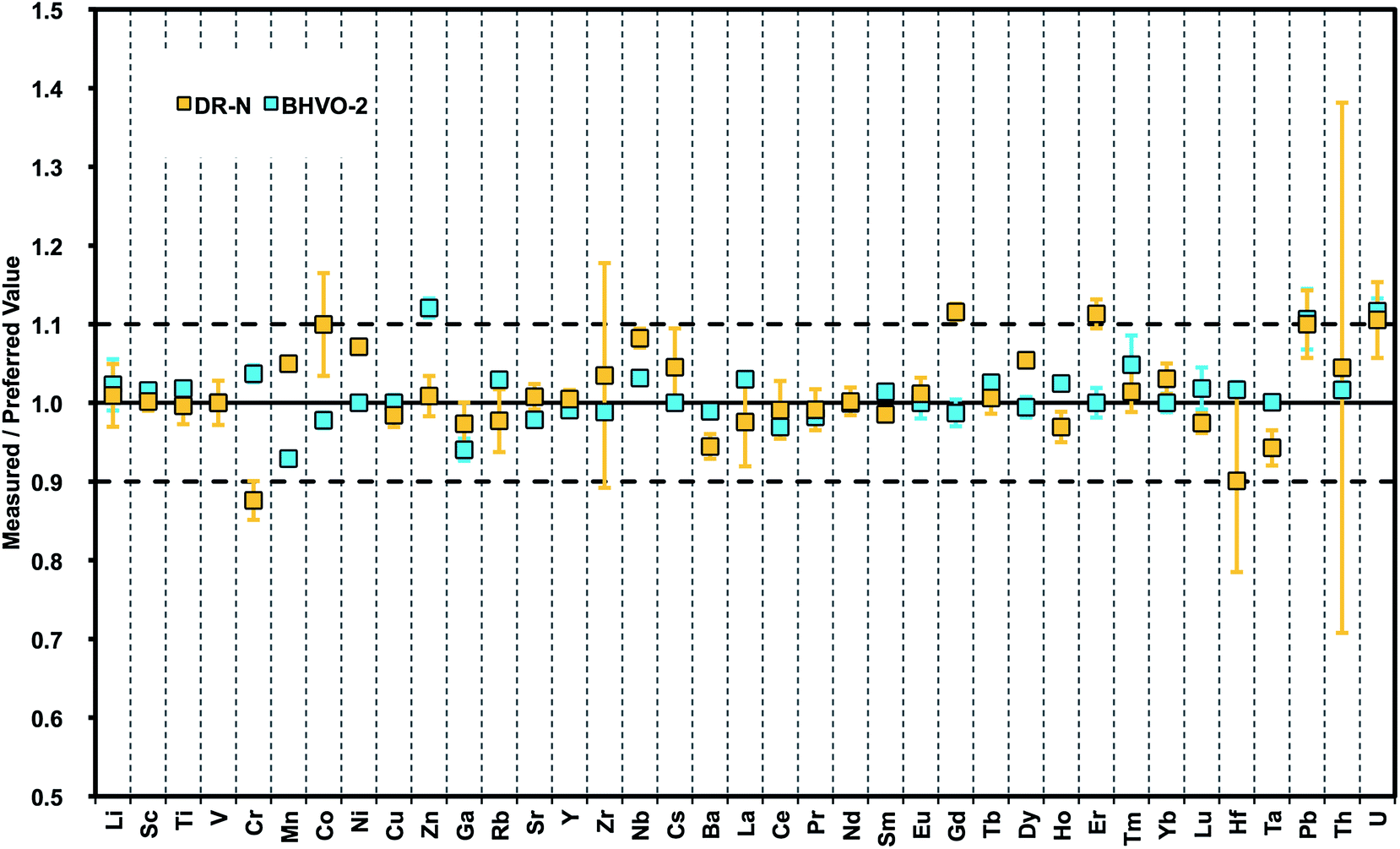 | ||
| Fig. 5 Plot of results from LA-ICPMS analysis of nano-particulate pressed powder tablets for 38 trace elements normalized to preferred values (GeoReM): basalt BHVO-2 and granodiorite DR-N. | ||
4 Conclusions
We developed, optimized, and applied an advanced method for manufacturing undiluted nano-particulate pressed powder tablets without addition of any binder by applying wet-milling protocols in aqueous suspension using a planetary ball mill and agate tools.58 The method was optimized using pulverized diorite DR-N with abundant zircon crystals that are especially difficult to grind and to digest in acid-digestion methods. The method produces nano-particulate powders with a typical grain size of d50 < 1.5 μm forming pressed powder tablets with excellent cohesion, homogeneity and mechanical stability suitable for laser ablation micro-analysis. The optimized protocol was further validated on a selection of other geochemical CRMs covering a wide range of natural rocks. Precision was found to be in the same range of <2–5%RSD as it is obtained from ablation of glasses (e.g., volcanic glasses and vitrified rocks i.e., glasses produced from melting of natural rock powders), and this is significantly better than data and errors reported from the analysis of pressed powder tablets in previous studies.59 Preferred values could be reproduced within their uncertainty for most of the 38 trace elements analyzed. Since no binder is needed for the preparation of the tablets detection limits in the low ppb range can be achieved that are comparable to those obtained with the analysis of undiluted rock glass materials. Moreover, volatile elements are still quantitatively present in the pressed powders.To obtain optimum performance during wet milling and maximum homogeneity in nano-particulate powders, milling protocols (ball-to-powder ratio, water-to-powder ratio, milling time etc.) have to be adapted to rock types having different mineralogical compositions and properties of minerals with respect to hardness, cleavage, water-uptake (clay minerals, phyllosilicates, serpentine) etc. For any material grain size distribution obtained from wet milling should be kept in a narrow, nano-particle size range d50 < 1 μm in order to obtain homogeneous and cohesive powder tablets.
Few elements showed not yet fully satisfying precision and accuracy (e.g., Zn, Mo, Sb, Sn, W) and need further attention in future studies. The observed discrepancies might be partly due to “fractionation” as a consequence of LA conditions that were not specifically optimized for more volatile elements (Sb, Sn). However, some of the inhomogeneity observed in our pressed powder tablets indicates contamination by particles that may have been introduced during the sample preparation procedure (Zn, Mo, W). One source of contamination could be the anvil and dies used in the tablet press that are usually made of alloyed steel or tungsten carbide.
Our new method opens up at least three new prospects: (i) preparation of refractory sample materials (e.g., oxides as in banded iron formation, ceramics), or materials containing refractory phases (sediments, granites and plutonic rocks in general) for subsequent direct bulk analysis of these materials by LA-ICPMS;22,60 (ii) preparation of mono-mineral RMs that are homogeneous on the micro-scale and suitable for LA-ICPMS micro-analysis. The manufactured highly cohesive tablets can be used for the matrix-matched calibration of in situ geochemical and isotopic analyses of (natural) minerals. Rather than a glass, mineral powder still contains the elements in their natural mineral lattice position. This opens up new approaches for interesting comparative experiments looking into matrix-dependent absorption of laser radiation in minerals and glass, respectively. Our initial and preliminary data with respect to matrix effects indicate significantly different ablation yields for basaltic and granitic matrices but these data are still too limited to be shown here; (iii) mixing and homogenization of different nano-particulate powders may also offer the opportunity for addition of internal standards or isotopic spikes in their solid form – a further step towards making LA-ICPMS a “stand-alone” technique.
Acknowledgements
We highly appreciate the skilled assistance of Dipl.-Ing. Ulrike Westernströer during ICP-MS set-up and lab operation. Simon Nordstad and Leewe Schönberg helped with careful sample preparation and laser ablation ICP-MS measurements, and Daniel Unverricht with grain size analysis. Thoughtful comments by Jan Košler to an earlier version of this manuscript, and from Wieland Hopfe (Fritsch GmbH, Idar Oberstein) are kindly acknowledged. This work was partly funded by a DFG research grant to DGS (no. Ga500/9-1).References
- J. Koch and D. Günther, Appl. Spectrosc., 2011, 65, 155A–162A CrossRef CAS PubMed.
- D. Profrock and A. Prange, Appl. Spectrosc., 2012, 66, 843–868 CrossRef PubMed.
- A. J. Edmund, S. D. Bergeson, M. Lyon, N. Taylor, I. Kalinitchenko and P. B. Farnsworth, Spectrochim. Acta, Part B, 2012, 76, 109–118 CrossRef CAS PubMed.
- B. Hattendorf and D. Günther, J. Anal. At. Spectrom., 2000, 15, 1125–1131 RSC.
- Z. Hu, S. Gao, Y. Liu, S. Hu, H. Chen and H. Yuan, J. Anal. At. Spectrom., 2008, 23, 1093–1101 RSC.
- M. Guillong, I. Horn and D. Günther, J. Anal. At. Spectrom., 2001, 17, 8–14 RSC.
- M. Guillong and C. A. Heinrich, J. Anal. At. Spectrom., 2007, 22, 1488–1494 RSC.
- J. Kosler, S. E. Jackson and Z. Yang, J. Anal. At. Spectrom., 2014, 29, 832–840 RSC.
- I. Kroslakova and D. Günther, J. Anal. At. Spectrom., 2006, 22, 51–62 RSC.
- L. Flamigni, J. Koch and D. Günther, Spectrochim. Acta, Part B, 2012, 76, 70–76 CrossRef CAS PubMed.
- A. El Korh, Chem. Geol., 2014, 371, 46–59 CrossRef CAS PubMed.
- M. Wiedenbeck, et al. , Geostand. Newsl., 1995, 19, 1–23 CrossRef CAS.
- J. Slama, J. Košler, D. J. Condon, J. L. Crowley, A. Gerdes, J. M. Hanchar, M. S. A. Horstwood, G. A. Morris, L. Nasdala, N. Norberg, U. Schaltegger, B. Schoene, M. N. Tubrett and M. J. Whitehouse, Chem. Geol., 2008, 249, 1–35 CrossRef CAS PubMed.
- C. C. Wohlgemuth-Ueberwasser, C. Ballhaus, J. Berndt, V. Stotter née Paliulionyte and T. Meisel, Contrib. Mineral. Petrol., 2007, 154, 607–617 CrossRef CAS.
- T. Ulrich and B. S. Kamber, Geostand. Geoanal. Res., 2013, 37, 169–188 CrossRef CAS.
- K. P. Jochum, M. Willbold, I. Raczek, B. Stoll and K. Herwig, Geostand. Geoanal. Res., 2005, 29, 285–302 CrossRef CAS.
- K. P. Jochum, B. Stoll, K. Herwig, M. Willbold, A. W. Hofmann, M. Amini, S. Aarburg, W. Abouchami, E. Hellebrand and B. Mocek, Geochem., Geophys., Geosyst., 2006, 7 DOI:10.1029/2005GC001060.
- J. S. Fedorowich, J. P. Richards, J. C. Jain, R. Kerrich and J. Fan, Chem. Geol., 1993, 106, 229–249 CrossRef CAS.
- B. Stoll, K. P. Jochum, K. Herwig, M. Amini, M. Flanz, B. Kreuzburg, D. Kuzmin, M. Willbold and J. Enzweiler, Geostand. Geoanal. Res., 2008, 32, 5–26 CrossRef CAS.
- F. Nehring, D. E. Jacob, M. G. Barth and S. F. Foley, Microchim. Acta, 2008, 160, 153–163 CrossRef CAS.
- L. Zhu, Y. Liu, Z. Hu, Q. Hu, X. Tong, K. Zong, H. Chen and S. Gao, Geostand. Geoanal. Res., 2013, 37, 207–229 CrossRef CAS.
- A. L. Gray, Analyst, 1985, 110, 551–556 RSC.
- A. A. Van Heuzen, Spectrochim. Acta, Part B, 1991, 46, 1803–1817 CrossRef.
- W. T. Perkins, R. Fuge and N. J. G. Pearce, J. Anal. At. Spectrom., 1991, 6, 445–449 RSC.
- W. T. Perkins, N. Pearce and T. E. Jeffries, Geochim. Cosmochim. Acta, 1993, 57, 475–482 CrossRef CAS.
- H. Cousin and B. Magyar, Microchim. Acta, 1994, 113, 313–323 CrossRef CAS.
- C. D. Garbe-Schönberg and G. M. McMurtry, Fresenius' J. Anal. Chem., 1994, 350, 264–271 CrossRef.
- C. A. Morrison, D. D. Lambert, R. Morrison, W. W. Ahlers and I. A. Nicholls, Chem. Geol., 1995, 119, 13–29 CrossRef CAS.
- E. R. Denoyer, J. Anal. At. Spectrom., 1992, 7, 1187–1193 RSC.
- A. Stankova, N. Gilon, L. Dutruch and V. Kanický, J. Anal. At. Spectrom., 2011, 26, 443–449 RSC.
- C. O'Connor, M. R. Landon and B. L. Sharp, J. Anal. At. Spectrom., 2007, 22, 273–282 RSC.
- M. E. Shaheen, M. E. Shaheen, B. J. Fryer and B. J. Fryer, Spectrochim. Acta, Part B, 2011, 66, 627–636 CrossRef CAS PubMed.
- Y. Zhu, Y. Zhu, A. Hioki, A. Hioki, K. Chiba and K. Chiba, J. Anal. At. Spectrom., 2013, 28, 301–306 RSC.
- M. Pakieła, M. Wojciechowski, B. Wagner and E. Bulska, J. Anal. At. Spectrom., 2011, 26, 1539–1543 RSC.
- M. Thompson, M. Thompson, J. E. Goulter, J. E. Goulter, F. Sieper and F. Sieper, Analyst, 1981, 106, 32–39 RSC.
- N. J. Pearce, W. T. Perkins and R. Fuge, J. Anal. At. Spectrom., 1992, 7, 595–598 RSC.
- C.-A. Craig, K. E. Jarvis and L. J. Clarke, J. Anal. At. Spectrom., 2000, 15, 1001–1008 RSC.
- J. G. Williams and K. E. Jarvis, J. Anal. At. Spectrom., 1993, 8, 25–34 RSC.
- M. Tibi and K. G. Heumann, J. Anal. At. Spectrom., 2003, 18, 1076–1081 RSC.
- S. F. Boulyga and K. G. Heumann, J. Anal. At. Spectrom., 2004, 19, 1501–1503 RSC.
- B. Fernández, F. Claverie, C. Pécheyran and O. F. Donard, J. Anal. At. Spectrom., 2008, 23, 367–377 RSC.
- N. Imai, Anal. Chim. Acta, 1990, 235, 381–391 CrossRef CAS.
- M. Motelica Heino, O. Donard and J. M. Mermet, J. Anal. At. Spectrom., 1999, 14, 675–682 RSC.
- L. Arroyo, T. Trejos, P. R. Gardinali and J. R. Almirall, Spectrochim. Acta, Part B, 2009, 64, 16–25 CrossRef PubMed.
- K. Ito, N. Hasebe, A. Hasebe and S. Arai, Geochem. J., 2011, 45, 375 CrossRef CAS.
- R. Arbain, M. Othmann and S. Palaniandy, Miner. Eng., 2011, 24, 1–9 CrossRef CAS PubMed.
- T.-H. Hou, C.-H. Su and W.-L. Liu, Powder Technol., 2007, 173, 153–162 CrossRef CAS PubMed.
- D. Tabersky, N. Luechinger, S. Halim, M. Rossier and D. Günther, Mineral. Mag., 2013, 77, 2297 CrossRef PubMed.
- J. Slama and J. Košler, Geochem., Geophys., Geosyst., 2012, 13 DOI:10.1029/2012GC004106.
- F. M. Etzler and M. S. Sanderson, Part. Part. Syst. Charact., 1995, 12, 217–224 CrossRef CAS.
- F. M. Etzler and R. Deanne, Part. Part. Syst. Charact., 1997, 14, 278–282 CrossRef CAS.
- M. B. Fricker, D. Kutscher, B. Aeschlimann, J. Frommer, R. Dietiker, J. Bettmer and D. Günther, Int. J. Mass Spectrom., 2011, 307, 39–45 CrossRef CAS PubMed.
- W. L. Griffin, W. J. Powell, N. J. Pearson and S. Y. O'Reilly, Laser Ablation-ICP-MS in the Earth Sciences, Mineral. Assoc. Can., Short Course Ser., 2008, 40, 204–207 Search PubMed.
- K. P. Jochum, U. Weis, B. Stoll, D. Kuzmin, Q. Yang, I. Raczek, D. E. Jacob, A. Stracke, K. Birbaum, D. A. Frick, D. Günther and J. Enzweiler, Geostand. Geoanal. Res., 2011, 35, 397–429 CrossRef CAS.
- K. Govindaraju, Geostand. Newsl., 1995, 19, 1–32 CrossRef CAS.
- K. P. Jochum, U. Nohl, K. Herwig, E. Lammel, B. Stoll and A. W. Hofmann, Geostand. Newsl., 2005, 29, 333–338 CrossRef CAS.
- N. Imai, S. Terashima, S. Itoh and A. Ando, Geostand. Newsl., 1995, 19, 135–213 CrossRef CAS.
- S. Müller and C. D. Garbe-Schönberg, Mineral. Mag., 2013, 77, 1806 Search PubMed.
- P. K. Mukherjee, P. P. Khanna and N. K. Saini, Geostand. Geoanal. Res., 2014 DOI:10.1111/j.1751-908X.2013.012015.x.
- B. J. Fryer, S. E. Jackson and H. P. Longerich, Can. Mineral., 1995, 33, 303–312 CAS.
| This journal is © The Royal Society of Chemistry 2014 |