Sweeping lymph node micrometastases off their feet: an engineered model to evaluate natural killer cell mediated therapeutic intervention of circulating tumor cells that disseminate to the lymph nodes†
Siddarth
Chandrasekaran
,
Matthew J.
McGuire
and
Michael R.
King
*
Department of Biomedical Engineering, Cornell University, Weill Hall, Ithaca, NY 14853, USA. E-mail: mike.king@cornell.edu; Tel: +1 607-255-9803
First published on 25th July 2013
Abstract
Approximately 90% of cancer related deaths are due to metastasis. Cells from the primary tumor can metastasize through either the vascular or lymphatic circulation. Cancer cells in circulation are called circulating tumor cells (CTCs) and it has been shown that bone marrow is a niche for homing of blood borne CTCs from several epithelial tumors. Cancer cells found in bone marrow are termed disseminated tumor cells (DTCs). Likewise, CTCs in the lymphatic circulation are more often seeded in the sentinel lymph nodes (SLN) that drain the tumor. Micrometastases (<2 mm) occur after the arrest and implantation of DTCs in lymph nodes over time. This paper presents a cell culture platform termed microbubbles formed in polydimethylsiloxane (PDMS) from a microfabricated silicon wafer for mimicking lymph node micrometastases. We cultured lymph node seeking cancer cells in microbubbles to evaluate the efficacy of natural killer (NK) mediated therapy for targeting lymph node micrometastasis. The microbubble platform consists of an array of microcavities that provides a unique microenvironment for mimicking the deep cortical unit of the lymph nodes. We show that cancer cells cultured in microbubbles with therapeutic NK cells undergo apoptosis after 24 h in culture. Since lymph node metastases are prevalent across several types of cancer, this platform may be useful for developing improved cancer therapies for targeting lymph node micrometastases.
1. Introduction
Metastatic disease is a result of dissemination of highly invasive cells from the primary tumor to a secondary site.1 The two major paths that lead to metastasis are the circulatory and the lymphatic systems.2 A cancer cell that leaves the primary tumor has distinct identities before it becomes a cell in the metastatic lesion. The cells that are found in circulation are called CTCs3 and cells that lodge in lymph nodes and bone marrow are called DTCs.2 The presence of DTCs in bone marrow and lymph nodes is a marker for haematogenous spread and lymphatic spread of cancer, respectively.4 Once cancer cells disseminate, the interplay of several factors in the microenvironment is required for these cells to establish at the secondary site. There is usually a period of dormancy associated with transformation of DTCs into occult metastases called micrometastases.5 Micrometastases are aggregates of tumor cells less than 2 mm in diameter, however some studies report isolated tumor cells in bone marrow and lymph nodes as micrometastasis.6 The presence of micrometastases in lymph nodes and bone marrow correlates with an unfavourable prognosis in cancer patients after surgical removal of the primary tumor.4,7,8Lymph nodes are structures that are dispersed along the lymphatic circulation that contain lymphocytes, macrophages and antigen presenting cells.9 The clear fluid that circulates in the lymphatic circulation called lymph holds important information about the local inflammatory response. Despite the presence of many immune cells that respond to tumor antigens, the lymph node remains the first organ of metastasis in several types of carcinomas10 and melanomas.11 This is largely because of the ability of cancer cells to evade the host immune response.12 Also, as suggested by our recent study, inflammatory cytokines such as IL-6 that are key players during host immunosurveillance can facilitate the metastatic cascade.13 The last decade has seen several immunotherapeutic approaches to engineer the host immune system to target cancer cells.14 Adoptive immunotherapy is a cell-based approach in which naturally occurring or engineered T-cells are infused into patients after expanding them in vitro to specifically target tumor cells.15 This method has been shown to reduce the metastatic lesions in the liver of a patient with pancreatic cancer16 and several secondary organ metastases in metastatic melanoma patients.17
NK cells are the most aggressive subset of T-cells representing 5–15% of total circulating lymphocytes.18 NK cells deliver lethal bursts of toxins that activate apoptotic pathways in cancer cells.19 One of the most important toxins is tumor necrosis factor-α related apoptosis inducing ligand (TRAIL), which is increasingly being explored as a potential chemotherapeutic agent.19 TRAIL binds to cognate death receptors (DR4 and DR5) expressed by cancer cells and triggers apoptotic signalling pathways.20 NK cells kill most of the tumor cells that enter the peripheral circulation,21 drastically reducing their number in whole blood which is why circulating tumor cells by definition are “rare cells”.22 However, NK cells in cancer patients have several abnormalities, ranging from reduced count, decreased cytotoxicity and poor tumor infiltrating capacity, to name a few.23 Most of these abnormalities are exaggerated in patients after surgical removal of a primary tumor due to immunosuppression from chemotherapy, which is often an adjuvant therapy after tumor resectioning.24,25 The goal of adjuvant chemotherapy is to eradicate micrometastases, but more often the treatment favors micrometastatic lesions. Thus, the development of more targeted adjuvant therapies has seen significant advancements in the past decade.26 In the last few years, technological advances in cancer detection have led to the identification of these occult metastatic lesions and their characterization.27 SLN are the first set of lymph nodes draining the primary tumor and there is a growing body of data that suggests the involvement of SLN in a variety of tumors that has led to the evolution of the term called “micrometastatic nodal disease”.28–33 Additionally, SLN in cancer patients show morphological and functional variation suggesting possible immune suppression, which may potentially result in failure to eliminate micrometastatic lesions.34,35 This could potentially explain why SLN is a niche for cancer cells in the lymphatic circulation despite being a home for immune cells. Thus, augmenting the host immune response can reverse the immune suppression in SLN. Recently, several targeted therapeutic approaches have shown reductions in the incidence of SLN metastases in animal models.36,37 Of the several nanoscale drug delivery systems developed, liposome based drug delivery has been successfully used and commercialized in the treatment of Kaposi's sarcoma (Doxil®) and breast cancer (Myocet®).38 Liposome based drug carriers are advantageous because of their small size, tunable membrane properties and long circulating times. In this study, we conjugate TRAIL-functionalized liposomes to NK cells for targeting lymph node micrometastases. NK cells are present in the paracortex of lymph nodes and studies suggest that NK cells are confined to the deep cortical unit in the paracortex of lymph nodes.39 Deep cortical units are semi-rounded structures containing T-cells40,41 (Fig. 1A). Immunohistochemical analyses of lymph node biopsies often map micrometastases to the paracortical region of lymph nodes.42 Also, more advanced studies based on spectral detection of lymph node micrometastases map small clusters of cancer cells with T-lymphocytes in the paracortex region.43 Given the co-existence of NK cells and cancer cells within the lymph node, manipulating NK cells for treating micrometastases is a rational approach.
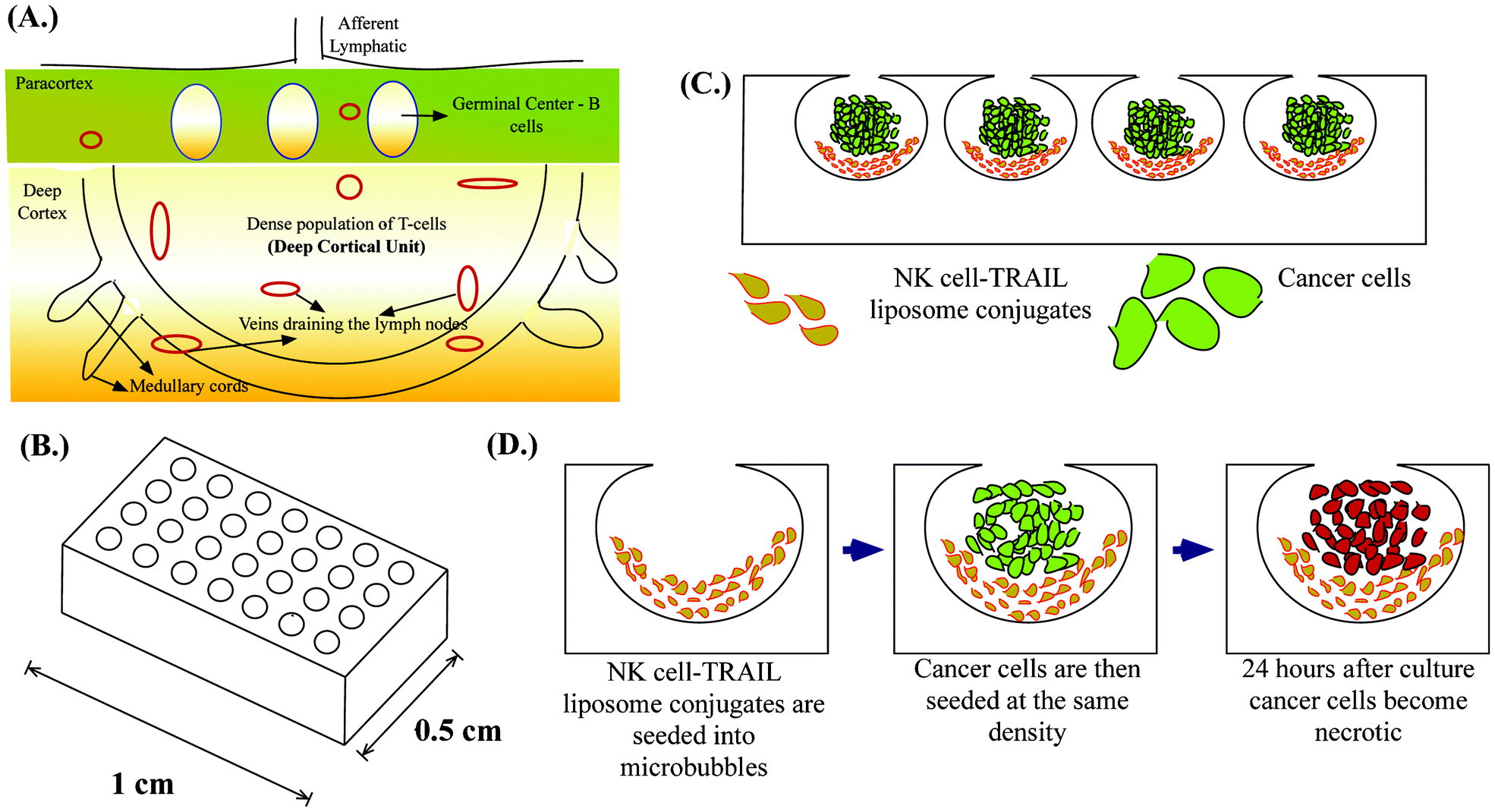 | ||
| Fig. 1 Schematic diagram of (A.) Deep cortical unit of a lymph node (B.) Array of MB on a single chip (C.) Cancer cells co-cultured with NK cell-TRAIL liposome conjugates in MB (D.) Single MB showing our proposed approach. | ||
Microfabrication has led to the development of several 3D cell culture platforms using bioinspired materials to study the important molecular signatures of cancer progression.44 An ideal in vitro tumor model should recapitulate the microenvironment of tumors by facilitating cell–cell interactions, cell–extracellular matrix (ECM) interactions, soluble factor accumulation and signalling, and physiologically relevant mechanical loading.45 We previously introduced deep reactive ion etched (DRIE) silicon wafers for forming spherical cavities termed microbubbles (MB) in PDMS by a process called gas expansion molding (GEM).46 It has been shown that the microbubble array on a chip (Fig. 1B) is well suited for studying the tumor microenvironment because of its unique geometry and ability to recapitulate the tumor microenvironment.47 We have previously shown that MB can be used for studying the clonogenic potential of cancer stem cells48 and can be used as a model for tumor spheroids when they are continuously perfused under physiologically relevant flow conditions.49 Here we demonstrate a unique application of MB to mimic lymph node micrometastases. Their semi-rounded geometry mimic the deep cortical unit of lymph nodes and culturing engineered NK cells with cancer cells in MB arrays (Fig. 1C) provides a platform to evaluate the efficacy of our proposed therapeutic approach to target lymph node micrometastases (Fig. 1D).
2. Materials and methods
2.1 Microfabrication of silicon wafers and microbubble formation
A lithography mask was designed using AutoCAD LT 2008 (Autodesk, Inc., USA). The mask included three different types arrays of rectangular trenches with 60 μm, 100 μm and 200 μm circular openings. Depth of the trenches was 150 μm. Silicon wafers were produced from this mask (MEMS and Nanotechnology Exchange, Virginia USA) using the Bosch deep reactive ion etching (DRIE) process. The hydrophobic coating produced during the Bosch process was left intact. The DRIE silicon wafers were used to produce microbubbles in PDMS. Dow Corning's Sylgard® 184 silicone elastomer kit in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 base to curing agent ratio (w/w) was manually mixed in a 50 mL tube for 2 min and poured onto silicon wafers. The mixture was allowed to spread out on the wafer for 30 min at room temperature and cured at 100 °C for 2 h. The PDMS was then peeled off from the wafers and the arrays were cut in small chips to be used in cell culture experiments. The experiments described in this work were all performed on microbubbles formed from the 100 μm circular openings on the wafer.
:1 base to curing agent ratio (w/w) was manually mixed in a 50 mL tube for 2 min and poured onto silicon wafers. The mixture was allowed to spread out on the wafer for 30 min at room temperature and cured at 100 °C for 2 h. The PDMS was then peeled off from the wafers and the arrays were cut in small chips to be used in cell culture experiments. The experiments described in this work were all performed on microbubbles formed from the 100 μm circular openings on the wafer.
2.2 Cell lines and culture conditions
Three lymph node seeking metastatic cell lines from different types of cancers were used in this study. LNCaP (prostate cancer), COLO 205 (colorectal cancer) and MDA-MB-231 (breast cancer) were purchased from American Type Culture Collection (ATCC) and maintained in culture at 37 °C with 5% CO2 in humidified incubator. LNCaP and COLO 205 cells were cultured in RPMI-1640 (Cellgro, 10-040-CV) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, S11050H), and 1% penicillin/streptomycin (PS) (Gibco®, 15140-122). MDA-MB-231 cells were cultured in high glucose DMEM (Gibco®, 11965-092) supplemented with 10% FBS and 1% PS. HNK-1, a hybridoma cell line (ATCC® TIB-200TM) was cultured in RPMI-1640 (ATCC® 30-2001TM) supplemented with 0.02 mM 2-mercaptoethanol (Gibco®, 21985-023) and 20% ultra low IgG FBS (Gibco®, 16250-078). The hybridoma line secretes anti-CD57 (IgM isotype), a monoclonal antibody against human natural killer cells.502.3 Preparation of TRAIL and anti-CD57 functionalized liposomes
Hydro Soy PC L-α-phosphatidylcholine (HSPC), cholesterol, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-mPEG2000) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol)-2000] (DSPE-Mal-mPEG2000) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Liposomes were composed of HSPC:cholesterol:DSPE-mPEG200:DSPE-Mal-mPEG2000 at a molar ratio of 2:1:0.08:0.02. Lipids were mixed to produce a total of 10 mM lipid in 1 mL and kept in a glass vial covered with parafilm inside a vacuum glass chamber overnight to remove residual organic solvent. The lipid cake was then hydrated in 1 mL of buffer composed of 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 150 mM sodium chloride in phosphate buffered saline (PBS) at pH 7.5. The hydrated lipids formed multi-lamellar vesicles which were then disrupted by 10–15 freeze (2 min)/thaw (3 min) cycles before extrusion. The lipid suspension was then forced through a polycarbonate filter with successively smaller pore size (400 nm, 200 nm and 100 nm) mounted on a holder in a heating block (Avanti®mini-extruder). The temperature was maintained at 60 °C, which is above the phase transition temperature of the lipids.
Anti-CD57 was isolated from hybridoma supernatant using a protein-L agarose column (Thermo Scientific, 20510) following a protocol supplied by the manufacturer for gravity-flow based isolation of proteins. Recombinant human TRAIL (Peprotech 310-04) and anti-CD57 were thiolated using 1 mM Traut's reagent (Thermo Scientific, 26101) added in 6-fold molar excess to the proteins in solution. Excess Traut's reagent was removed using a desalting column (Thermo Scientific, 89889) by spinning the reaction mixture three times for 2 min at 1000 × g. Thiolated proteins were then functionalized on the surface of liposomes overnight at 4 °C via maleimide-thiol chemistry by adding an appropriate amount of protein to set the concentration of maleimide in liposomes at 6-fold molar excess to the concentration of thiol groups in proteins. The protein- functionalized liposomes were stored at 4 °C for up to a week and prepared fresh whenever required. Successful functionalization of proteins to liposomes was determined by measuring the average particle size by dynamic light scattering using a Malvern Zetasizer nano ZS (Malvern Instruments Ltd., UK).
To produce fluorescent liposomes used in this study, cholesterol in the lipid composition was replaced with TopFluor cholesterol (Avanti polar lipids, 810225). The same protocol was used to make fluorescent liposomes by minimizing the exposure to light.
2.4 Conjugation of liposomes to natural killer cells
Natural killer cells were isolated from whole blood of healthy volunteers following informed consent using the RosetteSepTM human NK cell enrichment cocktail (Stemcell technologies, 15065) using a protocol supplied by the manufacturer. The Institutional Review Board for Human Participants (IRB) at Cornell University approved the protocol for drawing blood for inclusion of human participants in research. NK cells were maintained in culture using MyelocultTM (Stemcell technologies, H5100) supplemented with 1 mM hydrocortisone and 100 U/mL of human recombinant interleukin-2 (IL-2) (Millipore, IL002).For conjugating CD57-expressing NK cells to anti-CD57 and TRAIL functionalized liposomes, NK cells were pelleted and resuspended in 200 μL of 2% bovine serum albumin (BSA). They were then incubated with a specific volume of protein- functionalized liposomes at 4 °C for 1 h. The volume of liposomes to be added to 1 × 106 NK cells was determined by incubating 1 × 106 NK cells with 5, 10, 25 and 50 μL of fluorescent liposomes. The NK cells were then washed twice with PBS to remove any unbound liposomes by centrifuging them at 1000 rpm for 10 min. The fluorescent intensity histograms from flow cytometric analysis (BD AccuriTM C6 flow cytometer) were used to determine the optimum volume of liposomes to be added to 1 × 106 NK cells. Successful conjugation of liposomes to NK cells was verified using flow cytometry and fluorescent microscopy with appropriate filters on an Olympus IX81 microscope. NK cells conjugated to TRAIL-functionalized liposomes will be referred to as “super NK cells” in the rest of this paper. To ensure that there were no deleterious effects of conjugating TRAIL-functionalized liposomes to NK cells, conjugated cells were cultured in vitro for 24 h and stained for live cell marker Calcein-AM (Molecular Probes®, C3100MP).
2.5 Characterization of cell lines for death receptor expression
Cancer cells were analysed for the expression of death receptors (DR4 and DR5) using flow cytometry (Guava EasycyteTM flow cytometer). LNCaP, COLO 205 and MDA-MB-231 cells grown to confluence in 6-well plates were washed with PBS and cells were harvested using enzyme-free cell dissociation buffer (Gibco®, 13151-014). Approximately 0.5 × 106 cells were resuspended in 100 μL 2% BSA. Cells were then incubated with 10 μL of PE-conjugated monoclonal antibody against DR4 (R&D Systems, FAB347P) and DR5 (R&D Systems, FAB6311PS) or 10 μL of corresponding isotype control (R&D Systems, IC0041P) at 4 °C for 45 min. Following incubation, unreacted antibodies were removed by washing with PBS twice by centrifugation at 1000 rpm for 5 min. Cells were resuspended in 400–600 μL of 1% BSA for flow cytometric analysis.2.6 Cell culture experiments in MB arrays
Super NK cells were co-cultured with cancer cells in MB arrays at 37 °C for 24 h. For co-culturing super NK cells and cancer cells in MB arrays, we followed a protocol described in detail previously.48 Briefly, small chips (1 cm × 0.5 cm) containing arrays of MB were rinsed in 70% ethanol and distilled water and dried with nitrogen gas. The backside of the chips was rendered hydrophilic by etching them in a plasma chamber with atmospheric air for 10 min to keep the chips submerged in cell culture media. The chips were then blocked and subjected to negative pressure in a vacuum chamber to remove the air trapped inside the microbubbles during the curing process. NK cells were conjugated to TRAIL-functionalized liposomes as described in Section 2.4 and about 0.1 × 106 super NK cells were resuspended in 200 μL cell culture media composed of RPMI-1640 supplemented with 10% FBS and 1% PS. Approximately 100 μL of the cell stock solution was applied on top of the chip and incubated for 30 min at room temperature (25 °C) under sterile conditions. After incubation, the cell solution was removed and the chip was rinsed twice to remove any cells that may have deposited on the surface outside the spherical cavities. After seeding super NK cells, about 0.1 × 106 cancer cells (LNCaP, COLO 205 and MDA-MB-231) in 200 μL of cell culture media was applied to the top of the chip and incubated for 10–15 min. The chips were rinsed twice and then placed inside a single well of a 24-well plate filled with 1 mL of cell culture media. The 24-well plate containing the MB chips was placed inside a humidified cell culture chamber maintained at 37 °C with 5% CO2.The following control conditions were used in the cell culture experiments. Before seeding MB arrays with cancer cells, MB arrays were incubated with liposome buffer, naked liposomes (liposomes without TRAIL), TRAIL-functionalized liposomes or unmodified NK cells.
2.7 In situ detection of apoptotic cancer cells
To determine the therapeutic efficacy of the proposed approach, cells cultured in MB arrays were stained in situ using an apoptosis detection kit (Trevigen®, 4840-01-K). The kit is based on Annexin V-FITC to label early apoptotic cells and propidium iodide (PI) to label late apoptotic/necrotic cells. Incubation reagent was prepared using a protocol provided by the manufacturer. The reagent was further diluted 1:10 times in 2% BSA. The MB chips were removed from 24-well plates and 100 μL of diluted reagent was applied on top of the MB chips. The chips were incubated for 30 min at room temperature in the dark. After incubation, the chips were washed gently 5–10 times by immersing them in a well containing PBS. The chips were then imaged using an Olympus IX81 microscope with appropriate filters for FITC and PI. Images were taken at 12 h and 24 h after culture to label apoptotic cancer cells.
Digital images from fluorescence microscopy were processed using ImageJ after normalizing for background intensity (NIH, USA). A region of interest was defined around each microbubble for analysis of Annexin V-FITC and PI staining. Areas were analysed for the mean intensity and the values are reported as bar graphs for a quantitative measure of apoptotic cancer cells in MB arrays. The mean pixel intensity between different treatment groups was compared for statistical significance using unpaired student t-test.
2.8 Confocal microscopy
NK cells were labelled with CellTracker green (Molecular Probes®, C7025) and LNCaP cells were labelled with CellTracker red (Molecular Probes®, C34552) and seeded on MB arrays following the protocol mentioned in Section 2.6. The chips were then transferred into a glass bottomed 24-well plate and imaged using a Zeiss 710 laser scanning confocal microscope.3. Results and discussion
3.1 Microbubble geometry closely recapitulates the anatomy of deep cortical unit of a lymph node
In this work we were interested in developing a unique application of MB arrays as a model for mimicking lymph node micrometastases. Their semi-rounded geometry with a single circular opening on the top closely resembles the semi-rounded anatomy of a deep cortical unit of a lymph node centered on the top of an afferent lymphatic. PDMS is a commonly used polymer for cell culture applications. PDMS provides a realistic mechanical loading to cells. The elastic modulus of PDMS is 50–200 kPa,51 which is very close to the modulus of several soft tissues52 including lymph nodes.53 The top view and side view of MB formed from 100 μm circular openings are shown in Supplementary Fig. 1.†3.2 Characterization of TRAIL and anti-CD57 functionalized liposome
Successful functionalization of TRAIL and anti-CD57 on the liposome surface is mediated by maleimide-thiol conjugation as indicated in Fig. 2A and 2B. NK cells isolated from whole blood were maintained in culture (Supplementary Fig. 2A†) and flow cytometry histograms indicated that NK cells were positive for CD57 expression (Supplementary Fig. 2B†). CD57 is a marker for terminal differentiation in T-cells and it was recently shown that it is expressed by a distinctly mature subpopulation of human NK cells.54 The size distribution of naked liposomes and protein-functionalized liposomes was analysed using a zetasizer. The average size of naked liposomes was 112.17 ± 1.62 nm and TRAIL and anti-CD57 functionalized liposomes were roughly 45–55 nm larger than naked liposome (Fig. 2D). The increase in size of liposomes after protein incubation indicated that TRAIL and anti-CD57 have been functionalized on the surface of liposomes.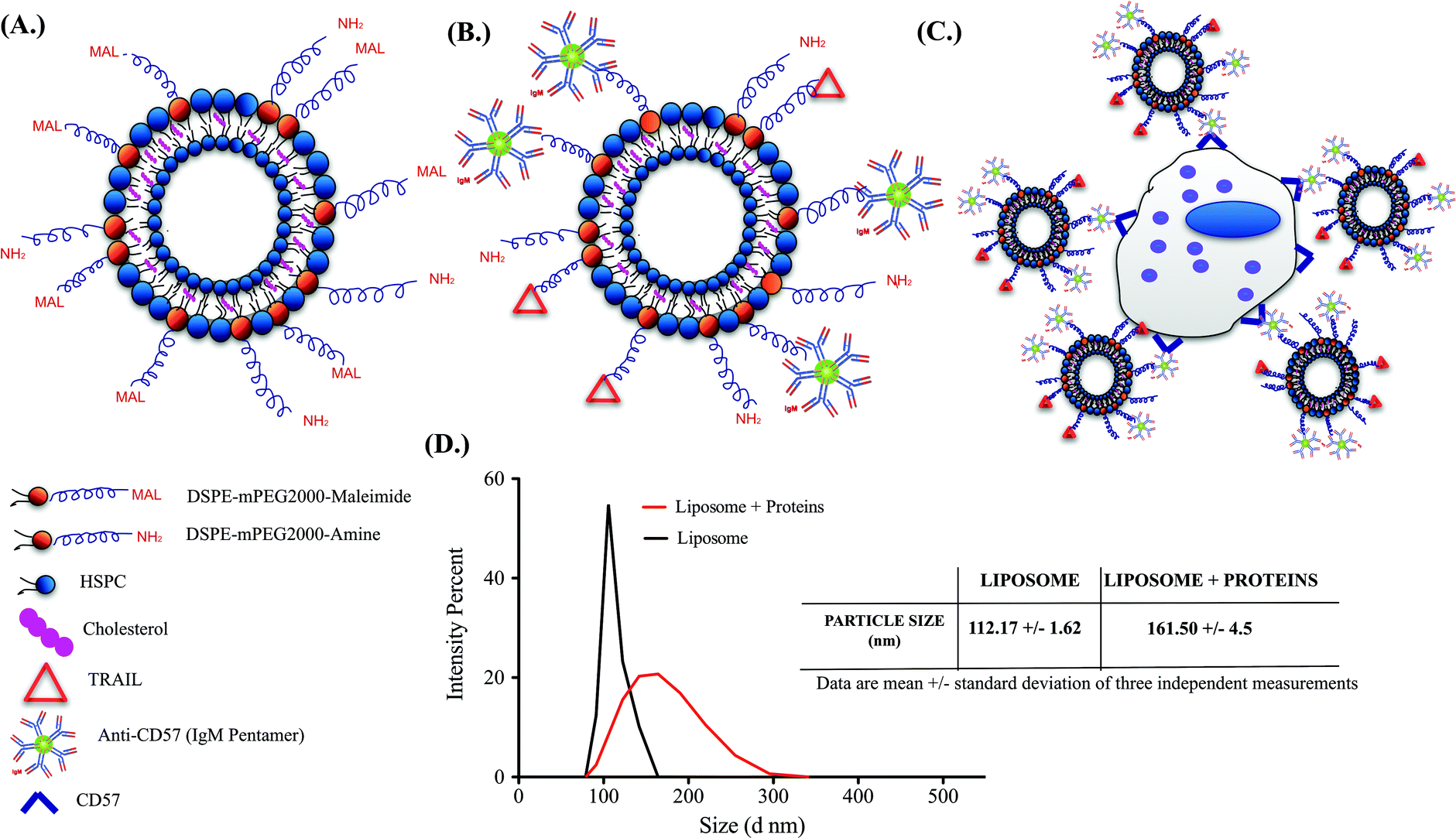 | ||
| Fig. 2 Schematics of a (A.) Naked liposome (B.) TRAIL and anti-CD57 functionalized liposome (C.) NK cell-liposome conjugate (Super NK cell) (D.) Size distribution of naked liposomes and protein-functionalized liposomes from Malvern zetasizer instrument. Table indicates the average size of particles in each group. | ||
3.3 Anti-CD57 on the liposome surface mediates conjugation of liposome to NK cells
Conjugation of TRAIL-functionalized liposomes to NK cells is mediated by CD57 expressed on the surface of NK cells and anti-CD57 functionalized on the liposome surface (Fig. 2C). Conjugating TRAIL-functionalized liposomes to NK cells did not cause any detrimental effects on NK cells (Supplementary Fig. 2C and 2D†). It has been shown that long-circulating liposomes can effectively target solid tumors in cancer patients.55 Conjugating liposomes to NK cells can further increase the circulating time, which is an important criterion for targeted drug delivery. Additionally, since the natural niche of NK cells is lymph nodes, conjugating therapeutic liposomes to NK cells can specifically target micrometastases in the lymph nodes. To ensure successful conjugation of TRAIL-functionalized liposomes to NK cells, liposomes composed of fluorescent TopFluor cholesterol were used. Fluorescent images indicated that liposomes were successfully conjugated to NK cells (Fig. 3A). Closer examination indicated that liposomes were evenly coated on the surface of NK cells (Fig. 3A inset). Flow cytometric analysis revealed that NK cells conjugated to fluorescent liposomes functionalized with anti-CD57 showed a shift in fluorescent intensity peak with respect to NK cells conjugated to non-fluorescent liposomes (Fig. 3B). To investigate any nonspecific interaction of liposomes with NK cells, NK cells were incubated with fluorescent liposomes without functionalized anti-CD57. Fluorescent intensity peak showed a small shift with respect to the control, indicating that there was minimal nonspecific interaction. Functionalizing more PEG on the liposome surface could further reduce nonspecific interactions.56 To determine the volume of liposomes needed to completely saturate the CD57 receptors on NK cells, 1 × 106 NK cells were incubated with different volumes of fluorescent liposomes. Flow cytometry histograms indicated increasing shift in fluorescent intensity peak for increasing volume of liposomes. For a treatment volume of 25 μL and 50 μL, there was no change in fluorescent intensity peak, indicating that a treatment volume of 25 μL saturated all the available CD57 receptors on the surface of NK cells (Fig. 3C).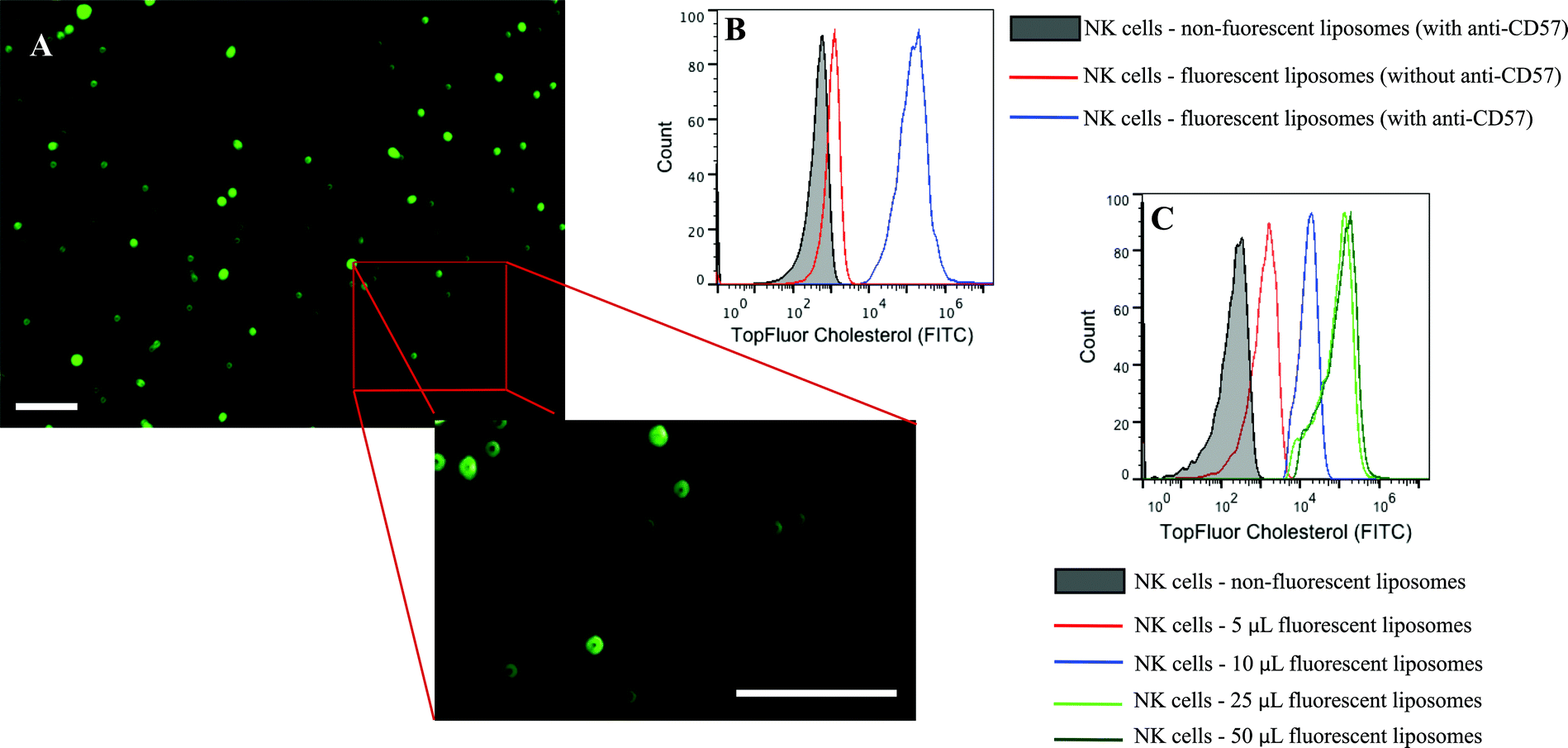 | ||
| Fig. 3 (A.) NK cells conjugated to fluorescent liposomes with inset showing even distribution of liposomes on NK cell surface. (B.) Flow cytometry histogram indicating that conjugation is anti-CD57 dependent. (C.) Flow cytometry histograms showing the fluorescent shift intensity for different treatment volume of liposomes. Scale bar = 100 μm. | ||
3.4 Cancer cell lines cultured in microbubbles form small micrometastasis-like spheroids
Micrometastases are small aggregates of cancer cells less than 2 mm in diameter and they can remain dormant for many years before forming overt metastases.57 Microfabrication has led to the development of 3D cell culture systems which have taken cancer cell lines a step closer to in vivo tumors.58 A model for micrometastasis should be able to recapitulate the in vivo microenvironment. Microenvironment plays an important role in driving cells in micrometastasis progressively towards becoming an established metastatic lesion ultimately leading to secondary metastases.5 We have shown that MB arrays can concentrate soluble factors secreted by cells to create a microenvironmental niche for cells cultured in them.47 Cancer cells seeded in MB arrays form micrometastasis-like aggregates 24 h after culture (Fig. 4). We seeded MDA-MB-231 (Fig. 4A), COLO 205 (Fig. 4B) and LNCaP (Fig. 4C) cells at similar seeding density and observed the formation of micrometastasis-like aggregates after 24 h in culture (Fig. 4D–4F).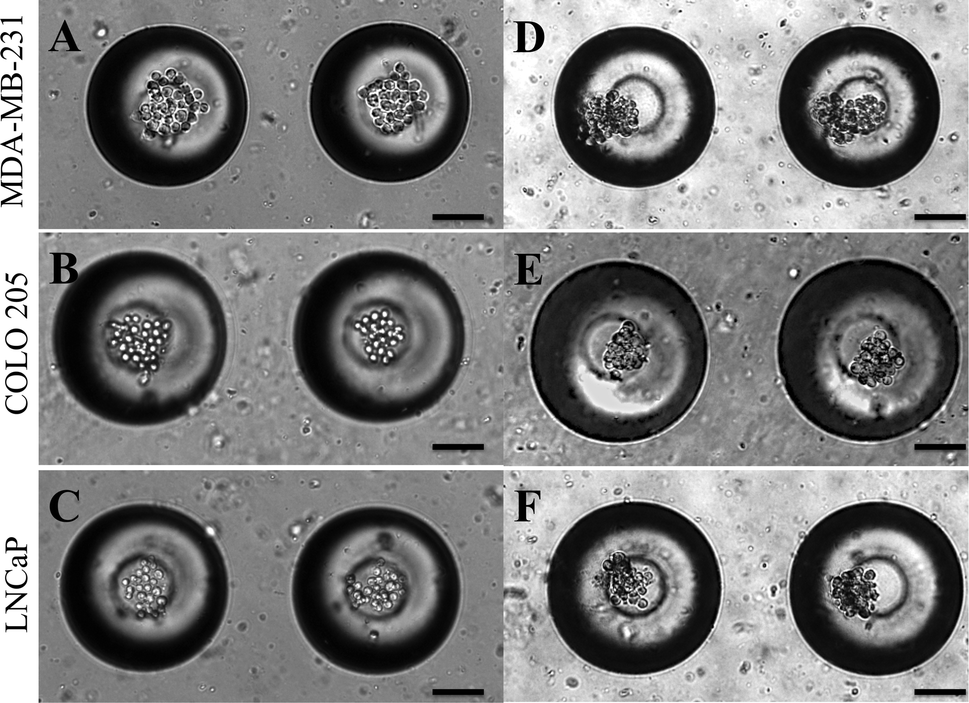 | ||
| Fig. 4 (A.) MDA-MB-231, (B.) COLO 205 and (C.) LNCaP cells seeded in MB arrays immediately after seeding and 24 h after seeding (D–F). Scale bar = 100 μm. | ||
3.5 Cancer cells progressively become apoptotic when cultured with super NK cells in MB
To determine the effectiveness of super NK cells in targeting cancer cells, super NK cells were seeded prior to seeding cancer cells in MB arrays. Confocal micrographs of LNCaP cells labelled with CellTracker red and super NK cells labelled with CellTracker green cultured in a single MB are shown in Supplementary Fig. 3.† The images shown are a superimposition of all the Z-plane images acquired with individual filters for LNCaP cells (Supplementary Fig. 3A†) and super NK cells (Supplementary Fig. 3B†), and both the channels to visualize the co-culture of LNCaP and super NK cells in a MB (Supplementary Fig. 3C†). The cell lines chosen for this study were based on reports indicating that these cells readily metastasize to the lymph nodes in experimental animal models (MDA-MB-231,59 COLO 20560 and LNCaP61). The cell lines were characterized for the expression of death receptors (DR4 and DR5). Flow cytometric analysis for surface expression of DR indicated that the three cell lines showed positive expression of death receptors (Supplementary Fig. 4†). TRAIL has been shown to induce apoptosis in cancer cells by binding to death receptors. TRAIL is a toxin that is involved in the body's natural defense mechanism that holds advantages over other chemotherapeutic drugs that can be toxic to non-cancerous cells.62 The first step taken by cells undergoing apoptosis is the loss of membrane integrity, which leads to the exposure of phospholipids. Annexin-V based apoptosis detection assay exploits this loss of membrane integrity. Annexin-V binds to exposed phospholipids in cells undergoing apoptosis and is a marker for early stages of apoptosis.63 Propidium iodide (PI) is a fluorescent membrane impermeable DNA binding stain that is a marker for dead cells those have completely lost membrane integrity.63 Fluorescein isothiocyanate (FITC) conjugated Annexin-V and PI were used to label apoptotic and dead cells in situ in MB arrays seeded with cancer cells and super NK cells. We noticed that cancer cells were progressively driven towards apoptosis. After 12 h of incubation of super NK cells with MDA-MB-231 (Fig. 5A), COLO 205 (Fig. 5B) and LNCaP cells (Fig. 8C), most of the cells were positive for Annexin-V staining (green) and very few cells stained positive for PI (red) (Fig. 5D–5F), indicating that the cells were primarily in early stages of apoptosis. After 24 h of incubation most of the cells formed a heterotypic aggregate and it was hard to distinguish between super NK cells and cancer cells (Fig. 5G–5I). Super NK cells seeded in MB arrays without cancer cells did not form aggregates but mostly remained as individual cells even 24 h after culture (Supplementary Fig. 5A†). By the end of 24 h, cells were positive for PI (red), indicating that most of the cells were dead (Fig. 5J–5L). The mean pixel intensity for Annexin-V FITC and PI positive cells was quantified using ImageJ software and the bar graphs indicate that by 12 h, most of the cells were positive for Annexin-V and by 24 h, most of the cells were positive for PI (Fig. 6A). We conclude that cells that stained positively for Annexin-V and PI in co-cultured MB arrays were cancer cells since in a few MB where cancer cells formed a separate aggregate away from super NK cells, only cancer cell aggregates stained positive for Annexin-V and PI (Supplementary Fig. 5B†). Additionally, in MB arrays with only super NK cells there was no staining for either Annexin-V or FITC (Supplementary Fig. 5B†). MB arrays incubated with liposome buffer prior to cancer cell seeding (Supplementary Fig. 6A and 6B†) were found to form healthy aggregates by the end of 24 h and did not stain positive for PI. Naked liposomes were not able to induce apoptosis in cancer cells (Supplementary Fig. 6C and 6D†) whereas liposomes functionalized with TRAIL were able to induce apoptosis in cancer cells (Supplementary Fig. 6E and 6F†). NK cells were able to mediate apoptosis in cancer cells but the response was not significant (Supplementary Fig. 6G and 6H†) in comparison to super NK cells (Supplementary Fig. 6I and 6J†). The mean intensity for PI positive cells after 24 h in culture indicates that super NK cells more significantly induced apoptosis in cancer cells in comparison to NK cells and TRAIL-functionalized liposomes (Fig. 6B).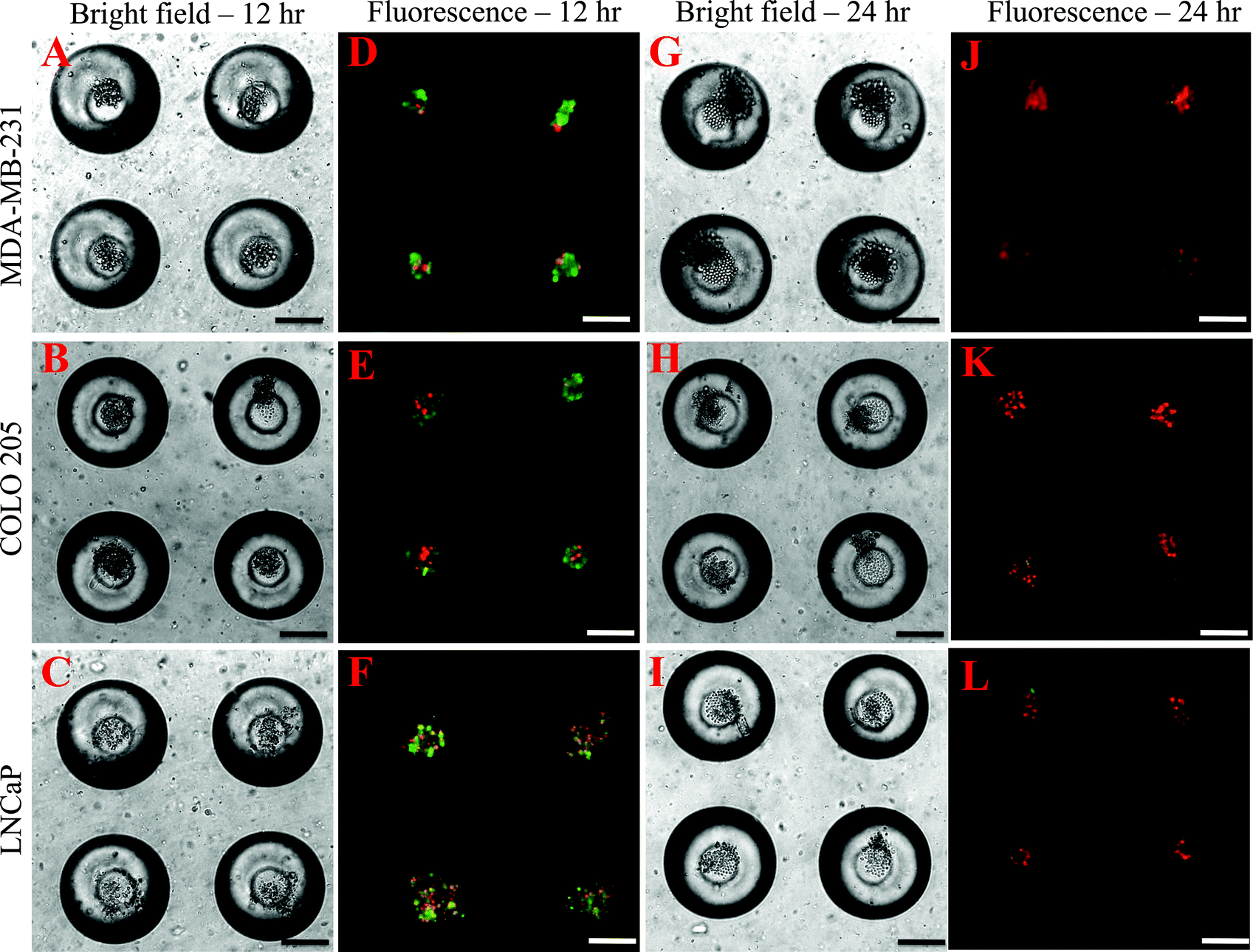 | ||
| Fig. 5 Bright field (A–C, G–I) and fluorescent (D–F, J–L) images of MDA-MB-231, COLO 205 and LNCaP cells labelled with Annexin-V FITC (green) and propidium iodide (red) at 12 h (A–F) and 24 h (G–L) respectively. Scale bar = 100 μm. | ||
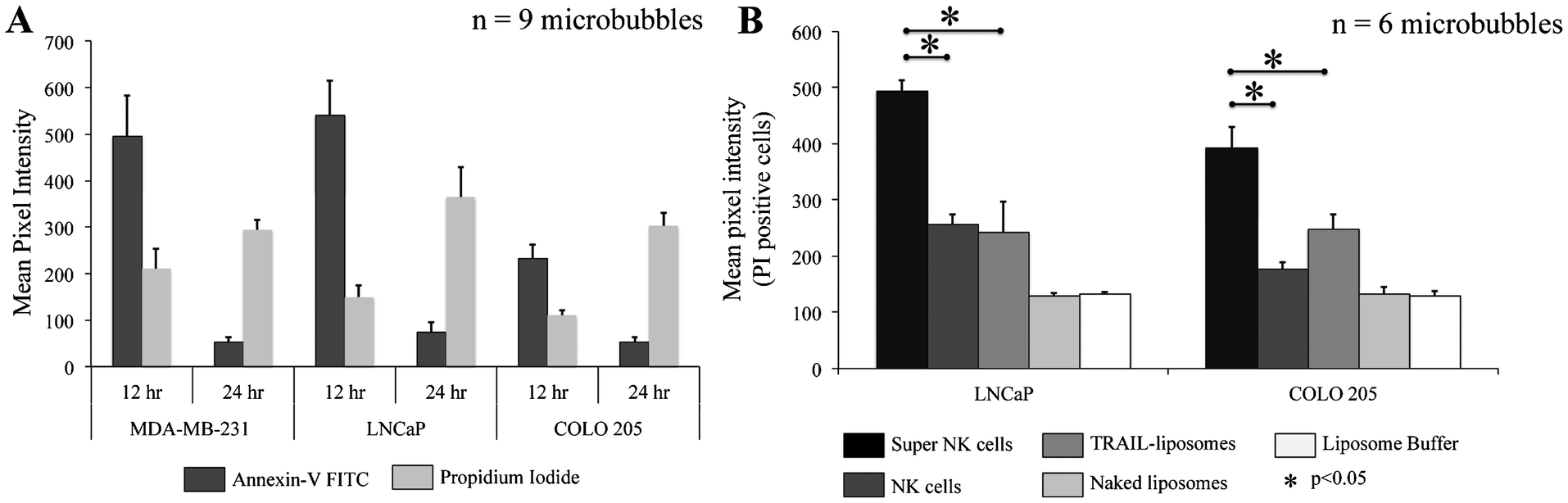 | ||
| Fig. 6 Bar graphs of mean pixel intensity of (A.) Annexin-V FITC and PI positive cells by the end of 12 h and 24 h (B.) PI positive cells for different culture conditions by the end of 24 h. | ||
4. Conclusion
Identification of DTCs in lymph nodes has served as a prognosticator of poor survival in several types of cancers. The results from several clinical studies suggest that DTCs can remain inactive for several years before a patient is diagnosed with overt metastasis. This has led to the development of several targeted therapies to target micrometastases within a relatively short span of time. However, to date, an adaptive immune therapy based approach to amplify the therapeutic potential of endogenous NK cells has not been proposed. The advent of efficient, high-throughput 3D cell culture platforms enables one to microengineer tumor models in vitro. In this work we showed a novel application for microbubble arrays formed on PDMS as an in vitro model for mimicking lymph node micrometastases. We showed that lymph node-seeking cancer cells cultured with NK cells conjugated to TRAIL-functionalized liposomes in MB arrays begin to undergo apoptosis after 24 h in culture. The ability of the proposed chip containing MB arrays to mimic the microenvironment of lymph node micrometastases could be applied to studies investigating targeted therapies for cancer cells metastasizing to lymph nodes. The proposed chip can also be used to culture tumor cells from surgical explants and study their sensitivity to a therapeutic approach to target lymph node micrometastases.Acknowledgements
The authors thank Jeff Mattison for his work with blood sample collection and donor recruitment. The authors also thank Jiahe Li for his prior work in liposome formation and protein functionalization. The authors acknowledge the Microscopy and Imaging Facility (MIF) at Weill Hall, Cornell University. The authors gratefully acknowledge the use of Zetasizer instrument at the Nanobiotechnology center (NBTC) at Cornell University. The work was supported by the Cornell Center on the Microenvironment & Metastasis through Award Number U54CA143876 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.References
- K. Pantel and R. H. Brakenhoff, Nat. Rev. Cancer, 2004, 4, 448–456 CrossRef CAS.
- S. Y. Wong and R. O. Hynes, Cell Cycle, 2006, 5, 812–817 CrossRef CAS.
- M. R. King, Front. Oncol., 2012, 2, 184 CrossRef.
- A. D. Hartkopf, M. Banys, N. Krawczyk, A. Staebler, S. Becker, J. Hoffmann, M. Hahn, M. Wallwiener and T. Fehm, Breast Cancer Res. Treat., 2012, 131, 501–508 CrossRef.
- H. Wikman, R. Vessella and K. Pantel, APMIS, 2008, 116, 754–770 CrossRef CAS.
- P. Hermanek, R. V. Hutter, L. H. Sobin and C. Wittekind, Cancer, 1999, 86, 2668–2673 CrossRef CAS.
- B. Gerber, A. Krause, H. Muller, D. Richter, T. Reimer, J. Makovitzky, C. Herrnring, U. Jeschke, G. Kundt and K. Friese, J. Clin. Oncol., 2001, 19, 960–971 CAS.
- B. Kubuschok, B. Passlick, J. R. Izbicki, O. Thetter and K. Pantel, J. Clin. Oncol., 1999, 17, 19–24 CAS.
- C. L. Willard-Mack, Toxicol. Pathol., 2006, 34, 409–424 CrossRef.
- J. P. Sleeman, in Lymphatic Metastasis and Sentinel Lymphonodectomy SE - 6, ed. P. Schlag and U. Veronesi, Springer Berlin Heidelberg, 2000, vol. 157, pp. 55–81 Search PubMed.
- B. Zbytek, J. A. Carlson, J. Granese, J. Ross, M. Mihm and A. Slominski, Expert Rev. Dermatol., 2008, 3, 569–585 CrossRef CAS.
- G. P. Dunn, A. T. Bruce, H. Ikeda, L. J. Old and R. D. Schreiber, Nat. Immunol., 2002, 3, 991–998 CrossRef CAS.
- Y. Geng, S. Chandrasekaran, J.-W. Hsu, M. Gidwani, A. D. Hughes and M. R. King, PLoS One, 2013, 8, e54959 CAS.
- A. Bot, Int. Rev. Immunol., 2011, 30, 235–7 CrossRef.
- N. P. Restifo, M. E. Dudley and S. A. Rosenberg, Nat. Rev. Immunol., 2012, 12, 269–281 CrossRef CAS.
- M. Kobari, S. Egawa, K. Shibuya, M. Sunamura, K. Saitoh and S. Matsuno, Br. J. Surg., 2000, 87, 43–8 CrossRef CAS.
- S. A. Rosenberg, J. C. Yang, R. M. Sherry, U. S. Kammula, M. S. Hughes, G. Q. Phan, D. E. Citrin, N. P. Restifo, P. F. Robbins, J. R. Wunderlich, K. E. Morton, C. M. Laurencot, S. M. Steinberg, D. E. White and M. E. Dudley, Clin. Cancer Res., 2011, 17, 4550–7 CrossRef CAS.
- K. Cheent and S. I. Khakoo, Immunology, 2009, 126, 449–57 CrossRef CAS.
- L. Zamai, C. Ponti, P. Mirandola, G. Gobbi, S. Papa, L. Galeotti, L. Cocco and M. Vitale, J. Immunol., 2007, 178, 4011–4016 CAS.
- B. Bonavida, C. P. Ng, A. Jazirehi, G. Schiller and Y. Mizutani, Int. J. Oncol., 1999, 15, 793–802 CAS.
- J. Li and M. R. King, Front. Oncol., 2012, 2, 79 Search PubMed.
- S. L. Stott, R. J. Lee, S. Nagrath, M. Yu, D. T. Miyamoto, L. Ulkus, E. J. Inserra, M. Ulman, S. Springer, Z. Nakamura, A. L. Moore, D. I. Tsukrov, M. E. Kempner, D. M. Dahl, C.-L. Wu, A. J. Iafrate, M. R. Smith, R. G. Tompkins, L. V. Sequist, M. Toner, D. A. Haber and S. Maheswaran, Sci. Transl. Med., 2010, 2, 25ra23 CrossRef.
- E. M. Levy, M. P. Roberti and J. Mordoh, J. Biomed. Biotechnol., 2011, 2011, 676198 CrossRef.
- R. Zagozdzon and J. Golab, Int. J. Oncol., 2001, 18, 417–424 CAS.
- L. Rasmussen and A. Arvin, Environ. Health Perspect., 1982, 43, 21–25 CrossRef CAS.
- M. Vanneman and G. Dranoff, Nat. Rev. Cancer, 2012, 12, 237–51 CrossRef CAS.
- K. Pantel, R. J. Cote and O. Fodstad, J. Natl. Cancer Inst., 1999, 91, 1113–1124 CrossRef CAS.
- H. Takeuchi, M. Kitajima and Y. Kitagawa, Cancer Sci., 2008, 99, 441–450 CrossRef CAS.
- N. Wada and S. Imoto, Int. J. Clin. Oncol., 2008, 13, 24–32 CrossRef.
- D. J. Dewar, B. Newell, M. A. Green, A. P. Topping, B. W. E. M. Powell and M. G. Cook, J. Clin. Oncol., 2004, 22, 3345–9 CrossRef CAS.
- S. Imoto, A. Ochiai, C. Okumura, N. Wada and T. Hasebe, Eur. J. Surg. Oncol., 2006, 32, 1175–1179 CrossRef CAS.
- K. Dowlatshahi, M. Fan, H. C. Snider and F. A. Habib, Cancer, 1997, 80, 1188–1197 CrossRef CAS.
- G. S. Herbert, V. Y. Sohn and T. A. Brown, Am. J. Surg., 2007, 193, 571–574 CrossRef.
- A. J. Cochran, R. R. Huang, J. Lee, E. Itakura, S. P. L. Leong and R. Essner, Nat. Rev. Immunol., 2008, 8, 901 CrossRef CAS.
- A. J. Cochran, R. R. Huang, J. Lee, E. Itakura, S. P. L. Leong and R. Essner, Nat. Rev. Immunol., 2006, 6, 659–670 CrossRef CAS.
- T. Saga, R. D. Neumann, T. Heya, J. Sato, S. Kinuya, N. Le, C. H. Paik and J. N. Weinstein, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 8999–9003 CrossRef CAS.
- Y. R. Miao, B. L. Eckhardt, Y. Cao, R. Pasqualini, P. Argani, W. Arap, R. G. Ramsay and R. L. Anderson, Clin. Cancer Res., 2013, 19, 2107–2116 CrossRef CAS.
- B. Čeh, M. Winterhalter, P. Frederik, J. J. Vallnerd and D. Lasicev, Adv. Drug Delivery Rev., 1997, 24, 165–177 CrossRef.
- M. Bajenoff, B. Breart, A. Y. Huang, H. Qi, J. Cazareth, V. M. Braud, R. N. Germain and N. Glaichenhaus, J. Exp. Med., 2006, 203, 619–631 CrossRef CAS.
- C. Belisle and G. Sainte-Marie, Anat. Rec., 1981, 199, 45–59 CrossRef CAS.
- C. Belisle and G. Sainte-Marie, Anat. Rec., 1981, 201, 553–561 CrossRef CAS.
- S. Noura, H. Yamamoto, Y. Miyake, B. N. Kim, O. Takayama, I. Seshimo, M. Ikenaga, M. Ikeda, M. Sekimoto, N. Matsuura and M. Monden, Clin. Cancer Res., 2002, 8, 759–767 Search PubMed.
- B. Bird, M. Miljkovic, N. Laver and M. Diem, Technol. Cancer Res. Treat., 2011, 10, 135–144 CAS.
- C. M. Ghajar and M. J. Bissell, Tissue Eng. A, 2010, 16, 2153–2156 CrossRef.
- E. Laconi, BioEssays, 2007, 29, 738–44 CrossRef CAS.
- U. B. Giang, D. Lee, M. R. King and L. A. DeLouise, Lab Chip, 2007, 7, 1660–1662 RSC.
- S. Chandrasekaran, U. B. T. Giang, M. R. King and L. A. DeLouise, Biomaterials, 2011, 32, 7159–7168 CrossRef CAS.
- S. Chandrasekaran and L. A. DeLouise, Biomaterials, 2011, 32, 9316–27 CrossRef CAS.
- S. Agastin, U. B. T. Giang, Y. Geng, L. A. Delouise and M. R. King, Biomicrofluidics, 2011, 5, 24110 CrossRef.
- K. Schroer, K. Kim and B. Prescott, J. Exp. Med., 1979, 155, 1–6 Search PubMed.
- D. Fuard, T. Tzvetkova-Chevolleau, S. Decossas, P. Tracqui and P. Schiavone, Microelectron. Eng., 2008, 85, 1289–1293 CrossRef CAS.
- R. N. Palchesko, L. Zhang, Y. Sun and A. W. Feinberg, PLoS One, 2012, 7, e51499 CAS.
- Q. Wing-Han Yuen, Y. P. Zheng, Y. P. Huang, J. F. He, J. Chung-Wai Cheung and M. Ying, Open Biomed. Eng. J., 2011, 5, 39–46 CrossRef.
- S. Lopez-Verges, J. M. Milush, S. Pandey, V. A. York, J. Arakawa-Hoyt, H. Pircher, P. J. Norris, D. F. Nixon and L. L. Lanier, Blood, 2010, 116, 3865–3874 CrossRef CAS.
- A. Gabizon, D. Goren, A. T. Horowitz, D. Tzemach, A. Lossos and T. Siegal, Adv. Drug Delivery Rev., 1997, 24, 337–344 CrossRef CAS.
- A. Gabizon, Ann. Biol. Clin., 1993, 51, 811–813 CAS.
- K. Pantel, C. Alix-Panabieres and S. Riethdorf, Nat. Rev. Clin. Oncol., 2009, 6, 339–351 CrossRef CAS.
- S. Chandrasekaran and M. R. King, J. Bioeng. Biomed. Sci., 2012, 2, e109, DOI:10.4172/2155-9538.1000e109.
- D. Olmeda, G. Moreno-Bueno, J. M. Flores, A. Fabra, F. Portillo and A. Cano, Cancer Res., 2007, 67, 11721–11731 CrossRef CAS.
- H. Sasaki, K. Miura, A. Horii, N. Kaneko, W. Fujibuchi, L. Kiseleva, Z. Gu, Y. Murata, H. Karasawa, T. Mizoi, T. Kobayashi, M. Kinouchi, S. Ohnuma, N. Yazaki, M. Unno and I. Sasaki, Cancer Sci., 2008, 99, 711–719 CrossRef CAS.
- X. Wang, Z. An, J. Geller and R. M. Hoffman, Prostate, 1999, 39, 182–186 CrossRef CAS.
- M. Nagane, H. J. Huang and W. K. Cavenee, Apoptosis, 2001, 6, 191–197 CrossRef CAS.
- M. van Engeland, L. J. Nieland, F. C. Ramaekers, B. Schutte and C. P. Reutelingsperger, Cytometry, 1998, 31, 1–9 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc50584g |
| This journal is © The Royal Society of Chemistry 2014 |