 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSlanted spiral microfluidics for the ultra-fast, label-free isolation of circulating tumor cells†
Majid Ebrahimi
Warkiani‡
a,
Guofeng
Guan‡§
ab,
Khoo Bee
Luan‡
c,
Wong Cheng
Lee
a,
Ali Asgar S.
Bhagat
d,
Parthiv
Kant Chaudhuri
c,
Daniel Shao-Weng
Tan
e,
Wan Teck
Lim
e,
Soo Chin
Lee
f,
Peter C. Y.
Chen
ab,
Chwee Teck
Lim
*abcg and
Jongyoon
Han
*ah
aBioSystems and Micromechanics (BioSyM) IRG, Singapore-MIT Alliance for Research and Technology (SMART) Centre, Singapore. E-mail: jyhan@mit.edu; ctlim@nus.edu.sg
bDepartment of Mechanical Engineering, National University of Singapore, Singapore
cMechanobiology Institute, National University of Singapore, Singapore
dClearbridge BioMedics Pte Ltd, Singapore
eDepartment of Medical Oncology, National Cancer Centre Singapore, Singapore
fDepartments of Hematology-Oncology and Pharmacology, National University Hospital, Singapore
gDepartment of Bioengineering, National University of Singapore, Singapore
hDepartment of Electrical Engineering and Computer Science, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA
First published on 3rd July 2013
Abstract
The enumeration and characterization of circulating tumor cells (CTCs), found in the peripheral blood of cancer patients, provide a potentially accessible source for cancer diagnosis and prognosis. This work reports on a novel spiral microfluidic device with a trapezoidal cross-section for ultra-fast, label-free enrichment of CTCs from clinically relevant blood volumes. The technique utilizes the inherent Dean vortex flows present in curvilinear microchannels under continuous flow, along with inertial lift forces which focus larger CTCs against the inner wall. Using a trapezoidal cross-section as opposed to a traditional rectangular cross-section, the position of the Dean vortex core can be altered to achieve separation. Smaller hematologic components are trapped in the Dean vortices skewed towards the outer channel walls and eventually removed at the outer outlet, while the larger CTCs equilibrate near the inner channel wall and are collected from the inner outlet. By using a single spiral microchannel with one inlet and two outlets, we have successfully isolated and recovered more than 80% of the tested cancer cell line cells (MCF-7, T24 and MDA-MB-231) spiked in 7.5 mL of blood within 8 min with extremely high purity (400–680 WBCs mL−1; ∼4 log depletion of WBCs). Putative CTCs were detected and isolated from 100% of the patient samples (n = 10) with advanced stage metastatic breast and lung cancer using standard biomarkers (CK, CD45 and DAPI) with the frequencies ranging from 3–125 CTCs mL−1. We expect this simple and elegant approach can surmount the shortcomings of traditional affinity-based CTC isolation techniques as well as enable fundamental studies on CTCs to guide treatment and enhance patient care.
Introduction
Circulating tumor cells (CTCs) are rare cancer cells found in the blood of metastatic cancer patients that potentially provide a convenient source for the detection, characterization and monitoring of non-hematologic cancers.1 They have been shown to resemble the primary tumor, carry similar genetic information and can thus act as surrogates for obtaining primary material from the initial tumor for monitoring of the tumor phenotype/genotype.2,3 Besides its prognostic significance, CTC enumeration can also be used to assess the efficacy of therapeutic treatment.4 The ability to isolate and enrich a large number of intact CTCs for analysis and characterization is pivotal to elucidating the mechanisms underlying the metastatic process.5 Additionally, the enrichment and culture of viable CTCs from blood of cancer patients will provide immense opportunities for the development of new anti-cancer drugs for future targeted therapies.6,7 However, the extremely low number of CTCs in the blood makes enumeration and characterization a huge technical challenge.8 Therefore, high enrichment yield and high detection accuracy are essential to maximize both assay sensitivity and reproducibility.Many current CTC enrichment techniques primarily involve affinity-based detection using antibodies against epithelial cell surface markers (e.g., EpCAM). For instance, the only US FDA-approved CTC diagnostic system, CellSearch® by Veridex LLC, is one such technique using epithelial antibodies conjugated to magnetic beads to enrich tumor cells from blood. Similar methods have been reduced to the microscale using antibody-coated micro-pillars,9,10 microchannels11,12 and nanotubes13 to capture CTCs from blood. Because these enrichment strategies rely on a single parameter, its efficiency can be compromised due to the downregulation of EpCAM in many aggressive CTC subpopulations that undergo an epithelial to mesenchymal transition (EMT). Moreover, since viable CTCs cannot be retrieved post isolation, these assays are unsuitable for the prediction of therapy response in order to guide treatment, a key hallmark of personalized medicine.
An alternative strategy for CTC enrichment is through leveraging differences in physical characteristics of tumor cells, such as size and deformability,14,15 density,16 optical, and magnetic properties,17,18 which can ultimately lead to target cell isolation. Over the past decade, a diverse suite of microfluidic-based technologies have been developed for CTC separation with an improved detection sensitivity, and a high separation efficiency and enrichment factor.19,20 However, most of these techniques have several drawbacks such as clogging, low recovery, complicated integration of external force fields, possible loss of cell viability21 and prolonged sample processing time (up to several hours) due to the relatively high fluidic resistance of their lateral fluidic structures.9,22
Recently, high-throughput passive particle sorting based on the inertial migration of particles inside curvilinear microchannels has been reported.23,24 Inertial microfluidic devices exploiting hydrodynamic forces for particle separation have distinct advantages over the aforementioned CTC isolation methods. Firstly, they do not require any integration of external force fields (e.g., electric/magnetic), but instead, they purely rely on microchannel dimensions, fluidic forces (i.e., inertial lift and Dean drag forces) and particle size to achieve separation.25,26 Secondly, this approach offers the ability to perform separation continuously without any clogging issues with extremely high throughput and efficiency.27,28 Our team has recently developed a spiral microfluidic device for high throughput size-based separation and retrieval of CTCs from blood using the Dean drag force coupled with the inertial microfluidics phenomenon.29 This device, named the “Dean Flow Fractionation” has been successfully employed for the isolation of CTCs from the blood of patients with advanced metastatic non-small cell lung cancer (NSCLC). We have shown that our spiral biochip with a rectangular cross-section is capable of processing blood with high hematocrit levels (∼20–25%), thus achieving enrichment of rare cells at a processing speed of 3 mL per hour, which is much higher than that of existing microfluidic devices. In this work, we further improve the throughput by designing a spiral channel with a trapezoidal cross-section.30 In contrast to the rectangular cross-section channels that require a sheath flow for particle dispersion inside the channels when the particle concentration is high, the trapezoidal channel requires only a single inlet for the sample and two outlets for waste and enriched cell collection during operation. Using a microfluidic device with this newly designed microchannel, we have successfully demonstrated the enrichment of a high number of CTCs (3–125 CTCs mL−1) from the peripheral blood of patients with metastatic breast and lung cancer. This device can process 7.5 mL of red blood cell lysed blood in about 8 min, allowing for the enrichment of viable CTCs with relatively high purity and yield. The trapezoidal spiral channels can be produced at extremely low cost and with high resolution using conventional micro-milling and PDMS casting, and can be operated using a single syringe pump, which facilitates automation. We strongly believe that this novel strategy can be utilized for large-scale processing of clinical samples in order to enrich CTCs sufficiently for various detailed molecular analyses as well as clinical monitoring of individual patients undergoing therapy. The device is well suited to process even larger quantities of blood if required (20 mL in ∼15 min), which is a growing need for obtaining large numbers of CTCs for multiple downstream tests.
Materials and methods
Device design and fabrication
The device design consists of an 8-loop spiral microchannel with one inlet and two outlets with the radius increasing from 8 mm to 24 mm for efficient cell migration and focusing. The width of the channel cross-section is 600 μm, and the inner/outer heights were optimized at 80 and 130 μm, respectively, for the trapezoid cross-section.30 The mold with its specific channel dimensions was designed using SolidWorks software and then fabricated by a conventional micro-milling technique (Whits Technologies, Singapore) on a polymethyl methacrylate (PMMA) sheet for subsequent PDMS casting. The microfluidic device was fabricated by casting degassed PDMS (mixed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of base and curing agent, Sylgard 184, Dow Corning Inc.) on the mold and subsequent baking in an oven for 2 h at 70 °C. After curing, the PDMS was peeled from the mold, access holes (1.5 mm) for fluidic inlets and outlets were punched with the Uni-Core™ Puncher (Sigma-Aldrich Co. LLC. SG), and the PDMS devices were irreversibly bonded to another layer of cured PDMS using an oxygen plasma machine (Harrick Plasma, USA) to complete the channels. The assembled device was finally placed in an oven at 70 °C for 30 min to further enhance the bonding.
:1 mixture of base and curing agent, Sylgard 184, Dow Corning Inc.) on the mold and subsequent baking in an oven for 2 h at 70 °C. After curing, the PDMS was peeled from the mold, access holes (1.5 mm) for fluidic inlets and outlets were punched with the Uni-Core™ Puncher (Sigma-Aldrich Co. LLC. SG), and the PDMS devices were irreversibly bonded to another layer of cured PDMS using an oxygen plasma machine (Harrick Plasma, USA) to complete the channels. The assembled device was finally placed in an oven at 70 °C for 30 min to further enhance the bonding.
Cell culture and sample preparation
Two commercially available green fluorescent protein (GFP)-tagged human cancer cell lines, namely breast adenocarcinoma (MCF-7 and MDA-MB-231), one bladder (T24; HTB-4 ATCC, USA) and one lung cancer cell line (H159; HTB-4 ATCC, USA) were used to mimic the CTC separation. The SKBR3 cell line was also employed as a control for DNA FISH analysis of HER2 in enriched CTCs. The cells were seeded into coated T25 flasks (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and cultured with high-glucose Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, USA) and 1% penicillin–streptomycin (Invitrogen, USA). The culture was kept in a humidified atmosphere at 37 °C containing 5% (v/v) CO2 and harvested at 80% confluence for spiking. Sub-confluent monolayers were dissociated using 0.01% trypsin and 5.3 mM EDTA solution (Lonza, Switzerland). For all experiments, unless otherwise mentioned, whole blood obtained from healthy donors and patient samples were lysed with RBC lysis buffer (G-Bioscience, USA) for 5 min at room temperature with continuous mixing. Lysis was stopped by dilution with PBS buffer and cell pellets were obtained after centrifugation at 1000g for 5 min. The cell pellets were resuspended to the desired concentrations with PBS.Device characterization
In all the experiments, the spiral biochips were initially mounted on an inverted phase contrast microscope (Olympus IX71) equipped with a high speed CCD camera (Phantom v9, Vision Research Inc., USA). The biochip was primed with a priming buffer (1× PBS, 2 mM EDTA supplemented with 0.5% BSA) using a syringe pump (PHD 2000, Harvard Apparatus, USA) for around 2 min at a flow rate of 2 mL min−1. During testing, cancer cells and blood samples were filled in a 10 mL syringe and pumped through the device using a syringe pump connected to the microchannel through flexible Tygon® tubing. The flow rate was set to 1700 μL min−1 for all the experiments. High speed videos were captured at the channel outlet using the Phantom Camera Control software and then analyzed using ImageJ® software.Immunofluorescence staining
To calculate the separation efficiency and enrichment ratio between the sample and sorted CTCs, flow cytometry using a BD Accuri™ C6 Flow Cytometer was employed for both the inlet and the CTC outlet. Immunofluorescence staining based on common markers for cancer cells and white blood cells (WBCs) was used for differentiation and quantification. The enriched cells from the CTC outlet were stained with fluorescein isothiocyanate (FITC) conjugated pan-cytokeratin (CK) (1:100, MiltenyiBiotec Asia Pacific, Singapore) and an allophycocyanin (APC) conjugated CD45 marker (1:100, MiltenyiBiotec Asia Pacific, Singapore) for 30 min to identify cancer cells and WBCs, respectively. For clinical samples, CTCs were identified by staining them with FITC-conjugated pan-cytokeratin (CK) (1:100, MiltenyiBiotec Asia Pacific, Singapore). Cells staining positively for pan-CK and Hoechst (nuclei stain) and negatively for CD45 with the characteristic morphology of cancer cells (i.e., high nucleus to cytoplasm ratio) were identified as CTCs. Cells staining positively for CD45 and Hoechst and negatively for pan-cytokeratin were identified as leukocytes.
Cell viability assay (using PI staining and culturing)
MDA-MB-231 and MCF-7 GFP-tagged cells mixed with blood from healthy donors were processed through the spiral microfluidic devices, and cell viability was assessed via the trypan blue (or propidium iodide (PI)) exclusion assay and through long-term re-culturing. Isolated CTCs were seeded onto polylysine-coated 2D cell culture substrates and cultured overnight as described. The cells were then stained with propidium iodide (PI) in situ. The cells were imaged and enumerated for PI positive staining to determine the percentage of cell viability after lysis and processing. The cell viability numbers were compared with cells obtained after lysis without spiral biochip processing.Clinical samples
Human whole blood samples were obtained from healthy donors and metastatic lung and breast cancer patients. This study was approved by our institutional review board and the local ethics committee according to a protocol permitted by the Institutional Review Board (IRB). A total of 10 blood samples from healthy donors were used as controls, and 10 samples from lung and breast cancer patients were processed for CTC enumeration. The blood samples were collected in vacutainer tubes (Becton-Dickinson, Franklin Lakes, NJ, USA) containing EDTA anticoagulant, and were processed within 2–4 h to prevent blood coagulation. For all the samples, 7.5 mL of whole blood was lysed initially using RBC lysis buffer and re-suspended in PBS prior to processing on chip.Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) was performed on the SKBR3 (amplified HER2 signals) and MDA-MB-231 (non-amplified HER2 signal) cell lines as well as isolated CTCs according to the manufacturer's protocol. The cells were spun onto slides using a Cytospin centrifuge (Thermo Scientific, USA) at 600 rpm for 6 min. The slides were fixed in 4% PFA at room temperature for 10 min and dehydrated via ethanol series (80%, 90%, and 100%). For FISH analysis, the slides were treated with RNase (4 mg mL−1) (Sigma, USA) for 40 min at 37 °C, washed with 1× PBS–0.2% Tween 20 (Sigma, USA) three times and denatured with 70% formamide–2× SCC (saline sodium citrate, Path Vysion, Abbott, USA) for 10 min at 80 °C. They were then quench-dehydrated again via ice-cold ethanol series. HER2/neu (Abbott Laboratories, Illinois, USA) probes were directly applied to slides maintained at 42 °C. The hybridization was continued at 42 °C under dark and humid conditions overnight. The slides were washed with 50% formamide–2× SSC and 2× SSC at 45 °C under shaking, counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and sealed with 50 × 50 mm2 coverslips (Fisher Scientific, USA).Immunofluorescence for assay characterization
To characterize the phenotypic ratio of the CTC cultures, the cells were incubated with a variety of antibodies, including fluorescein isothiocyanate (FITC) conjugated pan-cytokeratin (CK) (1:100, Miltenyi Biotec Asia Pacific, Singapore), fluorescein isothiocyanate (FITC) conjugated CD44 (1:100, Miltenyi Biotec Asia Pacific, Singapore), allophycocyanin (APC) conjugated CD24 (1:100, Miltenyi Biotec Asia Pacific, Singapore), allophycocyanin (APC) conjugated CD45 (1:100, Miltenyi Biotec Asia Pacific, Singapore), and with Hoechst (Invitrogen, USA). Staining was performed either by adding the staining reagents directly onto the assay, or to cell suspensions after trypsinization. Cells were permeabilized with 0.1% Triton 100× (Thermo Scientific, USA) for 30 min on ice after fixation with 4% paraformaldehyde (PFA) (Sigma Aldrich, USA).
Results and discussion
Design principle
When particles flow within a spiral microchannel, neutrally buoyant particles under the influence of inertial lift forces arising from the parabolic nature of the laminar velocity profile migrate across the streamlines to an equilibrium position away from the channel center towards the channel walls. At the same time, they also experience a drag force introduced by Dean vortices along the Dean flow arising due to the curvilinear geometry. The combination of inertial and Dean forces reduces the equilibrium positions to a single position at the inner microchannel wall within the channel width, inducing a continuous inertial focusing.28 Since both forces are functions of the particle size, particles of different size occupy distinct lateral positions near the channel wall and exhibit different degrees of focusing, allowing size-based separation. One major challenge in utilizing a spiral microchannel with a rectangular cross-section is that spacing between equilibrium positions of particles with varying diameter is narrow. This can affect the separation resolution. Previously, we have shown that by modifying the channel cross-section into a trapezoidal geometry, we can significantly enhance the separation resolution.31 This is mainly due to the fact that the asymmetry of trapezoid cross-sections influences the velocity profile and results in the formation of strong Dean vortex cores near the outer wall that has a larger channel depth (Fig. 1A). Therefore, in contrast to the rectangular cross-section channels where small particles are focused near the center of the channel width under the balance of inertial and drag forces, the modified velocity field of the trapezoidal spiral traps the smaller particles within the strong Dean vortex cores near the outer wall. With the large cells focused at the inner side, the spacing between the two cell streams is maximized, and high throughput high resolution sorting can be achieved. This unique design is ideally suited for enriching larger-diameter but lower-abundance target cells in smaller but higher-abundance background cell mixtures, such as isolating leukocytes from raw blood,31 or enriching ultra-low abundance CTCs from blood as demonstrated in this work. Fig. 1B shows a picture of our experimental setup during sample processing. The continuous collection of CTCs facilitates coupling of the device with a conventional 96-well plate or a membrane filter for subsequent downstream analysis such as immunostaining, qRT-PCR, FISH and sequencing.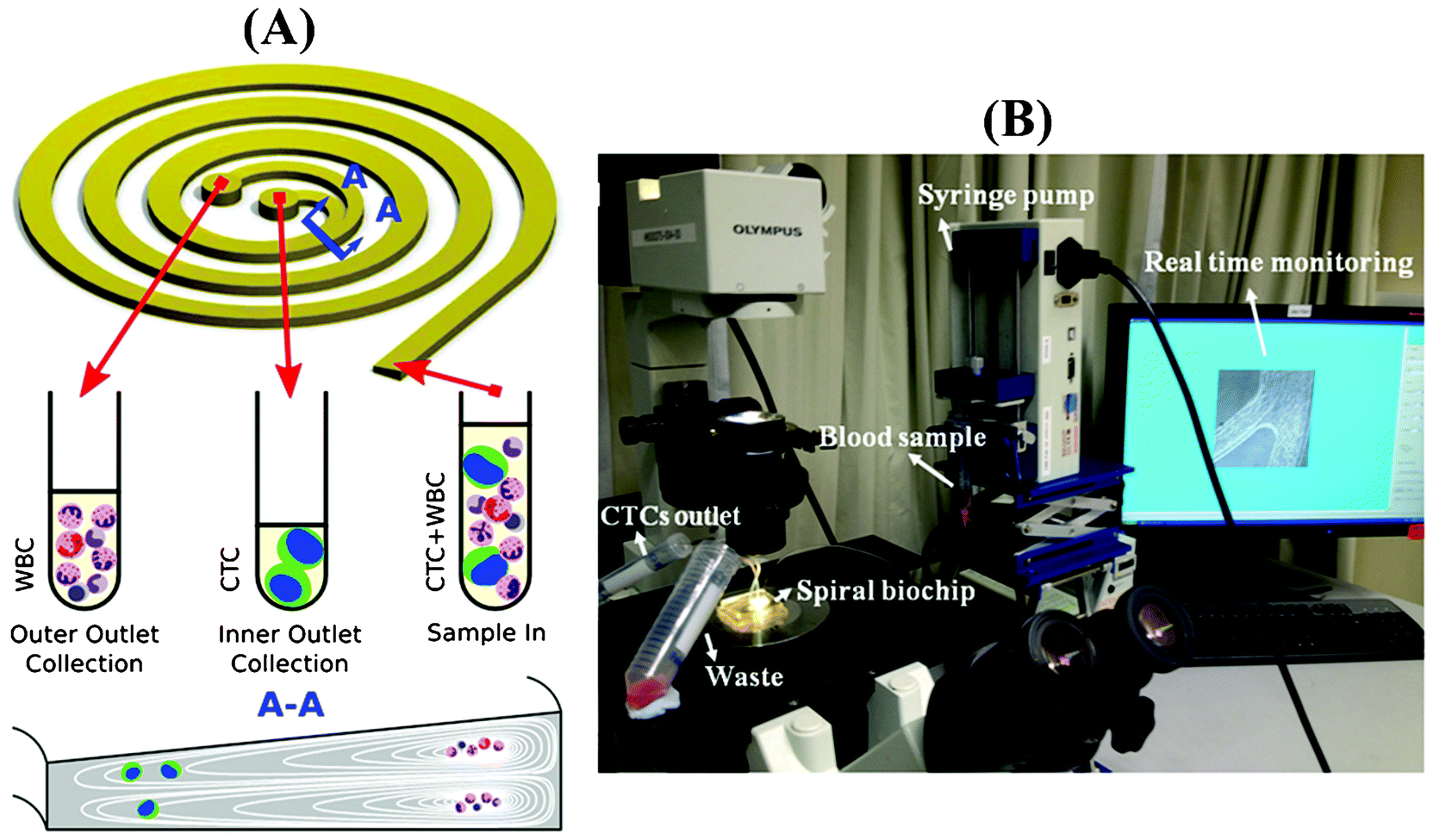 | ||
| Fig. 1 (A) The operating principle of CTC enrichment by a spiral channel with a trapezoid cross-section (80 and 130 μm in the inner and outer channel height, respectively). CTCs focused near the inner wall due to the combination of the inertial lift force and the Dean drag force at the outlet while white blood cells (WBCs) and platelets are trapped inside the core of the Dean vortex formed closer to the outer wall. (B) The workstation setup for the CTC separation. The lysed blood is pumped through the spiral chip using a syringe pump where CTCs are separated from other blood components rapidly and efficiently. | ||
Characterization of the spiral performance
An optimal technology for CTC isolation must aim to isolate the maximum number of viable cells with acceptable degrees of purity (i.e., depending upon the contamination tolerance of downstream molecular assays) without relying on specific markers (e.g., EpCAM) with minimum sample processing steps.32 To increase the purity and minimize the cellular components passing through the spiral chip, we utilized a conventional chemical RBC lysis approach to boost the throughput of our system while maximizing the number of enriched CTCs. Although it has been reported that the RBC lysis and density gradient centrifugation steps can lead to cell loss (10–30%),33 our experimental results show that the cell loss is less than 8% during the entire process. Furthermore, exposure to lysis buffer also did not alter the morphology and size of the cells (see Fig. S1, ESI†). Extensive characterization was performed to find the optimal device design by studying the effect of various parameters, including the channel aspect ratio, flow rate and sample concentration using both latex particles and healthy blood samples spiked with cancer cell lines (see movies S1,2†). As WBC and platelet concentrations are relatively high (>3%) in the lysed blood, their complete removal is pivotal for achieving meaningful enrichment. To investigate the impact of the input sample cell concentration on the output purity, we carried out the processing of blood under different nucleated cell concentrations. Initially, 7.5 mL of whole blood collected from healthy donors were lysed chemically using ammonium chloride, and the nucleated cell fraction was re-suspended back to 15 mL (0.5× concentration), 7.5 mL (1× concentration) and 5 mL (1.5× concentration) using PBS buffer for processing. The collected cells were stained using DAPI and CD45 antibodies to quantify the number of contaminated WBCs. Fig. 2A shows the total cell count (DAPI+/CD45+) collected from the CTC outlet at different sample concentrations. This graph shows that our device performs best when the cell concentration is below 1× (∼3.5–4 × 106 WBCs mL−1) where the minimum contamination of WBCs is observed (mean, 500 WBCs mL−1 of lysed blood; range, 400–680 WBCs mL−1). Hence, we decided to choose the 0.5× concentration as optimal for the processing of clinical samples, which translates to a total processing time of around 8 min for a 7.5 mL blood sample. To the best of our knowledge, this is the highest throughput achieved on a microfluidic platform for CTC isolation reported to date.34 In addition, the processing time can be further decreased by multiplexing of biochips together as we have demonstrated previously.14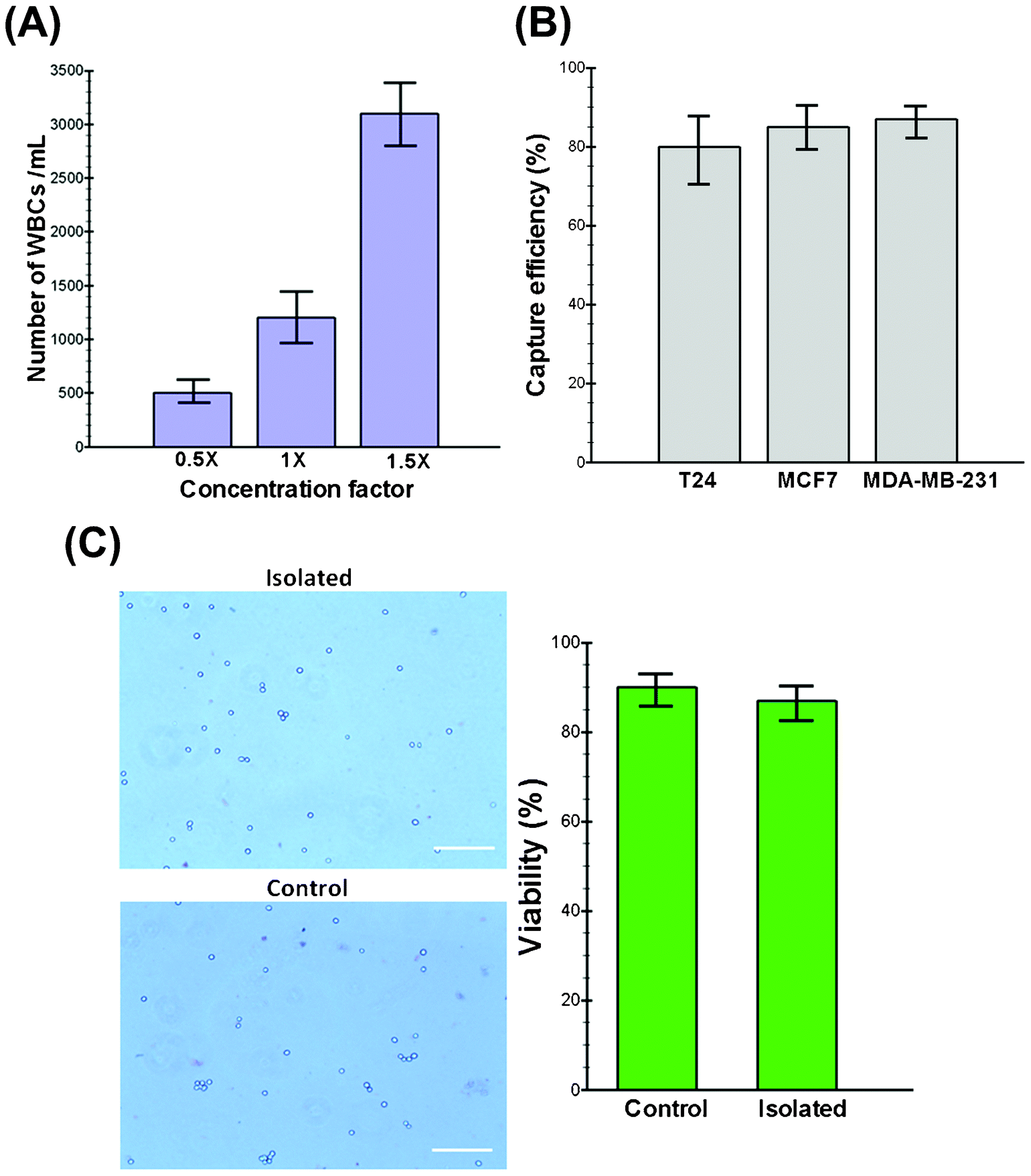 | ||
| Fig. 2 (A) The effect of the WBC concentration on the performance of the trapezoid spiral biochip and the final purity. (B) Histogram plot indicating a high separation efficiency of ∼85% for different cancer cell lines tested. (C) Phase contrast micrographs of control (unsorted) and sorted MDA-MB-231 cells stained using the trypan blue dye indicating high cell viability. The results confirm that the shear exerted on the cells during sample processing did not compromise their viability, retrieving >90% viable cells. | ||
Isolation efficiency and cell viability using cancer cell lines
Since CTCs are extremely rare in the blood stream, it is crucial to isolate a maximum number of target cells in a blood sample for various downstream assays. For this purpose, three different cell lines (i.e., MCF-7, T24 and MDA-MB-231) were employed in this study to quantify the performance of the trapezoid spiral biochip for CTC isolation and recovery. These cell lines were chosen to ascertain the versatility of the technique in the detection of CTCs. The model system is constructed by spiking a known number of cells (∼500 cells) into 7.5 mL of blood obtained from healthy donors. After RBC lysing and resuspension to the optimized concentration (∼0.5×), the sample was passed through the spiral chip for ultra-fast enrichment of spiked cells. Following enrichment, the cancer cells were identified by immunofluorescence staining by either enumerating them under an epi-fluorescence microscope or by flow cytometry analysis with common surface markers (CK+/CD45−). Fig. 2B shows a summary of the capture efficiency of tumor cells spiked into whole blood with average recoveries of 80% for the T24, 85% for the MCF-7 and 87% for the MDA-MB-231 cell line (n = 3). To validate the viability of the captured tumor cells using our device, the isolated cells were re-cultured onto 2-D culture substrates where they attached and proliferated under standard culture conditions (see Fig. S2, ESI†). The viability of the cells before and after processing was also validated by using functional assays including staining with propidium iodide (PI) and/or trypan blue. Our results demonstrate the high viability of the captured cells confirmed by their minimal staining (<10%) with trypan blue (Fig. 2C). Further morphological analysis of cancer cells also confirmed that the cells remain relatively unchanged during multiple steps of processing (data not shown).Clinical samples
To validate the clinical utility of the trapezoid chip, a 7.5 mL blood sample was obtained from each of (i) 5 healthy individuals (control), (ii) 5 patients with metastatic breast cancer (MBC) and (iii) 5 patients with non-small cell lung cancer (NSCLC) (Table S1, ESI†). The presence of isolated CTCs was determined by immunostaining with Hoechst (DNA), FITC–pan-cytokeratin (CK) antibodies (cancer/epithelial cell biomarker), and APC–anti-CD45 antibodies (hematologic biomarker) (see Fig. 3A). Hoechst+–pan-CK+–CD45− cells were scored as CTCs. The data presented in Table S1, ESI† display the clinico-pathological characteristics of the breast and lung cancer patients, as well as the CTC counts obtained from the spiral biochip. The CTCs were detected in 10 out of 10 patient samples (100% detection) with counts ranging from 6–57 CTCs mL−1 for MBC samples and 3–125 CTCs mL−1 for NSCLC samples (Fig. 3B). Cytokeratin-positive epithelial cells were also detected in healthy volunteers (1–4 per mL), but a distinct detection threshold can be drawn in comparison with that of the patient samples. The threshold analysis suggests 3–4 CTCs per 7.5 mL of blood sample as the optimal cut-off value for predicting metastatic disease.35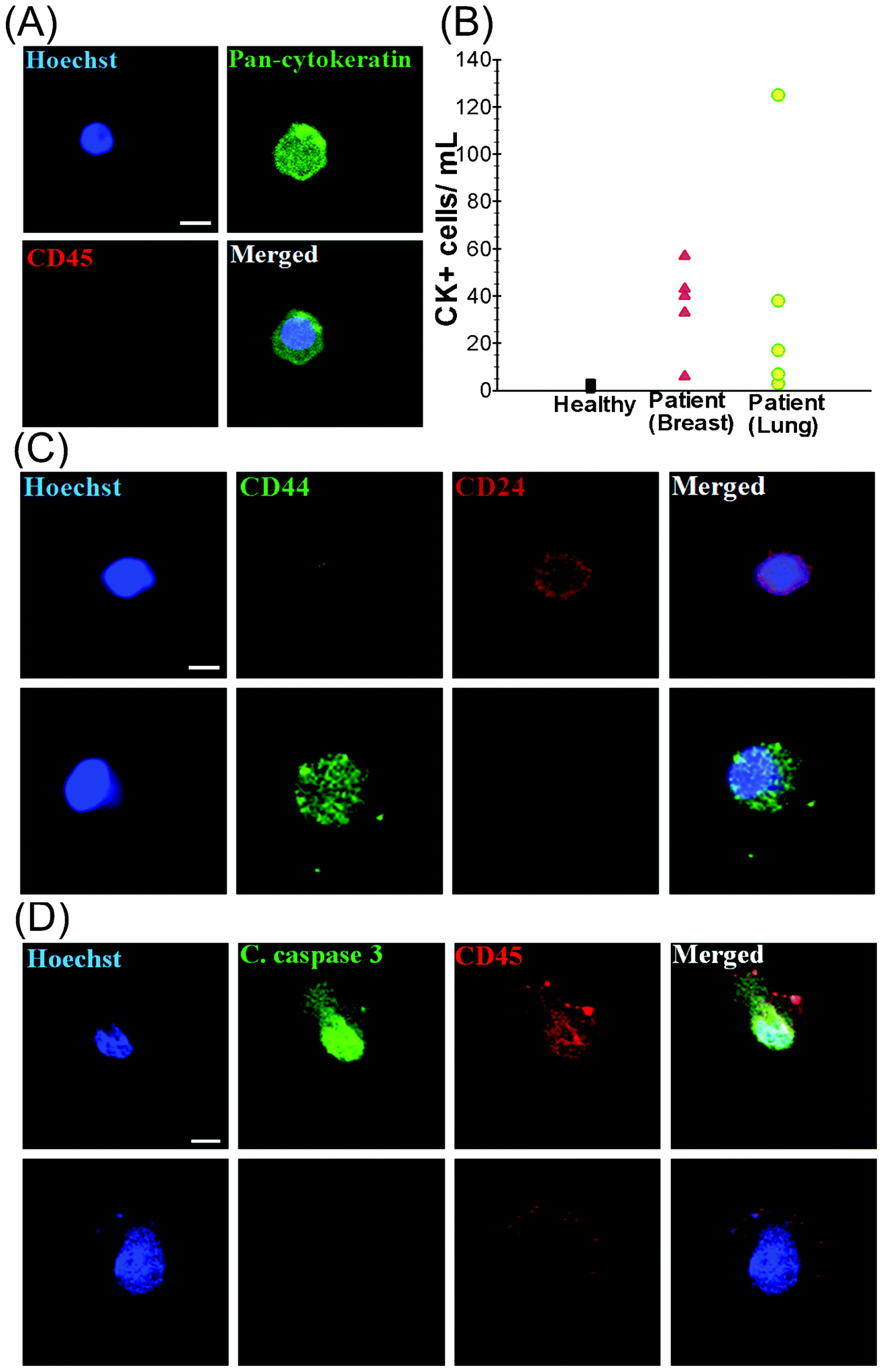 | ||
| Fig. 3 Blood from healthy donors (n = 5) as well as patients with metastatic breast and lung cancer (n = 10) was processed using the spiral chip. (A) Immunofluorescence staining of isolated CTC. CTC is identified by the following criteria: Hoechst positive, pan-cytokeratin positive and CD45 negative. (B) CTC enumeration plot for healthy donors (black), breast cancer patients (red), and lung cancer patients (green). (C) Identification of cancer stem cells (CSCs) in breast samples using standard markers. No CD44+/CD24+ cells were detected. It was found that CD44+ cells are larger than CD24+ cells. (D) Staining for apoptotic cells. Cleaved caspase-3 was absent in the isolated CTCs. The majority of the cells (>95%) expressing cleaved caspase-3 was CD45+. | ||
Enriched CTCs are highly heterogeneous as previously reported in various studies (Fig. S3, ESI†).36 The staining with the cancer stem cell markers CD44 and CD24 revealed distinct populations of CTCs which are mostly either CD44+/CD24− or CD44−/CD24+ (Fig. 3C). The CD44+/CD24− cells were evidently larger in size than the CD44−/CD24+ cells. It should also be noted that a proportion of CTCs were likely to be apoptotic. This is demonstrated by staining with the cleaved caspase-3 marker which plays an integral role in the apoptotic process of mammalian cells. The results of our analysis show that only 1–2% of the isolated CTCs were positive for cleaved caspase-3, indicating that most of the CTCs enriched with the spiral biochip do not display any characteristics that reflect apoptotic processes. Flow cytometry analysis also revealed that only 9.9% of the total isolated cells were positive for cleaved caspase-3, which could be most probably WBCs (Fig. S4, ESI†). These findings were confirmed by immunofluorescence staining of the cells as shown in Fig. 3D, which was also in close agreement with the previous findings.37 In addition, the presence of EpCAM−/pan-CK+ cells and EpCAM+/pan-CK+ cells was detected in the isolated CTCs. The population of EpCAM−/pan-CK+ cells was much lower among lung cancer samples as compared to breast samples, indicating the significant limitation of EpCAM based approaches for accurate detection and enrichment of putative CTCs. The ability to capture viable CTCs was demonstrated by overnight culture of the isolated cells. In this study, isolated cells were seeded onto polylysine coated well culture plates overnight under culture conditions as described (see Fig. S5, ESI†). Viable cells were able to spread onto the substrates and appeared negative for propidium iodide (PI) when stained.
FISH analysis
Tumor cell heterogeneity in respect to many different aspects has been apparent and widely reported previously.38 Herein, we show the variation of HER2 expression in CTCs isolated with the trapezoid chip, using samples from patients with HER2- tumors. Pantel et al. have shown that the HER2 status in CTCs varies with respect to the primary tumor.39 Specifically, HER2+ CTCs may be observed in ∼30% of samples obtained from HER2- origin.40 DNA fluorescence in situ hybridization (FISH) was carried out to evaluate the HER2 status of isolated CTCs. HER2 signals in isolated CTCs were compared against the control breast cancer cell lines MDA-MB-231 (non-amplified HER2 signal) and SKBR3 (amplified HER2 signals) as shown in Fig. 4. Amplified HER2 expression is present when the ratio of the HER2/centromere of chromosome 17 (Cen-17) signals in single nuclei is >2. A range of HER2/Cen-17 signals was observed. Cells displaying a ratio of HER2/Cen-17 = 1 are likely to be WBCs or non-amplified HER2 CTCs, and can be distinguished by further immunostaining. However, cells with amplified HER2 signals were also detected, indicating the definite presence of CTCs. This is in accordance with previous findings that heterogeneity of the HER2 status is evident in CTCs as compared to the primary tumor.39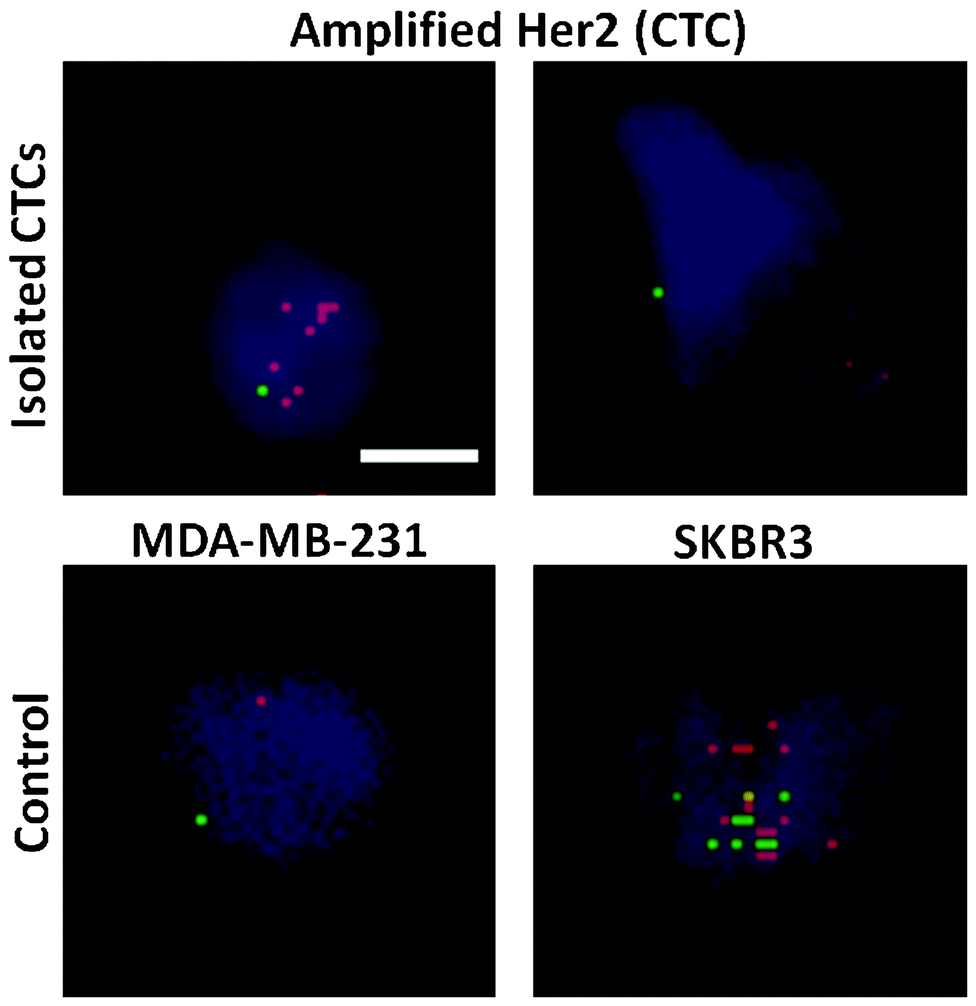 | ||
| Fig. 4 Detection of the centromere of chromosome 17 (Cen-17) and HER2 of enriched CTCs. Cells were amplified for HER2 when the HER2/Cen-17 ratio was >2. MDA-MB-231 and SKBR3 breast cancer cell lines were used as controls. Merged images (DAPI, orange spectrum: HER2 signal, green spectrum: Cen-17) are under 20× magnification. Scale bar: 10 μm. | ||
Conclusions
The “Holy Grail” of cancer medicine is the establishment of personalized therapies, where treatments shift from fixed regimes to therapies tailored to individual patient's tumor conditions.41 Circulating tumor cells have been shown to be a good alternative to primary tumor biopsies, as they carry similar genetic information.2 The detection of CTCs in the peripheral blood of cancer patients at different disease stages has been shown to be a promising prognostic marker for treatment efficacy and patient survival, indicating strong clinical relevance.42 However, systematic characterization of CTCs in vitro via downstream assays has been delayed by the lack of reliable and sensitive methods to detect and enrich these cells. Despite the rapid advances in microfluidic technologies, the isolation of CTCs with high throughput, high purity and high cell viability remained elusive. In this work we demonstrated the application of a novel microfluidic platform for ultra-fast enrichment of putative CTCs using inertial microfluidics in spiral microchannels with a trapezoid cross-section. This improved device achieved higher (blood) volume processing, and increased CTC capture efficiency and yield. In addition to the rapid blood processing speed of 1.7 mL min−1, the simple yet efficient trapezoidal spiral channels greatly facilitate scaled-up device production, and will thus enable larger-scale clinical studies. In this initial proof of principle investigation, the trapezoid chip successfully isolated CTCs from 10 out of 10 (100%) patients with advanced stage metastatic breast and lung cancer (3–125 CTCs mL−1), and allowed extensive heterogeneity studies via immunostaining and DNA FISH analysis. The viability of the isolated CTCs was also retained after processing, which will allow potential culture and expansion studies. The majority of the isolated CTCs from the peripheral blood of breast cancer patients is viable but non-proliferative after days in culture, suggesting the requirement of new therapeutic approaches that target cells in dormancy. In addition, the continuous collection of CTCs facilitates coupling of the device with conventional 96-well plates or a membrane filter for subsequent downstream analysis such as immunostaining, qRT-PCR, FISH and sequencing.The precision and recovery rates at low cell spiking levels given by the inertial microfluidic system were high. Because this approach does not require initial cell surface biomarker selection, it is suitable for use in different cancers of both epithelial and non-epithelial origin. CTCs are reported to be highly heterogeneous and variable in their expression of EpCAM and cytokeratin,43 which are biomarkers used for CTC enrichment in many microfluidic devices. The selection criteria of cell size will overcome this limitation and capture a wider proportion of CTCs. Cells with lower expression of specific cytokeratins will still be identified via immunostaining with pan-cytokeratin antibodies. We chose to analyze the sensitivity of the system by determining the recovery rate of GFP-tagged breast (MCF-7 and MDA-MB-231) and bladder (T24) cancer cell lines spiked into blood obtained from healthy volunteers at a concentration of 500 cells per 7.5 mL of blood. General limitations of various model systems were as described by Ring et al.44 The capture efficiency of cancer cells was high, ranging from 80–90%. This variability can partly be attributed to differences in cell size between cancer types. Nevertheless, the flexibility and simplicity of the system allows for maximum cell isolation from different cancer types by moderate alterations of the channel design. Our results demonstrate the versatility of the system for enriching CTCs of different cancer types, thus acting as a potential tool for the continuous assessment of CTCs and reliable CTC count in patients’ samples. The system was further validated by clinical trials on 10 blood samples from advanced stage metastatic breast and lung cancer patients. CTCs were detected in 10 out of 10 samples, which clearly demonstrates the sensitivity of the system. However, inter-patient variability was observed, a trend reported previously.42,45 This variability does not reflect the analytic performance of the system; rather it depends on various factors such as the stage of the disease and patient condition that may influence the number of CTCs present in the blood.
The application of standard histopathology and immunostaining procedures is essential to the understanding of the role of CTCs in cancer metastasis and potential drug treatment. To date, very low CTC counts were isolated by commercial platforms, which limits these downstream procedures. The improvement of CTC isolation will facilitate the use of standardized procedures for characterization, such as the cytological examination by Papanicolaou (PAP) stains and DNA FISH. CTCs isolated with the trapezoid chip were treated with the PAP stain, which revealed a high nuclear to cytoplasmic N:C ratio, which is characteristic of cancer cells. Some CTCs are HER2+ when treated with respective DNA FISH probes. The presence of HER2+ CTCs varies across samples and is also observed in samples derived from patients with HER2- tumors (2 out of 5). This supports previous finding that the heterogeneity of HER2 status is evident in CTCs as compared to the primary tumor.36 The detection of HER2 amplification in CTCs may identify high-risk breast cancer patients who may benefit from HER2 associated therapeutic strategies.46
In conclusion, our study shows that the examination of blood samples for CTCs with the inertial microfluidic system is possible in clinical trials. The technology we have described here requires further optimization before it can be considered for point-of-care applications. Nonetheless, the convenience and flexibility of the trapezoid chip system potentially promotes the progression and eventual clinical deployment of microfluidic CTC technologies. The next step will also involve the thorough characterization of primary human CTCs and the clinical validation of the prognostic claim for trapezoid chips in terms of treatment efficacy and patient response. The further characterization of these CTCs and demonstration of their prognostic potential are currently being pursued.
Acknowledgements
This research was supported by the National Research Foundation Singapore through the Singapore MIT Alliance for Research and Technology's BioSystems and the Micromechanics Inter-Disciplinary Research programme. This work is also supported by the use of the NTU's Micro-Machine Center (MMC) facilities for wafer fabrication and the lab facilities at the Mechanobiology Institute (MBI) and the Nano Biomechanics Laboratory at the National University of Singapore. The clinical samples and data collection were supported by the Singapore National Medical Research Council grant NMRC 1225/2009.References
- S. Mocellin, D. Hoon, A. Ambrosi, D. Nitti and C. R. Rossi, Clin. Cancer Res., 2006, 12, 4605–4613 CrossRef CAS.
- S. Maheswaran, L. V. Sequist, S. Nagrath, L. Ulkus, B. Brannigan, C. V. Collura, E. Inserra, S. Diederichs, A. J. Iafrate and D. W. Bell, N. Engl. J. Med., 2008, 359, 366–377 CrossRef CAS.
- I. Desitter, B. S. Guerrouahen, N. Benali-Furet, J. Wechsler, P. A. Jänne, Y. Kuang, M. Yanagita, L. Wang, J. A. Berkowitz and R. J. Distel, Anticancer Res., 2011, 31, 427–441 Search PubMed.
- D. F. Hayes, M. Cristofanilli, G. T. Budd, M. J. Ellis, A. Stopeck, M. C. Miller, J. Matera, W. J. Allard, G. V. Doyle and L. Terstappen, Clin. Cancer Res., 2006, 12, 4218–4224 CrossRef CAS.
- V. Zieglschmid, C. Hollmann and O. Böcher, Crit. Rev. Clin. Lab. Sci., 2005, 42, 155–196 CrossRef CAS.
- K. Ameri, R. Luong, H. Zhang, A. Powell, K. Montgomery, I. Espinosa, D. Bouley, A. Harris and S. Jeffrey, Br. J. Cancer, 2010, 102, 561–569 CrossRef CAS.
- M. Al-Hajj, M. S. Wicha, A. Benito-Hernandez, S. J. Morrison and M. F. Clarke, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3983–3988 CrossRef CAS.
- K. Pantel, R. H. Brakenhoff and B. Brandt, Nat. Rev. Cancer, 2008, 8, 329–340 CrossRef CAS.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS.
- J. P. Gleghorn, E. D. Pratt, D. Denning, H. Liu, N. H. Bander, S. T. Tagawa, D. M. Nanus, P. A. Giannakakou and B. J. Kirby, Lab Chip, 2010, 10, 27–29 RSC.
- S. L. Stott, C.-H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas and G. K. Korir, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS.
- Z. Du, K. Cheng, M. Vaughn, N. Collie and L. Gollahon, Biomed. Microdevices, 2007, 9, 35–42 CrossRef CAS.
- N. Shao, E. Wickstrom and B. Panchapakesan, Nanotechnology, 2008, 19, 465101 CrossRef.
- A. A. S. Bhagat, H. W. Hou, L. D. Li, C. T. Lim and J. Han, Lab Chip, 2011, 11, 1870–1878 RSC.
- S. J. Tan, L. Yobas, G. Y. H. Lee, C. N. Ong and C. T. Lim, Biomed. Microdevices, 2009, 11, 883–892 CrossRef.
- R. Gertler, R. Rosenberg, K. Fuehrer, M. Dahm, H. Nekarda and J. R. Siewert, in Molecular Staging of Cancer, Springer, 2003, pp. 149–155 Search PubMed.
- A. L. Allan, S. A. Vantyghem, A. B. Tuck, A. F. Chambers, I. H. Chin–Yee and M. Keeney, Cytometry, Part A, 2005, 65A, 4–14 CrossRef.
- M. Yu, S. Stott, M. Toner, S. Maheswaran and D. A. Haber, J. Cell Biol., 2011, 192, 373–382 CrossRef CAS.
- J. Sun, M. Li, C. Liu, Y. Zhang, D. Liu, W. Liu, G. Hu and X. Jiang, Lab Chip, 2012, 12, 3952–3960 RSC.
- A. A. S. Bhagat, H. Bow, H. W. Hou, S. J. Tan, J. Han and C. T. Lim, Med. Biol. Eng. Comput., 2010, 48, 999–1014 Search PubMed.
- J. Chen, J. Li and Y. Sun, Lab Chip, 2012, 12, 1753–1767 RSC.
- I. Cima, C. W. Yee, F. S. Iliescu, W. M. Phyo, K. H. Lim, C. Iliescu and M. H. Tan, Biomicrofluidics, 2013, 7, 011810 CrossRef.
- W. C. Lee, A. A. S. Bhagat, S. Huang, K. J. Van Vliet, J. Han and C. T. Lim, Lab Chip, 2011, 11, 1359–1367 RSC.
- S. S. Kuntaegowdanahalli, A. A. S. Bhagat, G. Kumar and I. Papautsky, Lab Chip, 2009, 9, 2973–2980 RSC.
- A. A. S. Bhagat, S. S. Kuntaegowdanahalli and I. Papautsky, Microfluid. Nanofluid., 2009, 7, 217–226 CrossRef CAS.
- D. Di Carlo, D. Irimia, R. G. Tompkins and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18892–18897 CrossRef CAS.
- D. R. Gossett and D. D. Carlo, Anal. Chem., 2009, 81, 8459–8465 CrossRef CAS.
- A. A. S. Bhagat, S. S. Kuntaegowdanahalli and I. Papautsky, Lab Chip, 2008, 8, 1906–1914 RSC.
- H. W. Hou, M. E. Warkiani, B. L. Khoo, Z. R. Li, R. A. Soo, D. S.-W. Tan, W.-T. Lim, J. Han, A. A. S. Bhagat and C. T. Lim, Sci. Rep., 2013, 3, 1259 CAS.
- G. Guan, L. Wu, A. A. Bhagat, Z. Li, P. C. Chen, S. Chao, C. J. Ong and J. Han, Sci. Rep., 2013, 3, 1475 CrossRef CAS.
- L. Wu, G. Guan, H. W. Hou, A. A. S. Bhagat and J. Han, Anal. Chem., 2012, 84, 9324–9331 CAS.
- B. N. G. Sajay, C.-P. Chang, H. Ahmad, W. C. Chung, P. D. Puiu and A. R. A. Rahman, Biomed. Microdevices, 2013, 1–11 Search PubMed.
- X. Tong, L. Yang, J. C. Lang, M. Zborowski and J. J. Chalmers, Cytometry, Part B, 2007, 72B, 310–323 CrossRef CAS.
- E. Ozkumur, A. M. Shah, J. C. Ciciliano, B. L. Emmink, D. T. Miyamoto, E. Brachtel, M. Yu, P.-i. Chen, B. Morgan and J. Trautwein, Sci. Transl. Med., 2013, 5, 179ra47 CrossRef CAS.
- M. G. Krebs, R. Sloane, L. Priest, L. Lancashire, J. M. Hou, A. Greystoke, T. H. Ward, R. Ferraldeschi, A. Hughes and G. Clack, J. Clin. Oncol., 2011, 29, 1556–1563 CrossRef.
- S. Riethdorf, H. Fritsche, V. Müller, T. Rau2, C. Schindlbeck, W. J. Brigitte Rack6, C. Coith, K. Beck, F. Jänicke, S. Jackson, T. Gornet, M. Cristofanilli and K. Pantel, Clin. Cancer Res., 2007, 13, 920 CrossRef CAS.
- T. J. Metzner, K. Bethel, E. H. Cho, M. S. Luttgen, D. C. Lazar, M. L. Uson, J. J. Nieva, L. Bazhenova, A. Kolatkar and P. Kuhn, Cancer Res., 2012, 72(8 Supplement), 3619 Search PubMed.
- A. A. Powell, A. H. Talasaz, H. Zhang, M. A. Coram, A. Reddy, G. Deng, M. L. Telli, R. H. Advani, R. W. Carlson and J. A. Mollick, PLoS One, 2012, 7, e33788 CAS.
- B. R. BAS Jaeger, U. Andergassen, J. K. Neugebauer, C. A. Melcher, C. Scholz, C. Hagenbeck, K. Schueller, R. Lorenz, T. Decker, G. Heinrich, T. Fehm, A. Schneeweiss, W. Lichtenegger, M. W. Beckmann, K. Pantel, H. L. Sommer, K. Friese and W. Janni, presented in part at the Thirty-Fifth Annual CTRC-AACR San Antonio Breast Cancer Symposium, San Antonio, TX, 2012.
- L. M. Flores, D. W. Kindelberger, A. H. Ligon, M. Capelletti, M. Fiorentino, M. Loda, E. S. Cibas, P. A. Jänne and I. E. Krop, Br. J. Medicine, 2010, 102, 1495–1502 CAS.
- E. Heitzer, M. Auer, C. Gasch, M. Pichler, P. Ulz, E. M. Hoffmann, S. Lax, J. Waldispuehl-Geigl, O. Mauermann and C. Lackner, Cancer Res., 2013 Search PubMed.
- M. Cristofanilli, T. Budd, M. J. Ellis, A. Stopeck, J. Matera, M. C. Miller, J. M. Reuben, G. V. Doyle, J. Allard, L. W. M. M. Terstappen and D. F. Hayes, N. Engl. J. Med., 2004, 351, 781–791 CrossRef CAS.
- D. R. Parkinson, N. Dracopoli, B. Gumbs Petty, C. Compton, M. Cristofanilli, A. Deisseroth, D. F. Hayes, G. Kapke, P. Kumar and J. Lee, J. Transl. Med., 2012, 10, 138 CrossRef.
- A. Ring, I. E. Smith and M. Dowsett, Lancet Oncol., 2004, 5, 79–88 CrossRef.
- W. J. Allard, J. Matera, M. C. Miller, M. Repollet, M. C. Connelly, C. Rao, A. G. J. Tibbe, J. W. Uhr and L. W. M. M. Terstappen, Clin. Cancer Res., 2004, 10, 6897 CrossRef.
- R. Y. Tsang and R. S. Finn, Br. J. Cancer, 2012, 106, 6–13 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc50617g |
| ‡ Authors contributed equally. |
| § Current address: Clearbridge BioMedics Pte Ltd, Singapore. |
| This journal is © The Royal Society of Chemistry 2014 |