Technologies for detection of circulating tumor cells: facts and vision
Catherine
Alix-Panabières
ab and
Klaus
Pantel
*c
aUniversity Medical Centre, Saint-Eloi Hospital, Department of Cellular and Tissue Biopathology of Tumors, Laboratory of Rare Human Circulating Cells (LCCRH), Montpellier, France
bUniversity Institute of Clinical Research UM1-EA2415-Epidemiology, Biostatistics & Public Health, Montpellier, France
cInstitute of Tumor Biology, University Medical Center Hamburg-Eppendorf, Martinistrasse 52, 20246 Hamburg, Germany. E-mail: pantel@uke.de; Fax: +49 40 7410 55379; Tel: +49 40 7410 53503
First published on 23rd September 2013
Abstract
Hematogeneous tumor cell dissemination is a key step in cancer progression. The detection of CTCs in the peripheral blood of patients with solid epithelial tumors (e.g., breast, prostate, lung and colon cancer) holds great promise, and many exciting technologies have been developed over the past years. However, the detection and molecular characterization of circulating tumor cells (CTCs) remain technically challenging. The identification and characterization of CTCs require extremely sensitive and specific analytical methods, which are usually a combination of complex enrichment and detection procedures. CTCs occur at very low concentrations of one tumor cell in the background of millions of normal blood cells and the epithelial–mesenchymal plasticity of CTCs can hamper their detection by the epithelial markers used in current CTC assays. In the present review, we summarize current methods for the enrichment and detection of CTCs and discuss the key challenges and perspectives of CTC analyses within the context of improved clinical management of cancer patients.
Introduction
The detection of circulating tumor cells (CTCs) in the peripheral blood of cancer patients holds great promise but remains technically challenging. Over the past few years, there has been a lot of interest in the CTC field with rapid growth of developing new biotechnologies to identify CTCs.CTCs occur at very low concentrations of one tumor cell in the background of millions of blood cells. Their identification and characterization require therefore extremely sensitive and specific analytical methods, which are usually a combination of enrichment and detection procedures. Since the detection of rare events is always hampered by the problems of the Poisson statistics, the analysis of a larger blood volume (at least 7.5 mL or more) is preferable, in particular, in early-stage cancer patients with a small burden of CTCs.
Aims of research on CTCs include (i) estimation of the risk for metastatic relapse or progression (i.e., prognostic information), (ii) stratification and real-time monitoring of therapies, (iii) identification of therapeutic targets and resistance mechanisms, and (iv) understanding metastatic development in cancer patients.
This review will focus on the current methodological challenges and perspectives of detection and characterization of CTCs, which might contribute to the development of improved individualized targeted treatment for cancer patients.
CTC enrichment
The CTC enrichment includes a large panel of technologies based on the different properties of CTCs that must differentiate them from the normal hematopoietic cells: (i) physical properties (e.g., size, density, electric charges and deformability) and (ii) biological properties (e.g., surface protein expression and invasion capacity) (Fig. 1).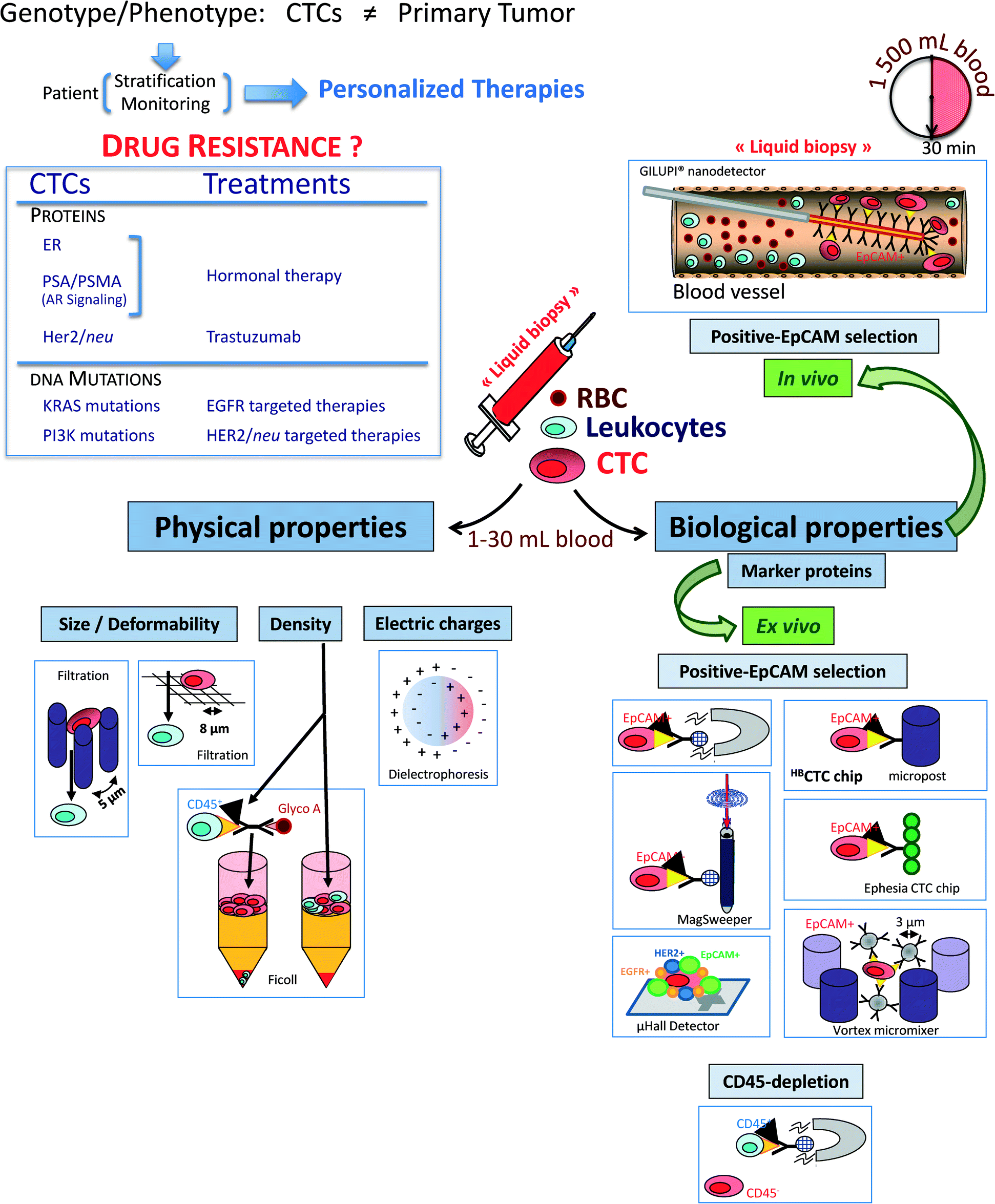 | ||
| Fig. 1 Circulating tumor cells (CTCs) as a real-time liquid biopsy after different enrichment methods from the bloodstream of cancer patients. (a) Physical properties include size, deformability, density, and electric charge. (b) Biological properties are based on the expression of cell surface markers, including an epithelial cell adhesion molecule (EpCAM) for positive selection and CD45 for negative selection. All these technologies have been used on blood samples ex vivo, however, recently, a new in vivo nanodetector allows the enrichment of CTCs directly in the arm vein of the patients, which enables the enrichment of CTCs from approximately 1500 mL of blood, allowing to collect larger quantities of CTCs.As the CTCs can be derived from different sources (e.g., primary tumor or metastatic organs such as liver, lung and BM) depending on the stage of the disease, they serve as a liquid biopsy of cancer and reveal important information on the therapeutic targets and/or resistance mechanisms, which might be used in the future to stratify patients to targeted therapies such as inhibition of EGFR/HER2 or hormonal therapy (i.e., ER, AR) and monitor the efficacy of the treatment and the development of resistance in real-time.Abbreviations: glyco A, glycophorin A (a 131-amino-acid protein present at the extracellular surface of the human red blood cell); RBC, red blood cells; ER, estrogen receptor; PSA, prostate specific antigen; PSMA, prostate specific membrane antigen; AR, androgen receptor, EGFR, epithelial growth factor receptor. This figure has been modified from the article Alix-Panabières et al., Ann Rev Med, 2012 and from the article Alix-Panabières et al., Clin Chem, 2013. | ||
Physical properties allowing for separation without labelling are used in (i) density gradient centrifugation (Ficoll, OncoQuick™), in which a certain amount of CTCs is known to be lost with these systems; (ii) filtration through special filters, ISET (isolation by size of epithelial tumor cells)1,2 or a novel three dimensional microfilter,3 in which small CTCs can be lost and large hematopoietic cells can be retained by the pores of the filters; (iii) a new versatile label free biochip using the unique differences in size (larger) and deformability (stiffer) of cancer cells from blood cells;4,5 (iv) a microfluidic device combining multi-orifice flow fractionation (MOFF) and dielectrophoretic (DEP) cell separation technique;6 and (v) a dielectrophoretic field-flow fractionation (DEP-FFF) device, allowing for the isolation of viable CTCs by different responses to DEP due to differences in size and membrane properties.7
Biological properties are mainly used in immunological procedures with antibodies against either tumour-associated antigens (positive selection) or common leukocytes antigen CD45 (negative selection). Immunomagnetic systems target an antigen with an antibody that is coupled to a magnetic bead. Then, the antigen–antibody complex is isolated via exposure to a magnetic field. Positive selection is usually carried out with antibodies against the epithelial cell adhesion molecule (EpCAM) and subsequent immunocytological detection of CTC is performed with antibodies to cytokeratins (CK), the intermediate filaments of epithelial cells.8 Among the current EpCAM-based technologies, the FDA-cleared CellSearch™ system has gained considerable attention over the past 7 years9 and is frequently compared for all new CTC detection methods.10,11 Other promising technologies using different ways to capture EpCAM-positive CTCs have been recently developed, including (i) a microfluidic platform called CTC-chip, which consists of an array of anti-EpCAM antibody-coated microposts and were presented and applied to the analysis of blood samples from patients with solid tumours.12–14 The high CTC counts in cancer patients, even in non-metastatic cancer patients, and the frequent detection of positive events in healthy controls warrant further investigations on the specificity of this assay. (ii) A high-throughput microfluidic mixing device, the herringbone-chip or “HB-Chip”, provides an enhanced platform for CTC isolation;15 (iii) a CTC-chip called “Ephesia”, which uses columns of biofunctionalized superparamagnetic beads self-assembled in a microfluidic channel onto an array of magnetic traps, was recently reported;16 (iv) high-throughput selection, enumeration, and electrokinetic manipulation of low-abundance CTCs using a microfluidic system was described recently;17 (v) another new automated immunomagnetic separation technology, the MagSweeper, positively enriches circulating epithelial cells (CepC), expressing EpCAM, from blood, and allows their subsequent molecular analysis;18,19 (vi) through the use of a 3D nanostructured substrate or nano “Fly Paper” Technology, a silicon-nanowire (SiNW) array coated with anti-EpCAM Ab–CTCs can be captured efficiently and specifically;20 and (vii) a Velcro-like microfluidic device can separate CTCs with an efficiency ranging from 40–70%.21 This is not a complete list of existing assays but just highlights current separation strategies.
Recently, combination assays using tumor-associated physical and biological properties have been published. For example, Ozkumur et al. used a combination of size-based hydrodynamic cell sorting and immunomagnetic selection (positive or negative) and called this new device “CTC-iChip”.22
Given their very small size, such microdevices require small sample volumes and cell numbers, thus minimizing time and the use of expensive staining reagents. However, analyzing such a small amount of blood could be a big limit for detecting rare CTCs, mostly at the early stage of cancer. An elegant way to overcome this limitation is to target the CTCs directly in vivo. This approach is now possible using the Gilupi nanodetector®. During the 30 min application of this device in the peripheral arm vein, up to 1.5 liters of blood, including the CTCs, pass the 2 cm functionalized area of the nanodetector and enable a high number of CTCs to be bound by the anti-EpCAM antibodies.23 An alternative approach is the development of leukapheresis, elutriation for subsequent ex vivo CTC analyses using flow cytometry and real-time PCR for molecular characterization.24
Recently, excellent overviews on the specific advantages, technical challenges and general drawbacks of CTCs assays (including microfluidic technologies) have been published.8,25–28 In particular, Parkinson et al. provides a comprehensive list of current CTC assays and the comments on each assay are the result of consensus of an international group of experts. Briefly, microfluidic systems have the advantage of having usually high yields due to very controlled flow and selection devices. However, these devices might be prone to factors disturbing the blood flow such as air bubbles or blood clots. At present, it is very difficult to predict which physical principle is superior for capturing CTCs in clinical samples. Many CTC technologies are based on the hypothesis that tumor cells are larger and stiffer4,5 than CTCs, but the size of CTCs in primary blood samples from cancer patients varies considerably and tumor cells undergoing an EMT might be as flexible as leukocytes. Dielectric and photoacoustic properties as well as membrane ruffling of tumor cells have been recently introduced but clinical validation of these technologies is still ongoing.29,30
As an alternative, CTC capture based on the expression of tumor-associated cell surface antigens (e.g., EpCAM, EGFR, HER2, MUC1) exists but this approach may also encounter the problem of tumor heterogeneity unless a cocktail of various antibodies targeting a broad spectrum of antigens is used, which, however, may limit the purity of the enriched CTC fraction. Negative selection of CTCs by depletion of leukocytes might be an alternative for an unbiased enrichment of CTCs but the depletion procedure needs to be performed carefully to avoid the loss of CTCs in the “sea” of normal blood cells. Recently, a combination of CTC enrichment by a sequential combination of size selection and antibody-based capture or negative depletion of CTCs has been introduced as “CTC-iChip”.22
CTC identification
Experiments with different cell line cells spiked into normal blood are a good start to evaluate a new CTC assay. However, these cells are selected to deliver optimal results and the yields obtained with primary blood samples from cancer patients are usually much lower. CTCs in cancer patients are more heterogeneous and usually less robust than cell line cells passaged over many years. Thus, new assays should be validated by the analysis of a sufficient number of primary samples from cancer patients.Immunological detection
Most CTC assays described above use the same identification step: cells are fluorescently stained for cytokeratins (positive marker for epithelial tumor cells) and for the common leukocyte antigen CD45 (exclusion marker) with a nuclear dye (4,6-diamino-2-phenylindole (DAPI)). Although some antigens used to capture CTCs are applicable to various different cancer types (e.g., EpCAM for breast, colon, lung and prostate cancer and other epithelial tumors), cancer-specific detection principles might also be suitable (e.g., PSMA or PSA for prostate cancer). Thus, it is naïve to believe that one test will fit all cancer types.In order to detect only viable CTCs, the functional EPithelial ImmunoSPOT (EPISPOT) assay, combined with any kind of enrichment step, was introduced for CTC analyses.31 Avoiding direct contact with the target cells, this technique assesses the presence of CTCs based on secreted, shed or released proteins during 24–48 hours of short-term culture.32 EPISPOT has been applied to blood and bone marrow (BM) samples of breast, colon and prostate cancer patients providing first clinical data.32–34
Furthermore, ultra high-speed automated digital microscopy using fiber-optic-array scanning technology has been developed to detect CTCs that are labelled by antibodies with fluorescent conjugates.35,36 Other slide-based automated scanning microscopes have been introduced for detecting CTCs and show promising results that still need to be validated in large clinical studies.37,38
Immunological detection offers the advantage of subsequent isolation of stained CTCs for subsequent molecular characterization. While manual isolation of CTCs is possible,39,40 it is rather cumbersome and time-consuming. Alternative automated single cell selection devices have been therefore developed (e.g., DEPArray™ technology based on dielectrophoresis).41 Molecular characterization of CTCs might also increase assay specificity because epithelial cells fulfilling the phenotypic criteria of CTCs have been found in patients with benign colon diseases.42
Molecular detection
Assays targeting specific mRNAs are the most widely used alternatives to immunological assays to identify CTCs. In breast cancer, the CK19 mRNA has been most frequently used in clinical studies.8 Many transcripts (e.g. encoding CK18, CK19, CK20, mucin-1, prostate-specific antigen and carcinoembryonic antigen), however, are also expressed at low levels in normal blood and BM cells,8 so quantitative RT-PCR assays with validated cut-off values are required to overcome this problem. Moreover, gene transcription might be downregulated in CTCs (e.g. in the course of EMT, see below), which argues in favour of multimarker RT-PCR approaches.43–45 Very recently, Markou et al. described a new liquid bead array hybridization assay: mRNA isolated from immunomagnetically enriched CTCs was subjected to multiplex PCR for the simultaneous measurements of the expression of 6 genes in CTCs.46In principle, the detection of tumour-specific DNA aberrations is the most specific approach to detect CTCs; however, the marked genetic heterogeneity of solid tumours poses a problem because different tumours of the same entity (e.g., breast carcinomas) and even cells within an individual tumour harbour different genetic aberrations. Thus, a large panel of probes against various genomic aberrations is required to establish a sensitive DNA-based detection system. Moreover, genetic analysis of the primary tumour may not predict the DNA aberrations on CTCs and isolation and direct genetic analysis of single CTCs are technically very challenging.39,40
Epithelial–mesenchymal plasticity as a challenge for CTC detection
Cell plasticity refers to the ability of some cells to take on the characteristics of other cells in an organism. Epithelial–mesenchymal transition (EMT) is a complex process leading to cell dedifferentiation and increased motility via rearrangements of cellular contact junctions and eventually loss of cell adhesion. The reverse process, called mesenchymal–epithelial transition (MET), is expected to play a fundamental role after CTCs have settled down in distant organs and start to form metastases in the new microenvironment. Thus far, little is known about the triggers that steer epithelial–mesenchymal plasticity, i.e., the fine balance between EMT and MET during the metastatic cascade.47Mesenchymal-like cancer cells are present in the bloodstream of cancer patients, and these cells are missed by most of the current assays based on epithelial markers such as EpCAM or cytokeratins. Recent work suggested that EMT might particularly affect tumor cells with stem cell-like properties.48 Therefore, in recent years, there has been great interest in investigating EMT markers in CTCs and recent reports suggest that current assays based on epithelial antigens may miss the most aggressive CTC subpopulation.47 Thus, there is an urgent need for optimizing CTC detection methods through the inclusion of markers that are not repressed during EMT but still allow the analysis to distinguish CTCs from the surrounding blood cells (e.g., plastin-3 in colorectal cancer).49 Recently, Yu et al. have used a combination of cell surface antigens to capture mesenchymal and epithelial CTCs in patients with metastatic breast cancer.50 However, CTCs were only detected in less than 50% of MBC patients, which is lower than the detection rate published by the same group with EpCAM-dependent CTC assays.12 The reason for this puzzling finding is unclear but it exemplifies the difficulties to develop sensitive assays that capture mesenchymal CTCs.
Finally, is there a potential role of platelets in tumor dissemination? Previous findings have indicated that platelet adhesion induces pro-survival and pro-angiogenic signaling in ovarian cancer cells.51 Moreover, experimental studies in mice have shown that platelets bound to CTCs induce EMT.52 Platelets have been shown on CTCs in breast cancer50 and it might be interesting to investigate if CTCs covered with platelets have an increased potential to colonize secondary organs and if they can be hidden during the CTC enrichment step.
Conclusions
CTCs can be currently enriched and detected through different strategies. However, in the future, we need to develop better strategies that are also able to isolate and identify (i) EMT-like sub-populations of tumor cells with down-regulated epithelial-specific protein expression and also (ii) the subset of CTCs with organ-specific metastatic signatures to identify their origin. At present, for the enrichment step, negative selection strategies based on the depletion of the leukocytes surrounding the CTCs might be feasible approaches for the unbiased selection of tumor cells, including those subsets of disseminating cells that lack current epithelial markers.At present, analysis of CTCs is still not part of the routine tumor staging in clinical practice. This is mainly due to the low number of detectable CTCs with the currently available methods, limiting their value as “real-time liquid biopsy” especially in patients with early stage tumors.26 Therefore, the new innovative approaches urgently have to be evaluated for reproducibility, sensitivity and specificity in order to become applicable for clinical practice. Information about the CTC status may be used to assess the individual prognosis of cancer patients and to stratify the patients at risk to systemic therapies aimed at preventing recurrences and metastatic relapses. Furthermore, CTC measurements within clinical trials might serve as an important biomarker for real-time monitoring of the efficacy of systemic therapies in individual cancer patients and might thereby support accelerating drug development and defining subpopulations of patients with the highest treatment benefit. Moreover, CTC analysis for downstream components within signal transduction pathways, which influence targeted therapies (e.g., K-ras mutations in EGFR-targeted therapies or PI3K mutations in HER2-targeted therapies), might provide new insights into the complex mechanisms of drug resistance.
Disclosure statement:
No potential conflicts of interest were disclosed.Grant support:
C. Alix-Panabières, the European Commission (DISMAL project, contract no. LSHC-CT-2005-018911, the INCa, the INSERM and the DGOS via SIRIC Montpellier Cancer, Grant « INCa-DGOS-Inserm 6045 »;K. Pantel, the European Commission (DISMALproject, contract no. LSHC-CT-2005-018911, European Research Council Investigator Grant DISSECT (no. 269081).
References
- G. Vona, A. Sabile, M. Louha, V. Sitruk, S. Romana, K. Schutze, F. Capron, D. Franco, M. Pazzagli, M. Vekemans, B. Lacour, C. Brechot and P. Paterlini-Brechot, Am. J. Pathol., 2000, 156, 57–63 CrossRef CAS PubMed.
- P. Pinzani, B. Salvadori, L. Simi, S. Bianchi, V. Distante, L. Cataliotti, M. Pazzagli and C. Orlando, Hum. Pathol., 2006, 37, 711–718 CrossRef CAS PubMed.
- S. Zheng, H. K. Lin, B. Lu, A. Williams, R. Datar, R. J. Cote and Y. C. Tai, Biomed. Microdevices, 2011, 13, 203–213 CrossRef PubMed.
- S. J. Tan, R. L. Lakshmi, P. Chen, W. T. Lim, L. Yobas and C. T. Lim, Biosens. Bioelectron., 2010, 26, 1701–1705 CrossRef CAS PubMed.
- A. A. Bhagat, H. W. Hou, L. D. Li, C. T. Lim and J. Han, Lab Chip, 2011, 11, 1870–1878 RSC.
- H. S. Moon, K. Kwon, S. I. Kim, H. Han, J. Sohn, S. Lee and H. I. Jung, Lab Chip, 2011, 11, 1118–1125 RSC.
- P. R. Gascoyne, J. Noshari, T. J. Anderson and F. F. Becker, Electrophoresis, 2009, 30, 1388–1398 CrossRef CAS PubMed.
- K. Pantel, R. H. Brakenhoff and B. Brandt, Nat. Rev. Cancer, 2008, 8, 329–340 CrossRef CAS PubMed.
- K. Pantel, C. Alix-Panabieres and S. Riethdorf, Nat. Rev. Clin. Oncol., 2009, 6, 339–351 CrossRef CAS PubMed.
- E. Andreopoulou, L. Y. Yang, K. M. Rangel, J. M. Reuben, L. Hsu, S. Krishnamurthy, V. Valero, H. A. Fritsche and M. Cristofanilli, Int. J. Cancer, 2012, 130, 1590–1597 CrossRef CAS PubMed.
- V. Hofman, M. I. Ilie, E. Long, E. Selva, C. Bonnetaud, T. Molina, N. Vénissac, J. Mouroux, P. Vielh and P. Hofman, Int. J. Cancer, 2011, 129, 1651–1660 CrossRef CAS PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- S. Maheswaran, L. V. Sequist, S. Nagrath, L. Ulkus, B. Brannigan, C. V. Collura, E. Inserra, S. Diederichs, A. J. Iafrate, D. W. Bell, S. Digumarthy, A. Muzikansky, D. Irimia, J. Settleman, R. G. Tompkins, T. J. Lynch, M. Toner and D. A. Haber, N. Engl. J. Med., 2008, 359, 366–377 CrossRef CAS PubMed.
- S. L. Stott, R. J. Lee, S. Nagrath, M. Yu, D. T. Miyamoto, L. Ulkus, E. J. Inserra, M. Ulman, S. Springer, Z. Nakamura, A. L. Moore, D. I. Tsukrov, M. E. Kempner, D. M. Dahl, C. L. Wu, A. J. Iafrate, M. R. Smith, R. G. Tompkins, L. V. Sequist, M. Toner, D. A. Haber and S. Maheswaran, Sci. Transl. Med., 2010, 2, 25ra23 Search PubMed.
- S. L. Stott, C. H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd Jr., A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS PubMed.
- A. E. Saliba, L. Saias, E. Psychari, N. Minc, D. Simon, F. C. Bidard, C. Mathiot, J. Y. Pierga, V. Fraisier, J. Salamero, V. Saada, F. Farace, P. Vielh, L. Malaquin and J. L. Viovy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14524–14529 CrossRef CAS PubMed.
- U. Dharmasiri, S. K. Njoroge, M. A. Witek, M. G. Adebiyi, J. W. Kamande, M. L. Hupert, F. Barany and S. A. Soper, Anal. Chem., 2011, 83, 2301–2309 CrossRef CAS PubMed.
- A. H. Talasaz, A. A. Powell, D. E. Huber, J. G. Berbee, K. H. Roh, W. Yu, W. Xiao, M. M. Davis, R. F. Pease, M. N. Mindrinos, S. S. Jeffrey and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3970–3975 CrossRef CAS PubMed.
- A. A. Powell, A. H. Talasaz, H. Zhang, M. A. Coram, A. Reddy, G. Deng, M. L. Telli, R. H. Advani, R. W. Carlson, J. A. Mollick, S. Sheth, A. W. Kurian, J. M. Ford, F. E. Stockdale, S. R. Quake, R. F. Pease, M. N. Mindrinos, G. Bhanot, S. H. Dairkee, R. W. Davis and S. S. Jeffrey, PLoS One, 2012, 7, e33788 CAS.
- S. Wang, G. E. Owens and H. R. Tseng, Methods Mol. Biol., 2011, 726, 141–150 CAS.
- S. Hou, H. Zhao, L. Zhao, Q. Shen, K. S. Wei, D. Y. Suh, A. Nakao, M. A. Garcia, M. Song, T. Lee, B. Xiong, S. C. Luo, H. R. Tseng and H. H. Yu, Adv. Mater., 2013, 25, 1547–1551 CrossRef CAS PubMed.
- E. Ozkumur, A. M. Shah, J. C. Ciciliano, B. L. Emmink, D. T. Miyamoto, E. Brachtel, M. Yu, P. I. Chen, B. Morgan, J. Trautwein, A. Kimura, S. Sengupta, S. L. Stott, N. M. Karabacak, T. A. Barber, J. R. Walsh, K. Smith, P. S. Spuhler, J. P. Sullivan, R. J. Lee, D. T. Ting, X. Luo, A. T. Shaw, A. Bardia, L. V. Sequist, D. N. Louis, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Sci. Transl. Med., 2013, 5, 179ra147 Search PubMed.
- N. Saucedo-Zeni, S. Mewes, R. Niestroj, L. Gasiorowski, D. Murawa, P. Nowaczyk, T. Tomasi, E. Weber, G. Dworacki, N. G. Morgenthaler, H. Jansen, C. Propping, K. Sterzynska, W. Dyszkiewicz, M. Zabel, M. Kiechle, U. Reuning, M. Schmitt and K. Lücke, Int. J. Oncol., 2012, 41, 1241–1250 Search PubMed.
- R. L. Eifler, J. Lind, D. Falkenhagen, V. Weber, M. B. Fischer and R. Zeillinger, Cytometry, Part B, 2011, 80, 100–111 CrossRef PubMed.
- D. R. Parkinson, N. Dracopoli, B. Gumbs Petty, C. Compton, M. Cristofanilli, A. Deisseroth, D. F. Hayes, G. Kapke, P. Kumar, J. S. Lee, M. C. Liu, R. McCormack, S. Mikulski, L. Nagahara, K. Pantel, S. Pearson-White, E. A. Punnoose, L. T. Roadcap, A. E. Schade, H. I. Scher, C. C. Sigman and G. J. Kelloff, J. Transl. Med., 2012, 10, 138 CrossRef PubMed.
- C. Alix-Panabieres and K. Pantel, Clin. Chem., 2013, 59, 110–118 CAS.
- E. S. Lianidou, Clin. Chem., 2012, 58, 805–807 CAS.
- M. Yu, S. Stott, M. Toner, S. Maheswaran and D. A. Haber, J. Cell Biol., 2011, 192, 373–382 CrossRef CAS PubMed.
- M. A. Juratli, M. Sarimollaoglu, E. Siegel, D. A. Nedosekin, E. Galanzha, J. Y. Suen and V. P. Zharov, Head Neck, 2013 DOI:10.1002/hed.23439.
- D. A. Nedosekin, M. A. Juratli, M. Sarimollaoglu, C. L. Moore, N. J. Rusch, M. S. Smeltzer, V. P. Zharov and E. I. Galanzha, J. Biophotonics, 2013, 6, 523–533 CrossRef CAS PubMed.
- H. Schwarzenbach, C. Alix-Panabieres, I. Muller, N. Letang, J. P. Vendrell, X. Rebillard and K. Pantel, Clin. Cancer Res., 2009, 15, 1032–1038 CrossRef CAS PubMed.
- C. Alix-Panabieres, J. P. Vendrell, M. Slijper, O. Pelle, E. Barbotte, G. Mercier, W. Jacot, M. Fabbro and K. Pantel, Breast Cancer Res., 2009, 11, R39 CrossRef PubMed.
- C. Alix-Panabieres, J. P. Vendrell, O. Pelle, X. Rebillard, S. Riethdorf, V. Muller, M. Fabbro and K. Pantel, Clin. Chem., 2007, 53, 537–539 CAS.
- E. Denève, S. Riethdorf, J. Ramos, D. Nocca, A. Coffy, J. P. Daurès, T. Maudelonde, J. M. Fabre, K. Pantel and C. Alix-Panabières, Clin. Chem., 2013, 59, 1384–1392 Search PubMed.
- R. T. Krivacic, A. Ladanyi, D. N. Curry, H. B. Hsieh, P. Kuhn, D. E. Bergsrud, J. F. Kepros, T. Barbera, M. Y. Ho, L. B. Chen, R. A. Lerner and R. H. Bruce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10501–10504 CrossRef CAS PubMed.
- G. Somlo, S. K. Lau, P. Frankel, H. B. Hsieh, X. Liu, L. Yang, R. Krivacic and R. H. Bruce, Breast Cancer Res. Treat., 2011, 128, 155–163 CrossRef CAS PubMed.
- T. G. Ntouroupi, S. Q. Ashraf, S. B. McGregor, B. W. Turney, A. Seppo, Y. Kim, X. Wang, M. W. Kilpatrick, P. Tsipouras, T. Tafas and W. F. Bodmer, Br. J. Cancer, 2008, 99, 789–795 CrossRef CAS PubMed.
- G. Deng, M. Herrler, D. Burgess, E. Manna, D. Krag and J. F. Burke, Breast Cancer Res., 2008, 10, R69 CrossRef PubMed.
- C. Gasch, T. Bauernhofer, M. Pichler, S. Langer-Freitag, M. Reeh, A. M. Seifert, O. Mauermann, J. R. Izbicki, K. Pantel and S. Riethdorf, Clin. Chem., 2013, 59, 252–260 CAS.
- E. Heitzer, M. Auer, C. Gasch, M. Pichler, P. Ulz, E. M. Hoffmann, S. Lax, J. Waldispuehl-Geigl, O. Mauermann, C. Lackner, G. Höfler, F. Eisner, H. Sill, H. Samonigg, K. Pantel, S. Riethdorf, T. Bauernhofer, J. B. Geigl and M. R. Speicher, Cancer Res., 2013, 73, 2965–2975 CrossRef CAS PubMed.
- F. Fabbri, S. Carloni, W. Zoli, P. Ulivi, G. Gallerani, P. Fici, E. Chiadini, A. Passardi, G. L. Frassineti, A. Ragazzini and D. Amadori, Cancer Lett., 2013, 335, 225–231 CrossRef CAS PubMed.
- K. Pantel, E. Deneve, D. Nocca, A. Coffy, J. P. Vendrell, T. Maudelonde, S. Riethdorf and C. Alix-Panabieres, Clin. Chem., 2012, 58, 936–940 CAS.
- T. Fehm, O. Hoffmann, B. Aktas, S. Becker, E. F. Solomayer, D. Wallwiener, R. Kimmig and S. Kasimir-Bauer, Breast Cancer Res., 2009, 11, R59 CrossRef PubMed.
- A. Strati, S. Kasimir-Bauer, A. Markou, C. Parisi and E. S. Lianidou, Breast Cancer Res., 2013, 15, R20 CrossRef CAS PubMed.
- S. Zhao, H. Yang, M. Zhang, D. Zhang, Y. Liu, Y. Liu, Y. Song, X. Zhang, H. Li, W. Ma and Q. Zhang, Cell Biochem. Biophys., 2013, 65, 263–273 CrossRef CAS PubMed.
- A. Markou, A. Strati, N. Malamos, V. Georgoulias and E. S. Lianidou, Clin. Chem., 2011, 57, 421–430 CAS.
- N. Bednarz-Knoll, C. Alix-Panabières and K. Pantel, Cancer Metastasis Rev., 2012, 31, 673–687 CrossRef CAS PubMed.
- S. A. Mani, W. Guo, M. J. Liao, E. N. Eaton, A. Ayyanan, A. Y. Zhou, M. Brooks, F. Reinhard, C. C. Zhang, M. Shipitsin, L. L. Campbell, K. Polyak, C. Brisken, J. Yang and R. A. Weinberg, Cell, 2008, 133, 704–715 CrossRef CAS PubMed.
- T. Yokobori, H. Iinuma, T. Shimamura, S. Imoto, K. Sugimachi, H. Ishii, M. Iwatsuki, D. Ota, M. Ohkuma, T. Iwaya, N. Nishida, R. Kogo, T. Sudo, F. Tanaka, K. Shibata, H. Toh, T. Sato, G. F. Barnard, T. Fukagawa, S. Yamamoto, H. Nakanishi, S. Sasaki, S. Miyano, T. Watanabe, H. Kuwano, K. Mimori, K. Pantel and M. Mori, Cancer Res., 2013, 73, 2059–2069 CrossRef CAS PubMed.
- M. Yu, A. Bardia, B. S. Wittner, S. L. Stott, M. E. Smas, D. T. Ting, S. J. Isakoff, J. C. Ciciliano, M. N. Wells, A. M. Shah, K. F. Concannon, M. C. Donaldson, L. V. Sequist, E. Brachtel, D. Sgroi, J. Baselga, S. Ramaswamy, M. Toner, D. A. Haber and S. Maheswaran, Science, 2013, 339, 580–584 CrossRef CAS PubMed.
- K. Egan, D. Crowley, P. Smyth, S. O'Toole, C. Spillane, C. Martin, M. Gallagher, A. Canney, L. Norris, N. Conlon, L. McEvoy, B. Ffrench, B. Stordal, H. Keegan, S. Finn, V. McEneaney, A. Laios, J. Ducree, E. Dunne, L. Smith, M. Berndt, O. Sheils, D. Kenny and J. O'Leary, PLoS One, 2011, 6, e26125 CAS.
- M. Labelle, S. Begum and R. O. Hynes, Cancer Cell, 2011, 20, 576–590 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |