Size-selective collection of circulating tumor cells using Vortex technology†
Elodie
Sollier‡
*ab,
Derek E.
Go‡
a,
James
Che‡
a,
Daniel R.
Gossett
ac,
Sean
O'Byrne
a,
Westbrook M.
Weaver
ab,
Nicolas
Kummer
d,
Matthew
Rettig
ef,
Jonathan
Goldman
fg,
Nicholas
Nickols
d,
Susan
McCloskey
df,
Rajan P.
Kulkarni
*h and
Dino
Di Carlo
*abcf
aDepartment of Bioengineering, University of California, 420 Westwood Plaza, 5121 Engineering V, P.O. Box 951600, Los Angeles, CA 90095, USA. E-mail: dicarlo@seas.ucla.edu
bVortex Biosciences Inc., 228 Hamilton Avenue, #3F, Palo Alto, CA 94301, USA. E-mail: elodie@netscientific.net
cCalifornia Nano Systems Institute, 570 Westwood Plaza, Building 114, Los Angeles, CA 90095, USA
dDepartment of Radiation Oncology, UCLA Medical Center, 200 UCLA Medical Plaza, Los Angeles, CA 90095, USA
eFrank Clark Urological Center, UCLA Medical Center, 200 UCLA Medical Plaza, Los Angeles, CA 90095, USA
fJonsson Comprehensive Cancer Center, Los Angeles, CA 90095, USA
gUCLA Santa Monica Hematology Oncology, 2020 Santa Monica Blvd, Suite 600, Santa Monica, CA 90404, USA
hDivision of Dermatology, UCLA Medical Center, 52-121 CHS, Los Angeles, CA 90095, USA. E-mail: rkulkarn@ucla.edu
First published on 15th August 2013
Abstract
A blood-based, low cost alternative to radiation intensive CT and PET imaging is critically needed for cancer prognosis and management of its treatment. “Liquid biopsies” of circulating tumor cells (CTCs) from a relatively non-invasive blood draw are particularly ideal, as they can be repeated regularly to provide up to date molecular information about the cancer, which would also open up key opportunities for personalized therapies. Beyond solely diagnostic applications, CTCs are also a subject of interest for drug development and cancer research. In this paper, we adapt a technology previously introduced, combining the use of micro-scale vortices and inertial focusing, specifically for the high-purity extraction of CTCs from blood samples. First, we systematically varied parameters including channel dimensions and flow rates to arrive at an optimal device for maximum trapping efficiency and purity. Second, we validated the final device for capture of cancer cell lines in blood, considering several factors, including the effect of blood dilution, red blood cell lysis and cell deformability, while demonstrating cell viability and independence on EpCAM expression. Finally, as a proof-of-concept, CTCs were successfully extracted and enumerated from the blood of patients with breast (N = 4, 25–51 CTCs per 7.5 mL) and lung cancer (N = 8, 23–317 CTCs per 7.5 mL). Importantly, samples were highly pure with limited leukocyte contamination (purity 57–94%). This Vortex approach offers significant advantages over existing technologies, especially in terms of processing time (20 min for 7.5 mL of whole blood), sample concentration (collecting cells in a small volume down to 300 μL), applicability to various cancer types, cell integrity and purity. We anticipate that its simplicity will aid widespread adoption by clinicians and biologists who desire to not only enumerate CTCs, but also uncover new CTC biology, such as unique gene mutations, vesicle secretion and roles in metastatic processes.
1. Introduction
Circulating tumor cells (CTCs) are cancer cells that are shed from primary tumors and travel through the bloodstream to other organs 1 CTCs may play a key role in metastasis, which is known to be the direct cause of 90% of cancer deaths.2,3 The isolation of these CTCs from the bloodstream, commonly termed a “liquid biopsy”, offers a minimally invasive method to obtain cancer cells for enumeration or molecular and cell analysis.3–5First, the number of CTCs has been shown to be predictive of cancer prognosis 6 and may also assist with characterizing treatment efficacy.7 Indeed, an increase in CTCs may indicate that a chemotherapy treatment is no longer effective or that there is a change in the cancer, rendering the tumor cells more resistant to current therapy. Biopsy of CTCs from blood would offer a more rapid method for treatment monitoring to potentially detect tumor resistance and regrowth at an earlier stage than traditional detection methods based on CT and/or PET scans 8 which can be conducted less frequently because of radiation concerns and cost. However, such routine prognostics would require a high-throughput, low-cost and user-friendly technique to be competitive and widely used in clinical settings.
Second, CTCs can also be used in cell and molecular analysis, especially for personalized cancer therapies.3 An emerging trend in cancer therapy is the use of pharmacological agents that target specific molecular pathways affected by common genetic lesions.9,10 These drugs are only effective in patients who have tumors with the specified molecular lesions, and drug efficacy can also drop due to the emergence of resistance associated with peripheral or secondary mutations. As a result, drug treatments must be individualized following the molecular analysis of the tumor, by detecting the presence of particular sensitivity mutations or drug-resistance mutations.9,11–13 In some cases the primary tumor can be biopsied and cancer cells analyzed, but often the primary tumor has already been surgically removed or is otherwise inaccessible. A commonly discussed issue is also the tumor heterogeneity, between a primary and metastasis, and among different metastasis, where secondary mutations can be found at some sites and not at others. Because of the impossibility to biopsy every site, the CTC liquid biopsy constitutes an ideal alternative, which may provide a more complete picture of the cancer's complete mutational makeup.
Third, CTC isolation and analysis is crucial for cancer research: towards understanding the mechanisms in which secondary tumor sites form.1–3 Greater understanding of cancer malignancy would impact the translation of cancer research toward the therapeutic benefit of patients, as it would enable new preventative approaches with the discovery of new biomarkers. Extensive studies on the mechanisms of metastasis suggest that a large molecular and cellular heterogeneity exists among CTCs, for different patients with the same type of cancer or even among the cancer cells of the same patient,14,15 and emphasize that metastasis is a complex multi-step process, still largely unknown.16 Some subpopulations of CTCs may undergo epithelial-to-mesenchymal transitions (EMT) and have little to no expression of epithelial markers, which are necessary for their detection by many currently used immunological methods. Thus, these assays may fail to recognize some of the most important CTCs and their role in cancer progression will go unexplored.17,18 There is also a crucial need for isolating a larger number of viable CTCs for research purposes,18 for example isolating CTCs from animal models towards to investigate the metastatic process and the relationship between tumor hypoxia and CTC dissemination.19 One approach to obtain sufficient viable cells would be to process large blood volumes, with higher throughputs and without immunological-based CTC capture.
However, CTCs are very rare, occurring at clinically relevant concentrations of 1 to 10 CTCs per mL of blood and dispersed among billions of red blood cells and millions of white blood cells.1 Isolating pure CTCs from this large background of contaminating blood cells still remains a significant challenge, especially since high purity is crucial for mutational analysis such as gene expression analysis and sequencing.3
Several approaches have been developed for isolating CTCs.3,18,20–22 The current gold standard and only FDA-approved device for CTC enumeration is CellSearch by Veridex. This system immunomagnetically separates target cells from blood using EpCAM-conjugated magnetic microparticles (EpCAM is presumed to be present on carcinomas, which arise from epithelial origins).23,24 Cells captured are then fixed and labeled with fluorescent antibodies to increase the specificity of the assay. Thus, these cells are dead and cannot be expanded. Cells are identified as CTCs if they stain positive for cytokeratin (epithelial cell marker) and negative for CD45 (leukocyte marker). Newer techniques optimize capture efficiency by improving magnetic bead binding or magnetic collection (AdnaGEN, MagSweeper, Fluxion),25–27 but still require manual pre-processing of the samples. Other affinity-based technologies are under investigation, which operate on the principles of affinity chromatography, such that cells with EpCAM coated surfaces preferentially adhere to the high device surface area. Anti-EpCAM is immobilized on the chip surfaces and blood is flowed through the device at a controlled rate to maximize specific binding. Among others, these technologies include the HB-CTC chip from Massachusetts General Hospital,5,28 GEDI from Cornell University,29 and OncoCell-CEE-Breast from Biocept.17 These approaches importantly provide the user with cells on a surface useful for traditional histological staining and cytomorphological analysis. However, the time necessary per run is at least several hours, and production may be costly due to the need for expensive antibodies and surface chemistry quality control.5,28 Furthermore, the cells cannot easily be removed from the microchannel or coated surfaces once attached,30 which limit further analysis for molecular signatures or downstream culture. Finally, affinity-based technologies do not capture all types of CTCs, as capture is based upon the expression of EpCAM, with the consequence that CTCs with little to no EpCAM expression are missed.17,18,20 Specific antibodies to other surface markers can be employed instead; however, prior knowledge of the surface expression of a protein may be unavailable and will likely vary significantly between patients.17,31
In addition, some techniques are based on high-throughput scanning of smeared slides (EpicScience and Scripps Research Institute).32 These approaches require less specialized devices to capture cells, and provide high-resolution pictures of CTCs and clusters, which enables visualization and investigation of cell-cell interactions or molecular pathways by FISH staining. However, the high concentration of contaminating red cells in blood samples, which would lengthen imaging area and time, necessitates some preliminary lysis and cells cannot easily be analyzed further for molecular markers or cultured.
Newer alternatives are based on cell properties other than membrane proteins, such as cell size, deformability or electric properties. Dielectrophoresis (ApoCell) and acoustophoresis (CellCare) make use of the electrical properties and compressibility, respectively, of cancer cells. However, the throughput of these approaches is still low, and both require preliminary lysis of blood.33,34 CTC diameters for a range of cancer types (carcinomas, sarcomas and melanoma) have been shown to be 2-3 times larger than normal red blood cells, and this has been exploited for some CTC isolation technologies.35,36 To take advantage of the larger size of CTCs, Cote et al. have used a filtration method to trap these larger cells using precisely microfabricated filters. Although it is a simple and efficient approach, CTCs are highly deformable and are able to squeeze through filter pores unless they are stiffened by chemical fixation.37,38 As a result of fixation, cells become difficult to elute from the filter once trapped and cannot be easily used for gene expression analysis, metabolomics or cell culture. Other filter alternatives, such as ScreenCell39 or ISET,36,40,41 do not require preliminary cell fixation and constitute an attractive and simple approach for CTC counting, but require a small blood volume and long post-processing for cell elution and downstream molecular analysis. To overcome these additional concerns for facile molecular analysis, as partly defined as one of the “Grand Challenges of the LOC Community” by J. den Toonder in 2011,42 a size-based purely hydrodynamic approach is still needed for high-purity and high-throughput extraction of viable CTCs from a large volume of blood.
Recent microfluidic devices proposed new promising approaches to meet this challenge. For instance, Tan et al. proposed and commercialized an appealing technology (Captor system, from Abnova) for the label-free isolation and enumeration of CTCs in a series of crescent-shaped microwells, but this approach was limited to < 2 mL of blood with the need for additional steps for CTC release afterwards.43,44 Toner et al. recently introduced a platform named CTC-iChip, combining magnetic-affinity-based capture for positive or negative depletion - with both deterministic lateral displacement and inertial focusing.45 This new approach greatly improved upon previous performance for CTC-chip in terms of throughput, efficiency, integrity of cells collected and applicability to non-epithelial cancers and larger sample volumes. However, the purity (0.1%) of negative depletion still needs to be increased, while the requirement of antibodies and the complexity of the integration of three technologies in one platform make this approach potentially expensive. Moreover, Hou et al. introduced the use of spiral channels and Dean vortices for CTC enrichment,46 and interestingly provided viable CTCs in suspension independently of marker expression as well. The throughput for continuous CTC collection is high compared to other techniques (3 mL h−1); however, the purity is still low (10%), and an additional step of CTC concentration by centrifugation is required downstream. Consequently, despite many recent advances in microfluidic-based technologies, there is still a need for a technology that provides continuous, high-throughput, simple and pure collection of viable CTCs from larger volume blood samples.
Some of the authors have previously developed a miniaturized microfluidic system that integrates the high-throughput operations of enrichment and concentration of a standard centrifuge, and possesses the ability to selectively enrich large cells from large volumes of complex fluids.47,48 Our approach makes use of micro-scale vortices and inertial focusing to passively and selectively isolate and concentrate larger cells while smaller cells are flushed out of the device. Here, we tailor a Vortex Chip specifically for the high-purity extraction of cancer cells from blood samples and assess its performance. First, we systematically varied parameters including channel dimensions and flow rates to arrive at an optimal device for maximum trapping efficiency. Second, we validated the final device for capture of cancer cell lines in saline buffer and blood, by considering several factors, including the effect of blood dilution, blood lysis, cell viability, deformability and EpCAM expression. Finally, as a proof-of-concept, circulating tumor cells were extracted and counted from the blood of patients with different primary cancers.
2. Particle trapping mechanism
2.1. Inertial focusing and high-throughput particle alignment
As first experimentally visualized by Segre and Silberberg in 1961 and more recently engineered by Di Carlo and others, inertial focusing uses the effects of fluid inertia in shaped microchannels to align microparticles and cells at high flow rates.49–51 (Fig. 1) When randomly dispersed particles flow through a channel with a particle Reynolds number, ReP, of order 1 or greater (Fig. 1C), they are subjected to two counteracting inertial lift forces: (i) a shear-gradient lift force FLS, scaling as FLS = fLρUm2a3/WC that directs particles toward the channel walls, and (ii) a wall effect lift force FLW, scaling as FLW = fLρUm2a6/WC4, that is due to the presence of the wall and repels the particles toward the channel centerline52 (Fig. 1D). ReP= Re(a/WC)2 where a/WC is the ratio of particle diameter to the channel's smaller width, and Re is channel Reynolds number Re = ρUmD/μ. Here D is the hydraulic diameter of the channel, Um is the maximum velocity of the fluid with density ρ and dynamic viscosity μ, while fL is the dimensionless lift coefficient. In square or rectangular channels, the combination of these forces leads to the migration of particles to two to four dynamic equilibrium positions (Xeq) located between the channel centerline and the wall.53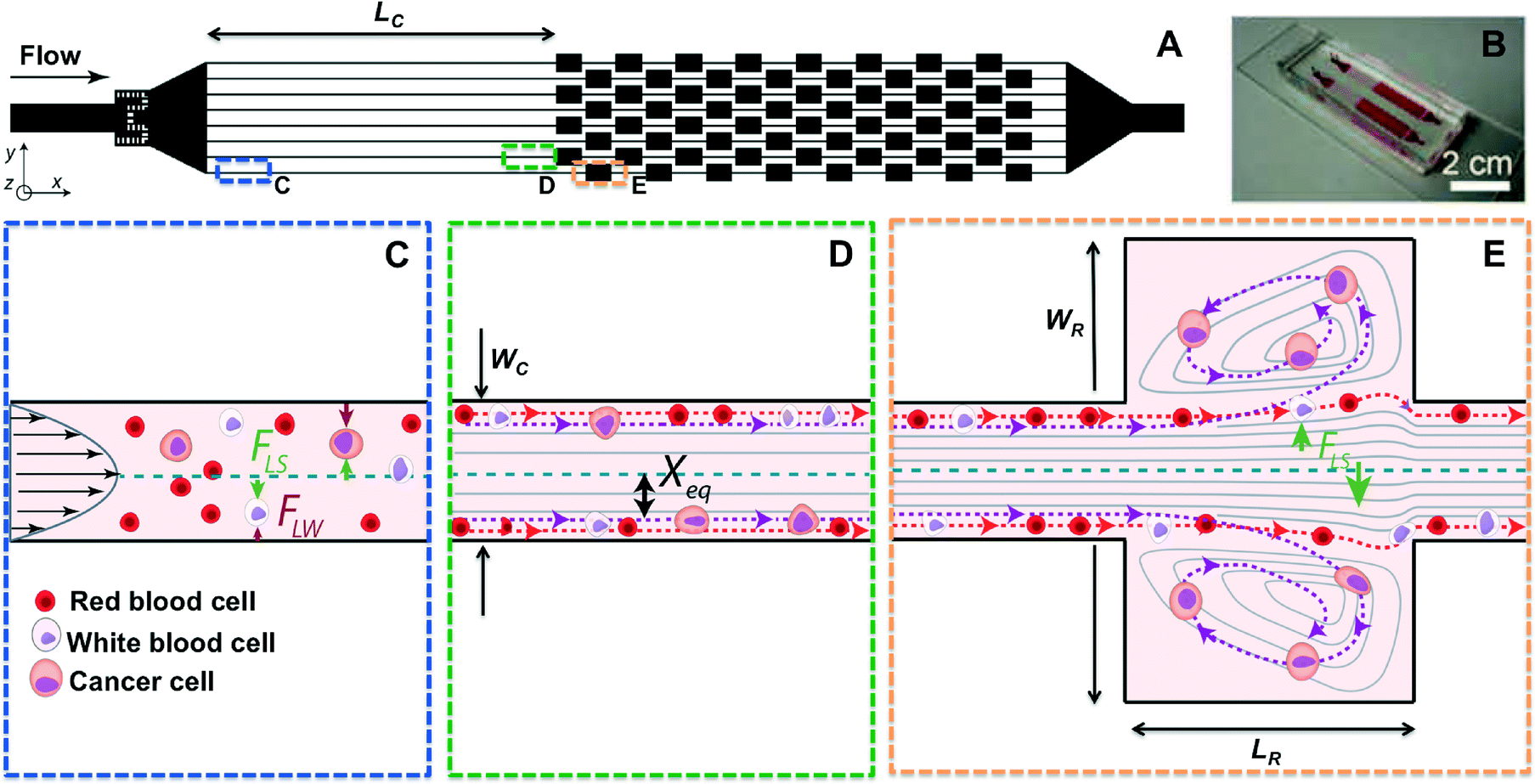 | ||
| Fig. 1 Vortex chip design and principle. (A, B) The Vortex chip used for cell trapping consists of 8 channels (WC = 40 μm, H = 80–85 μm, LC = 4 mm) in parallel with 8 reservoirs (WR = 480 μm, LR = 720 μm) per channel. (C) At the channel inlet, cells are randomly distributed and experience two opposing lift forces, the wall effect FLW and the shear-gradient lift force FLS. (D) As a result, particles migrate to dynamic lateral equilibrium positions, Xeq, depending on the channel cross section. (E) Upon entrance in to the reservoir, the wall effect is reduced. Larger cells (violet dashed line) still experience a large FLS and are pushed away from the channel center through the separatrix and into the vortices, where they are stably trapped. Smaller cells (red dashed line) do not experience enough FLS to cross the separatrix and remain in the main flow. | ||
Following focusing, by subsequently dividing channel into multiple outlets, focused cell scan be collected in a smaller volume and significantly concentrated in a size dependent fashion.54 Besides using the overall particle diameter, inertial lift combined with other lift forces has been applied to separate particles based on their deformability55 and their shape.56 Hur et al. used inertial focusing effects combined with deformability-based separation to extract cancer cells from blood.55 Although these inertial techniques can separate cells at very high throughput, target cells were not significantly concentrated at the outlet (enrichment of 5.4), which is necessary for high purity CTC analysis.
2.2. Laminar microvortices and particle trapping
The second part of this technology makes use of multiple expansion-contraction reservoirs placed in series and parallel, which generate multiple vortices when flow of a sample occurs at high Q and Re (Fig. 1E). An inertial boundary layer separation accounts for these predictable laminar vortices (i.e. Moffatt corner eddy flow) that occur at sudden expansions in microscale channels.57,58 Particle motion in the vicinity of these recirculating flow regions has been investigated and widely used for cell separation,59–61 while Hur et al. first demonstrated that these vortices could be used for size-based cancer cell concentration.47The use of straight inertial focusing channels upstream from the vortex reservoirs allows cells to dynamically focus to equilibrium positions in proximity to the channel walls, aiding in entry into vortices downstream.50 As discussed above when the cells enter an expanding reservoir, the wall effect becomes negligible but cells are still exposed to FLS, which is directed toward the vortex center. Since the shear-gradient lift force scales with a3, larger particles experience a larger lateral lift force, which directs them towards the vortex core after crossing fluid streamlines and passing through the separatrix (i.e. the boundary between the main flow and the secondary vortex patterns). Cells larger than a defined size cut-off can be selectively isolated in reservoirs and continuously orbit stably in the micro-vortices generated, while smaller particles remain in the main stream. After a wash step that removes any remaining small particles, large particles may then be released simply by lowering the flow rate. The microvortices generated in this geometry are different from the vortices generated in the main flow direction, such as Dean vortices created in curved channels62 or due to asymmetrically structured microchannels.63
3. Materials and methods
3.1. Microfluidic device design and fabrication
Microfluidic devices were fabricated using common polydimethylsiloxane (PDMS) replica molding processes.64 Briefly, standard lithographic techniques were used to produce a mold from a silicon master wafer spin-coated with KMPR 1050 (Microchem). PDMS chips were produced from this mold using Sylgard 184 Elastomer Kit (Dow Corning Corporation) and a cross-linker to polymer ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. To enclose the channels, PDMS and glass were both activated by O2 plasma (Technics Micro-RIE series 800, 500 mTorr, 80 W, 30 s) before being bonded together.
:10. To enclose the channels, PDMS and glass were both activated by O2 plasma (Technics Micro-RIE series 800, 500 mTorr, 80 W, 30 s) before being bonded together.
The Vortex chip used for cell trapping is composed of 64 reservoirs, with 8 paths in parallel and 8 reservoirs per path (Fig. 1A–B). Each path is comprised of a single straight high-aspect ratio channel (WC = 40 μm, H = 80–85 μm, LC = 4 mm) followed by 8 reservoirs (WR = 480 μm, LR = 720 μm) located every 1 mm downstream. Filters located at the inlet prevent channel clogging by bead or cell aggregates or debris present in blood samples. To identify the best device geometry, we also used devices with H = 55 μm, WC = 50 μm, and LC = 100, 500 and 1000 μm.
3.2. Preparation of particles, cells and blood
15 and 19 μm polystyrene particles (Thermo Scientific, CV 1%, 3000 particles per mL ± 10% and CV ≤ 16%, 10% w/w, respectively) were diluted in PBS at a defined spiking concentration. All cell lines were cultured in the growth media suggested by American Type Culture Collection (ATCC) and incubated at 37 °C in 5% CO2 to 30–40% confluence. MCF7 (breast carcinoma, ATCC) were cultured in medium containing DMEM supplemented with 10% FBS and 1% penicillin–streptomycin (Invitrogen). OVCAR-5 (ovarian carcinoma), M395 (melanoma), PC-3 (prostate adenocarcinoma) cell lines were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin–streptomycin (Invitrogen). A549 (lung carcinoma) cell lines were cultured with F12-K medium, 10% FBS and 1% penicillin–streptomycin (Invitrogen). Blood specimens were collected from healthy volunteers under IRB-approved protocol (UCLA-IRB #11-001120), into EDTA tubes (BD Vacutainer) by trained physicians, stored at room temperature, and processed within 24 h.Our experiments required spiking known amounts of beads/cancer cells in PBS or blood samples. Particle number was determined by transferring a 100-μL volume of particle solution into a 96-well plate. The well was then imaged using microscopy to count the number of particles. For low spiking ratios, three wells were imaged and averaged to give the cell spiking concentration. 100 μL of particle solution was spiked into each PBS/blood sample for the experiments described below.
3.3. Particle injection, trapping, release and imaging
For experiments using flow-rate control: a simple two-inlet/two-syringe pump system enables rapid solution exchange within the device. One inlet was connected to a syringe containing the sample (beads or cells, spiked in PBS or diluted blood) while the second inlet was connected to a syringe containing PBS as a washing buffer. All solutions were injected into the devices through PEEK tubing (Upchurch Scientific Product No. 1569) using syringe pumps (Harvard Apparatus PHD 2000) and plastic syringes for flow rates ranging from 2 to 12 mL min−1. The sample was continuously agitated by actuated magnetic stir bar during the injection to maintain a uniform concentration throughout the experiment. For clinical experiments using a computer-controlled system the device is connected to a custom-made pressure system that delivers samples from pressurized glass bottles through the Vortex Chip. The LabVIEW (National Instruments) controlled system contains a pair of air regulators, air valves and liquid valves that brings compressed air into the bottles and drives fluid through the microchip device.For both set-ups, the protocol to process samples remains the same. Before cell/particle injection, the vortices are developed and stabilized in each reservoir by flowing PBS at 4 mL min−1 (Priming Step). The cell suspension is then injected into the device to trap particles/cells in the reservoirs (Capture Step). In parallel, maintaining the pressure inside the buffer reservoir prevents back-flow of the sample into the buffer solution. After sample injection, PBS buffer is introduced into the device at 4 mL min−1 to wash out untrapped blood cells (Washing Step). Finally, cells trapped in the vortices are released on-demand by lowering the buffer flow rate, escape from reservoirs, and are collected in a 96-well plate (Release Step).
During each experiment, on-chip particle/cell injection, capture and release were recorded using Phantom v7.3 high-speed camera and Phantom Camera Control and Software (Vision Research Inc.). Live observations allow us to check for the presence of debris or bubbles, which could perturb the trapping experiments. Each well of collected particles was imaged using a Photometrics CoolSNAP HQ2 CCD camera mounted on a Nikon Eclipse Ti microscope, with an ASI motorized stage operated with Nikon NIS-Elements AR 3.2 software. Images were automatically captured with 10× magnification for up to four configurations: brightfield, DAPI, FITC, and TRITC filter sets. Capture efficiency is defined as the number of cells/particles collected off-chip over the number of particles injected (i.e. spiked at the inlet). Capture reproducibility is estimated using the average of 3 independent experiments, i.e. using 3 different replicas of the same microfluidic device.
3.4. Cell recovery and culture after collection
MCF7 cancer cells were used to assess cell viability after collection with vortex chips. MCF7 cells in PBS were injected through the chip at 4 mL min−1, captured, collected off-chip, fluorescently stained with a live/dead assay (CalceinAM and ethidium homodimer) and counted immediately after their release. Viability % is defined as the number of live cells over the total number of cells collected (live and dead cells). Initial cell samples were used as a comparison. For statistical significance, 3 independent experiments were performed, injecting 3 different cell suspensions through 3 different replicas of the same microfluidic device.For cell culture studies, A549 cells in PBS were captured using the Vortex chip in a biosafety cabinet and collected into 96- and 48-well plates. Cells were cultured over the course of 3 days in F12 media supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin. Each day, cells were counted and images were taken in the 48-well plate. Viability assays were performed each day in the 96 well plate, with same protocol as described above.
3.5. Cancer cell isolation from spiked blood samples
For spiking experiments, cells were released with trypsin and incubated in media at room temperature on a shaker plate for 20 min to allow for ample recovery from trypsin treatment. Cells were spiked in blood and injected immediately. A Coulter counter (Beckman Coulter Z2, Cell and Particle Counter) was used to measure average cell size before each injection. Blood was diluted with PBS at dilution factors of 5×, 10×, 20× or 40× and spiked with a known number of cells. Typically, there were about 200–700 cells spiked in 1 mL of whole human blood diluted with 9 mL PBS for 10× dilution. For experiments with lysed blood, red cells were selectively lysed (Roche, RBC Lysis Buffer) following the manufacturer's protocol, before spiking of cancer cells and dilution with PBS to achieve an equivalent dilution rate as for the non-lysed blood.Isolated cells were collected into a 96-well plate for fixation, permeabilization, and immunolabeling: cells in suspension were treated with 4% v/v paraformaldehyde for 15 min, permeabilized with 0.4% v/v Triton X-100 (Sigma-Aldrich) for 7 min, and incubated with anti-CK-PE (BD), DAPI (Invitrogen) and anti-CD45-FITC (Invitrogen) in 2% w/v BSA for leukocyte, epithelial, and nuclear stains, with PBS washing between each step. After staining, the cells were imaged and enumerated to quantify capture efficiency, enrichment, and purity. Capture efficiency for cancer cells is defined as the number of cancer cells captured in the 96-well-plate (CK+, DAPI+ and CD45−) over the total number of cells spiked into diluted human blood. Capture purity is defined as the ratio of cancer cells captured to the total number of captured cells (all nucleated cells: DAPI+). Enrichment ratio is defined as the purity of the released sample divided by the purity of the sample before processing, i.e. ER = (cancer cells/nucleated cells output)/(cancer cells/nucleated cells input).
3.6. Cancer cell isolation from patient samples
Patients with advanced cancer were recruited according to a protocol approved by the Institutional Review Board (UCLA-IRB #11-001798), from the Department of Radiation Oncology and the Department of Hematology and Oncology of the Geffen School of Medicine at UCLA. Blood samples were drawn from 12 patients, diagnosed with breast (N = 4, 1 non-metastatic, 3 metastatic) and lung cancers (N = 8, metastatic non-small-cell lung cancer, NSCLC) seen at the clinics, as well as 4 healthy volunteers, and processed using the Vortex Chip. Team members processing blood and counting cells within samples were told that all samples (including healthy samples) were metastatic breast or lung patients to prevent any bias in operation or counting. For clinical samples, cells were fixed and permeabilized as described previously, but stained with DAPI (Invitrogen), anti-CD45-PE (BD), anti-Pan-CK-FITC (Miltenyi Biotech.) and anti-CK CAM5.2-FITC (BD) to target CK 7, 8, 18 and 19. To evaluate capture purity, leukocytes and CTCs were enumerated by 2 different persons, where leukocytes were identified as CD45+/DAPI+ cells while CTCs were defined as circular, CK+/DAPI+/CD45− cells, that ranged from 10 to 30 μm in diameter. To estimate the size of cells collected off-chip, we used a manual measurement tool on NIS Elements AR software to average the length across cells based on each cell's longest and shortest axis.3.7. Comparison with deformability cytometry
Previously documented guidelines for the design and operational specifications of Deformability Cytometry (DC) were followed.65 The chip used in this study had cross-sectional dimensions 1.5 times those of the previously reported DC chip, in order to accommodate larger cells. The extensional flow is generated at the junction of opposing flows, carried by W = 105 μm by H = 45 μm channels (Fig. SI5†). Perpendicular outlet channels had the same dimensions. DC device fabrication was conducted with the same materials (PDMS, Sylgard 184 Elastomer Kit) as that of the Vortex Chip.Cultured cancer cells were prepared in the same fashion as for blood spiking experiments but spiked into PBS. The methods used to measure cell mechanical properties were previously described.65 Briefly, the harvested cells were diluted with PBS to achieve a cell concentration within the range of 100000 and 200000 cells per mL. Cell suspensions were introduced into the DC chip to achieve a mean velocity of 4 m s−1. Using the Phantom v711 high-speed camera (Vision Research), video data were collected at a rate of 500k fps with a 300 ns exposure achieving 30 frames per cell. Image and data analysis was processed with a custom MATLAB script. The size metric was extracted upstream of the junction by averaging the whole cell diameter, while the deformability metric was defined as the maximum aspect ratio achieved in the extensional flow junction as observed over the whole cell time series. Further details about this technique can be found in prior reports.65
4. Results and discussion
4.1. Vortex chip design and optimization
To identify the optimal device for particle trapping, different geometric parameters were systematically compared with a set of experiments: channel length before the first reservoir (LC = 100, 500 and 1000 μm) and channel aspect ratio (HC = 55 and 80 μm, WC = 40 and 50 μm), were combined with different flow rates (from 2 to 11 mL min−1), with the expectation that these parameters would influence the inertial focusing and vortex flow patterns.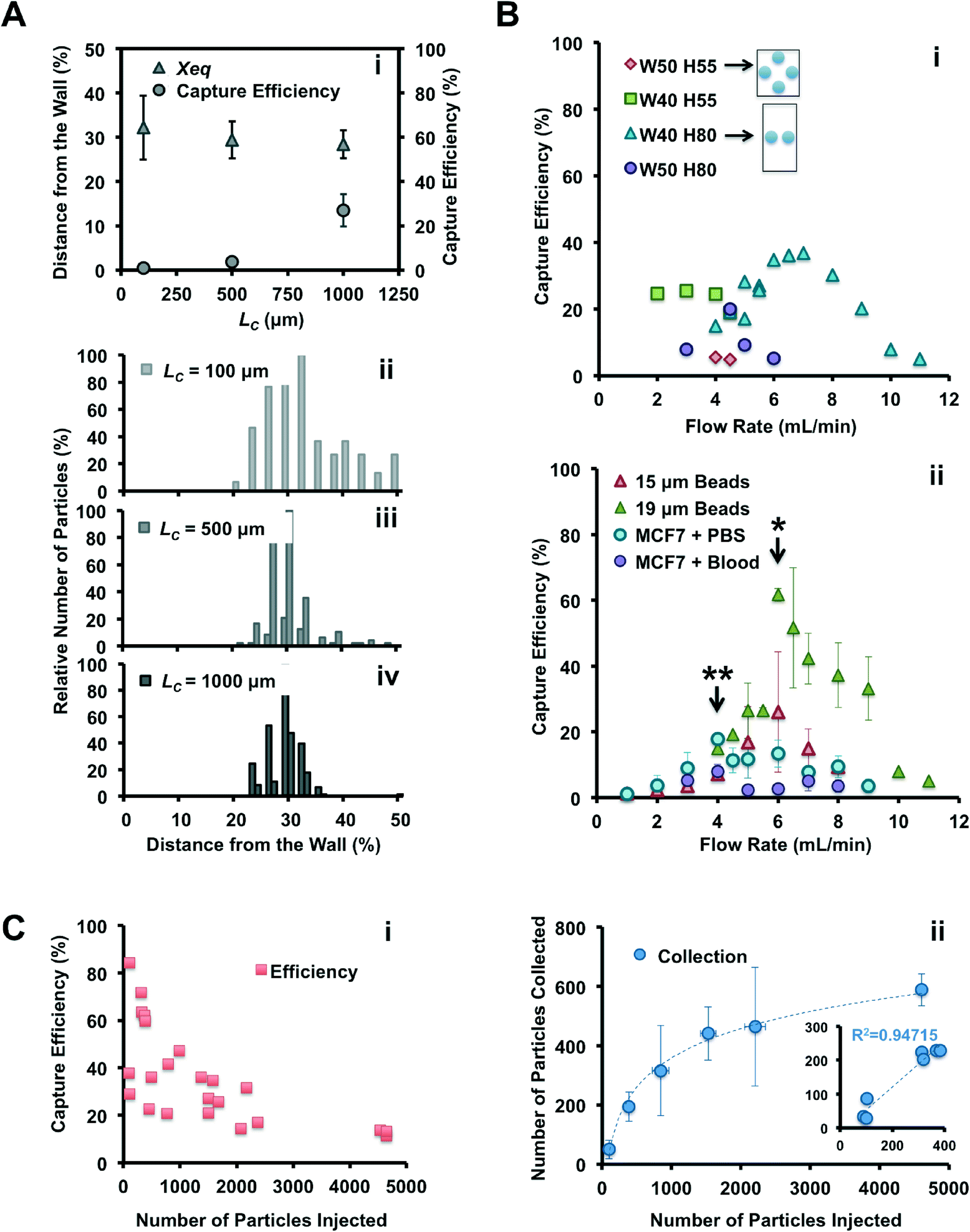 | ||
| Fig. 2 Optimization of microfluidic device for improved capture of 19 μm particles. (A) Importance of channel length upstream from the reservoirs for accurate inertial focusing. Particles were injected (400 μL min−1) in a device with WC = 40 μm, H = 55 μm, WR = 480 μm, LR = 720 μm and LC = 100 (ii), 500 (iii) and 1000 μm (iv). The average particle equilibrium position, Xeq, was calculated by measuring the distance between the particle center and the channel wall, with Xeq being equal to 0% and 50% indicating that the particle center is located at the channel wall and channel center, respectively. Histograms represent the position of particles along the channel width, as they progressively focus with increasing channel length. (i) Effect of channel length on Xeq and capture efficiency. Longer channels allow migration of particles closer to the channel walls and higher capture efficiency. (B) Capture efficiency versus flow rate. (i) Effect of channel aspect ratio and flow rate on capture efficiency (LC = 1000 μm). Channels with higher aspect ratio (i.e. taller and narrower) provide an improved capture and at higher flow rates. Two schematics illustrate the particle repartition and their expected focusing positions in a square (50 × 55 μm) and rectangular (40 × 80 μm) channel cross-section. (ii) Capture efficiency for different particles and different flow rates in the optimal device (WC = 40 μm, H = 80 μm, LC = 1000 μm). Asterisks indicate the optimal flow rate for 15 or 19 μm beads (*), and for MCF7 cells (**), in PBS or in blood. (C) Particle trapping as a function of the number of particles injected and demonstration of vortex saturation, for 19 μm beads injected in the optimal device (WC = 40 μm, H = 80 μm, LC = 1000 μm). (i) Capture efficiency decreases with higher number of particles injected. (ii) The capture is linear with injected amount for lower numbers of particles, but saturates at around 500 particles trapped within the reservoirs. | ||
In conclusion, vortex trapping depends on various geometric and system parameters in a complex and non-linear fashion. The observations and conclusions presented here require further study of the entrance and especially the maintenance of particles in microvortices, which is currently in progress in our laboratory.
4.2. Collection of viable cancer cells from healthy blood samples
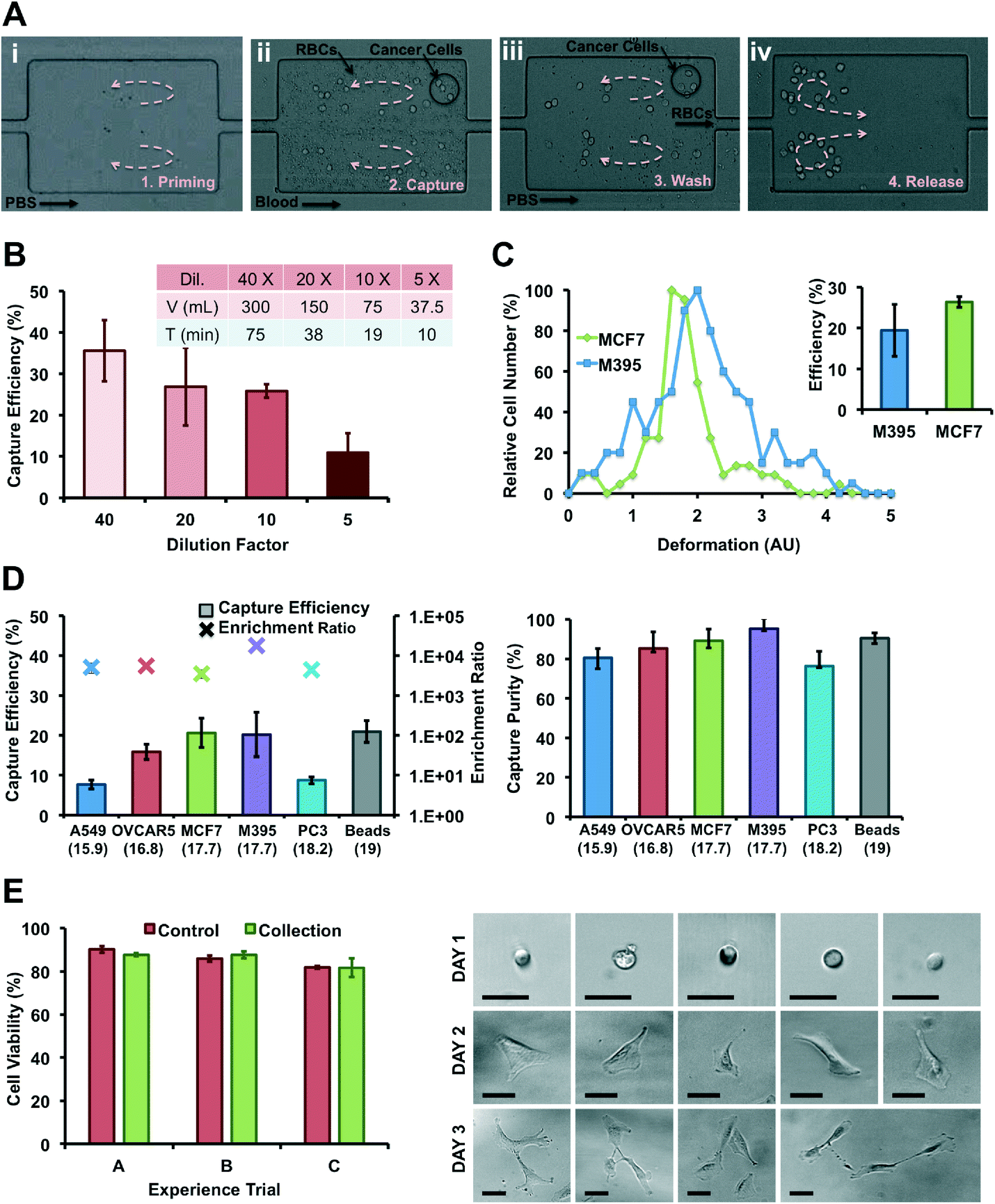 | ||
| Fig. 3 Collection of viable cancer cells from blood. (A) High-speed images illustrate blood processing through the Vortex Chip. (i) First, the vortices are formed and stabilized in each reservoir by flowing PBS (Priming). (ii) Blood and cancer cells are injected in the device to isolate cancer cells in reservoirs (Capture). (iii) After sample injection, PBS is introduced into the device to wash out untrapped blood cells (Wash). (iv) Finally, cells trapped in the vortices are released on-demand by lowering the PBS flow rate. Cells escape from the reservoirs, and are collected in a 96-well plate (Release). (B) Effect of blood dilution. Capture efficiency for MCF7 cells spiked in 5×, 10×, 20× and 40× diluted blood, with around 500 cells spiked in 0.5 mL of whole blood diluted with PBS. N = 3. The table (inset) indicates the corresponding volume and the time required for the processing of 7.5 mL sample. Higher dilution provides better capture efficiency but requires longer processing. 10× dilution has a comparable efficiency and throughput with the 20× dilution but half of the processing time. (C) The difference in cell deformation as measured by Deformability Cytometry may be related with (inset) the difference between capture efficiencies of similarly sized M395 and MCF7 cells. (D) Capture efficiency and purity for various cancer cell lines (MCF7 breast carcinoma, OVCAR5 ovarian carcinoma, M395 melanoma, PC3 prostate adenocarcinoma, A549 lung carcinoma), independently of EpCAM expression. Cell size as measured by Coulter Counter is indicated below each cell line. (E) Cell viability after collection. Left: MCF7 breast cancer cells injected through the chip and collected off-chip, maintain membrane integrity at similar levels to the unprocessed control as determined by a live/dead assay. Right: A549 cells processed with the Vortex Chip and collected in a well-plate proliferate over 3 days. | ||
000 antigens per cell),5,68 prostate PC3 carcinoma for medium expression (50000 antigens per cell),5 lung A549 carcinoma for low expression,69 and finally melanoma M395, which are not of epithelial origin. Melanoma M395 and breast MCF7 have similar sizes (17.7 and 17.7 μm) and are both captured well, with >20% efficiency. Ovarian OVCAR5 are smaller (16.8 μm) but still yield an efficiency of 15.9%. Lung A549 have a size (15.9 μm) closest to the 15 μm size cut-off, which may explain the lower efficiency of 7.7%. Finally, despite their larger size (18.2 μm), prostate PC3 are extracted at efficiencies below 9%. Interestingly, independent of cell line and capture efficiency, the purity of cells enriched is higher than 75%. Thus, Vortex technology is not restricted to epithelial cells only, and does not need costly antibodies or antibody cocktails. However, it would require that cancer cells possess larger diameters than normal blood cells.35
In addition, work by Hur et al. showed that more deformable particles are inertially focused at equilibrium positions closer to the channel center55 and this could address the inconsistent correlation between capture efficiencies of similarly sized cells. More deformable cells will be focused further away from the vortices and may not experience shear gradient lift forces large enough to cross the separatrix. Such an effect should also occur in other recent inertial microfluidic approaches.45,46 To test this hypothesis, cancer cells were processed in the Vortex Chip in parallel with Deformability Cytometry65 (Fig. 3C). Deformability Cytometry is a high-throughput, inertial microfluidic technique, which utilizes hydrodynamic forces to quantify deformability of cell populations (Fig. SI5†). As expected, MCF7 cells (18.9 μm) yielded the greatest capture efficiency (26.4%) and were the least deformable (1.6 median deformability). M395 cells of similar size (19.3 μm) but greater deformability (1.9 median deformability) were isolated at a lower rate (19.4%). As illustrated in ref. 70 for LNCaP prostate cell lines and prostate patient CTCs, cell lines and CTC populations exhibit considerable differences, in level of protein expression but also size, nuclear structure, cytoplasmic area, and consequently deformability. These differences, which result from, among others, the culturing conditions used for cell lines, are critical to consider when translating from cell lines to isolating CTCs from patients.
4.3. Automated CTC isolation from cancer patient blood
We validated the optimized Vortex Chip with clinical samples from patients with malignant disease of breast and lung origins. CTCs were successfully captured from all cancer patients at high purity, with 5 or more CTCs from 9 of the 12 patient samples (75%) (Fig. 4A). Fig. 4D–F present a gallery of the CTCs extracted. 3.3 to 6.9 CTCs per mL were collected from 4 breast cancer samples, 3.1 to 42.4 CTCs per mL from 8 NSCLC (non-small-cell lung cancer) samples, and 1.8 to 4.6 CTCs per mL from 4 healthy controls (Fig. 4B). As seen with spiked blood samples, purity also remained high for these samples (Fig. 4C). Indeed, leukocyte background is negligible, with 0.5 to 12.7 WBCs per mL for malignant samples, equivalent to a purity of 57–95%, which is higher than for previously reported technologies and well-suited for molecular analysis. We also observe CTC clusters from patient samples. As explained by Kuhn et al. among others,72,73 CTC clusters are a subject of growing interest for their increased potential for metastasis and venous thromboembolism in comparison to single cells.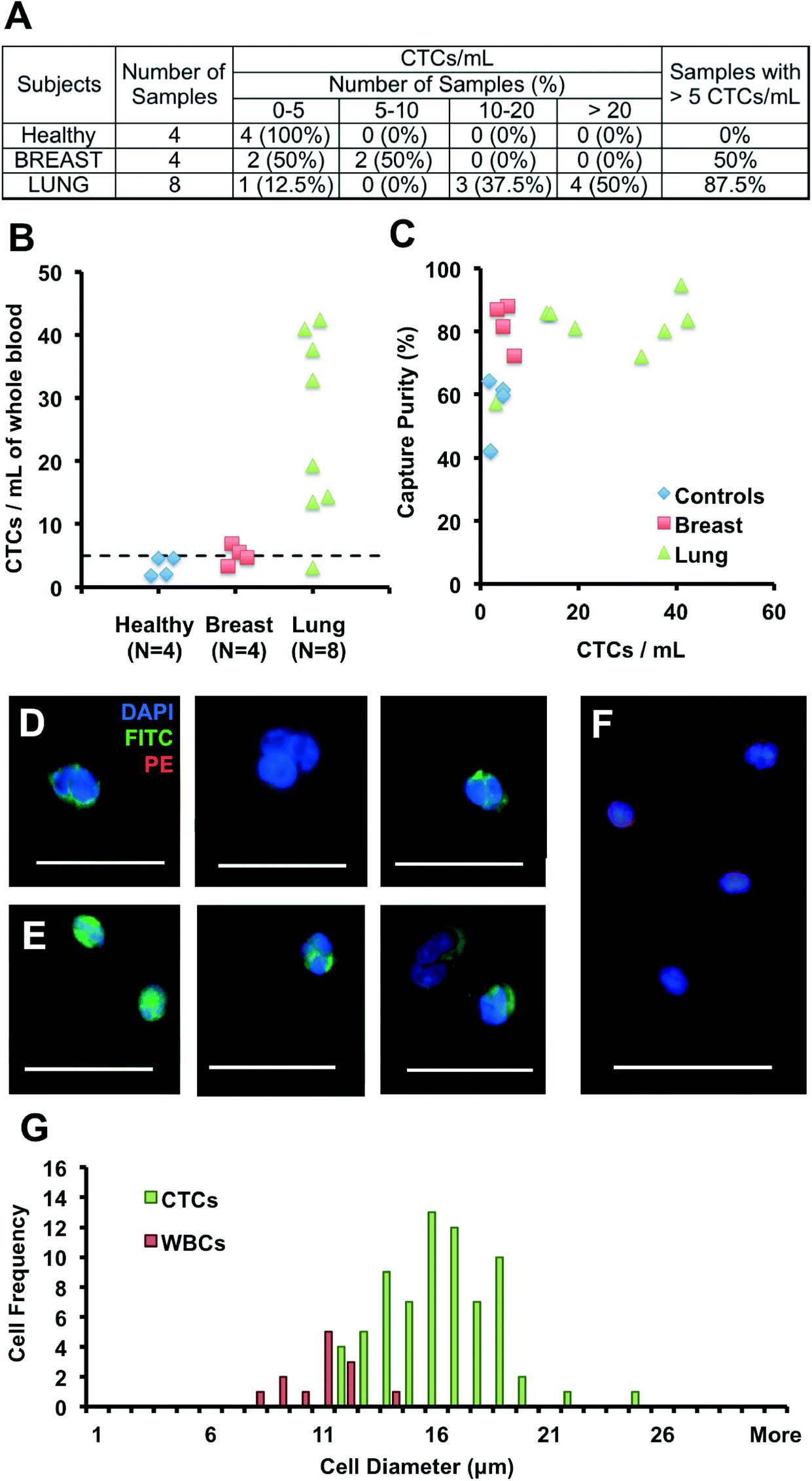 | ||
| Fig. 4 Collection and enumeration of circulating tumor cells from cancer patients. (A) Table summarizing CTC counts for 4 healthy patients, 8 patients with non-small-cell lung cancer (8 metastatic) and 4 patients with breast cancer (3 metastatic and 1 non-metastatic). (B) CTC count per mL of whole blood, for healthy patients (blue diamonds), breast cancer patients (red squares) and lung cancer patients (green triangles). A threshold of 5 CTCs per mL is defined with a dashed line based on healthy controls. (C) Capture purity for all patient samples processed as a function of the number of CTCs collected. (D, E, F) Picture gallery of cells collected with the Vortex Chip and fluorescently stained with DAPI, CK-FITC and CD45-PE, with examples of breast cancer CTCs (D), NSCLC CTCs (E) and white blood cells (F). Scale bar is 37 μm. (G) Histogram representing the size distribution of WBCs (DAPI+, CD45+, CK−) and CTCs (DAPI+, CD45−, CK+) collected by the Vortex chip for a metastatic NSCLC sample, with 71 CTCs, 13 WCs, and a purity of 84.5%. | ||
Although the size distribution of cancer cell lines are well known, there is little data on the sizes of CTCs. We measured the sizes of WBCs and CTCs collected from a metastatic NSCLC patient (Fig. 4G). After processing, WBCs in the sample ranged from 8 to 14 μm, while CTCs ranged from 12 to 25 μm, with a CTC purity of 84%. These results indicate that WBCs larger than 10 μm are extremely rare (9 of 13 captured WBCs, versus 71 CTCs), while a limited number of smaller WBCs, although not trapped by the Vortex chip, likely result from flow line contamination. Previous reports by G.W. Schmid-Schonbein et al.74 have confirmed the size of leukocytes in suspension (< 10 μm in diameter), which is smaller than sizes reported for cells on blood smears; the smear process causes cells to be squashed which overestimates the true size in solution. Although our trapping cut-off likely under-represents smaller cell populations, cells identified as CTCs were larger than ~12 μm (N = 71). In the future, it will be of interest to see how the size distribution of CTCs may depend on patient cancer type and stage.
Several CK+/DAPI+/CD45− cells were collected in healthy samples as well, however at lower levels than for malignant samples (< 5 CTCs per mL). The presence of CK+ cells in healthy patients or in patients with benign diseases has been previously reported but not explained.28,32,75 Other staining approaches will be necessary to determine the origin of these larger CK+ cells circulating in the blood and whether they reflect other cell types (e.g. endothelial cells or endothelial progenitor cells) that may in some cases express cytokeratins.76 Similarly, some DAPI+ cells were collected that expressed neither CK nor CD45, or that expressed both of these markers simultaneously. These cells were not included in counts and were on average 30% of total cells. Additional staining should help in their identification in the future.
Conclusions
A number of promising CTC technologies are currently under investigation, including affinity-capture, size-based microfiltration, high-throughput imaging, passive microfluidics or microfluidics combined with an additional field. The vortex trapping approach, validated in this work with clinical samples, has particular advantages. A short processing time, high concentration ratio, applicability to various sample and cancer types, cell integrity and high-purity are especially important for clinical adoption and use in downstream assays of mutational status. Processing time: a 7.5 mL sample can be processed in less than 20 min, as compared to 4 h for cell separation with CellSearch, 1 h for MagSweeper, 2–3 h with the CTC-HB Chip, and >2.5 h for Abnova among others. Wide applicability: vortex trapping is not restricted to blood samples and other bodily fluids can be processed, as emphasized by our recent work with isolating malignant cells from pleural effusions.77 Cancer cells can also be isolated independently of marker expression, suggesting our approach can handle a large range of tumor cell types, including cells of non-epithelial origin like melanoma or cells undergoing epithelial to mesenchymal transition. Purity: high purity is generally challenging because of a high background of white blood cells adhering to coated surfaces or filter membranes. When using spiked samples, we can routinely achieve purity of 80–100%, compared to 14% for CTC-HB, 51% for MagSweeper and 68% for GEDI. Efficiency: on the other hand, our capture efficiency can be considered low compared to other techniques but can be improved with serial injections of the sample (data not shown), with the caveat that this increases processing time. Additionally, capture efficiency can be compensated for by processing a larger blood volume (compared to limitations of ~1 mL for some current filter-based approaches). Concentration and viability: importantly, no matter what volume is processed, the enriched cells are free, non-fixed, viable, and collected in suspension in a small 300 μL final volume.Automation and cost will also be a key consideration for successful adoption and implementation in a clinical setting. Vortex trapping employs passive fluid physics, to selectively collect larger cells in laminar fluids microvortices at high rates, without complex high-resolution features, instrumentation or surface chemistry. As a consequence, the cost of an injection-molded chip and the per-sample cost for the pressure control system to run samples is expected to be much lower than ~$500 per test for the CellSearch system. Further, cyto-technicians can process samples with the Vortex chip without significant additional training and can perform standard staining on pure collected cells for routine cytomorphological analysis by a cytopathologist, such that this key stakeholder is intellectually invested.
Our preliminary results for pure CTC collection from NSCLC samples may also address an unmet clinical need, as CellSearch is not FDA-approved for determining lung cancer prognosis while lung cancer being the leading cause of cancer death in both men and women, worldwide, with 80% of all lung cancers being attributed to NSCLC cancer. Additionally, treatment outcomes for NSCLC remain unsatisfactory, with low survival rates and late-stage detection.10,73,78 Significant efforts have been made over the last years to identify novel targeted therapies interfering with lung cancer cell pathways,10 and a high purity “liquid biopsy” approach could help understand NSCLC cancer biology, and the development of new personalized treatments. In this direction, some of the authors have demonstrated the vortex-based collection of cancer cells from pleural effusions and the detection of KRAS mutations.77
Considering the set of capabilities discussed, we believe the simplicity of vortex trapping can aid widespread adoption from clinicians and cancer biologists who desire to not only enumerate CTCs, but also uncover new CTC biology, such as unique gene mutations, vesicle production, or the roles of circulating cells in the metastatic process.
Acknowledgements
The authors acknowledge funding for this work from a Coulter Translational Research Award (Wallace H. Coulter Foundation), the Office of Naval Research Young Investigator Program (grant #N000141210847), and a sponsored research grant from NetScientific. The authors would like to thank Dr. S. Claire Hur for her technical assistance and advice on her vortex setup and past experiments (Hur et al., Biomicrofluidics 2011), Coleman Murray for his technical advice on the automated pressure set-up, Dr. Henry Tse for advice on cell line passaging and deformability measurements, Ricky Chiu for instructions on the Coulter Counter system, Dr. Emilie Perre and Janay Kong for helpful inputs, Viviana Ogawa and Melanie Triboulet for their help on sample processing. The authors are also grateful to Dr. Oladunni Adeyiga for healthy blood collection, Melissa Matsumoto for the coordination of clinical blood sample collection, the phlebotomists Sara Soto, Nazy Zomorodian, Vicky Padilla, H. Emma Wang, and the other physicians who helped with patient recruitment: Dr. Julie Jung Kang, Dr. Michael Steinberg, Dr. Patrick Kupelian, Dr. Niraj Mehta and Dr. Phillip J. Beron. Finally, we would like to thank all the patients who donated their blood to make this study possible.References
- T. R. Ashworth, A case of cancer in which cells similar to those in the tumours were seen in the blood after death, Med. J. Aust., 1869, 14, 146–147 Search PubMed.
- S. Paget, The distribution of secondary growths in cancer of the breast, Lancet, 1889, 133(3421), 571–573 CrossRef.
- K. Pantel and C. Alix-Panabieres, Circulating tumor cells in cancer patients: challenges and perspectives., Trends Mol. Med., 2010, 16, 398–406 CrossRef PubMed.
- C. Alix-Panabieres and K. Pantel, Circulating tumor cells: liquid biopsy of cancer, Clin. Chem., 2013, 59(1), 110–118 CAS.
- S. Nagrath, et al., Isolation of rare circulating tumor cells in cancer patients by microchip technology, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- M. Cristofanilli, et al., Circulating tumor cells in metastatic breast cancer: biologic staging beyond tumor burden, Clin. Breast Cancer, 2007, 7(6), 471–479 Search PubMed.
- J. S. de Bono, et al., Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer, Clin. Cancer Res., 2008, 14, 6302–6309 CrossRef CAS PubMed.
- G. T. Budd, et al., Circulating tumor cells versus imaging—predicting overall survival in metastatic breast cancer, Clin. Cancer Res., 2006, 12(21), 6403–6409 CrossRef CAS PubMed.
- S. Riethdorf, et al., Detection and HER2 expression of circulating tumor cells: prospective monitoring in breast cancer patients treated in the neoadjuvant GeparQuattro trial, Clin. Cancer Res., 2010, 16(9), 2634–2645 CrossRef CAS PubMed.
- M. Méndez, A. Custodio and M. Provencio, New molecular targeted therapies for advanced non-small-cell lung cancer, J. Thorac. Dis., 2011, 3, 30–56 Search PubMed.
- P.L. Paris, et al., Functional phenotyping and genotyping of circulating tumor cells from patients with castration resistant prostate cancer, Cancer Lett., 2009, 227(2), 164–173 CrossRef PubMed.
- M. Pestrin, et al., Correlation of HER2 status between primary tumors and corresponding circulating tumor cells in advanced breast cancer patients, Breast Cancer Res. Treat., 2009, 118(3), 523–530 CrossRef CAS PubMed.
- N. Xenidis, et al., Cytokeratin-19 mRNA-positive circulating tumor cells after adjuvant chemotherapy in patients with early breast cancer, J. Clin. Oncol., 2009, 27(13), 2177–2184 CrossRef CAS PubMed.
- A. Marusyk, V. Almendro and K. Polyak, Intra-tumour heterogeneity: a looking glass for cancer, Nat. Rev. Cancer, 2012, 12, 323–334 CrossRef CAS PubMed.
- A. A. Powell, et al., Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines, PLoS One, 2012, 7(5), e33788 CAS.
- M. Labelle and R. O. Hynes, The initial hours of metastasis: the importance of cooperative host–tumor cell interactions during hematogenous dissemination, Cancer Discovery, 2012, 2, 1091–1099 CrossRef CAS PubMed.
- C. V. Pecot, et al., A novel detection platform for detection of CK+ and CK− CTCs, Cancer Discovery, 2011, 1(7), 580–586 CrossRef CAS PubMed.
- J. Kling, Beyond counting tumor cells, Nat. Biotechnol., 2012, 30(7), 578–580 CrossRef CAS PubMed.
- K. Ameri, et al., Circulating tumor cells demonstrate an altered response to hypoxia and an aggressive phenotype, Br. J. Cancer, 2010, 102, 561–569 CrossRef CAS PubMed.
- P. Paterlini-Brechot and N. L. Benali, Circulating tumor cells (CTC) detection: clinical impact and future directions, Cancer Lett., 2007, 253, 180–204 CrossRef CAS PubMed.
- J. Chen, J. Lib and Y. Sun, Microfluidic approaches for cancer cell detection, characterization, and separation, Lab Chip, 2012, 12, 1753–1767 RSC.
- Y. F. Sun, et al., Circulating tumor cells: advances in detection methods, biological issues, and clinical relevance, J. Cancer Res. Clin. Oncol., 2011, 137, 1151–1173 CrossRef PubMed.
- M. Craig Miller, G. V. Doyle and L. W. M. M. Terstappen, Review article: Significance of Circulating Tumor Cells Detected by the Cell Search System in Patients with Metastatic Breast, Colorectal and Prostate Cancer, J. Oncol., 2010, 617421 Search PubMed.
- S. Riethdorf, H. Fritsche and V. Muller, et al., Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the cell search system, Clin. Cancer Res., 2007, 13(3), 920–928 CrossRef CAS PubMed.
- A. A. H. Talasaz, et al., Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(10), 3970–3975 CrossRef CAS PubMed.
- E. Andreopoulou, et al., Comparison of assay methods for detection of circulating tumor cells in metastatic breast cancer: AdnaGenAdnaTestBreastCancer Select/Detect™ versus VeridexCellSearch™ system, Int. J. Cancer, 2012, 130, 1590–1597 CrossRef CAS PubMed.
- J. H. Kang, et al., A combined micromagnetic–microfluidic device for rapid capture and culture of rare circulating tumor cells, Lab Chip, 2012, 12, 2175–2181 RSC.
- S. L. Stott, et al., Isolation of circulating tumor cells using a microvortex-generating herringbone-chip, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(43), 18392–7 CrossRef CAS PubMed.
- J. P. Gleghorn, et al., Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody, Lab Chip, 2010, 10, 27–29 RSC.
- A. M. Shah, et al., Biopolymer system for cell recovery from microfluidic cell capture devices, Anal. Chem., 2012, 84, 3682–3688 CrossRef CAS PubMed.
- J. A. Mayer, et al., FISH-based determination of HER2 status in circulating tumor cells isolated with the microfluidic CEE platform, Cancer Genet., 2011, 204, 589–595 CrossRef CAS PubMed.
- D. Marrinucci, et al., Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers, Phys. Biol., 2012, 9, 016003 CrossRef PubMed.
- V. Gupta, et al., ApoStream(tm), a new dielectrophoretic device for antibody independent isolation and recovery of viable cancer cells from blood, Biomicrofluidics, 2012, 6, 024133 Search PubMed.
- P. Augustsson, et al., Microfluidic, label-free enrichment of prostate cancer cells in blood based on acoustophoresis, Anal. Chem., 2012, 84(18), 7954–7962 CrossRef CAS PubMed.
- A. Williams, M. Balic, R. Datar and R. Cote, Size-based enrichment technologies for CTC detection and characterization, Recent Results Cancer Res., 2012, 195, 87–95 Search PubMed.
- G. Vona, et al., Isolation by size of epithelial tumor cells: a new method for the immunomorphological and molecular characterization of circulating tumor cells, Am. J. Pathol., 2000, 156(1), 57–63 CrossRef CAS PubMed.
- H. K. Lin, et al., Portable filter-based microdevice for detection and characterization of circulating tumor cells, Clin. Cancer Res., 2010, 16(20), 5011–5018 CrossRef CAS PubMed.
- S. Zheng, et al., Membrane microfilter device for selective capture, electrolysis and genomic analysis of human circulating tumor cells, J. Chromatogr., A, 2007, 1162(2), 154–61 CrossRef CAS PubMed.
- I. Desitter, et al., A new device for rapid isolation by size and characterization of rare circulating tumor cells, Anticancer Res., 2011, 31, 427–442 Search PubMed.
- V. De Giorgi, et al., Application of a filtration- and isolation-by-size technique for the detection of circulating tumor cells in cutaneous melanoma, J. Invest. Dermatol., 2010, 130, 2440–2447 CrossRef CAS PubMed.
- J. M. Hou, et al., Circulating tumor cells as a window on metastasis biology in lung cancer, Am. J. Pathol., 2011, 178(3), 989–996 CrossRef PubMed.
- J. den Toonder, Circulating tumor cells: the Grand Challenge, Lab Chip, 2011, 11, 375–377 RSC.
- S. J. Tan, et al., Microdevice for the isolation and enumeration of cancer cells from blood, Biomed. Microdevices, 2009, 11, 883–892 CrossRef PubMed.
- S. J. Tan, et al., Versatile label free biochip for the detection of circulating tumor cells from peripheral blood in cancer patients, Biosens. Bioelectron., 2010, 26, 1701–1705 CrossRef CAS PubMed.
- E. Ozkumur, et al., Inertial focusing for tumor antigen dependent and independent sorting of rare circulating tumor cells, Sci. Transl. Med., 2013, 5, 179 Search PubMed.
- H.W. Hou, et al., Isolation and retrieval of circulating tumor cells using centrifugal forces, Sci. Rep., 2013, 3, 1259, DOI:10.1038/srep01259.
- S. C. Hur, A. J. Mach and D. Di Carlo, High-throughput size-based rare cell enrichment using microscale vortices, Biomicrofluidics, 2011, 5, 022206 Search PubMed.
- A. J. Mach, J. H. Kim, A. Arshi, S. C. Hur and D. Di Carlo, Automated cellular sample preparation using a Centrifuge-on-a-chip, Lab Chip, 2011, 11, 2827–2834 RSC.
- G. Segre and A. Silberberg, Radial particle displacements in Poiseuille flow of suspensions, Nature, 1961, 189, 209–210 CrossRef.
- D. Di Carlo, Inertial microfluidics, Lab Chip, 2009, 9, 3038–3046 RSC.
- A. A. S. Bhagat, et al., Inertial microfluidics for continuous particle filtration and extraction, Microfluid. Nanofluid., 2008, 7, 217–226 CrossRef.
- D. Di Carlo, J. F. Edd, K. J. Humphry, H. A. Stone and M. Toner, Particle segregation and dynamics in confined flows, Phys. Rev. Lett., 2009, 102, 094503 CrossRef PubMed.
- S. C. Hur, H. T. K. Tse and D. Di Carlo, Sheathless inertial cell ordering for extreme throughput flow cytometry, Lab Chip, 2010, 10, 274–280 RSC.
- A. J. Mach and D. Di Carlo, Continuous scalable blood filtration device using inertial microfluidics, Biotechnol. Bioeng., 2010, 107(20), 302–311 CrossRef CAS PubMed.
- S. C. Hur, N. K. Henderson-MacLennan, E. R. B. McCabe and D. Di Carlo, Deformability-based cell classification and enrichment using inertial microfluidics, Lab Chip, 2011, 11, 912–920 RSC.
- M. Masaeli, et al., Continuous inertial focusing and separation of particles by shape, Phys. Rev. X, 2012, 2, 031017 Search PubMed.
- H. Moffatt, J. Fluid Mech., 1964, 18(1), 1–18 CrossRef.
- W. Cherdron, F. Durst and J. Whitelaw, J. Fluid Mech., 1978, 84, 13 CrossRef CAS.
- E. Sollier, M. Cubizolles, Y. Fouillet and J.L. Achard, Fast and continuous plasma extraction from whole human blood based on expanding cell-free layer devices, Biomed. Microdevices, 2010, 12(3), 485–97 CrossRef PubMed.
- J. Park and H. Jung, Anal. Chem., 2009, 81, 8280 CrossRef CAS PubMed.
- M. Khabiry, et al. , Small, 2009, 51186 Search PubMed.
- D. Di Carlo, D. Irimia, R. G. Tompkins and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18892–18897 CrossRef CAS PubMed.
- K. A. Hyun, K. Kwon, H. Han, S. I. Kim and H. I. Jung, Microfluidic flow fractionation device for label-free isolation of circulating tumor cells (CTCs) from breast cancer patients, Biosens. Bioelectron., 2013, 40(1), 206–12 CrossRef CAS PubMed.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Rapid prototyping of microfluidic systems in poly(dimethylsiloxane), Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- D. R. Gossett, Hydrodynamic stretching of single cells for large population mechanical phenotyping, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(20), 7630–7635 CrossRef CAS PubMed.
- E. Sollier, et al., Rapid prototyping polymers for microfluidic devices and high pressure injections, Lab Chip, 2011, 11, 3752–3765 RSC.
- H. Amini, et al., Engineering fluid flow using sequenced microstructures, Nat. Commun., 2013, 4, 1826 CrossRef PubMed.
- R. Strauss, et al., Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotype heterogeneity and plasticity, PLoS One, 2011, 6(1), e16186 CAS.
- Y. Kim, et al., Clinicopathological implications of EpCAM expression in adenocarcinoma of the lung, Anticancer Res., 2009, 29(5), 1817–1822 CAS.
- D. C. Lazar, et al., Cytometric comparisons between circulating tumor cells from prostate cancer patients and the prostate-tumor-derived LNCaP cell line, Phys. Biol., 2012, 9, 016002 CrossRef PubMed.
- S. K. Lee, et al., Nanowire substrate-based laser scanning cytometry for quantitation of circulating tumor cells, Nano Lett., 2012, 12, 2697–2704 CrossRef CAS PubMed.
- E. H. Cho, et al., Characterization of circulating tumor cell aggregates identified in aptietns with epithelial tumors, Phys. Biol., 2012, 9, 016001 CrossRef PubMed.
- M. Wendel, et al., Fluid biopsy for circulating tumor cell identification in aptietns with early and late stage non small cell lung cancer: a glimpse into lung cancer biology, Phys. Biol., 2012, 9, 016005 CrossRef PubMed.
- G.W. Schmid-Schonbein, et al., Morphometry of human leukocytes, Blood, 1980, 56, 866–875 CAS.
- M. C. Miller, G. V. Doyle and L. W. M. M. Terstappen, Significance of circulating tumor cells detected by the cell search system in patients with metastatic breast, colorectal and prostate cancer, J. Oncol., 2010, 2010, 617421 Search PubMed.
- K. Spanel-Borowski, A. M. Ricken and W. F. Patton, Cytokeratin-positive and cytokeratin-negative cultured endothelial cells from bovine aorta and vena cava, Differentiation, 1994, 57(3), 225–234 CrossRef CAS PubMed.
- J. Che, et al., Microfluidic purification and concentration of malignant pleural effusions for improved molecular and cytomorphological diagnostics, PLoS One, 2013 Search PubMed , accepted.
- J. Nieva, et al., High-definition imaging of circulating tumor cells and associated cellular events in non-small cell lung cancer patients: a longitudinal analysis, Phys. Biol., 2012, 9, 016004 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc50689d |
| ‡ E. Sollier, D. E. Go and J. Che contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |