DOI:
10.1039/C3LC50805F
(Paper)
Lab Chip, 2014,
14, 244-251
Real-time detection of an airborne microorganism using inertial impaction and mini-fluorescent microscopy
Received
7th July 2013
, Accepted 30th September 2013
First published on 2nd October 2013
Abstract
To achieve successful real-time detection of airborne pathogenic microorganisms, the problem must be considered in terms of their physical size and biological characteristics. We developed an airborne microorganism detection chip to realize the detection of microorganisms, ensuring compactness, sensitivity, cost-efficiency, and portability, using three key components: an inertial impaction system, a cartridge-type impaction plate, and a mini-fluorescent microscope. The inertial impaction system was used to separate microorganisms in terms of their aerodynamic particle size, and was fabricated with three impaction stages. Numerical analysis was performed to design the system; the calculated cutoff diameter at each impaction stage was 2.02 (first stage), 0.88 (second stage), and 0.54 μm (third stage). The measured cutoff diameters were 2.24, 0.91, and 0.49 μm, respectively. A cartridge-type impaction plate was used, composed of molded polydimethylsiloxane (PDMS) and an actual impaction region made of a SYBR green I dye-stained agar plate. A mini-fluorescent microscope was used to distinguish microbes from non-biological particles. Images of the microorganisms deposited at the impaction zone were obtained via mini-fluorescent microscopy, and fluorescent intensities of the images were calculated using in-house image-processing software. The results showed that the developed system successfully identified aerosolized biological particles from non-biological particles in real time.
Introduction
Airborne microorganisms, known as bioaerosols, including viruses, bacteria, and fungal spores, are major components of atmospheric aerosols. Their size distribution ranges from 20 nm to 100 μm.1 Generally, small bioaerosols (e.g., viruses and bacteria) are suspended in the atmosphere for a long time due to their low settling velocities, leading to an increased possibility of inhaling them into the respiratory system.1,2 There have been many studies on the adverse effects of inhalation of pathogenic bioaerosols on human health, such as infectious diseases, pneumonia, and allergies.3,4 Thus, it is important to be able to detect these pathogenic bioaerosols rapidly and effectively.
To date, several methods to detect airborne microorganisms have been proposed.5–11 Polymerase chain reaction (PCR) techniques have been used extensively to detect sampled airborne microbes because PCR can provide a highly quantitative analysis.12–14 However, these techniques are not adequate for real-time detection because they take over 1 h to analyze the microbes. Although antibody-based detection methods have been used due to their sensitivity, additional pretreatment processes (e.g., particle condensation/purification) are necessary to transfer bioaerosols from an airborne to a colloidal state. Thus, they cannot guarantee portability for an integrated “μ-total analysis system” (μ-TAS). One of the most frequently used real-time detection techniques with an air-based platform exploits the autofluorescence of microorganisms by exerting UV light on them. Autofluorescence is caused by the metabolites and structural components of living cells.15 Although this technique allows continuous real-time monitoring/detection of bioaerosols directly in the air stream, it suffers from low fluorescent intensity, leading to poor detection limits and the requirement for precise optical systems for measurements.
To detect atmospheric bioaerosols in real time, it is necessary to identify bioaerosols from two perspectives, size and biological characteristics. Different bioaerosols have different size distributions, although agglomeration among them and shielding by water may distort their size. For example, Staphylococcus epidermidis (S. epidermidis; bacterial group) and influenza A H1N1 (viral group) have size distributions of 0.5–2 μm and 0.07–0.5 μm, respectively.16,17 Also, non-biological particles, suspended together with bioaerosols in the atmosphere, can have similar size distributions as biological particles.
To detect airborne microorganisms directly, considering the perspectives above and seeking to overcome the disadvantages of previous systems, in the present study we demonstrate the real-time detection of airborne microorganisms directly in the air stream. The system is composed of three major parts: an inertial impaction system, a cartridge-type impaction plate, and a mini-fluorescent microscope. Numerical analyses and several experiments were performed to obtain optimum design parameters and to evaluate the feasibility of the system. An inertial impaction technique was used to separate airborne particles in terms of their aerodynamic diameter. A disposable cartridge-type impaction plate was used to provide real-time fluorescent staining of the airborne microbes and to address the saturation problem during long sampling times. To minimize system dimensions – for portability and cost-efficiency – a mini-fluorescent microscope, remodeling a commercially available webcam, was used. Biological particles were identified from non-biologicals by fluorescent intensity detection. The proposed system is intended for early alarm/warning/detection of hazardous biochemical components. Once the system recognizes the emergence of a bioaerosol, more quantitative analyses of the microbes at the DNA level could be performed with qualified instruments such as PCR methods.
Theory and numerical analysis
The working principle of inertial impaction of the particles on a curved channel can be explained with a simple theory. When particles suspended in a working fluid (here, air) are injected from an inlet towards a flat impaction plate and the streamlines of the working fluid are deflected around the edge of the corner towards an outlet, particles larger than the cutoff diameter do not follow the deflected streamlines due to their higher inertia. Instead, they collapse on the impaction plate. On the other hand, particles smaller than a threshold follow the streamline and stay suspended in the working fluid. Thus, particles are separated in terms of their sizes, or more specifically, their aerodynamic diameter, defined as:| | 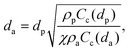 | (1) |
where dp is the particle diameter, ρp is the particle density, χ is the dynamic shape factor, ρa is the standard particle density (i.e., 1000 kg m−3), and Cc(dp) is the Cunningham slip correction factor, based on particle diameter, defined as:| | | Cc = 1 + 0.5Knp[2.34 + 1.05 exp(−0.195Knp)], | (2) |
where Knp is the particle Knudsen number, defined as 2λ/dp, where λ is the mean free path of the working fluid. Cc(da) is the Cunningham slip correction factor based on aerodynamic diameter.
To find the optimal design parameters for the device, flow field and particle motions were simulated in three dimensions using commercial software (CFD-ACE; ESI, USA). The behavior of particles in the curved channel was characterized by the following dimensionless parameters: Stokes number, Reynolds number, and Dean number:18
| | 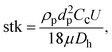 | (3) |
| | 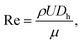 | (4) |
| | 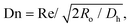 | (5) |
where
μ is the air viscosity,
U is the air velocity at the inlet,
Dh is the hydraulic diameter, and
Ro is the radius of curvature.
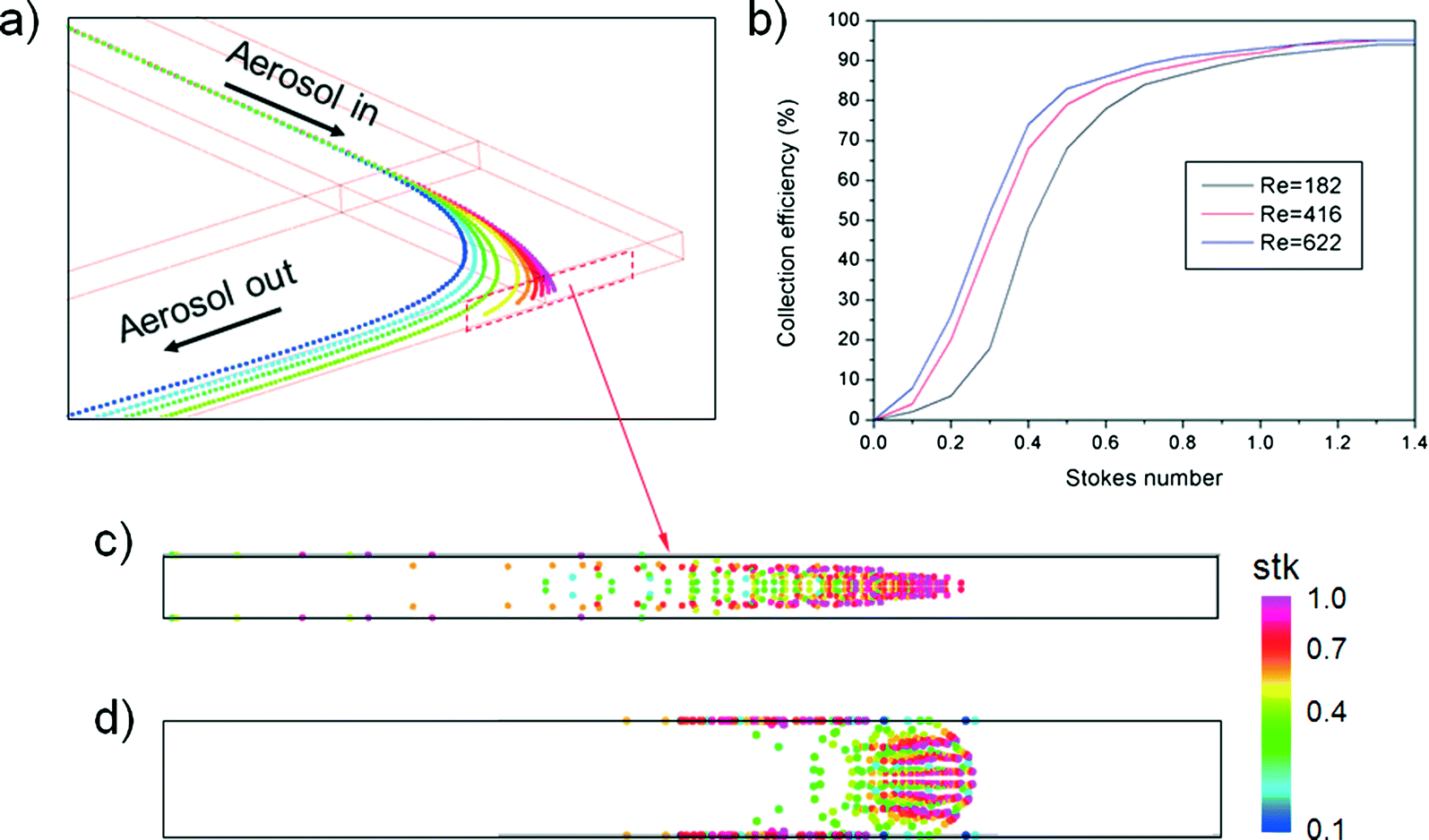
Fig. 1(a) shows the typical curvilinear motions of particles injected at the same initial position as a function of Stokes number. Because the Stokes number represents the dimensionless particle diameter,19 particles with smaller Stokes numbers more closely follow the original streamlines of the working fluid under the given flow conditions. Reynolds number affects the particle collection efficiency, (Nd/Nt) × 100, where Nd is the number of particles that collapse on the impaction plate and Nt is the total number of particles at the inlet. Fig. 1(b) shows the particle collection efficiency curves for different flow conditions. As the Reynolds number decreased in laminar flow, the particle collection efficiency was reduced due to the increase in the flow boundary layer in the channel.20 This phenomenon became remarkable for the low Stokes number region (stk < 0.4), and was attributed to the combined effects of (1) the low Reynolds number that enhanced the flow boundary layer and (2) particles having a sufficiently low Stokes number to follow the streamlines of the secondary flow (i.e., Dean flow21) being deposited at the top/bottom channel walls on the curved channel. The deposition pattern of the particles and collection efficiency were also affected by the secondary flow, the so-called Dean vortex.22 Because a strong Dean vortex causes unwanted particle deposition at the top/bottom walls of the channel, not at the detection zone, flow conditions for a weak Dean vortex are preferable. Although reduction of the Reynolds number or increasing the ratio of curvature at the curved region could prevent a strong Dean vortex effect, they also increase the boundary layer and restrict a sudden deflection of the axial streamlines, respectively. An alternative method to suppress the formation of a strong Dean vortex at the curved region is to reduce the aspect ratio (height/width) of the channel.23Fig. 1(c) and (d) show the particle deposition patterns as a function of the Stokes number for the same Dean number. The aspect ratios of the simulated channels were 0.1 and 1, respectively. It is clear that a channel with a low aspect ratio had advantages over one with a high aspect ratio because a large proportion of the particles were attached to the top/bottom walls of the channel with the high aspect ratio.
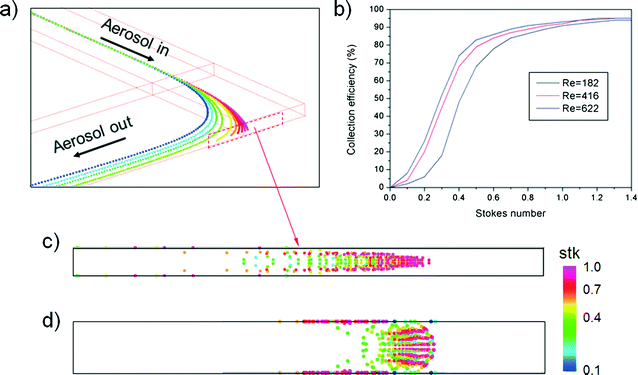 |
| | Fig. 1 Numerical results of the inertial impaction of particles in the curved channel: (a) trajectories of airborne particles at the curved region (Re = 506, Dn = 292) as a function of Stokes number, (b) particle collection efficiency curve for different flow conditions, (c) particle deposition pattern in a channel with an aspect ratio of 0.1, and (d) particle deposition pattern in a channel with an aspect ratio of 1. | |
Materials and methods
Design of the proposed device
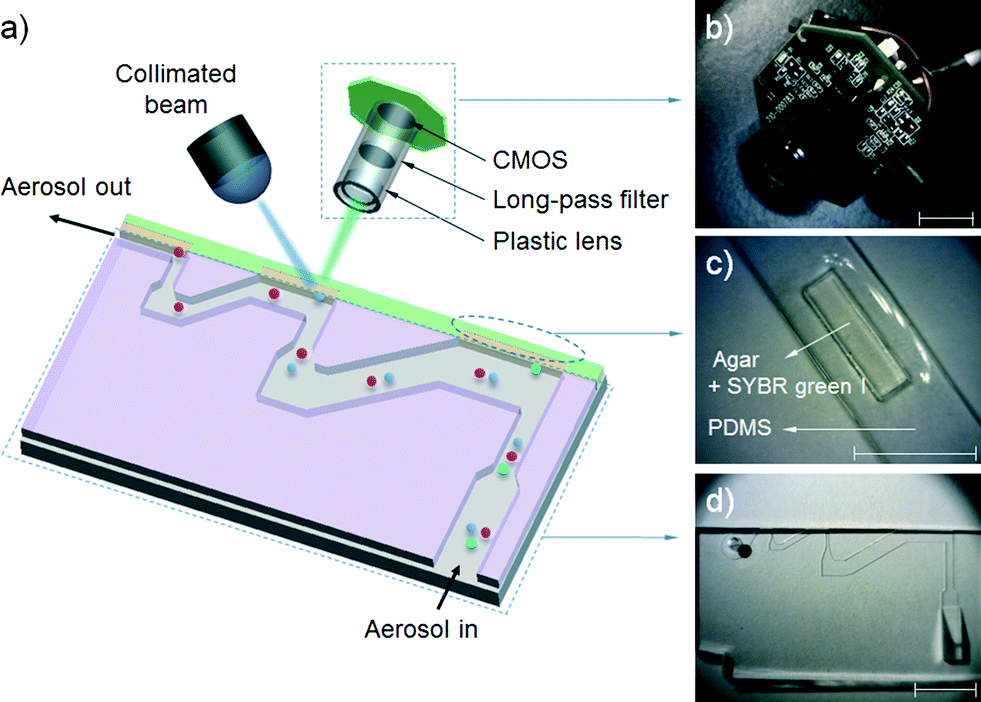
Fig. 2(a) shows a schematic diagram of the proposed airborne microbe detection chip, composed of a main inertial impaction part, a cartridge-type impaction plate, and mini-fluorescent microscope. The cutoff diameter that yields a 50% collection efficiency at each impaction stage can be calculated from eqn (3):| | 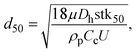 | (6) |
where stk50 denote the Stokes number that corresponds to 50% particle collection efficiency, which can be determined based on the numerical analysis (Fig. 1(b)). Fig. 2(d) shows the main inertial impaction part with three stages, fabricated using a conventional soft-lithography process.24 The sample flow rate and channel height were set to 0.12 L min−1 and 160 μm, respectively, to prevent a strong Dean vortex effect. The d50 at each stage was 2.02 μm (first stage), 0.88 μm (second stage), and 0.54 μm (third stage), because the target bioaerosol element, S. epidermidis, has a size distribution from 0.5 to 2 μm. Relatively large bioaerosols are removed at the first stage, and the target bioaerosols are collapsed and detected at the second and third stages. By adjusting the design parameters, bioaerosols with specific diameters can be collapsed and detected. The channel width at each stage was 1287 μm (first stage), 472 μm (second stage), and 263 μm (third stage). The ratio of the distance between the impaction plate, the channel wall before deflection, and the channel width was two, ensuring adequate sharpness of the collection efficiency curve.20 To prevent particle loss, the inlet was designed to have a tapered region in the vertical direction because the sudden deflection of the streamlines at the inlet makes it act like an impaction stage.25 When the target particles/microbes were collapsed at the impaction region, we could detect them in real time from the fluorescent signal induced by the interaction between the microbes and fluorescent dye (details of the principles of the fluorescent signal production are provided in the cartridge-type impaction plate section). To detect differences at the impaction region, a state-of-the-art mini-fluorescent microscope was used, ensuring compactness and portability. Details of the mini-fluorescent microscope are provided in the next section. To our knowledge, this is the first reported description of a mini-microscopy system used to detect a fluorescent signal.
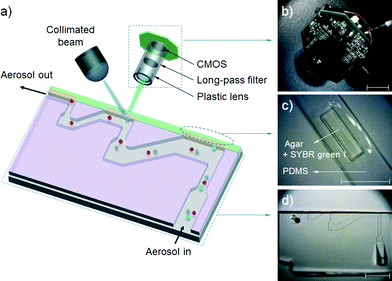 |
| | Fig. 2 (a) Schematic diagram of the present airborne microbe detection chip, (b) mini-fluorescent microscope, (c) disposable cartridge-type PDMS impaction plate with an impaction zone, and (d) microchannel with three impaction stages (all scale bars indicate 1 cm). | |
Mini-fluorescent microscope
Fig. 2(b) shows a photograph of the mini-fluorescent microscope. It consisted of three major parts: a plastic lens (×20), a long-pass filter, and a complementary metal-oxide semiconductor (CMOS) module. The long-pass filter (cutoff wavelength = 500 nm, diameter = 12.5 mm, Edmund Optics, USA) was placed between the plastic lens and the CMOS module. The CMOS module and plastic lens were obtained from a commercially available webcam (Logitech, C-250). The webcam lens was used to demagnify an object; magnification of the object was accomplished by reversing the lens.26 Because the total magnification of the microscopy system could be adjusted by varying the distance between the plastic lens (objective lens) and CMOS module, we designed the plastic lens to move back and forth along a screw inside the lens housing. The travel distance of the plastic lens was 15 mm, and the working distance of the microscope was determined (2–5 mm) by the total magnification of the microscope. The system had dimensions of 3 × 3.5 × 2 cm3 (height × width × depth), and was equipped with an xy stage for translation and focusing. Collimated blue light (470 nm) was exerted from the top of the impaction plate, and the mini-fluorescent microscope was located perpendicular to the light source and in front of the particle deposition plate.
Cartridge-type impaction plate
Fig. 2(c) shows a cartridge-type impaction plate with an impaction zone. We used SYBR green I (Life Technologies, USA) to distinguish between biological and non-biological particles on the impaction zone. The SYBR green I nucleic acid stain increases the fluorescent signal that is induced when it is bound to the double-stranded DNA of a microorganism.27 Due to its high sensitivity, it has been widely studied for detecting and enumerating various microorganisms in diverse environmental samples.28–30 Because the molecules of the dye readily penetrate microbial cell walls via membrane diffusion and bind with the DNA, a membrane permeabilization process is not required for rapid staining.31,32 The dye protocol recommends a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10000 dilution ratio of dye to sample solution. In the present study, we used a higher dye concentration than this to achieve more rapid staining. The peak excitation and emission wavelengths of SYBR green I are 495 nm and 525 nm, respectively.
:10000 dilution ratio of dye to sample solution. In the present study, we used a higher dye concentration than this to achieve more rapid staining. The peak excitation and emission wavelengths of SYBR green I are 495 nm and 525 nm, respectively.
A PDMS mold with a well (dimensions: width × height × depth = 3 × 10 × 0.6 mm3) in the center region was fabricated using a soft-lithography process, to be filled with agar (the impaction zone). Distilled water with 3 wt% agar powder (Becton Dickinson, USA) was autoclaved for 1 hour, and SYBR green I was mixed with the autoclaved agar at 0.05% v/v. The mixture was poured into the PDMS mold before starting gelation. The semi-solidified impaction region allowed the reagent molecules to move freely into the DNA when a microorganism was inserted into the agar. Since the agar plate was slightly translucent, it was made thin to allow fluorescent signal detection. The thickness of the impaction plate was 2 mm, taking into account the working distance of the mini-fluorescent microscope. It was designed to be disposable for convenient long-term usage. The microchannel and cartridge-type impaction plate, shown in Fig. 2(c) and (d), were packed in a stainless steel housing for mechanical sealing.
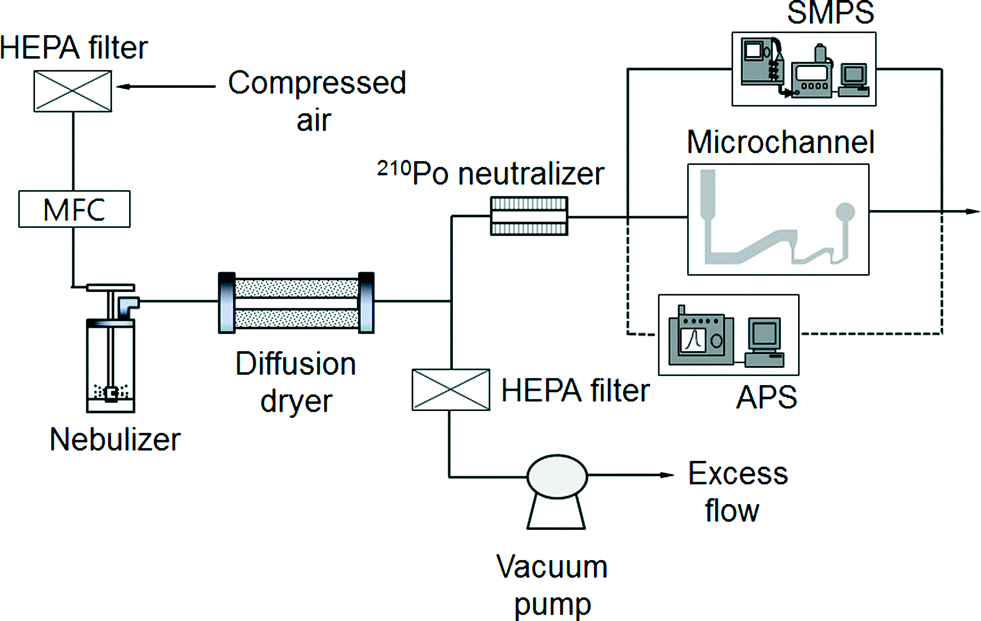
Experimental
Fig. 3 shows a schematic diagram of the experimental setup for performance evaluation of the present bioaerosol detection chip. Filtered compressed air, at a flow rate of l L min−1, entered the collision nebulizer (BGI Corp., USA), which was used for the aerosolization of particles/bioaerosols. Two different particles were used for the experiments. Polystyrene latex (PSL) particles with diameters of 0.173, 0.222, 0.265, 0.305, 0.426, 0.482, 0.523, 0.598, 0.652, 0.72, 0.806, 0.913, 1.00, 1.11, 1.53, 2.1, 2.50, 3.00, and 4.00 μm (Duke Scientific, USA) were used for calibration, and S. epidermidis was used as an actual target bioaerosol. The number concentration of the aerosolized particles/microbes was set to approximately 20 particles cm−3, higher than that in the atmosphere because the proposed system is intended for early alarm/warning regarding hazardous biochemical components. A diffusion dryer was added after the nebulizer to remove moisture from the aerosol stream. The aerosol stream, at a flow rate of 0.12 L min−1, entered a 210Po neutralizer to remove the electrical charge of the particles. Excess flow was vented to the atmosphere. Particles and bioaerosols with diameters smaller and larger than 0.55 μm were measured using a scanning mobility particle sizer (SMPS; Model 3081, TSI Inc., USA) and an aerodynamic particle sizer (APS; Model 3321, TSI Inc.), respectively.
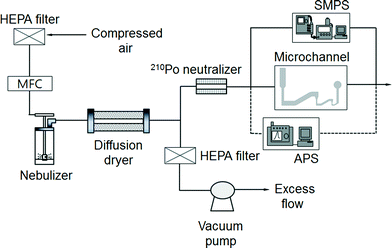 |
| | Fig. 3 Schematic diagram of the experimental setup. | |
Results and discussion
Particle collection efficiency
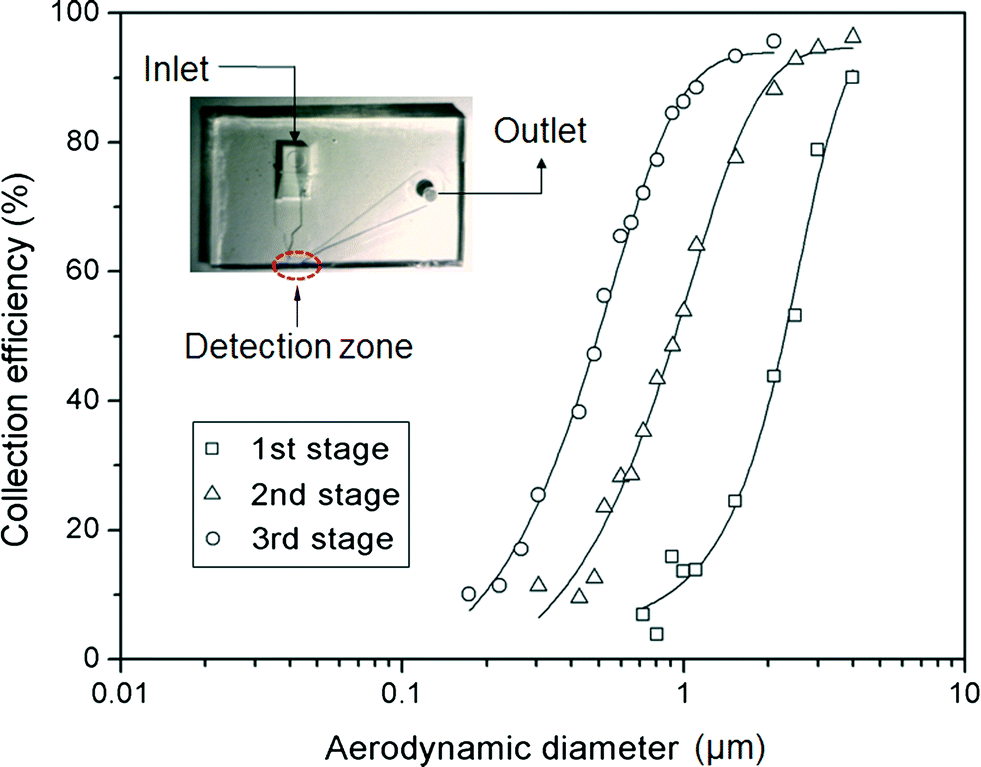
Fig. 4 shows the collection efficiency curves of PSL particles at each impaction stage. To measure the collection efficiency at each stage, three different devices with single stages were fabricated, having designed cutoff diameters of 2.02 μm, 0.88 μm, and 0.54 μm (as shown in the inset of Fig. 4). The collection efficiency of a given particle size is calculated using the following equation:| | 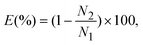 | (7) |
where N1 and N2 are the particle number concentrations at the inlet and outlet, respectively. The measured cutoff diameter at each stage was 2.24 μm (first stage), 0.91 μm (second stage), and 0.49 μm (third stage). The measured cutoff diameter values were slightly different from those designed based on the numerical analysis, but within 11%. The differences between the theoretical values and experimental results were attributed to particle loss because some of the particles were deposited on other regions of the microchannel instead of the impaction zone. Some of the particles with larger diameters (dP > 2 μm) settled at the bottom of the channel due to gravitational force or stuck in the particle acceleration region,33 while the loss of some smaller particles was caused by diffusional deposition. Particle losses in the acceleration region may be reduced by aerosol focusing techniques.34 In the case of solid particles, some of the deposited particles were bounced from the impaction zone, leading to distortion of the collection efficiency curve.35 To prevent this phenomenon, some researchers have coated the impaction zone with highly viscous liquids (e.g., silicon oil, grease).35,36 In the present system, an agar plate, instead of a PDMS plate, was used as an impaction zone, because the agar plate could hold a large amount of humidity, reducing the particle bounce effect. Furthermore, although particles with large inertia bounce more frequently at the impaction zone, they may also become embedded deeply into the agar plate.37
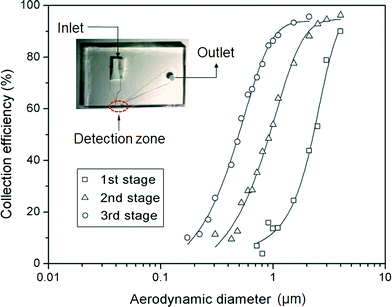 |
| | Fig. 4 Collection efficiency of PSL particles at each impaction stage. The inset shows the device for measuring the collection efficiency at each impaction zone. | |
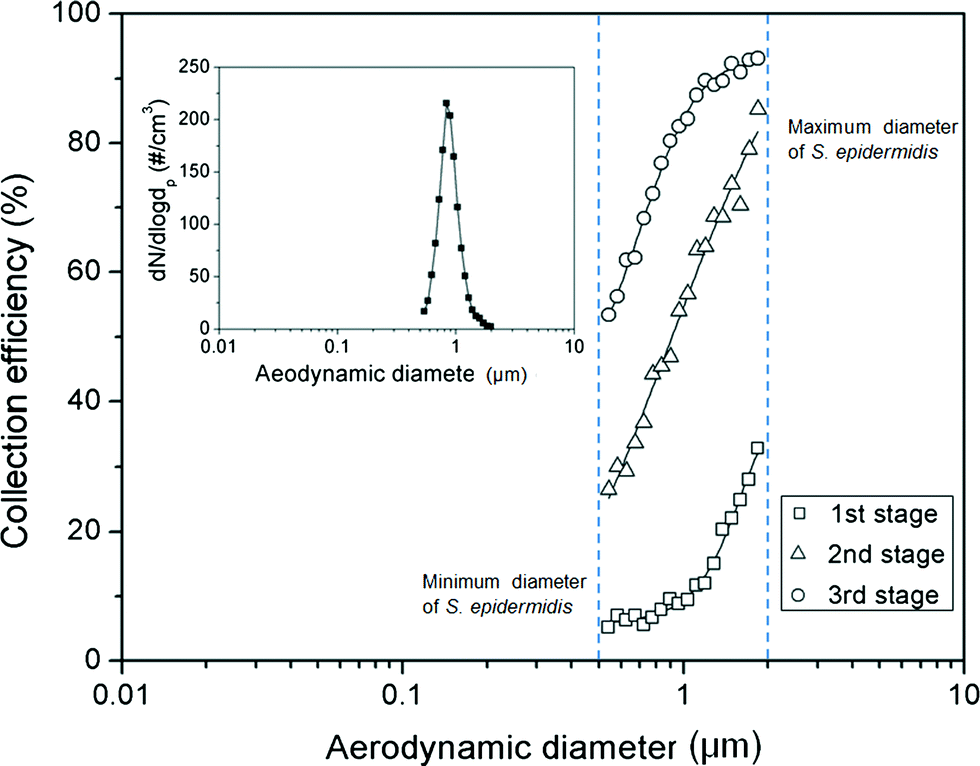
Fig. 5 shows the collection efficiency curve of the aerosolized S. epidermidis at each impaction stage. The measured size distribution of the S. epidermidis is shown in the inset of Fig. 5. The peak size and geometric standard deviation (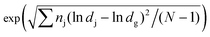 , where dj is the diameter of an individual particle, nj is the number of particles in the jth group, N is the total number of particles, and ln dg is the natural logarithm of the geometric mean diameter of the particles, defined as ∑nj ln dj/N) of S. epidermidis were 0.84 μm and 1.33, respectively. The maximum and minimum aerodynamic diameters of S. epidermidis were 0.55 μm and 2 μm, respectively. The collection efficiency curve of S. epidermidis showed similar characteristics to that of the PSL particles. The cutoff diameter of S. epidermidis at the second stage was 0.9 μm, similar to that of the PSL particles (0.91 μm). The measured collection efficiencies of the peak size (0.84 μm) at each impaction stage were 9% (first stage), 44% (second stage), and 77% (third stage), while the collection efficiencies of the PSL particles with a diameter of 0.84 μm at each impaction stage were 10% (first stage), 45% (second stage), and 81% (third stage), respectively.
, where dj is the diameter of an individual particle, nj is the number of particles in the jth group, N is the total number of particles, and ln dg is the natural logarithm of the geometric mean diameter of the particles, defined as ∑nj ln dj/N) of S. epidermidis were 0.84 μm and 1.33, respectively. The maximum and minimum aerodynamic diameters of S. epidermidis were 0.55 μm and 2 μm, respectively. The collection efficiency curve of S. epidermidis showed similar characteristics to that of the PSL particles. The cutoff diameter of S. epidermidis at the second stage was 0.9 μm, similar to that of the PSL particles (0.91 μm). The measured collection efficiencies of the peak size (0.84 μm) at each impaction stage were 9% (first stage), 44% (second stage), and 77% (third stage), while the collection efficiencies of the PSL particles with a diameter of 0.84 μm at each impaction stage were 10% (first stage), 45% (second stage), and 81% (third stage), respectively.
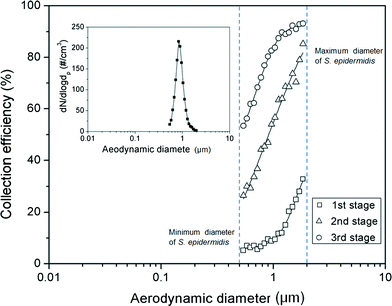 |
| | Fig. 5 Collection efficiency of S. epidermidis at each impaction stage. The inset shows the size distribution of the aerosolized S. epidermidis. | |
Fluorescent image and intensity
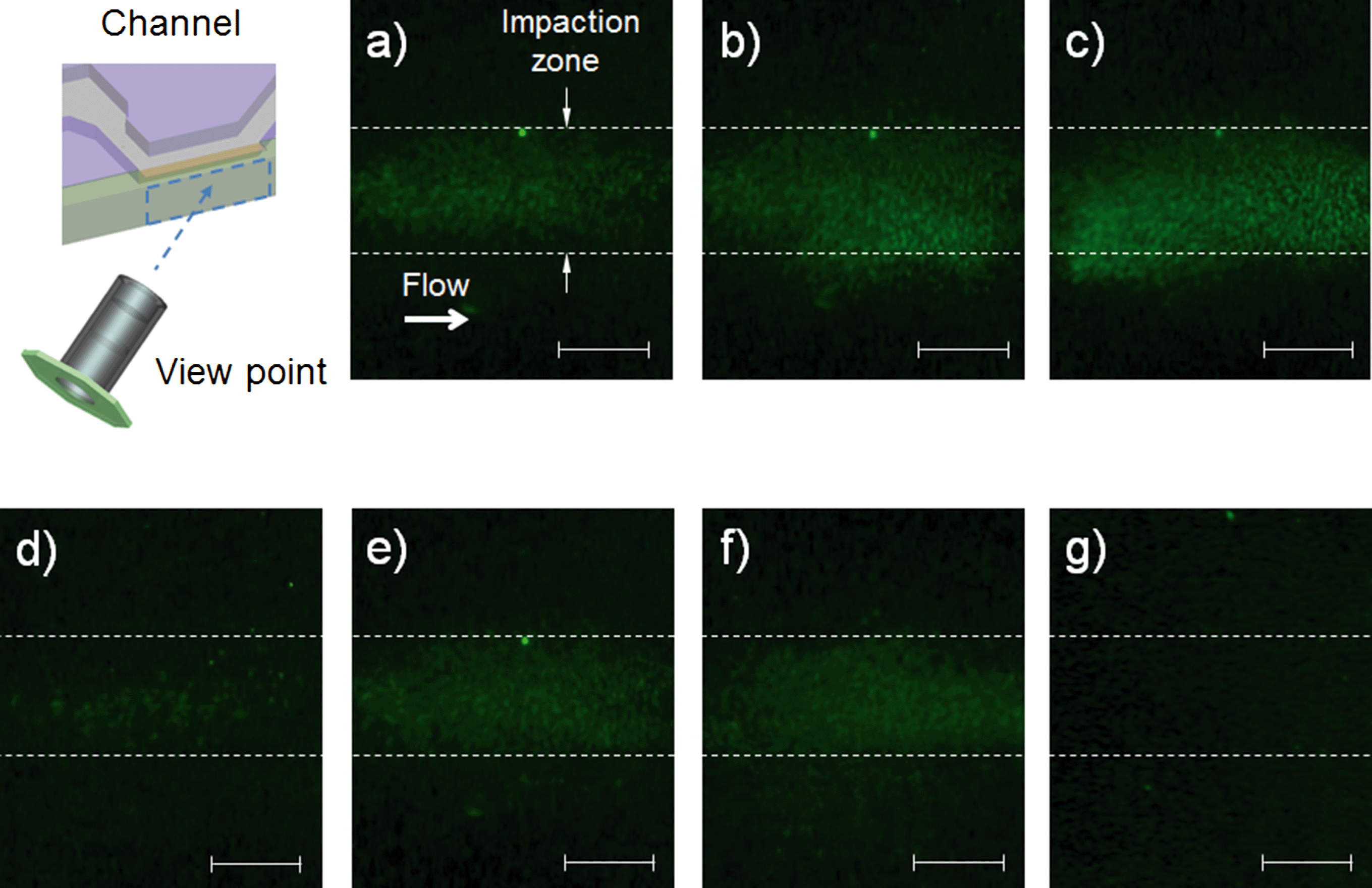
Fig. 6 shows fluorescent images of S. epidermidis and PSL particles at the impaction zone. The images were obtained from a device with three impaction stages (Fig. 2(d)). The magnification of the images was 200×, and the working distance of the microscope was 3 mm. The images show that the collected S. epidermidis could be stained directly on the impaction plate in real time, without a permeabilization process. The mini-fluorescent microscope provided appropriate resolution for the imaging and detection of stained S. epidermidis. When microorganisms are collapsed at the impaction zone, they land smoothly on it or are embedded deeply into it, depending on their inertia.37 Of these two collection processes, the fluorescent staining of the bioaerosol was mainly the result of the latter, since SYBR green I molecules do not move freely in landed bioaerosol particles.
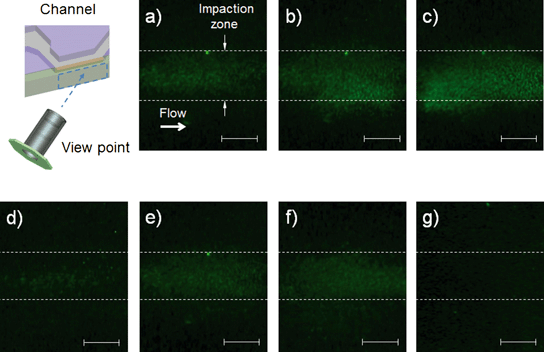 |
| | Fig. 6 Fluorescent images at different sampling times and impaction stages. The schematic diagram indicates the viewpoint of the mini-fluorescent microscope; (a), (b), and (c) show photos at the second impaction zone with sampling times of 20 s, 40 s, and 60 s, respectively; and (d), (e), and (f) show photos at the first, second, and third stages, respectively, with a sampling time of 30 s. (g) shows an image of PSL particles (diameter: 1 μm) at the second impaction zone with a sampling time of 30 s (all scale bars indicate 100 μm). | |
Fig. 6(a), (b), and (c) show fluorescent images of the second impaction zone with respective sampling times of 20, 40, and 60 s. As the sampling time increased, more particles were attached to the impaction zone, leading to an increase in fluorescent intensity. Some of the particles shown in Fig. 6(c) deviated from the impaction zone (bottom wall region of the microchannel). Since the agar plate was in a deformable semi-solid state, it exhibited concave deformation caused by the pressure of the airflow. Fig. 6(d), (e), and (f) show fluorescent images of the first, second, and third impaction zones, respectively, with a sampling time of 30 s. Because the collection efficiencies of the particles differed at each of the impaction zones in terms of aerodynamic particle diameter, the fluorescent intensities also differed at each stage. The fluorescent intensity at the first stage was quite low compared with the intensities at the second and third stages. Fig. 6(g) shows an image of the PSL particles at the second impaction zone with 30 s sampling time. The non-biological particles (which do not contain DNA) exhibited a negligible green fluorescent signal compared with microorganisms on the impaction zone, since double stranded DNA-bound dye has a fluorescent intensity 1000 times greater than that of plain dye.38
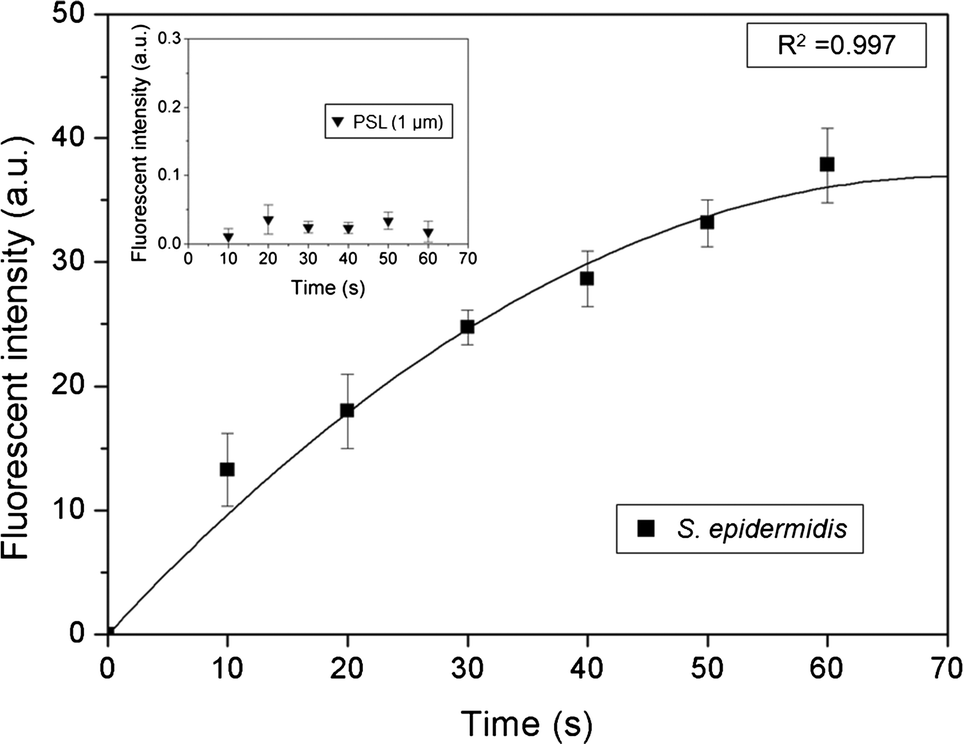
Fig. 7 shows the fluorescent intensity at the second stage with varying sampling times. The green fluorescent intensity was calculated from an in-house image processing code programmed using MATLAB. Because the agar plate initially showed a low level of green signal, the initial fluorescent intensity was subtracted from the fluorescent intensity at certain later sampling times.
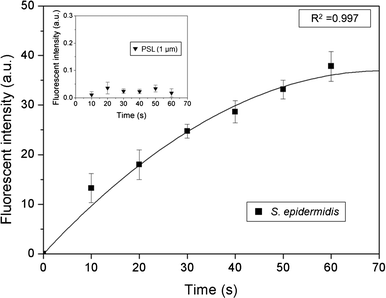 |
| | Fig. 7 Fluorescent intensity of S. epidermidis at the detection zone as a function of sampling time. The inset shows the fluorescent intensity of the PSL particles with a diameter of 1 μm. The fluorescent intensity on each graph is the same. | |
Generally, the fluorescent intensity increased with sampling time. However, although the number of microorganisms on the impaction zone increased linearly with sampling time, the fluorescent intensity did not show a linear relationship. Instead, the slope became more gradual with longer sampling times. This tendency was attributed primarily to the partial staining of the deposited microorganisms by SYBR green I because of the mountain-shaped deposition pattern, so that some of them did not make contact with the SYBR green I-containing agar, as well as a bleaching effect of the stain.39
To address this problem, a cartridge-type impaction plate was used. From our calculations, we could detect biological particles in the standard atmosphere over about 2 h, because the number concentration used in the present experiment was about 100 times higher than that of standard atmospheric conditions. The inset in Fig. 7 shows the fluorescent intensity of the PSL particles with a diameter of 1 μm at various sampling times. The fluorescent intensity (arbitrary units) in the inset was the same as that shown in the main part of Fig. 7. The fluorescent intensity of the PSL particles was extremely low compared with that of the bioaerosols, indicating that the proposed system could identify biological particles from non-biological particles.
Conclusions
We demonstrated the real-time detection of an airborne microorganism using inertial particle separation and mini-fluorescent microscopy. The system showed reasonable performance with both biological and non-biological particles. The cutoff diameter at each impaction stage was 2.24, 0.91, and 0.49 μm, respectively. The deviations in the cutoff diameter between the designed values and experimental results were within 11%. The fluorescent images for the different experimental conditions showed that the airborne microorganism was stained directly by SYBR green I at the impaction zone. The cost-efficient mini-fluorescent microscope showed excellent resolution for microorganism imaging and detection. A disposable cartridge-type impaction plate was used to prevent saturation of the fluorescent signal and to provide real-time fluorescent staining of the airborne microbes. The fluorescent intensity of the microorganism and PSL particles allowed us to identify microbes from non-biological particles. The proposed device could be used for an early alarm/warning system for pathogenic bioaerosols in real time using fluorescence acting on the bioaerosol components, before analyzing the exact properties of the bioaerosol at the DNA level using qualified instruments such as a PCR technique.
Acknowledgements
This research was supported by the Converging Research Center Program funded by the Ministry of Education, Science, and Technology (2012K001370) and was partially supported by the KIST Institutional Program (2E23972).
References
-
W. C. Hinds, Aerosol Technology, John Wiley and Sons, New York, 1999 Search PubMed.
- J. Gralton, E. Tovey, M. Mclaws and W. D. Rawlinson, J. Infection, 2011, 62, 1–13 CrossRef PubMed.
- J. Douwel, P. Thorne, N. Pearce and D. Heederik, Ann. Occup. Hyg., 2003, 47, 187–200 CrossRef.
- F. M. Blachere, W. G. Lindsley, T. A. Pearce, S. E. Anderson, M. Fisher, R. Khakoo, B. J. Meade, O. Lander, S. Davis, R. E. Thewlis, I. Celik, B. T. Chen and D. H. Beezhold, Clin. Infect. Dis., 2009, 48, 438–440 CrossRef PubMed.
- O. V. Pyankov, I. E. Agranovskl, O. Pyankova, E. Mokhonova, V. Mokhonov, A. S. Safatov and A. A. Khromykh, Environ. Microbiol., 2007, 9, 992–1000 CrossRef CAS PubMed.
- E. V. Usachev and I. E. Agranovski, J. Environ. Monit., 2012, 14, 1631–1637 RSC.
- B. J. Hindson, M. T. McBride, A. J. Makarewicz, B. D. Hemderer, U. S. Setlur, S. M. Smith, D. M. Gutierrez, T. R. Metz, S. L. Nasarabadi, K. S. Venkateswaran, S. W. Farrow, B. W. Colston Jr. and J. M. Dzenitis, Anal. Chem., 2005, 77, 284–289 CrossRef CAS PubMed.
- I. Oita, H. Halewyck, B. Thys, B. Rombaut, Y. V. Heyden and D. Mangelings, Anal. Bioanal. Chem., 2010, 398, 239–264 CrossRef CAS PubMed.
- D. N Stratis-Cullum, G. D. Griffin, J. Mobley, A. A. Vass and T. Vo-dinh, Anal. Chem., 2003, 75, 275–280 CrossRef CAS.
- D. K. Farmer and J. L. Jimenez, Anal. Chem., 2010, 82, 7879–7884 CrossRef CAS PubMed.
- B. U. Lee, J. H. Jung, S. H. Yun, G. B. Hwang and G. N. Bae, J. Aerosol Sci., 2010, 41, 694–701 CrossRef PubMed.
- A. Oppliger, N. Charrière, P. Droz and T. Rinsoz, Ann. Occup. Hyg., 2008, 52, 405–512 CrossRef CAS PubMed.
- D. Hospodsky, N. Yamamoto and J. Peccia, Appl. Environ. Microbiol., 2010, 76, 7004–7012 CrossRef CAS PubMed.
- E. V. Usachev and I. E. Agranovski, J. Environ. Monit., 2012, 14, 1631–1637 RSC.
- N. Bao, B. Jagadeesan, A. K. Bhunia, Y. Yao and C. Lu, J. Chromatogr., A, 2008, 1181, 153–158 CrossRef CAS PubMed.
- J. H. Jung, G. B. Hwang, J. E. Lee and G. N. Bae, Langmuir, 2011, 27, 10256–10264 CrossRef CAS PubMed.
- I. Lee, H. J. Kim, D. H. Lee, G. B. Hwang, J. H. Jung, M. Lee, J. Lim and B. U. Lee, Aerosol Air Qual. Res., 2010, 11, 230–237 Search PubMed.
- D. Y. H. Pui, F. Romay-Novas and B. Y. H. Liu, Aerosol Sci. Technol., 1987, 7, 301–315 CrossRef CAS.
- R. Chein and J. N. Chung, Int. J. Multiphase Flow, 1987, 13, 785–802 CrossRef CAS.
- V. A. Marple and K. Willeke, Atmos. Environ., 1976, 10, 891–896 CrossRef.
- W. R. Dean, Proc. R. Soc. London, Ser. A, 1928, 121, 402–420 CrossRef.
- Y. S. Cheng and C. S. Wang, Atmos. Environ., 1981, 15, 301–306 CrossRef.
- P. B. Howell, D. R. Mott, J. P. Golden and F. S. Ligler, Lab Chip, 2004, 4, 663–669 RSC.
- D. Qin, Y. Xia and G. M. Whitesides, Nat. Protoc., 2010, 5, 491–502 CrossRef CAS PubMed.
- S. A. Grinshpun, G. Mainelis, M. Trunov, R. R. Górny, S. K. Sivasubramani, A. Adhikari and T. Reponen, J. Aerosol Sci., 2005, 36, 575–591 CrossRef CAS PubMed.
- S. B. Kim, K. Koo, H. Bae, M. R. Dokmeci, G. A. Hamilton, A. Bahinski, S. M. Kim, D. E. Ingber and A. Khademhosseini, Lab Chip, 2012, 12, 3976–3982 RSC.
- D. Lee, S. H. Park, H. Yang, K. Chung, T. H. Yoon, S. Kim, K. Kim and Y. T. Kim, Lab Chip, 2004, 4, 401–407 RSC.
- H. J. Tobias, M. P. Schafer, M. Pitesky, D. P. Fergenson, J. Horn, M. Frank and E. E. Gard, Appl. Environ. Microbiol., 2005, 71, 6068–6095 CrossRef PubMed.
- R. T. Noble and J. A. Fuhrman, Aquat. Microb. Ecol., 1998, 14, 113–118 CrossRef.
- M. G. Weinbauer, C. Beckmann and M. G. Hofle, Appl. Environ. Microbiol., 1998, 64, 5000–5003 CAS.
- E. Manini and R. Danovaro, FEMS Microbiol. Ecol., 2005, 55, 416–423 CrossRef PubMed.
- P. Lebaron, P. Catala and N. Parthuisot, Appl. Environ. Microbiol., 1998, 64, 2697–2700 CAS.
- P. Demokritou, S. J. Lee, S. T. Ferguson and P. Koutrakis, J. Aerosol Sci., 2004, 35, 281–299 CrossRef CAS PubMed.
-
S. Seshadri, S. Hari and A. R. McFarland, Proc. 7th International Aerosol Conf., St. Paul, MN, 2006 Search PubMed.
- S. S. Pak, B. Y. H. Liu and K. L. Rubow, Aerosol Sci. Technol., 1992, 16, 141–150 CrossRef CAS.
- J. S. Kang, K. S. Lee, K. H. Lee, H. J. Sung and S. S. Kim, Aerosol Sci. Technol., 2012, 46, 966–972 CrossRef CAS.
- S. L. Stewart, S. A. Grinshpun, K. Willeke, S. Terzieva, V. Ulevicius and J. Donnelly, Appl. Environ. Microbiol., 1995, 61, 1232–1239 CAS.
- J. Wilhelm and A. Pingoud, ChemBioChem, 2003, 4, 1120–1128 CrossRef CAS PubMed.
- Y. Nishimura, T. Higashiyama, L. Suzuki, O. Misumi and T. Kuroiwa, Eur. J. Cell Biol., 1998, 77, 124–133 CrossRef CAS.
Footnote |
| † Authors equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 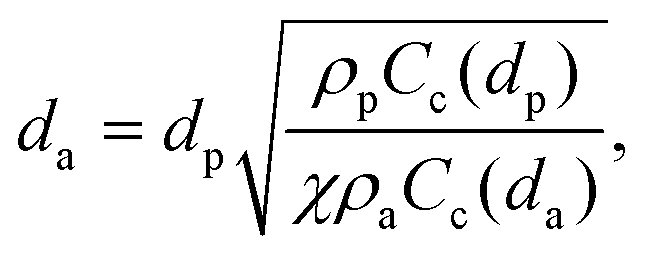
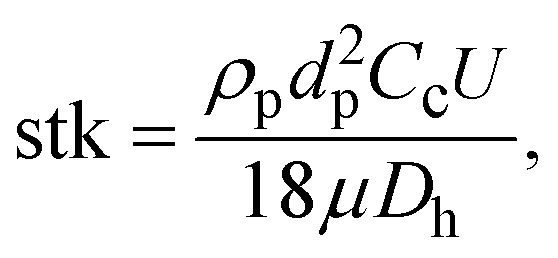
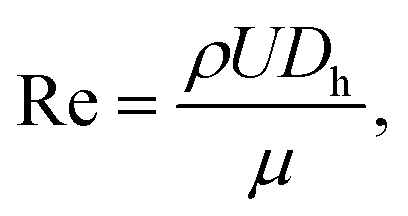
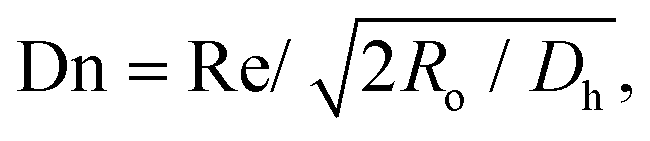
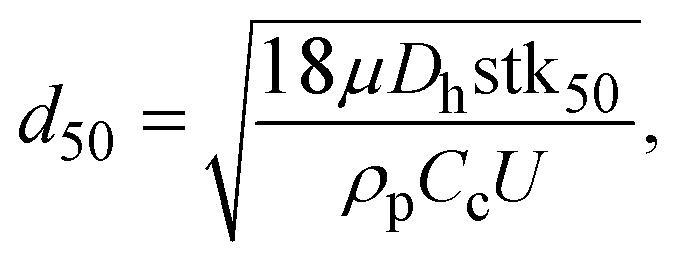
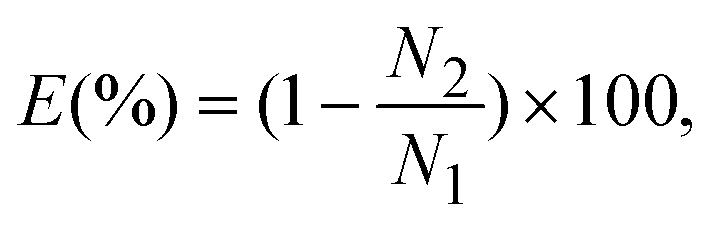
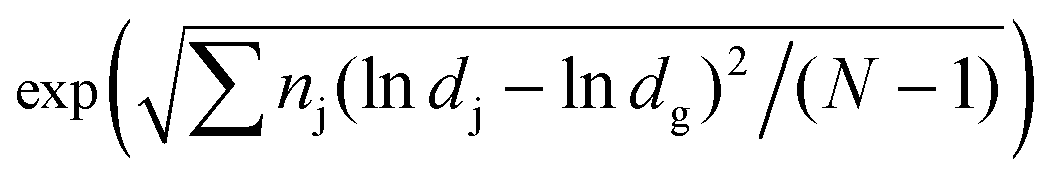 , where dj is the diameter of an individual particle, nj is the number of particles in the jth group, N is the total number of particles, and ln dg is the natural logarithm of the geometric mean diameter of the particles, defined as ∑nj ln dj/N) of S. epidermidis were 0.84 μm and 1.33, respectively. The maximum and minimum aerodynamic diameters of S. epidermidis were 0.55 μm and 2 μm, respectively. The collection efficiency curve of S. epidermidis showed similar characteristics to that of the PSL particles. The cutoff diameter of S. epidermidis at the second stage was 0.9 μm, similar to that of the PSL particles (0.91 μm). The measured collection efficiencies of the peak size (0.84 μm) at each impaction stage were 9% (first stage), 44% (second stage), and 77% (third stage), while the collection efficiencies of the PSL particles with a diameter of 0.84 μm at each impaction stage were 10% (first stage), 45% (second stage), and 81% (third stage), respectively.
, where dj is the diameter of an individual particle, nj is the number of particles in the jth group, N is the total number of particles, and ln dg is the natural logarithm of the geometric mean diameter of the particles, defined as ∑nj ln dj/N) of S. epidermidis were 0.84 μm and 1.33, respectively. The maximum and minimum aerodynamic diameters of S. epidermidis were 0.55 μm and 2 μm, respectively. The collection efficiency curve of S. epidermidis showed similar characteristics to that of the PSL particles. The cutoff diameter of S. epidermidis at the second stage was 0.9 μm, similar to that of the PSL particles (0.91 μm). The measured collection efficiencies of the peak size (0.84 μm) at each impaction stage were 9% (first stage), 44% (second stage), and 77% (third stage), while the collection efficiencies of the PSL particles with a diameter of 0.84 μm at each impaction stage were 10% (first stage), 45% (second stage), and 81% (third stage), respectively.