Research highlights
Justin R.
Smith
a,
Imee G.
Arcibal
a,
Alessandro
Polini
bc,
Mehmet R.
Dokmeci
bc and
Ali
Khademhosseini
*bcde
aUS Army Corps of Engineers Construction Engineering Research Laboratory, Champaign, IL 61822, USA
bCenter for Biomedical Engineering, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Cambridge, Massachusetts 02139, USA. E-mail: alik@rics.bwh.harvard.edu
cHarvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
dWyss Institute for Biologically Inspired Engineering, Harvard University, Boston, Massachusetts 02115, USA
eWorld Premier International – Advanced Institute for Materials Research (WPI-AIMR), Tohoku University, Sendai 980-8577, Japan
First published on 19th November 2013
Attomolar detection with microfluidic ELISA
In recent years there has been a significant investment to develop technologies able to quantitatively detect low abundant proteins in order to analyze protein variations in single cells and to investigate signaling pathways. Understanding these variations and pathways is a key step in the early detection of diseases such as Alzheimer's disease and cancer to begin treatment at an early stage. Typically, enzyme-linked immunosorbent assays (ELISA) are used to probe these proteins, employing a capture antibody to bind to the target protein and a secondary antibody fluorescently tagged to provide a color change to measure the amount of protein present. However, traditional ELISA is limited by the need for large sample size, has issues with reproducibility, and lacks high throughput capability. To circumvent these issues, researchers have combined ELISA technology with microfluidics, which has led to sensitive detection in the femto-to-picomolar range.1 Digital immunoassays, able to manipulate samples in small, discrete volumes, have also been employed to achieve low limits of detection, but are hampered by a limited dynamic range because they focus on single molecule detection.2 Moreover, these assays use manual off-chip operations to capture target molecules, introducing variance in analysis, rather than employing a reliable automated process.In efforts to overcome these limitations, Dreher and colleagues3 have recently integrated a microfluidic pneumatically actuated microwell-patterned assay for quantitative and sensitive detection of proteins. The monolithic design of the pumps combined with the actuatable assay chamber provided programmable and reproducible fluid delivery and mixing. The assay by Zeng et al. consisted of a poly(dimethylsiloxane) (PDMS) chip with three-valve pumps coordinating fluid flow to four parallel channels, which house an array of femtoliter microwells within the assay chamber (Fig. 1). The pneumatic control of the assay chamber reduced the volume of detection reagent, which helped improve the sensitivity and the speed of ELISA. The reduction in volume allowed the fluorescently tagged secondary antibody to more rapidly attach to the target protein. Furthermore, the closed well also effectively increased the relative concentration of the target molecules.
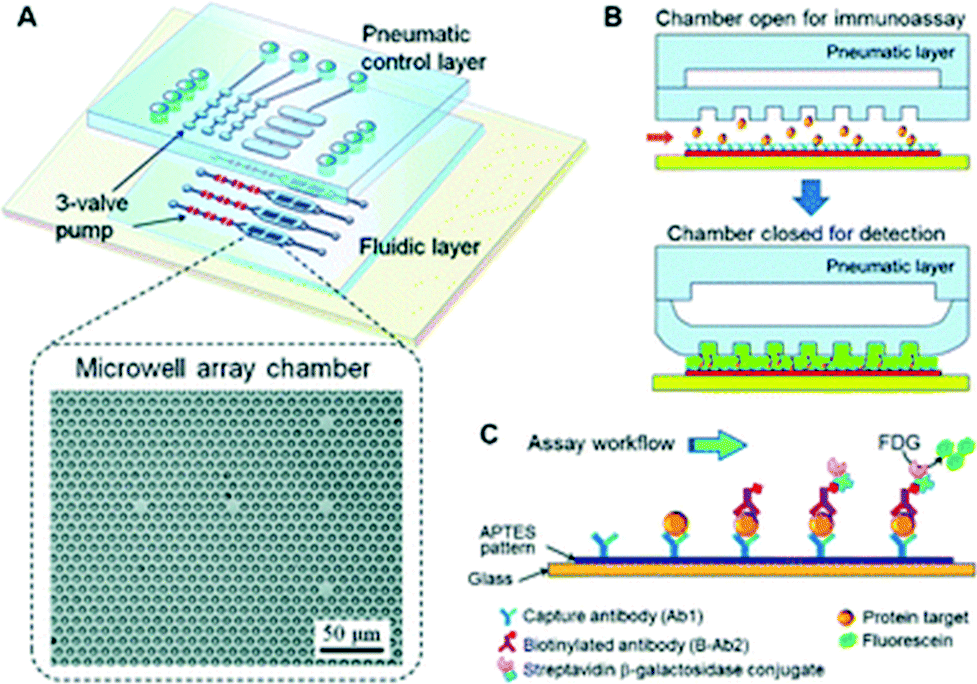 | ||
| Fig. 1 Pneumatically actuated microfluidic ELISA. (A) Schematic of the three-layer device with a pneumatic control layer and fluidic assay chamber. (B) The state of the pneumatic actuator during target protein capture (top) and during chemifluorescent detection (bottom). (C) Magnified view of the immunoassay occurring in each microwell. Figure reprinted with permission from Zeng et al.3 | ||
A streptavidin–biotin assay was first employed to assess the microwell-assisted microfluidic chemifluorescence detection capabilities with biotinylated β-galactosidase (BβG). Line scans across the microwells of the device showed constant fluorescence intensity over a five-fold concentration range. The authors also demonstrated that a uniform and reproducible thin layer of liquid remained between the membrane and the glass substrate, which was essential for quantitative detection and reproducibility by removing the variations in measurements from well to well. The microwell structure increased detection sensitivity by more than two-fold due to the gain in optical depth and also offered higher substrate capacity for chemifluorescent detection. A four step pumping sequence was used to ensure full closure and both the pulse time between cycles and the closing pressure was optimized to maximize the signal over the shortest timescale. Subsequently, the model biomarker insulin-like growth factor 1 receptor (IGF-1R) was employed with the assay because of its importance in IGF-1 signaling pathways and its implication in various cancers. When compared to traditional ELISA, the amount of reagents used and the time required was reduced by an order of magnitude for the microfluidic assay. IGF-1R was quantitatively detected from 10 fg mL−1 to 1 ng mL−1 with a limit of detection of 3.5 fg mL−1 (21.8 aM).
The authors demonstrated that their actuatable microwell patterned microfluidic chip was capable of performing ELISA assays with smaller volumes with a low limit of detection. While the limit of detection was comparable to other biosensors, the integrated pneumatic actuator combined with the femtoliter microwells provided a new platform capable of trace analysis of proteins over a wide dynamic range. The possibility for expanding the automated platform for an individual microwell and single molecule detection will be useful for quantitative study of low-level biomarkers. This device could potentially be used to investigate early disease diagnosis, drug efficacy, and aid in the advancement in the field of proteomics.
Highly tunable plasmofluidic lens
The field of plasmonics has exploded as a research area in recent years, yielding advances in super-resolution imaging, plasmonic circuitry, energy harvesting, biochemical sensing, and metamaterials due to the unique properties of surface plasmon polaritons (SPPs).4–6 Despite their ability to confine light in dimensions smaller than the diffraction limit and achieve high degrees of field enhancement, plasmonic devices fabricated on metals and solid state dielectrics have been limited by their lack of tunability and reconfigurability. In efforts to circumvent these limitations, researchers have sought to combine the properties of SPPs with optofluidics, itself an amalgamation of optics and microfluidics. Optofluidics allows for readily tunable and reconfigurable plasmonic devices due to the ease with which optical properties can be altered by precisely manipulating flow rates at low volumes.Recently, Huang and his group7 have exploited the combination of plasmonics and optofluidics to generate a hybrid plasmofluidic platform. The simple platform employs two lasers and thin gold films topped by microfluidic chambers to generate bubbles on the film surfaces. These vapour bubbles, created by a blue-violet laser, function as completely tunable plasmofluidic lenses. The lenses can subsequently alter the propagation of an incident SPP beam generated by a short IR laser pulse on the gold surface. In this scheme, the gold film serves two purposes: (1) to act as a heater for bubble formation, and (2) as a substrate capable of supporting the SPPs on its surface. The system is monitored carefully as the presence of a bubble alters the propagation of the SPPs on the metal surface.
Guided by theoretical simulations, the authors first interrogated their system with gold films having straight grating structures. Initially, they determined that the generated bubble diameter and its location remained constant on the film surface given steady laser power and system equilibrium. This finding illustrates the stability of the lens and the ease of the SPP beam modulation available simply by moving the laser spot on the film surface and/or by the changing the laser power. Therefore, in turn, the polariton beam could be easily manipulated and modulated on the platform. Using the straight grating, incident SPPs appeared to be plane wave-like, refracting near the boundaries of the bubble and diverging as they passed through the lens.
Arc-shaped (concave) gratings on gold films were also employed to examine SPP propagation. With the concave gratings, the incident polaritons were found to be focused as they passed through the bubble lens. Yet, if the beam passed just outside the bubble, the lens was able to collimate the SPP beam. These results further demonstrate that changing focal point can yield enhanced tuning of the bubble location and size. Subsequently, they also determined that the SPPs were able to propagate 15 μm farther when collimated as opposed to those only travelling at the water–gold interface. The intensity of these beams away from their initial point remained constant despite their traveling distance (Fig. 2).
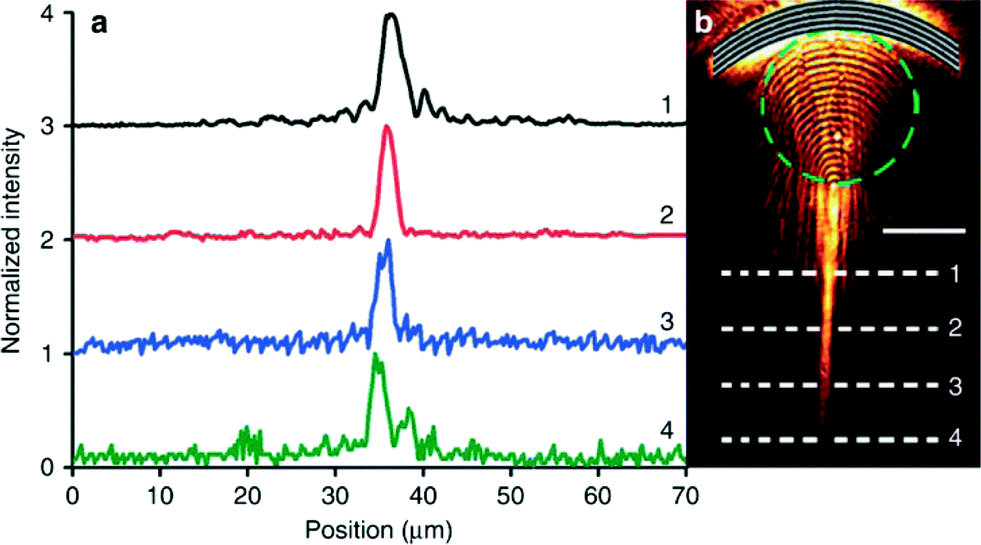 | ||
| Fig. 2 SPP beam intensity following collimation by the plasmofluidic lens. (a) Intensity profiles for line scans across the polariton beam. The numbers correlate to the scans in (b). (b) Microscopy image of collimated SPPs after passing through a surface bubble (represented by the dashed green circle). Figure reprinted by permission from Macmillan Publishers Ltd: Zhao et al.,7 copyright 2013. | ||
In their paper, Zhao et al. have seamlessly integrated the fields of plasmonics and optofluidics to generate an innovative plasmofluidic lens. This novel work not only allows for the creation of a tunable plasmonic lens capable of long-range SPP beam propagation, but also allows for flexible control of beam divergence, collimation, and focusing on a single device. The authors allude to the potential to further tune the lenses through the generation of different bubble shapes and the inclusion of solutions with different refractive indices. This increased tunability greatly enhances the utility of plasmonic devices for applications in microscopy, biosensing, energy, and materials. Furthermore, this platform serves as a foundation for subsequent studies in the emerging field of plasmofluidics.
Microfluidic membrane-assisted co-culture system
Human and mouse embryonic stem cells (hESCs and mESCs, respectively) are successfully derived from the inner cell mass of blastocyst stage embryos and show the capability to generate cell types from all three germ layers in vitro and in vivo. These properties make hESCs potentially unlimited source cells useful for regenerative medicine as well as drug discovery and toxicology tests. They can be harvested for over 250 population doublings (about one year) preserving their karyotype and phenotype and are typically co-cultured with a feeder layer, i.e., a cellular layer from primary mouse embryonic fibroblasts (mEFs) able to properly stimulate/interact with the ESCs and preserve their self-renewal properties and undifferentiated state.8 In the absence of feeder fibroblasts, ESCs spontaneously differentiate into cell types from different organs.To obtain a pure stem cell population, fibroblast layers are rendered mitotically inactive by physical (γ-irradiation) or chemical (mytomycin C) treatments. These procedures present several important challenges: (a) they are time consuming and expensive, (b) residual products can remain after treatment or released upon mEFs apoptosis, influencing stem cell viability and differentiation, and (c) mEFs are able to survive for days after the inactivation, making a purification step necessary for subsequent experiments with ESCs.
Few strategies have been developed to overcome these issues. For example, the addition of leukemia inhibition factor (LIF) to the growth medium does have feeder layer-like effects on mESCs (but not on hESCs).9 hESCs cultured on Matrigel or laminin substrates can also maintain pluripotency properties when medium conditioned by mEF is used, showing similar results to hESCs cultured on feeders.10 Recently, Lin and colleagues11 proposed a novel microfabricated approach to ensure cell purity using a feeder layer physically separated from the mESCs (Fig. 3). A 10 μm thick PDMS porous membrane, having through-holes (φ = 11.2 μm), was created by photolithography and replica molding, pouring a PDMS prepolymer mixture on a wafer having 40 μm-tall SU-8 microposts and peeling it off upon curing. This membrane separated two microchannels, conferring an ability to grow two different cell lines (mESCs and mEFs) on two separate compartments. In the approach taken by Chen et al., the membrane acted as a physical barrier to the cells preventing their mixing and preserving an efficient exchange of gas, small molecules and cytokines between the two compartments. This kept the mESCs at an undifferentiated state comparable to conventional co-culture methods, as demonstrated by the expression of Nanog, octamer binding protein 4 (Oct-4) and alkaline phosphatase (ALP) after 5 days of culture. In addition, this microfluidic chip facilitated the recovery of pure single cell lines (up to 89% for mESCs) by direct trypsin digestion in the microchannels.
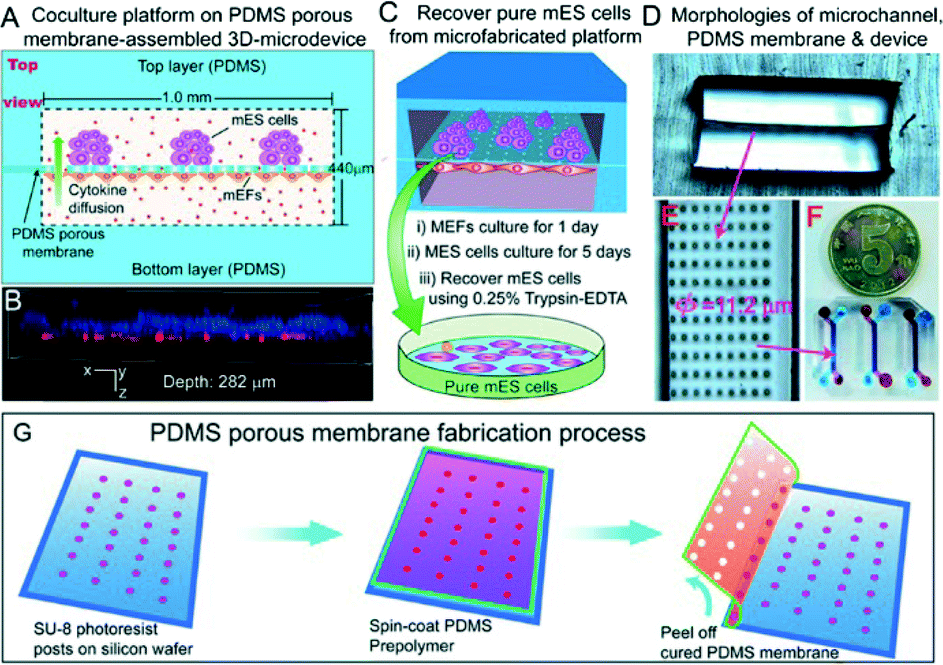 | ||
| Fig. 3 Microfluidic co-culture device. (a) Top view of the device. (b) Confocal image showing the stem cell layer (stained with Hoechst 33342) present on the top microchannel, and the feeder layer (red fluorescent protein-expressing mEFs) adhered on the bottom side of the membrane. (c) Schematic of the mESCs' recovery procedure. (d–f) Images of microchannels, PDMS porous membrane and the microfabricated device. (g) Fabrication steps of the PDMS membrane involving photolithography and replica molding. Figure reprinted by permission from Macmillan Publishers Ltd: Chen et al.,11 copyright 2013. | ||
Although more tests are necessary to assess its long-term potential use, the proposed approach presents several advantages: (1) mEFs can grow and remain viable for several days without going through apoptosis because no inactivation treatment is used, (2) downstream experiments can be performed without any further purification or separation step, reducing reagent consumption, (3) reduction of time was achieved for unnecessary preliminary (i.e., inactivation of the feeder layer) and post-culture (i.e., purification) steps, and (4) it is amenable to high throughput studies and automation through a more complex microfluidic design. Moreover, the long-term genetic stability of ESCs is guaranteed by the use of mitotically active mEFs, making this procedure a promising method for stem cells studies as well as tissue engineering applications.
Notes and references
- M. Adler, R. Wacker and C. M. Niemeyer, Analyst, 2008, 133, 702–718 RSC.
- D. M. Rissin, D. R. Fournier, T. Piech, C. W. Kan, T. G. Campbell, L. Song, L. Chang, A. J. Rivnak, P. P. Patel, G. K. Provuncher, E. P. Ferrell, S. C. Howes, B. A. Pink, K. A. Minnehan, D. H. Wilson and D. C. Duffy, Anal. Chem., 2011, 83, 2279–2285 CrossRef CAS PubMed.
- T. Wang, M. Zhang, D. D. Dreher and Y. Zeng, Lab Chip, 2013, 13, 4190–4197 RSC.
- J. A. Schuller, E. S. Barnard, W. Cai, Y. C. Jun, J. S. White and M. L. Brongersma, Nat. Mater., 2010, 9, 193–204 CrossRef CAS PubMed.
- F. Wei and Z. Liu, Nano Lett., 2010, 10, 2531–2536 CrossRef CAS PubMed.
- K. F. MacDonald and N. I. Zheludev, Laser Photonics Rev., 2010, 4, 562–567 CrossRef CAS.
- C. Zhao, Y. Liu, Y. Zhao, N. Fang and T. J. Huang, Nat. Commun., 2013, 4, 2305 Search PubMed.
- M. Amit, M. K. Carpenter, M. S. Inokuma, C. P. Chiu, C. P. Harris, M. A. Waknitz, J. Itskovitz-Eldor and J. A. Thomson, Dev. Biol., 2000, 227, 271–278 CrossRef CAS PubMed.
- B. E. Reubinoff, M. F. Pera, C. Y. Fong, A. Trounson and A. Bongso, Nat. Biotechnol., 2000, 18, 399–404 CrossRef CAS PubMed.
- C. Xu, M. S. Inokuma, J. Denham, K. Golds, P. Kundu, J. D. Gold and M. K. Carpenter, Nat. Biotechnol., 2001, 19, 971–974 CrossRef CAS PubMed.
- Q. Chen, J. Wu, Q. Zhuang, X. Lin, J. Zhang and J. M. Lin, Sci. Rep., 2013, 3, 2433 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |