A simple route towards graphene oxide frameworks†
Luna
Imperiali
a,
Christian
Clasen
a,
Jan
Fransaer
b,
Christopher W.
Macosko
c and
Jan
Vermant
*a
aDepartment of Chemical Engineering, KU Leuven, University of Leuven, W. de Croylaan 46, B-3001 Leuven, Belgium. E-mail: Jan.Vermant@cit.kuleuven.be
bDepartment of Metallurgy and Materials Engineering, KU Leuven, University of Leuven, Kasteelpark Arenbergpark 44, B-3001 Leuven, Belgium
cDepartment of Chemical Engineering and Materials Science, University of Minnesota, Minneapolis, Minnesota 55455, USA
First published on 11th October 2013
Abstract
Lightweight graphene structures have promising applications, for example as separation media, as microreactor environments, for use in energy storage or in microelectronics. The assembly of graphene or its derivatives into such frameworks typically requires rather elaborate and expensive methods which are moreover difficult to scale up. In the present work we propose a simpler route which is based on arresting the temperature induced phase separation in a water–lutidine mixture containing graphene oxide particles. The 2D nature of graphene oxide sheets, combined with an adequate surface chemistry simply controlled by their carbon to oxygen ratio, causes them to collect and assemble at the interfaces and create highly elastic layers which arrest the phase separation process. Soft solid graphene oxide frameworks are created which have sufficient mechanical stability to allow removal of the solvents by freeze drying. The advantage of the method is that the structure and mesh size of the frameworks can be easily controlled, providing direct access to the graphene-oxide surfaces while creating a mechanically robust and percolating network of graphene oxide tiles. This could be used, for example, as electrodes for supercapacitors or fuel cells.
Graphene and its derivatives such as graphene oxide have been identified as a class of materials with great potential in a variety of applications.1 Its 2D single-layer sheet structure and the resulting unique physicochemical properties make it a promising material for applications in areas such as photovoltaics, biosensors, supercapacitors, super adsorbents and fuel cells.2 There are different methods to produce graphene, for example by mechanical cleavage or chemical exfoliation.3,4 Amongst the derivatives of graphene, graphene oxide is a material which can be produced at low-cost and has the potential for large scale production, as its manufacturing is even simpler than pristine graphene. Graphene oxide also possesses the advantage of having many surface reactive oxygen groups making it a useful growth medium for creating all kinds of composite materials.5,6 Moreover, by tailoring the carbon to oxygen ratio, the wetting properties can be tuned and GO sheets are able to absorb strongly to a variety of liquid–liquid interfaces, assembling into percolated networks of nanometer thickness,7–9 resulting in interfacial assemblies which can have exceptional mechanical properties.10
Recently, some reports have appeared which focus on creating graphene and graphene-oxide frameworks.11,12 Three-dimensional graphene networks have been grown by methods such as hydrothermal gel synthesis, operating at temperatures of 1000 °C and more12,13 or by template-directed chemical vapor deposition, which requires often rather harsh environments.11 Whereas ultralight structures with exceptional properties can be obtained in this manner, the preparation methods are laborious and control over the microstructures is not straightforward. In the present work we present a simpler route to creating 3D graphene oxide frameworks, based on the arrest of spinodal decomposition of a critical mixture by the graphene oxide particles. The graphene oxide makes it easy to create so-called bi-continuous emulsions stabilized solely by colloidal particles. These ‘bijels’ were first created in large scale simulations,14 where it was suggested that the bi-continuous interface which forms through a binary liquid demixing via spinodal decomposition could be arrested using spherical particles that are neutrally wetted by both liquids. The experimental realisation hereof was not without difficulty as even a small degree of unequal wettability of the particles leads to ill-characterized, lumpy interfacial layers. Carefully chosen systems were required with carefully tuned surface chemistry.15 In the case of spherical particles the arrest is attributed to a colloidal jamming at interfaces.14 More recent work showed the role of interparticle interactions at the interface in dictating bijel stability,16 where the best stability is achieved when particles show strong attraction. This suggests that not only a jamming of the particles is required, but a more delicate control using tailored interfacial rheological properties may be possible.
Complex 3D structures can hence be created by assembly at interfaces, provided the particles are able to effectively sequester to interfaces and assemble into strong interfacial layers. Graphene oxide sheets form ideal candidates for interface templated assembly.7,10 Thanks to their sheet-like nature and wetting properties, GO sheets are able to absorb strongly to liquid interfaces, assembling into percolated networks of nanometer thickness.8 Recently, we have investigated the rheology of GO interfacial assemblies showing that these networks behave essentially as a viscoelastic solid, characterised by high moduli (both in shear and compression) and by the presence of a yield stress.10 In the present work we will exploit these properties.
When water and 2,6-lutidine are mixed at room temperature they form a homogenous liquid with a lower critical solution temperature (LCST) of about 34 °C.19,20 Water dispersible GO sheets were produced by a modified Hummers method,21 with a carbon to oxygen ratio close to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The sheets are one atomic layer thick and have an average lateral dimension of 1 μm as characterized in ref. 22 and 23. A uniform suspension of GO in water–lutidine was obtained which undergoes phase separation upon heating. Optical observations showed that the GO sheets do not significantly alter the phase diagram, which confirms their close to neutral wetting behavior (see Methods). Fig. 1 shows the microstructures which are obtained when an initially homogeneous mixture is heated into the two-phase region, and at varying compositions. The top row images were obtained when water–lutidine mixtures containing 0.8 wt% GO are heated from room temperature to 40 °C. The image on the left is obtained for a volume fraction of lutidine of vL = 0.19, the image on the right was obtained at vL = 0.55. Both compositions yielded droplet-matrix morphologies, and the growth of the droplet size was arrested by the GO sheets after about 1 minute and stable emulsions were obtained. Interestingly, the GO sheets used here were able to stabilize both lutidine-rich droplets in a water-rich matrix as well as the inverse systems. The droplet sizes for the case when the water-rich phase is the matrix yielded smaller droplets (of around 10 μm), suggesting that the GO sheets have a slightly higher affinity for the aqueous phase.24
:1. The sheets are one atomic layer thick and have an average lateral dimension of 1 μm as characterized in ref. 22 and 23. A uniform suspension of GO in water–lutidine was obtained which undergoes phase separation upon heating. Optical observations showed that the GO sheets do not significantly alter the phase diagram, which confirms their close to neutral wetting behavior (see Methods). Fig. 1 shows the microstructures which are obtained when an initially homogeneous mixture is heated into the two-phase region, and at varying compositions. The top row images were obtained when water–lutidine mixtures containing 0.8 wt% GO are heated from room temperature to 40 °C. The image on the left is obtained for a volume fraction of lutidine of vL = 0.19, the image on the right was obtained at vL = 0.55. Both compositions yielded droplet-matrix morphologies, and the growth of the droplet size was arrested by the GO sheets after about 1 minute and stable emulsions were obtained. Interestingly, the GO sheets used here were able to stabilize both lutidine-rich droplets in a water-rich matrix as well as the inverse systems. The droplet sizes for the case when the water-rich phase is the matrix yielded smaller droplets (of around 10 μm), suggesting that the GO sheets have a slightly higher affinity for the aqueous phase.24
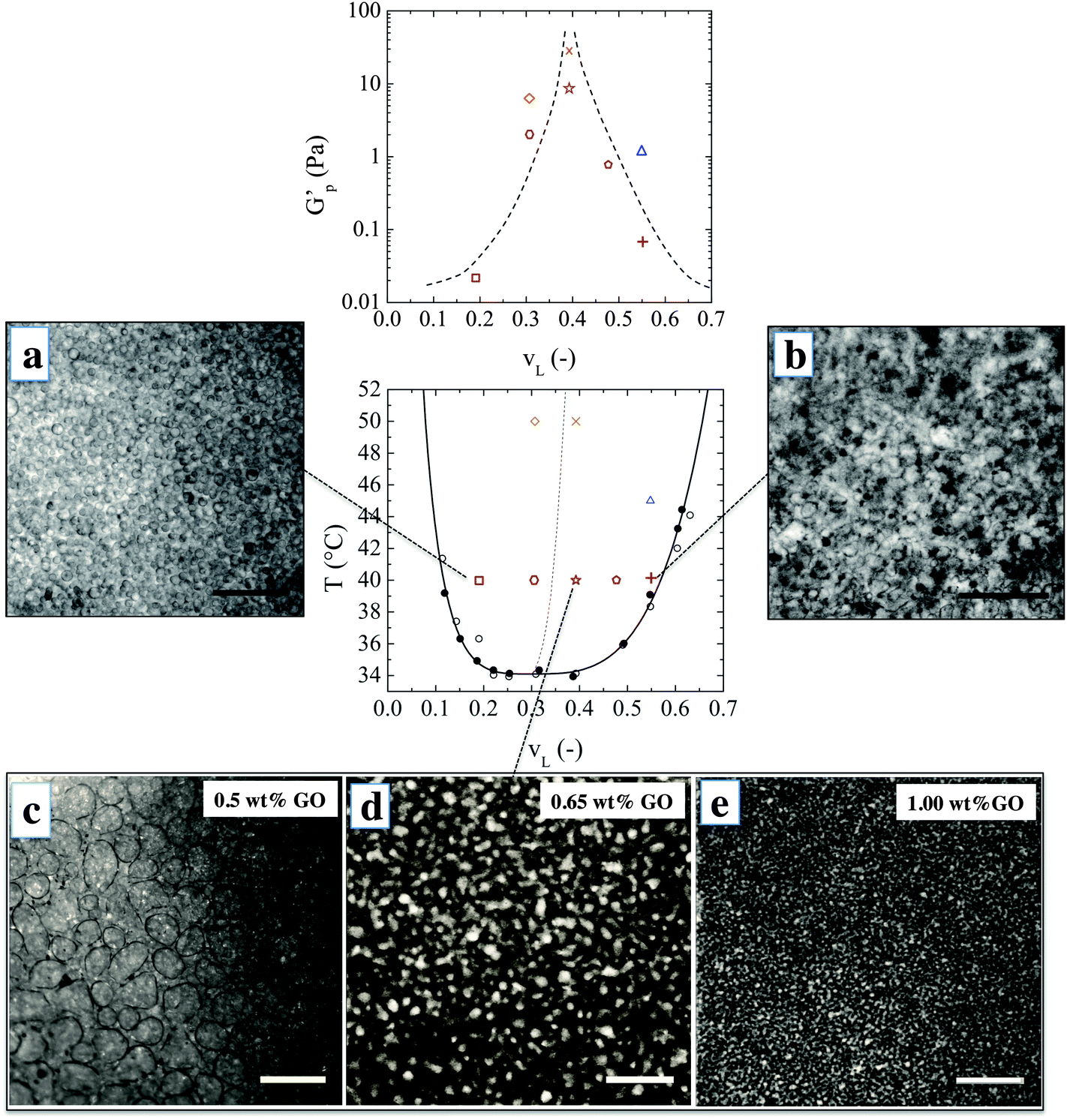 | ||
| Fig. 1 Phase behaviour and mechanical properties: water–lutidine mixtures with GO sheets at different lutidine volume fractions vL, GO concentration and temperature. (a and b) Optical micrographs of droplet matrix structures obtained at 0.8 wt% GO, T = 40 °C and at vL indicated on the phase diagram. Scale bar: 100 μm. (c–e) Microscopic observations of the arrested spinodal decomposition for mixtures at vL = 0.39 and different GO concentrations (as indicated). Scale bar: 300 μm. The phase diagram is shown in the middle. The full black line is the binodal derived from optical observation of mixtures without GO (•) and with 0.8 wt% GO (∘). The dotted line is the geometric locus where the volume ratio of the coexisting phases is 50/50 and the maximum degree of co-continuity is expected. The bulk plateau storage modulus G′p is plotted on the upper graph for mixtures with a fixed GO content of 0.8 wt%, at vL and temperatures indicated on the phase diagram (the same legend). The maximum values of G′p are measured for the co-continuous morphologies (the line is a guide to the eye). | ||
The bottom row of Fig. 1 shows the morphologies obtained after heating vL = 0.39 to 40 °C. Volume fractions 0.3 < vL < 0.6 are expected to undergo spinodal decomposition. In this series of experiments, the concentration of GO was varied from 0.5 wt% to 1 wt% and the microstructure was imaged after the structural evolution was arrested (about 1 minute after the onset of phase separation). At the lowest GO concentration of 0.5 wt%, the particle concentration was too low to fully arrest the spinodal decomposition and the co-continuous structure coarsened into a droplet-matrix morphology within a minute from the onset of phase separation. Higher concentrations of GO yielded a co-continuous structure which was fully arrested after spinodal decomposition. This could be inferred by the observation of elongated channels and saddle-like interfaces by bright field microscopy (see ESI†).
With increasing GO concentration, the spinodal decomposition was arrested at a finer morphology. The characteristic length scale of the arrested microstructure changed by approximately one order of magnitude, starting to form a stable co-continuous structure at 0.65% by weight and a mesh size of 100 μm, down to a minimum size of 10 μm when increasing the concentration of GO to 1% by weight. This could possibly be further optimised by tuning the size and concentration of the GO sheets.
Fig. 2a shows in more detail how the phase separation is arrested and graphene oxide frameworks are formed at the interface between the water-rich and the lutidine-rich phase. The structure is arrested because the GO sheets accumulate at the interface and create a rheologically active layer. The arrest is not so much due to the jamming of the particles at the interface, as is observed for spheres,14,15 but can be attributed here to the formation of a highly elastic interface as the interfacial area is reduced and the adsorbed GO sheets percolate laterally.10 GO sheets can have a surface area of the order of 1000 m2 g−1,25 which, for the typical particle concentrations used, results in a particle surface area per mL of the suspension ∼10 m2 mL−1. This value is two orders of magnitude higher than the interfacial area A in a bi-continuous morphology with domain size ∼10 μm, with A ∼ 1/10 μm. A similar effect was observed for GO sheets that were directly deposited onto a planar air–water interface, where only 1% of the added amount effectively adsorbed onto the interface.10
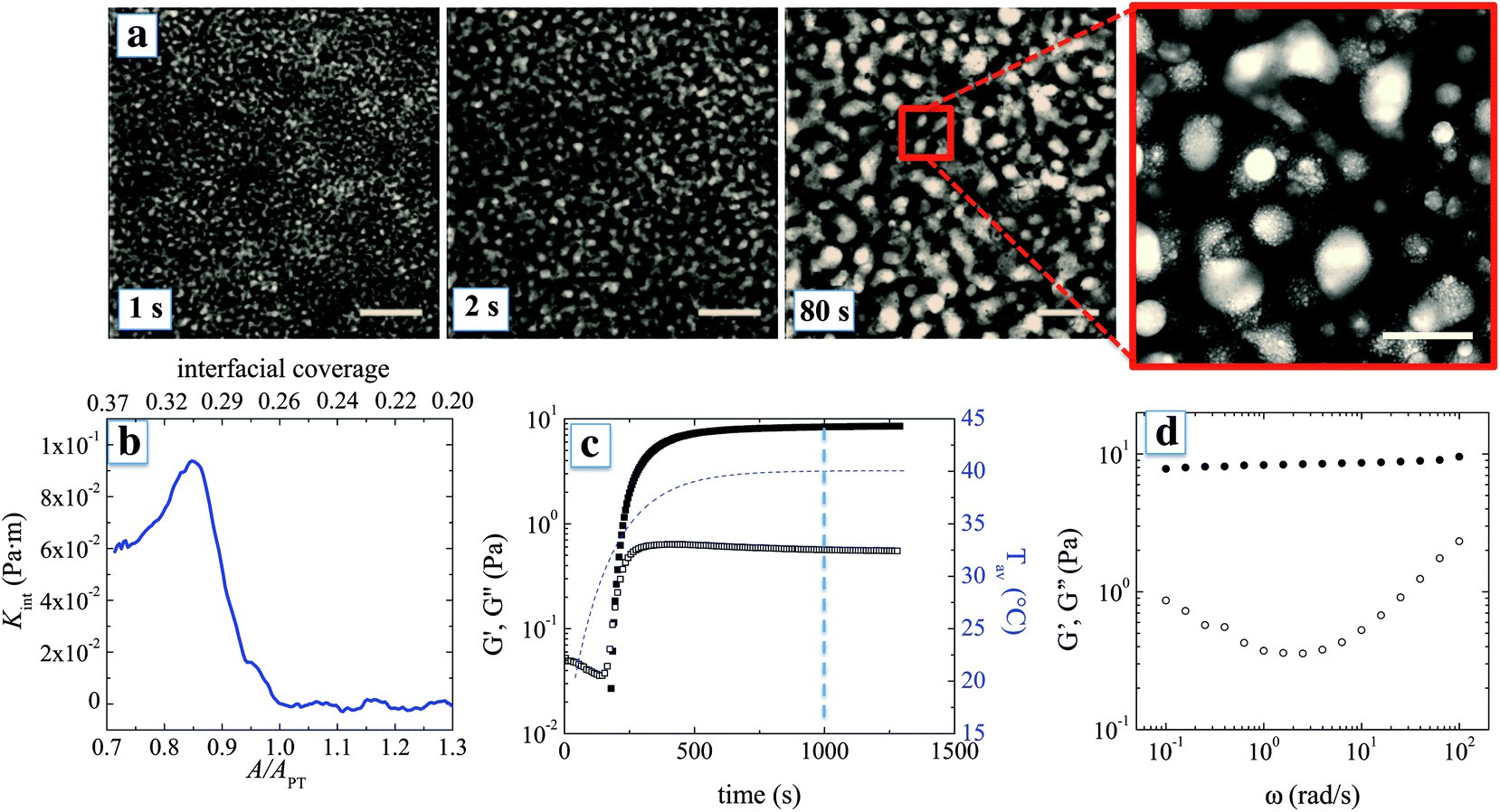 | ||
| Fig. 2 Time evolution of the graphene-oxide frameworks (a) microscopic observations of the microstructure as a function of time (scale bar: 300 μm). The inset on the right shows a zoom of the final microstructure (scale bar: 100 μm). (b) The compressional modulus Kint of a planar air–water interface covered with GO sheets as a function of reduction in interfacial area A/APT and interfacial coverage, with APT the interfacial area at percolation threshold. (c) Time evolution of the bulk dynamic moduli as the temperature is raised within the 2-phase region (•: elastic modulus G′, ∘: loss modulus G′′, and dotted line: average temperature) at a frequency of 10 rad s−1 and a strain amplitude of 0.5%. (d) Frequency response at a time of 1000 s from the start up of heating for a strain amplitude of 0.5%. | ||
During the phase separation, the interfacial coverage can be expected to increase for two reasons. First, the adsorption of sheets and second, at the later stages, the decrease of the total interfacial area due to coarsening. As interfacial coverage increases, the sheets percolate laterally and the layer compresses until the interface builds up a compressional stress that is able to arrest phase separation. This interfacial compressional stress can be quantified by interfacial rheology. Direct interfacial rheological measurements at the interface between the water-rich and the lutidine-rich phase were not possible, mainly due to problems with strong evaporation. However, data are available for planar water–air and water–decane interfaces. Fig. 2b shows the compressive modulus Kint and the estimated interfacial coverage of a planar interface covered with GO sheets as the interfacial area A is reduced beyond APT, the area at which sheets percolate laterally (Kint re-calculated from ref. 10, see ESI†). As the interfacial area is reduced, the interfacial coverage increases up to percolation, beyond which the interface builds up Kint that reaches a maximum of 10−1 Pa m. This value is much higher than interfacial tension between the liquid phases, which at 40 °C is on the order of 0.1 mN m−1, or 1000 times lower. Hence during phase separation, the interfaces convectively pick up the sheets from the suspension and become mechanically robust, up to the point that further reduction of the interfacial area is hindered. For the mixtures such as the ones used here, sharp interfaces during spinodal decomposition typically form at a domain size of around 5 μm.26 Considering that, in our mixtures, the coarsening is typically arrested at a domain size between 10 and 100 μm, the interfacial area can be expected to be reduced by a factor 20 from the point where particles are picked up until separation is arrested. This should be sufficient for the adsorbed particles to reach the onset of percolation and be sufficiently compressed, possibly forming mechanically robust multilayers. As a result, Kint becomes high enough so that the coarsening of the liquid domains is effectively slowed down, with formation of a co-continuous structure of GO sheets.
The bulk rheological properties of the phase separated mixtures in the presence of GO provide proof for the creation of a percolating graphene oxide framework. Fig. 2c shows the evolution of the linear viscoelastic dynamic moduli at a frequency of 10 rad s−1 as a function of time after the temperature is changed. The lutidine–water system in the absence of particles has too low an interfacial tension to yield significant bulk elasticity. Moreover, coalescence and creaming prevent any meaningful rheological measurement. When GO sheets are present, a significant increase in both the viscous and elastic moduli is observed. After an initial period, where the viscoelastic response cannot be detected, the storage modulus rapidly increases. The frequency response of the structure after about 10 minutes is consistent with a soft solid, with a plateau modulus of about 10 Pa and an overall signature of soft glassy behaviour.27 Considering that water and lutidine are Newtonian fluids and do not contribute to the elasticity of the arrested morphology, the mechanical response can be attributed to the percolating graphene oxide frameworks, with their high elastic surface moduli.
A scaling argument can be used to link the high frequency plateau elastic modulus G′p of the material to the contribution of the interfaces assuming affine deformations within the linear viscoelastic regime. For a system dominated by surface tension a simple scaling argument states that G′p ≃ γ/6d,28 where γ is the interfacial tension and d is the typical size of the co-continuous domains. However, in our case, using the interfacial tension of 0.2 mN m−1 which is the upper limit of the surface tension for the bare interface, underpredicts the modulus by an order of magnitude. Hence, interfacial rheology plays a key role in determining the mechanical response. To introduce the effect of interfacial rheology on G′p, we consider that the main contribution to the oscillatory stress is given by the compression of the interfaces while the contribution coming from interfacial shear can be neglected, in line with the approach used by Palierne29 for emulsions. Indeed in our case, Kint/Gint ∼ 10−3, where Gint is the shear modulus of a water–air interface measured at maximum packing of GO sheets.10 Therefore G′p ≃ Kint/6d. For a typical domain size d of 10 to 100 μm, we calculate that a Kint between 10−3 and 10−2 Pa m is sufficient to give 1 < G′p < 100 Pa, which is in reasonable agreement with the experimentally observed values. Therefore we argue that interfacial rheology, on top of being responsible for arresting the coarsening, is also the main contributor to the structural rigidity of the arrested morphology.
The observed bulk elasticity of the phase separated structure is predominantly a measure of the amount of the interface created by the phase separation process. Fig. 1 includes the effect of lutidine volume fraction and temperature on the plateau elastic modulus G′p as a function of vL, which can be solely attributed to the presence of GO–laden interfaces. As shown in Fig. 1, the highest moduli are achieved for vL where the structure is co-continuous. For a given vL, at higher temperatures higher elastic moduli are achieved, suggesting a finer morphology.
The graphene oxide frameworks, embedded in the water–lutidine matrix, are not directly useful. However, because of the mechanical strength of the GO framework16 and the possibility for the GO sheets to undergo hydrogen bonding, as has been observed when making GO paper,17,18 we used freeze drying of the co-continuous structures. The subsequent GO foam is coherent and strong enough to be sliced for further analysis. The SEM pictures of a cross-section of the foam (Fig. 3) show porosity with a variable diameter from 10 to 100 μm, comparable to the size of the liquid domains in the co-continuous structure. Interestingly, in the SEM pictures it is possible to distinguish thin graphene-oxide assemblies constituting the structure. This important evidence indicates that the GO sheets remain well dispersed also upon phase separation of the liquids, allowing to achieve highly porous structures. The density of the GO foam is on the order of 10 mg cm−3, comparable to polymeric foams, which is higher compared to the frameworks prepared using a hydrothermal method or CVD synthesis,11–13 but using a relatively simple water based process. The possibility to isolate the obtained GO microstructures from the liquids opens up a whole new range of possible applications. For example, a reduction step30 allows us to readily tune the electrical properties of the foam to obtain extremely efficient ultra-capacitors.31,32
 | ||
| Fig. 3 Graphene oxide foams: graphene oxide foam and SEM images of a cross-section (b, scale bar: 100 μm). (c) shows a magnified view of (b), where thin GO sheets are visible; scale bar: 5 μm. | ||
In conclusion, lightweight graphene structures have been produced using a simple route, arresting the temperature-induced phase separation in a water–lutidine mixture containing graphene oxide particles. Similar to bijels, the GO particles collect at the interface and create highly elastic layers which arrest the phase separation process. Different from bijels, the arrest arises from the development of an elastic layer with specific interfacial rheological properties, rather than a jamming transition. The subsequent soft solid graphene oxide frameworks have sufficient mechanical stability, which, combined with the co-continuous nature of the scaffolds, provides the possibility to remove the solvents by freeze drying. The mesh size of the frameworks can be easily controlled by varying the temperature (history) and the concentration of GO. The method relies on the GO sheets not sliding over each other and will hence be limited to relatively low C/O ratios. The method could be easily generalized to different types of mixtures and holds promise for polymeric compounds and GO based polymer nanocomposites.
Materials
GO was obtained and characterized as described in detail by Liao et al.22 Briefly, GO was produced by a modified Hummers method21 to obtain a carbon to oxygen ratio C/O of close to 2:1. The obtained powder was repeatedly washed with deionized water and diethyl ether and left damp to prevent restacking of the sheets. The powder was analyzed by X-ray photoelectron spectroscopy and showed no detectable amounts of the chemicals used in the Hummers procedure. The damp GO powders were dispersed in deionized water (pH 7, conductivity = 0.55 μS cm−1) that was added to achieve a desired particle concentration. The suspension was sonicated (150 W) for about 1 hour at room temperature in an ultrasonic bath (Syskin–Elmasonic) and yielded exfoliated graphene oxide sheets as a brown aqueous solution at pH 3. The sheets in the suspension are mostly monolayers and are polydisperse in lateral size, ranging from 200 nm to 5 μm as measured by atomic force microscopy.23
For the water–lutidine mixtures, an aqueous solution of GO sheets was prepared (using water at pH 7 and conductivity = 0.55 μS cm−1) and then 2,6-lutidine (Sigma Aldrich) was carefully added.
Methods
Determination of the phase diagram
To determine the coexistence curve of the liquid mixtures, a Linkam cell (Linkam Scientific Instruments - CSS450) was used in combination with a transmission microscope (Leitz Laborlux 12) connected to a digital camera (Hamamatsu C8800). The temperature was measured with an accuracy of 0.01 °C and raised from 30 °C at 1 °C min−1 while the mixture within the cell was imaged. When the mixture reaches the point of phase separation, it becomes cloudy: this point is referred to as the cloud point. The cloud point temperature was selected as the temperature at which the light transmission through the sample was reduced by 40%. For the mixtures loaded with particles, graphene oxide with a concentration of 8 mg mL−1 was used. In this case, phase separation occurs much slower than for pure mixtures. To make sure that our detection of the cloud point was not affected by the finite heating rate, the mixtures were first heated up to the cloud point and then cooled down again at a rate of 1 °C min−1 to detect the temperature at which the light transmission of the homogeneous mixture is restored. No systematic deviation of the cloud and de-cloud point was observed. Within the accuracy of our measurement techniques, we observed no difference between the transition temperature of pure mixtures (indicated by full symbols on the phase diagram in Fig. 1) and mixtures loaded with GO sheets (empty symbols).Optical microscopy
The liquid mixtures were placed in a rectangular glass vial (Starna Scientific) with a volume of 0.4 mL and an optical path length of 1 mm, sealed by a polytetrafluoroethylene (PTFE) stopper. The mixture is heated within the two-phase region by inserting it in a pre-heated aluminum block. The aluminum block measures 4 cm × 7 cm × 3 cm and possesses 2 heating units and a thermometer embedded in the metal body. These are connected to a temperature controller (KSV Instruments Ltd). The vial containing the sample is inserted in the heating block. A window through the block allows to access the glass vial by optical microscopy. In this way, the water–lutidine mixtures can be heated from 20 °C to 40 °C at a heating rate of 10 °C min−1.Rheology
Rheological measurements were performed using a stress-controller rheometer (Physica MCR 501, Anton Paar). A double wall Couette geometry was used with a double lock solvent trap. The temperature was controlled by a Peltier module that surrounds the cup of the Couette and heats it up from the bottom and the outside wall. The thermal inertia of the geometry cannot be neglected when performing experiments with fast temperature changes. Moreover, the Peltier system reads the temperature at the outer wall of the cup, hence its reading can be far from the real temperature of the sample. For these reasons, and since the measurements we intend to perform are extremely sensitive on temperature, preliminary tests were carried out to measure the temperature inside the geometry during heating. A silicone oil with a heat conductivity close to that of 2,6-lutidine (0.1 W m−1 K−1) was heated from 20 to 40 °C and subjected to a shear stress. From the measured viscosity η of the oil, which is a known function of temperature, the average temperature of the sample was derived. We propose that the sample can be heated from 20 °C to 40 °C at an average rate of 2 °C min−1. To perform the rheological characterization of water–lutidine mixtures, the sample was first cooled to 20 °C and sheared for 1 minute at a shear rate of 10 s to erase any mechanical and thermal history. The sample was then heated to the desired temperature. For frequency sweep experiments, it was checked that the applied strain was within the linear viscoelastic regime. To check for reproducibility, the mixture was cooled back to 20 °C while sheared at a shear rate of 100 s−1 for 10 minutes, after which the mixture was again heated up to 40 °C and the rheological tests repeated.Freeze drying
Liquid mixtures at critical composition vL = vL,c = 0.31 containing 0.8 wt% GO were inserted in a round glass cuvette of 1 cm diameter and heated up within the 2-phase region by directly submerging them in a water bath at 40 °C. After 2 minutes, the vials were removed from the bath, quickly frozen in liquid nitrogen and readily placed in a vacuum chamber at a temperature of 20 °C and a pressure of 0.1 mbar for 36 hours. Upon drying, GO foams were obtained. To image the inner structure by scanning electron microscopy (SEM), a section of the GO foam was sliced, glued to a metal stub and sputter-coated with gold. The samples were imaged using a SEM (FEI XL30 FEG) with an acceleration voltage of 10 kV.Acknowledgements
The European Union – FP7-project Nanodirect (Grant no. CP-FP213948-2) and the FWO Vlaanderen – projects G.0554.10 and G.0697.11 are gratefully acknowledged for their financial support. The research was performed as part of the IAP programme MICROMAST financed by BELSPO and financial support of the Hercules foundation is acknowledged.References
- A. K. Geim and K. S. Novoselov, The rise of graphene, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, A roadmap for graphene, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- J. N. Coleman, et al. Two-dimensional nanosheets produced by liquid exfoliation of layered materials, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Chemical methods for the production of graphenes, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Processable aqueous dispersions of graphene nanosheets, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- G. Eda, G. Fanchini and M. Chhowalla, Large-area ultrathin films of reduced graphene oxide as a transparent and flexible electronic material, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- L. J. Cote, F. Kim and J. Huang, Langmuir-Blodgett assembly of graphite oxide single layers, J. Am. Chem. Soc., 2009, 131, 1043–1049 CrossRef CAS PubMed.
- F. Kim, L. J. Cote and J. Huang, Graphene oxide: surface activity and two-dimensional assembly, Adv. Mater., 2010, 22, 1954–1958 CrossRef CAS PubMed.
- L. J. Cote, J. Kim, Z. Zhang, C. Sun and J. Huang, Tunable assembly of graphene oxide surfactant sheets: wrinkles, overlaps and impacts on thin film properties, Soft Matter, 2010, 6, 6096–6101 RSC.
- L. Imperiali, K.-H. Liao, C. Clasen, J. Fransaer, C. W. Macosko and J. Vermant, Interfacial rheology and structure of tiled graphene oxide sheets, Langmuir, 2012, 28(21), 7990–8000 CrossRef CAS PubMed.
- Z. Z. Chen, W. W. Ren, L. L. Gao, B. B. Liu, S. S. Pei and H.-M. H. Cheng, Three-dimensional flexible and conductive interconnected graphene networks grown by chemical vapour deposition, Nat. Mater., 2011, 10(6), 424–428 CrossRef CAS PubMed.
- Y. Y. Zhao, C. C. Hu, Y. Y. Hu, H. H. Cheng, G. G. Shi and L. L. Qu, A versatile, ultralight, nitrogen-doped graphene framework, Angew. Chem., Int. Ed., 2012, 51(45), 11371–11375 CrossRef CAS PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, Self-assembled graphene hydrogel via a one-step hydrothermal process, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- K. Stratford, R. Adhikari, I. Pagonabarraga, J.-C. Desplat and M. E. Cates, Colloidal jamming at interfaces: a route to fluid bicontinuous gels, Science, 2005, 309, 2198–2201 CrossRef CAS PubMed.
- E. M. Herzig, K. A. White, A. B. Schofield, W. C. K. Poon and P. S. Clegg, Bicontinuous emulsions stabilized solely by colloidal particles, Nat. Mater., 2007, 6, 966–971 CrossRef CAS PubMed.
- M. N. Lee, J. H. J. Thijssen, J. A. Witt, P. S. Clegg and A. Mohraz, Making a robust interfacial scaffold: bijel rheology and its link to processability, Adv. Funct. Mater., 2013, 23, 417–423 CrossRef CAS.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Preparation and characterization of graphene oxide paper, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- N. V. Medhekar, A. Ramasubramaniam, R. S. Ruoff and V. B. Shenoy, Hydrogen Bond Networks in Graphene Oxide Composite Paper: Structure and Mechanical Properties, ACS Nano, 2010, 4, 2300–2306 CrossRef CAS PubMed.
- C. A. Grattoni, R. Dawe, C. Seah and J. Gray, Lower critical solution coexistence curve and physical properties (density, viscosity, surface tension, and interfacial tension) of 2,6-lutidine + water, J. Chem. Eng. Data, 1993, 38(4), 516–519 CrossRef CAS.
- J. D. Cox and E. F. G. Herington, The coexistence curve in liquid-liquid binary systems, Trans. Faraday Soc., 1956, 52, 926–930 RSC.
- W. S. Hummers, Jr and R. E. Offeman, Preparation of graphitic oxide, J. Am. Chem. Soc., 1958, 6, 1331–1339 Search PubMed.
- K.-H. Liao, A. Mittal, S. Bose, C. Leighton, K. A. Mkhoyan and C. W. Macosko, ACS Nano, 2011, 5, 1253–1258 CrossRef CAS PubMed.
- K.-H. Liao, Y.-S. Lin, C. W. Macosko and C. L. Haynes, Cytotoxicity of graphene oxide and graphene in human erythrocytes and skin fibroblasts, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CAS.
- B. P. Binks and S. O. Lumsdon, Influence of particle wettability on the type and stability of surfactant-free emulsions, Langmuir, 2000, 16, 8622–8631 CrossRef CAS.
- S. Park and R. S. Ruoff, Chemical methods for the production of graphenes, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- J. K. G. Dhont, An Introduction to Dynamics of Colloids, Elsevier Science, 1996 Search PubMed.
- P. Sollich, F. Lequeux, P. Hebraud and M. E. Cates, Rheology of soft glassy materials, Phys. Rev. Lett., 1997, 78, 2020–2023 CrossRef CAS.
- M. Doi and T. Ohta, Dynamics and rheology of complex interfaces, J. Chem. Phys., 1991, 95, 1242–1248 CrossRef CAS.
- J.-F. Palierne, Linear Rheology of viscoelastic emulsions with interfacial tension, Rheol. Acta, 1991, 29(3), 204–214 CrossRef.
- S. Stankovich, D. Dikin, R. Piner, K. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. Nguyen and R. S. Ruoff, Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Graphene-Based Ultracapacitors, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Laser Scribing of High-Performance and Flexible Graphene-Based Electrochemical Capacitors, Science, 2012, 335, 1326–1330 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mh00047h |
| This journal is © The Royal Society of Chemistry 2014 |