Supramolecular host–guest polymeric materials for biomedical applications
Xian Jun
Loh
*abc
aInstitute of Materials Research and Engineering (IMRE), 3 Research Link, Singapore 117602, Singapore. E-mail: lohxj@imre.a-star.edu.sg; XianJun_Loh@scholars.a-star.edu.sg
bDepartment of Materials Science and Engineering, National University of Singapore, 9 Engineering Drive 1, Singapore 117576, Singapore
cSingapore Eye Research Institute, 11 Third Hospital Avenue, Singapore 168751, Singapore
First published on 1st October 2013
Abstract
The bottom–up synthesis of highly complex functional materials from simple modular blocks is an intriguing area of research. Driven by the chemistry of supramolecular assembly, modules which self-assemble into intricate structures have been described. These hierarchically assembled systems extend beyond the individual molecule and rely on non-covalent interactions in a directed self-assembly process. The intrinsic properties of the materials can be modified by exploiting the dynamic and specific uni-directional interactions among the building. This also allows the building of novel supramolecular structures such as hydrogels, micelles and vesicles. These aqueous supramolecular networks belong to a novel category of soft biomaterials exhibiting attractive properties such as stimuli-responsiveness and self-healing properties derived from their dynamic behavior. These are important for a wide variety of emerging applications. In this review, the latest literature describing the formation of dynamic polymeric networks through host–guest complex formation will be summarised. These approaches carried out in the aqueous medium have unlocked a versatile toolbox for the design and fine-tuning of supramolecular self-assembled materials.
 Xian Jun Loh | Xian Jun Loh is currently a Research Scientist at the Institute of Materials Research and Engineering (IMRE) and an Assistant Professor at the National University of Singapore (NUS). He is also concurrently an adjunct scientist at the Singapore Eye Research Institute. He graduated with a PhD in 2009 from the National University of Singapore (NUS). He was awarded the National Science Talent Search Award in 1999, Agency for Science, Technology and Research (A*STAR) National Science Scholarship in 2002 (undergraduate studies) and A*STAR Graduate Scholarship in 2007 (postgraduate studies). He obtained a B.Appl.Sc. (1st Class Hons) in Applied Chemistry from NUS in 2006. After a one year attachment stint at IMRE, he continued his PhD studies in 2007. In 2011, he was elected into the Fellowship at Fitzwilliam College, Cambridge. He is the co-editor of an RSC-published book entitled “Polymeric and self-assembled hydrogels: from fundamental understanding to applications”. His main interests are in the design of supramolecular and stimuli-responsive polymers and hydrogels for biomedical applications. Currently, he is the author and co-author of 48 journal papers and 5 patents, publishing mainly in the area of biomaterials. |
Introduction
The rapid growth of supramolecular chemistry has led to the development of novel materials which are built based on the theory of self-assembly on the molecular level. Whitesides and Grzybowski defined self-assembly as “the autonomous organization of components into patterns or structures without human intervention”.1 The idea of building up an ordered structure from a mass of disordered individual subunits is immensely intriguing. From the building of order from disorder, to the emergence of novel properties from self-assembled structures not observed in individual subunits, there are interesting scientific questions that draw researchers to this field. The science of supramolecular chemistry is intrinsically reliant on the connecting moieties, their specific association/dissociation and the thermodynamics of the process. The bulk properties can be greatly affected by the individual components, either having an environmental response, varied mechanical properties as well stability. Self-assembled built-up materials have been extensively reported for several years; these exist in the form of vesicles, micelles and hydrogels.Vesicles are molecular containers made up of a thin membrane encasing a volume. Vesicles have attracted great research interest as they are important building blocks resembling the cellular units that make up most organisms and are useful in chemistry, biology, and materials science.2–4 They have wide applicability and can be used as drug/gene delivery systems, for the study of the function of ion channels and also for light-harvesting systems. Synthetic vesicles are typically derived from amphiphiles with different polar or hydrophilic head groups and hydrophobic tails. Polymeric micelles are yet another type of encapsulating agents that are built up from the self-assembly of amphiphilic polymers.5 Polymeric micelles have been used in a variety of applications including the purification of wastewater,6 templating agents for materials synthesis, biologically relevant nanobioreactors,7 and as phase-transfer catalysts.8 They have been widely investigated for their potential biomedical applications, particularly in drug delivery.9 Micelles, with their hydrophobic cores and hydrophilic coronas, solubilize hydrophobic compounds, encapsulating them within the micelle core. Bulkier materials such as hydrogels can also be assembled from small molecules. Hydrogels are crosslinked, three-dimensional macromolecular networks of hydrophilic copolymers. Hydrogels are widely used as wound dressings, soft contact lenses and soft tissue substitutes. The physical or chemical crosslinking of hydrogels have been described. Chemical crosslinks can take place with the incorporation of functional crosslinkers with more than two reactive groups. Physical crosslinks can be formed via hydrophobic interactions between polymer chains, host–guest complexation, metallo-mediated binding, or electrostatic approaches.
Macrocyclic host–guest complexation
Macrocyclic host–guest complexation is an interesting and widely exploited non-covalent interaction that can be used to bind two chemical entities together. A wide variety of macrocycles have been synthesized over the past three decades including crown ethers, cyclophanes, catenanes, cavitands (such as cyclodextrins, calixarenes and cucurbit[n]urils), porphyrins, cryptophanes and carcerands. The use of host–guest chemistries involving cyclodextrins, sulfonatocalixarenes, and cucurbiturils has been increasing in popularity. Part of the reason is that these macrocycles have been shown to be biocompatible.13–15 The host–guest complex formation involving such macrocycles usually takes place in aqueous media. The formation of supramolecular amphiphiles from these macrocycles is thus fundamentally interesting for biomedical applications. Supramolecular amphiphiles are constructed by non-covalent interactions or dynamic covalent bonds. These structures show a facile preparation and can be made responsive to various external stimuli.10,11 Hydrogen-bonding, charge-transfer, π–π interactions and host–guest strategies are used to build supramolecular structures.12 In this review, only the cavitands such as cyclodextrins, calixarenes and cucurbiturils will be considered. The cavitands form inclusion complexes, where a guest molecule is locked within the cavity of the host. In these cases, the ‘host’ typically has external features that interact with a solvent and internal features that foster the binding of a ‘guest’ through either a specific shape or a favourable environment. This is especially seen in cases where stronger binding occurs between a hydrophobic guest sequestered into the hydrophobic inner cavity of a host, such as cyclodextrins (α-, β- and γ-CD) or cucurbit[n]urils (CB[n], n = 5–8 and 10), in water, through favourable solvophobic interactions. For comparison, simple host molecules, such as crown ethers, have demonstrated Keq values between 102 to 104 M−1 in polar organic solvents and are highly dependent on solvent and temperature. The widely studied CD host family reports higher Keq values of up to 105 M−1 in water. The CB[n] host family is capable of using multiple, concerted, non-covalent interactions and exhibit much higher Keq values of up to 1015 M−1 in water.Cyclodextrin-based assemblies (CDs)
Cyclodextrins (CDs) are cyclic oligosaccharides made of D-glucose repeating units coupled through α-1,4-glucosidic linkages. The most commonly used CDs are α-, β-, γ-CDs containing 6, 7, and 8 D-glucose repeating units, respectively (Fig. 1). The three-dimensional molecular structure resembles a truncated cone with the primary and secondary hydroxyl groups on the narrower cone rim exposed to the solvent. The interior of the CD is less hydrophilic when compared to the aqueous media and allows hydrophobic molecules to be included within the cavity. Hydrophobic and van der Waals interactions, along with other factors such as the easing of the CD ring strain, changes in solvent–surface tensions, and hydrogen bonding with the hydroxyl groups of the CD drive the formation of the host–guest inclusion complex. The new entity demonstrates an enhanced water solubility, due to the hydrophilic exterior of the CD. The cost effectiveness and advantageous properties like the ability to form highly water soluble inclusion complexes, has seen CDs being used in different applications for analytical science, pharmaceutical technologies, separation sciences, catalysis, and food, textile and cosmetic industries.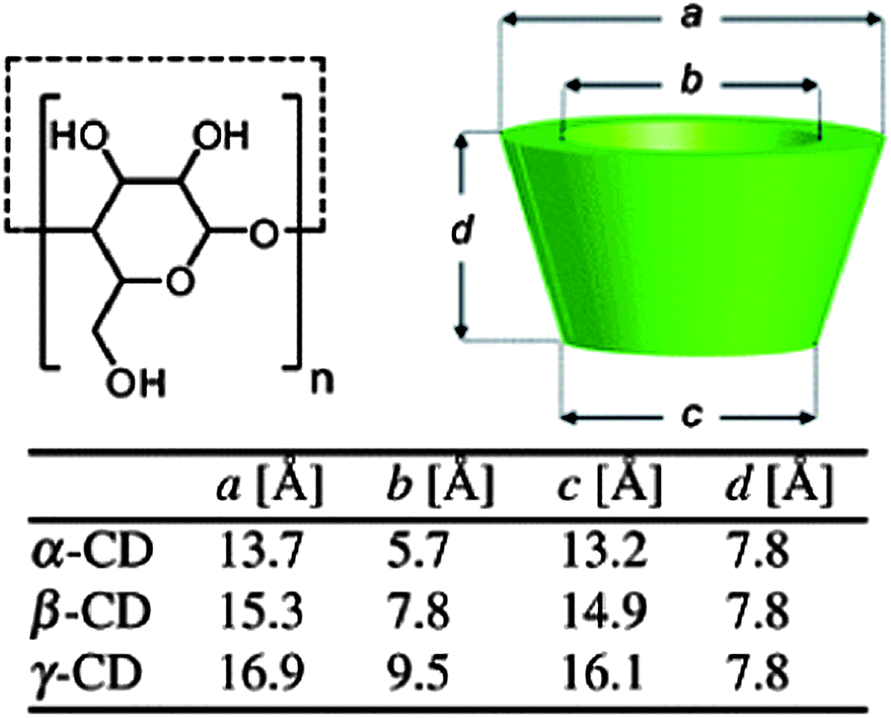 | ||
| Fig. 1 Schematic representation of the cyclodextrin (CD) family.12 Reprinted with permission from Chem. Soc. Rev., 2012, 41, 6195–6214. Copyright (2012) Royal Society of Chemistry. | ||
The inclusion complex formation ability of CDs has been used as a non-covalent binding motif for the development of a wide variety of dynamic polymeric networks and assemblies in aqueous media. These polymeric systems have been explored for biomedical applications such as the sustained and targeted release of therapeutics, biocompatible scaffolds for tissue engineering and medical diagnostics. Research in supramolecular CD polymers and associated self-assembled structures has been broadly developed since the 1990s and has been reviewed in several reports.16–20 The following section will cover the most recent developments in utilizing CDs for the fabrication of vesicles, micelles and self-healing materials, as well as photo-responsive hydrogels.
Capsule formation with cyclodextrins
A novel supramolecular Janus hyperbranched polymer (JHBP) was recently reported and the photo-controlled assembly into vesicles was studied.21 A hydrophilic hyperbranched polyglycerol with a β-cyclodextrin core (CD-g-HPG) was first prepared. The supramolecular JHBP was prepared by the complexation between the CD-g-HPG and hyperbranched poly(3-ethyl-3-oxetanemethanol) with an azobenzene core group, through specific host–guest interactions. When exposed to visible light, the supramolecular polymer assembled into vesicles in water. When exposed to UV light, the vesicles disassembled into unimers. CD-g-HPG can be made to form supramolecular amphiphilic structures by mixing it with a hydrophobic polymer.22 Polystyrene as a hydrophobic linear polymer with aromatic rings was chosen as the backbone to prepare a supramolecular linear-dendritic copolymer. Host–guest interactions between the aromatic rings of the polystyrene and the cyclodextrin cavity of β-CD-HPG in an aqueous solution lead to the formation of a supramolecular amphiphile. The supramolecular assembly was used to encapsulate a hydrophobic anti-cancer drug, paclitaxel, for drug delivery applications. Supramolecular polymers derived from amphiphilic dextran-β-CD and poly(ε-caprolactone) coupled with benzimidazole was prepared through a host–guest recognition between terminal benzimidazole and β-CD.23 At the pH 7.4, the benzimidazole is hydrophobic and binds to the β-CD molecule via host–guest interactions. Under acidic conditions (pH < 6), benzimidazole becomes protonated, leading to a great decrease in the binding constant between benzimidazole and β-CD resulting in the dissociation of the complex. Utilizing this interesting characteristic, these pH-responsive supramolecular amphiphilic micelles could be utilized in applications requiring the delivery of a drug cargo to the endosomal/lysosomal compartments. Doxorubicin was loaded into the micelles and showed a faster release in HepG2 cells than pH-insensitive micelles. In another approach to the formation of micelles, a series of double hydrophobic azobenzene-containing diblock copolymers of poly[6-(4-(phenyldiazenyl)phenoxy)hexyl acrylate]-b-polystyrene were prepared.24 By using the β-CD–azobenzene host–guest interaction, the diblock copolymers could be made to self-assemble into micelles in water. Photo-controlled supramolecular self-assembly and disassembly of the polymer complexes in aqueous solution were studied. Trans-azobenzene has a stronger binding affinity with β-CD in aqueous solution, while a weaker binding force exists between cis-azobenzene and β-CD. By exposing the solution to light of different wavelengths, the self-assembled micelles can be disassociated and reassembled under a different light irradiation. This concept is interesting and more work needs to be performed to enhance the magnitude of the change in size. Inclusion complexation of β-CD and adamantane can also be used to prepare a supramolecular star-shaped ABC terpolymer.25 First, β-CD with two different functional moieties was prepared. A diblock copolymer grafted β-CD host was prepared via a “click” reaction with alkynyl-poly(ethylene glycol) and the atom transfer radical polymerization (ATRP) of dimethylaminoethyl methacrylate (DMAEMA). The adamantane-modified polymeric guest was obtained by the ATRP of methyl methacrylate (MMA) using an AD-modified initiator. Based on the molecular recognition between the β-CD and adamantyl moieties, the polymeric host and guest formed a star-shaped ABC miktoarm terpolymer via a simple mixing procedure. The amphiphilic ABC miktoarm terpolymer self-assembled into micelles in aqueous solution, and the reversible transition between the assembly and disassembly of this supramolecular ABC miktoarm star terpolymer could be readily controlled by adding a competitive host or guest .β-CD modified with an anthraquinone moiety was found to self-assemble into vesicles in aqueous solution (Fig. 2).26 The vesicles were responsive to Cu2+ and pH. The vesicles can efficiently load paclitaxel within the membrane with functional macrocyclic cavities available, which can further carry small molecules, such as ferrocene. Such drug-loaded vesicles appeared to have good potential for use against cancer cells.
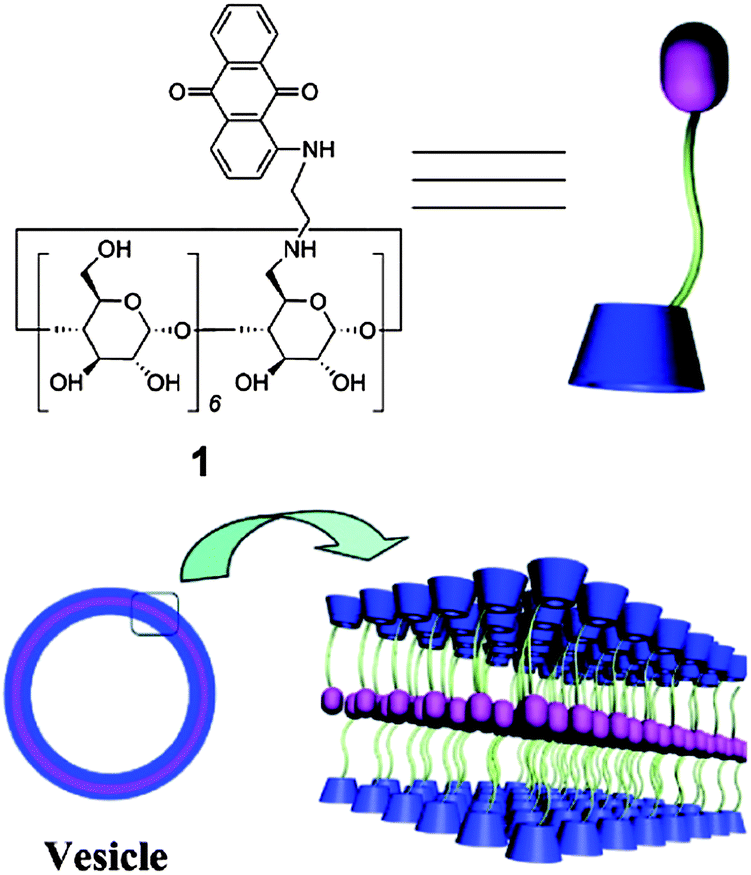 | ||
| Fig. 2 Illustration of the formation of the vesicles from the anthraquinone-functionalized β-CD.26 Reprinted with permission from Langmuir, 2012, 28, 8625–8636. Copyright (2012) American Chemical Society. | ||
Bulk modification of soft materials with cyclodextrin
DNA self-assembles in nature due to the pairing of complementary base pairs. Borrowing a leaf from nature, Harada et al. utilized synthetic molecular recognition principles to prepare supramolecular materials that are able to self-assemble in the bulk state (Fig. 3).27 Acrylamide-based gels functionalized with either cyclodextrin host rings or hydrocarbon guest moieties were prepared. The host and guest gels are shown to adhere to one another through the mutual molecular recognition of the cyclodextrins and hydrocarbon groups on their surfaces. When the size and shape of the host and guest units are changed, the different gels can be selectively assembled and sorted into distinct macroscopic structures. This report showed that it was possible to utilize host–guest interactions for the self-assembly of bulk objects.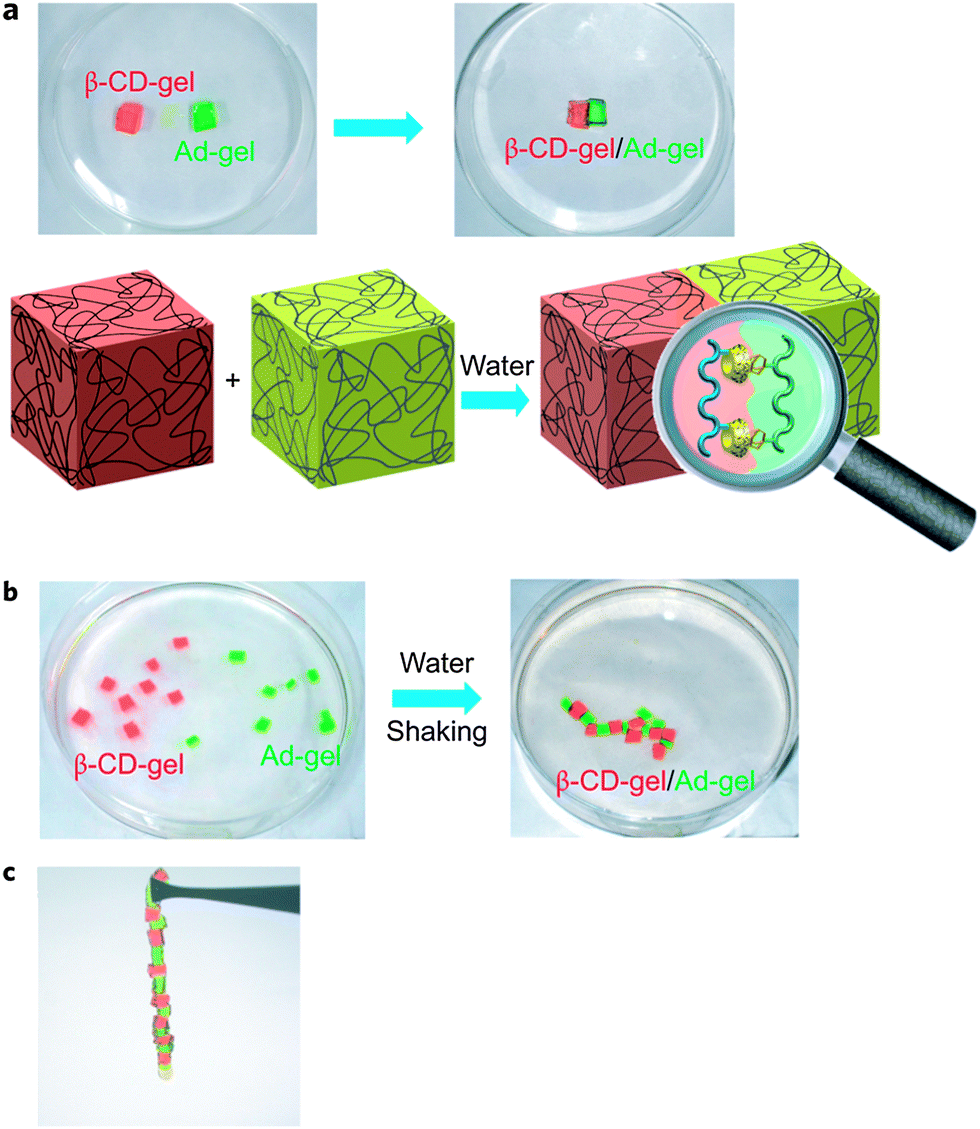 | ||
| Fig. 3 Macroscopic self-assembly between CD host gels and guest gels. (a) A β-CD-gel (red gel) was brought in contact with an Ad-gel (light green gel). A schematic of the molecular recognition responsible for the two gels sticking together is shown below. (b) Pieces of β-CD-gel (red) and Ad-gel (light green) were mixed and shaken in water resulting in a self-assembled structure. (c) β-CD-gel adheres firmly to Ad-gel, and alternating self-assembled structures were formed in accordance with the high complementarity of the host and guest gels.27 Reprinted with permission from Macmillan Publishers Ltd: Nat. Chem., 2011, 3, 34–37. Copyright (2011) Macmillan Publishers Ltd. | ||
Taking this concept one step further, a redox responsive gel assembly was reported in a follow-up paper.28 Poly(acrylic acid), which was modified with cyclodextrins, was used as a host polymer and pAA with ferrocene (Fc) was used as a guest polymer. The cyclodextrin-Fc inclusion complexes are redox-responsive. Upon the manipulation of the redox potential, a reversible sol–gel phase transition in a supramolecular hydrogel formed by β-CD modified pAA and Fc modified pAA can be observed. These materials also showed self-healing and self-repairing properties. Building on this, a photoresponsive macroscopic gel assembly was reported.29 The behavior of this hydrogel was regulated through the photoisomerization of the attached azobenzene moiety that interacts differently with two host molecules. Here, polyacrylamide-based hydrogels functionalized with azobenzene (guest) or cyclodextrin (host) moieties were first prepared. The reversible adhesion and dissociation of the host gel from the guest gel may be controlled by photoirradiation. The differential affinities of α-cyclodextrin or β-cyclodextrin for the trans-azobenzene and cis-azobenzene are employed in the construction of a photoswitchable gel assembly system. An assembly of the azo-gel with the α-CD-gel separated into separate gels on irradiation at 365 nm, and the gels reassembled on irradiation at 430 nm. The reversible dissociation and adhesion of host–guest gels on the macroscopic scale could be controlled by photoirradiation. This gel assembly system could be manipulated by the differential binding affinities of the α- and β-CDs towards the trans-/cis-azo moieties bound to the macroscopic gels. These gels could be used as photo-regulated artificial muscles.30 The expansion–contraction behaviour of the α-CD–azo gels could be controlled by photo-stimuli. The exposure of the gel to an ultraviolet light of wavelength 365 nm causes the isomerization of the trans-azo group into the cis-azo group. This can be reversed by exposing the gel to light of a wavelength of 430 nm. Ultraviolet irradiation of α-CD–azo gels increases the weight of the hydrogels, whereas exposure to light at 430 nm to the α-CD–azo gels causes the gel to contract and restore its initial weight and volume. It was hypothesized that the difference in the association constants of α-CD for the azo isomers creates the expansion and contraction behaviour in α-CD–azo gels upon irradiation. These volume changes of α-CD–azo gels are correlated with the inclusion complex formation between the α-CD and azo units. As the association constant of α-CD for trans-azo is larger than that for cis-azo, the exposure of the UV light causes gel expansion due to the inclusion complexes being less tightly bound. When it reverts back to the trans state, the binding tightens and the gel shrinks. The above examples all relied on chemically crosslinked hydrogels. Recently, a totally supramolecularly crosslinked self-healing CD–guest gel crosslinked between poly(acrylamide) chains with inclusion complexes was reported.31 Three monomers, acrylamide, a CD monomer and a guest monomer were mixed and polymerized in the presence of ammonium persulfate. Here, the inclusion complexes of either α-CD with a butyl group, or β-CD with an Ad group were used as two different supramolecular crosslinking systems for gel formation. The association constant of α-CD with an nBu group is Ka = 57 M−1, whereas that of β-CD with an Ad group is Ka = 1500 M−1. The elastic modulus of the CD–guest gels increases as the association constant of the CD with guest units increase, showing the facile tunability of the mechanical properties of these materials. These gels possess self-healing properties due to a reversible host–guest interaction between the side chains of the polymers. When the gels were cut in half, the fragments adhered together. While the β-CD–Ad gel mends immediately, the α-CD–nBu gel sample does not mend immediately.
Cucurbit[n]urils-based assemblies
Cucurbit[n]urils (CB[n], n = 5–8, 10) are macrocyclic oligomers based on repeating monomer units of glycoluril (Fig. 4). The main characteristics of CB[n]s are the hydrophobic cavity and polar carbonyl groups surrounding the portals. The cavity sizes and portal diameters range from 4.4–8.8 Å (for CB[n], n = 5–8) and 2.4–6.9 Å, respectively. This corresponds to a cavity volume range of 82–479 Å3. This difference causes visible differences in the host–guest chemistry of the different CB[n] homologues. The water solubility of CB[n]s had an odd–even variation across families, which could be due to the altered arrangement of H-bonding water clusters between the homologues in aqueous solution. CB[5] and CB[7] dissolve at a concentration of ∼20–30 mM in neutral water, while CB[6] and CB[8] have a solubility of 0.018 and <0.01 mM. All the CB[n]s are soluble both in acidic water and aqueous solutions of alkali metals because of either protonation or coordination of the metal ions to the carbonyl oxygen atoms. Thus, host–guest chemistry of CB[n]s has primarily been studied in aqueous media as the solubility of CB[n]s in common organic solvents is typically less than 10−5 M. Kim et al. reported an oxidative method to introduce hydroxyl moieties at the equatorial positions of CB[6].32 This was used in a range of supramolecular systems from synthetic pores to surface-modified vesicles. Although multivalent CB[6] has been used for different applications, the derivation of a facile protocol for the introduction of a single point of chemical modification on the parent CB molecule would allow for a high degree of control over structures and topologies at the molecular level. It is possible to use host–guest interactions to control the oxidation mechanism in the synthesis of monohydroxylated CB[6] from regular CB[6] in aqueous solution.33 Alternatively, a methylene bridged glycoluril hexamer can be prepared to form monofunctionalised cucurbituril derivatives in a subsequent step.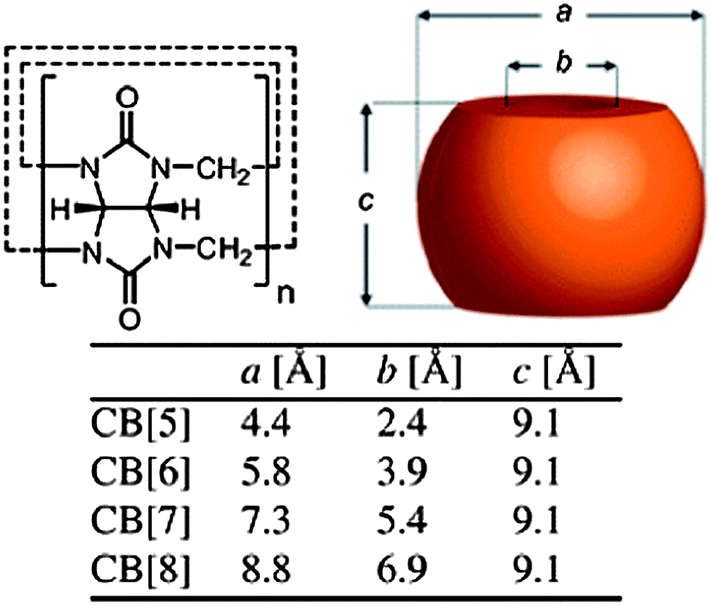 | ||
| Fig. 4 Schematic representation of the cucurbit[n]uril (CB[n]) family.12 Reprinted with permission from Chem. Soc. Rev., 2012, 41, 6195–6214. Copyright (2012) Royal Society of Chemistry. | ||
CB[6] forms remarkably stable complexes with protonated aminoalkanes, as the hydrophobic oligomethylene chain can partially fill the cavity while the ammonium cation can simultaneously bind to one of the portals through ion–dipole interactions. Highly stable host–guest complexes can be formed with CB[6] tightly binding protonated polyamines, such as 1,6-diaminohexane or spermine (Keq ∼ 1012 M−1). CB[7] forms strong complexes with positively charged amphiphilic guests, such as AD, ferrocene, p-xylene and trimethylsilyl derivatives containing one or two amino groups, as well as viologen derivatives. CB[7] exhibits association constants for some selected guests that equal or surpass that of the avidin–biotin interaction (Keq = 1015 M−1) and represent the strongest non-covalent interactions reported for a synthetic system. The reason for the extremely high affinity of these rigid, near-perfect complementary structures complexes, can be attributed to a large enthalpic gain, originating from the tight fit of the guest to the rigid CB[7] cavity. This is assisted by the entropic gain arising from the dehydration of the CB[7]. CB[8] also displays a remarkable binding affinity towards positively charged and relatively large guests, such as AD derivatives, cyclen, cyclam macrocyles and long alkylammonium aliphatic chains. The cavity of CB[8] is large enough to accommodate two organic guests simultaneously, thus forming highly stable ternary complexes. The formation of a stable ternary complex between CB[8] and two doubly-charged 2,6-bis(4,5-dihydro-1H-imidazol-2-yl)naphthalene molecules has been reported.34 The same authors also reported the selective formation of a complex between CB[8], viologen (paraquat) and 2,6-dihydroxynaphthalene, which results in an enhanced charge–transfer complex within the entity.35
Capsule formation with cucurbiturils
Nanoparticles can be formed from a CB[6] derivative by precipitation and loaded with hydrophobic drugs with a high loading capacity and efficiency. (3-(6-Hydroxyhexanethio)propan-1-oxy)nCB[6] (where n = 11) was synthesized by a thiol–ene reaction between (allyloxy)12CB[6] and 6-mercaptohexanol in methanol.36 The surface of the nanoparticles can be modified by simply mixing the nanoparticles with polyamine conjugates which can be used as a fluorescent probe or targeting ligand. The selective cellular uptake of targeting ligand-decorated nanoparticles was demonstrated by receptor-mediated endocytosis. An enhancement of the efficacy of paclitaxel delivered via this vehicle was also shown using the ligand-functionalized nanoparticles. A vesicular structure was formed by an amphiphilic CB[6] derivative.37 The surface of the vesicle has a strong affinity towards polyamines, which allows it to be easily modified via host–guest interactions. An amphiphilic CB[6] derivative was synthesized by a reaction between (allyloxy)12CB[6] and 2-[2-(2-methoxyethoxy)ethoxy]ethanethiol. This molecule assembles into a vesicle in aqueous solution. The treatment of the vesicle with a fluorescein isothiocyanate (FITC)–spermine conjugate ligand, in which spermine serves as a binding motif to CB[6] and FITC as a fluorescent tag, produced a surface-modified vesicle, which was visualized by a confocal microscope. The treatment of the CB[6]-based vesicle with a tag attached to polyamine allows for the decoration of the vesicle surface. Sugar-decorated vesicles were prepared by the supramolecular attachment and the interactions of the vesicles with concanavalin A (ConA) were studied. The easy surface modification of the vesicles opens doors to practical applications, such as targeted drug delivery. A CB[6] derivative that contains disulfide bonds between hexaethylene glycol units and a CB[6] core was designed as a reduction-sensitive and biocompatible vesicle.38 These vesicles could be easily surface-modified, possess good stability, are easily internalized into cells and able to release drugs efficiently in the cytoplasm. The unique features of the surface mean that the vesicle can be decorated with targeting and imaging moieties for the study of anti-cancer drugs uptake. This material is a highly versatile multifunctional platform for targeted drug delivery, which may find useful applications in cancer therapy. Scherman et al. demonstrated that when the first and second guests of CB[8] are present on the same polymer chain, even at very high molecular weights, single-chain polymeric nanoparticles can be formed at sufficiently low concentrations.39 A series of experiments were carried out that demonstrate stimuli-responsive transitions between ‘natured’ and ‘denatured’ states, similar to many biomacromolecules, such as proteins. CB[8] could be used as a supramolecular capping agent for the modification of the properties of a polymer.40 Poly(N-isopropylacrylamide) (PNIPAM), end functionalized with a dibenzofuran unit was complexed with a CB[8]-viologen complex.40 Traditionally, PNIPAM has been used in temperature-responsive drug delivery systems. Methods of modifying the system in a simple and non-covalent manner are very much sought after. In this paper, the supramolecular complex formation results in a shift in its lower critical solution temperature (LCST) by over 5 °C, suggesting that the amphiphilic nature of this polymer has been changed by the formation of the complex. An instantaneous phase change of the thermally responsive polymer beyond its LCST can be induced by the addition of a CB[8]-viologen binary complex to the polymer, to induce the formation of the ternary complex. Subsequent decomplexation upon addition of a competitive guest releases CB[8] from the PNIPAM terminus and triggers complete reversibility to the original state. Loh et al. report the synthesis of a supramolecular double hydrophilic block copolymer (DHBC) linked by CB[8] ternary complexation.41 Its subsequent self-assembly into micelles is described and found to be responsive to triggers such as temperature, pH and the addition of a competitive guest, such as adamantaneamine. The supramolecular block copolymer assembly consists of PNIPAM end-functionalized with viologen as a thermoresponsive block and poly(dimethylaminoethylmethacrylate) (PDMAEMA) with a naphthalene unit at the end group as a pH-responsive block. The encapsulation and controlled drug release was demonstrated with this system using the chemotherapeutic drug doxorubicin (DOX). This triple stimuli responsive DHBC micelle system represented an evolution over conventional double stimuli responsive covalent diblock copolymer systems and displayed a significant reduction in the viability of HeLa cells upon triggered release of DOX from the supramolecular micellar nanocontainers. This was followed up with another report of the synthesis of a supramolecular glucose-sensitive DHBC held together by CB[8] ternary complexation and its micellar behaviour.42 The supramolecular block copolymer assembly consists of PNIPAM and poly(acrylamidophenyl boronic acid) (PAAPBA) as temperature and glucose responsive blocks, respectively, and poly(dimethylacrylamide) (PDMAAm) as a hydrophilic block. These supramolecular micelles were used to encapsulate insulin and the release profile of insulin was responsive to temperature variation, glucose concentration variation or by adding a competitive guest for CB[8]. This system offers good control over the release of insulin under physiological conditions (pH 7.4, 37 °C). The results suggest that this system could be used as a model for a clinically relevant drug delivery vehicle for diabetes treatment. Supramolecular peptide amphiphiles have been prepared by the host–guest complexation of pyrene-labeled peptides and viologen lipids with CB[8].43 These structures self-assemble into stimuli-responsive vesicles, responding to competitive guests for CB[8]. Both “switching on” and “switching off” of fluorescence and cytotoxicity is demonstrated in vitro. The results showed that these vesicles could potentially be used for imaging and delivery applications in the biological setting. A stable ternary inclusion complex comprising CB[8], dihydroxynaphthalene, and a long alkyl tail end-functionalized with viologen forms giant vesicles spontaneously.44 The ternary complex behaves as a supramolecular amphiphile with a large polar head group and a single hydrophobic tail. The two guests which reside inside the cavity of CB[8] are redox active. The oxidation of dihydroxynaphthalene or reduction of viologen resulted in destruction of the ternary complex as a result of the loss of charge–transfer interaction between the two guests. This causes the collapse of the vesicles. Upon treatment with cerium(IV) ammonium nitrate (CAN) which can oxidize DHNp to naphthoquinone, the vesicles were disrupted.Hydrogel formation with cucurbiturils
Hydrogels prepared by the supramolecular assembly of the cucurbituril host and polymers functionalized with guest moieties have been reported. Scherman et al. presented the first report of a supramolecular polymeric hydrogel based on CB[n] host–guest inclusion complexes (Fig. 5).45 A pair of multivalent side-chain functional polymers bearing either viologen (a good first guest for CB[8]) or naphthoxy (a good second guest for CB[8]) moieties were first prepared. These were water-soluble and existed as a free-flowing solution. When CB[8] was added, supramolecular cross-linking and subsequent hydrogel formation was observed. The dissociation kinetics of the ternary complex with Kd = 1200 s−1 were based on rheological measurements. These hydrogels at 5 wt% concentration in water had a plateau modulus = 350–600 Pa and a zero-shear viscosity = 5–55 Pa s. The control of the microstructure of the system could be achieved by a variation of the amount of CB[8] added to the system.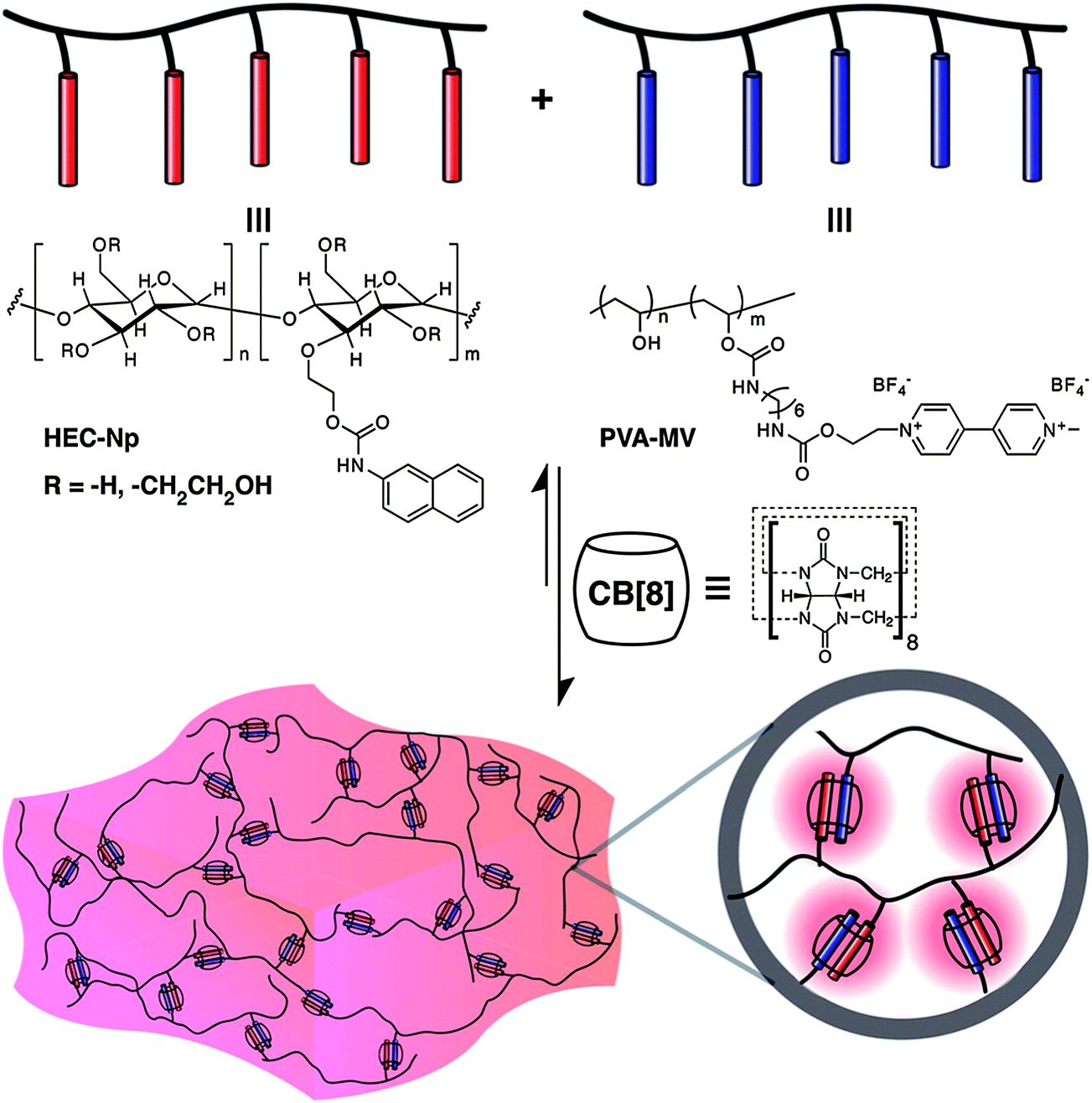 | ||
| Fig. 5 Schematic representation of a supramolecular hydrogel prepared through the addition of cucurbit[8]uril to a mixture of multivalent first- and second-guest-functionalized polymers in water.46 Reprinted with permission from J. Am. Chem. Soc., 2012, 134, 11767–11773. Copyright (2012) American Chemical Society. | ||
In further reports, Scherman et al. prepared hydrogels with an extremely high water-content (up to 99.75% water) prepared derived from renewable cellulosic polymers.46,47 The materials showed highly tunable mechanical properties that spanned three orders of magnitude. This was controlled simply by the relative loadings of the three components. These gels were responsive to different stimuli such as temperature, chemical potential and competing guests. The rapid formation of the CB[8] ternary complex lead to an instantaneous recovery of the mechanical properties after shear-induced deformation. The shear-thinning, fast recovery properties and cell compatibility of these hydrogels makes them very exciting candidates for use in various biomedical and industrial applications. The simple preparation process, their availability from inexpensive renewable resources, and the tunability of their mechanical properties are important considerations for a wide variety of applications. These gels could be used to provide an extremely sustained release of bioactive bovine serum albumin and lysozyme (used as a model protein for protein therapeutics) from supramolecular hydrogels containing only 1.5 wt% polymeric constituents, lasting over 160 days. Commercially available and natural amino acids such as phenylalanine and tryptophan could also be useful as guest moieties for CB[8].48 Hydrogels derived from these amino acids are purported to have a reduced toxicity profile and also simplifies the preparation of the hydrogels from a three component to a two component system. Recently, pH and thermo-responsive supramolecular hydrogels made from CB[8] and poly(N-(4-vinylbenzyl)-4,4′-bipyridinium dichloride-co-acrylamide) (P4VBAM) were prepared.49 The viologen units of P4VBAM and CB[8] formed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 CB[8]/viologen units binary complex upon the addition of NaOH. This led to the formation of a three-dimensional network and gel formation. These gels were also shear-thinning and showed gel-to-sol transition upon heating. The solutions became a gel again upon cooling. Lin et al.50 reported the synthesis of N-(4-diethylaminobenzyl) chitosan and its subsequent hydrogel formation with CB[8]. This supramolecular gel system showed thermo- and pH-responsive properties. In this report, the authors show the release profile of 5-fluorouracil encapsulated within the gel matrix, lasting for just about 10 hours.
:2 CB[8]/viologen units binary complex upon the addition of NaOH. This led to the formation of a three-dimensional network and gel formation. These gels were also shear-thinning and showed gel-to-sol transition upon heating. The solutions became a gel again upon cooling. Lin et al.50 reported the synthesis of N-(4-diethylaminobenzyl) chitosan and its subsequent hydrogel formation with CB[8]. This supramolecular gel system showed thermo- and pH-responsive properties. In this report, the authors show the release profile of 5-fluorouracil encapsulated within the gel matrix, lasting for just about 10 hours.
Kim et al. showed that CB[6] can be used to prepare hydrogels by using a hydroxy-functional CB[6] moiety with alkylammonium guests derived from 1,6-diaminohexane (DAH) and spermine (SPM).51,52 Firstly, hyaluronic acid functionalized with CB[6] was prepared by grafting (allyloxy)12CB[6] to a thiol-functionalised hyaluronic acid through a thiol–ene reaction. When the CB[6]-containing polymer and a DAH-containing hyaluronic were mixed, gelation occurred after 2 min. The addition of excess SPM to the hydrogel resulted in a phase transition from gel to sol within 10 min, suggesting that the supramolecular crosslinks formed from the specific host–guest interactions. A relatively high storage moduli in the region of kPa was measured for a 2 wt% hydrogel comprising a mixture of spermine and CB[6]-functionalised hyaluronic acid. Cytocompatibility studies demonstrated the high cell viability, enzymatic degradability and negligible cytotoxicity of gels. The presence of free alkylammonium guest in the polymeric network allows for the conjugation of targeting agents to the gel. The in situ formation of the hydrogel under the skin of mice was demonstrated by sequentially injecting solutions of the complementary polymers, CB[6]- and DAH-containing hyaluronic acid. The stability of the hydrogel was longer than 2 weeks. It was even possible to modified the hydrogels in vivo by injection of a solution of FITC-CB[6]. Tan et al. described the preparation of thermo-responsive supramolecular hydrogels consisting of CB[6] and butan-1-aminium 4-methylbenzenesulfonate (BAMB).53 The assembly of these moieties relies on hydrophobic interactions, π–π stacking and van der Waals interactions. These materials were found to be temperature responsive, turning into the sol state as the temperature is raised above 25 °C. The CB[6] was threaded in the butyl amine units of BAMB, and the self-assembly of the pseudorotaxane was essential for the hydrogel formation. Hydrogels with a three-dimensional network structure could only be formed at critical concentrations of both BAMB and CB[6].
Calix[n]arenes-based assemblies
Calixarenes are obtained by phenol and formaldehyde condensation. Calixarenes can be functionalized at either the upper and/or lower rim of the molecular skeleton among potential building blocks. A large number of cavities of different shapes and sizes can be involved in the molecular recognition as calixarenes have several conformational isomers. Calixarenes have become important synthetic receptors and have found applications as supramolecular platforms for molecular recognition, sensing and self-assembly, catalysis, nanotechnology and drug discovery. The formation of host–guest complexes with biological compounds are interesting applications of the functionalized calixarenes. The development of the water soluble calixarenes as receptors for biomolecules has become important in view of their potential medical applications. The binding properties of calixarenes and water-soluble calixarenes towards organic cationic substrates have been studied in the solid state, gas phase, and in solution by different techniques. Calixarene complexes are significantly stronger than the corresponding inclusion complexes of cyclodextrins and crown ethers with amino acids. In spite of their small cavity, a calixarene scaffold can be functionalized with moieties such as sulfonate, phosphate or ammonium groups on the upper rim. This allows the preparation of special receptors for the recognition of charged and ionic species, mimicking biological processes. As such, the investigation of the interactions between functionalized calix[4]arenes and different compounds is of fundamental interest to give new insights into molecular recognition, self-assembly processes, and a better understanding of the recognition processes running in biological systems. Functionalized calixarenes can act as carriers for membrane transport and incorporated into channeling systems, with analytical and therapeutic applications.Capsule formation with calixarenes
Calixarenes can be easily modified at their upper and lower rims, and have been used as versatile platforms in supramolecular chemistry. The modification of calixarene by hydrophilic long-chain poly(ethylene glycol) (PEG) at the lower rims gives a star-like molecule with good water-solubility (Fig. 6).54 The host–guest complex of this functionalized calixarene with a hydrophobic drug will lead to a formation of a supramolecular amphiphilic polymer. The supramolecular polymer self-assembles into polymeric micelles of about 200 nm in diameter in aqueous solution. The cellular uptake and photodynamic therapy (PDT) efficacy were studied using HeLa cells. These hosts are efficient nanocarriers for hydrophobic drugs, and a host–guest interaction leads to the formation of a supramolecular system with a stimuli-responsive ability. Liu et al. reported a phosphatase-responsive supramolecular amphiphilic assembly based on the host–guest complexation of amphiphilic calixarene with adenosine triphosphate (ATP).55 This was used as a targeted drug delivery agent. The complexation of calixarene with ATP greatly decreases its critical aggregation concentration, leading to the formation of hollow spherical nanoparticles. The self-assembled nanoparticles are responsive to phosphatase that is overexpressed in many tumor cells, leading to potential applications in drug delivery systems. The direct modification of calixarenes with poly(ε-caprolactone) (PCL) and PEG has been used to develop amphiphilic A2B2 miktoarm star copolymers with a calixarene core.56 These copolymers self-assembled into various morphologies in aqueous solution. The presence of the calix[4]arene moieties can be used for the encapsulation of drugs within the core of the micelles. P-sulfonatocalix[4]arene (SC4) with tetradecyltrimethylammonium bromide (TTABr) were mixed and the self-assembly behavior was studied.57 When SC4 is mixed with TTABr, a white dispersion forms at TTABr concentrations between 0.1 mM and 50 mM. When a milky dispersion of 50 mM SC4–TTABr with a molar ratio 1:2.5 was examined under light microscopy, giant vesicles with a diameter of 0.5–5 μm was observed. These vesicles could be stored upon lyophilization and rehydrated without change in the size or shape.
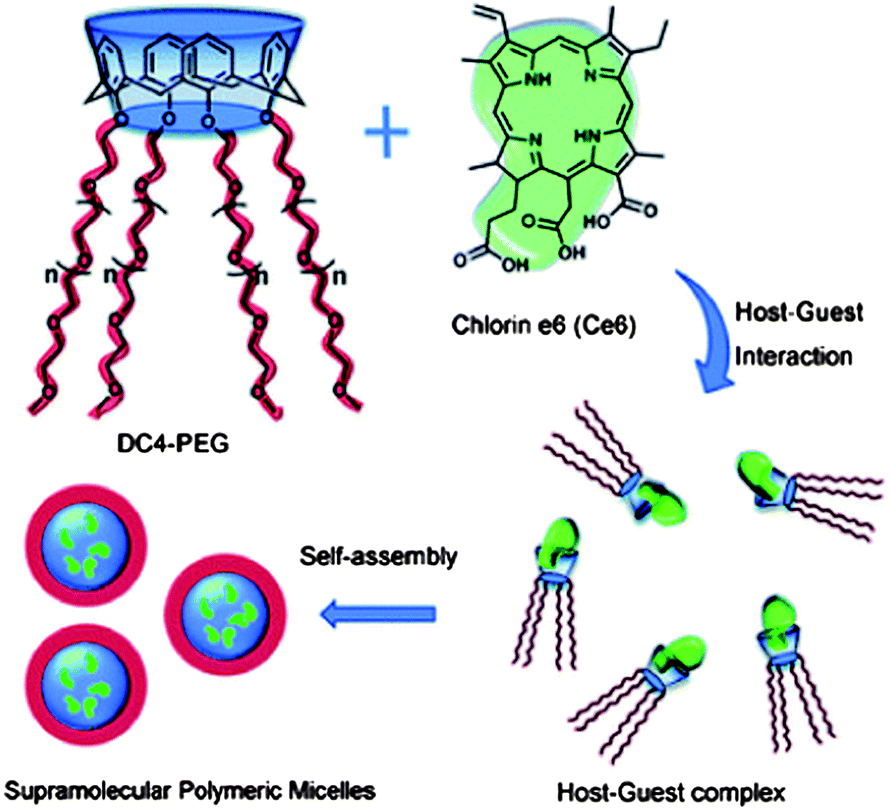 | ||
| Fig. 6 Representation of the structure of star-like calix[4]arene host, chlorin e6 guest molecule, and formation of the supramolecular polymeric micelles based on host–guest interaction.54 Reprinted with permission from Chem. Commun., 2011, 47, 6063–6065. Copyright (2011) Royal Society of Chemistry. | ||
Liu et al. reported the fabrication of enzyme-responsive supramolecular vesicles as a drug delivery system (Fig. 7).58p-Sulfonatocalix[4]arene (SC4A) and natural myristoylcholine were used as the macrocyclic host and enzyme-cleavable guest, respectively. The complexation of SC4A with myristoylcholine led to the formation of a binary vesicle. By using cholinesterases (acetylcholinesterase (AChE) and butyrylcholinesterase (BChE)), myristoylcholine is converted to myristic acid and choline, leading to a dissociation of the vesicle. Pillararenes are another class of macrocyclic hosts. They have a unique rigid pillar architecture made up of repeating units which are connected by methylene bridges at the para-positions. They are different from the basket-shaped structure of meta-bridged calixarenes. The unique structure and easy functionalization of pillararenes allow these molecules be used for the construction of various interesting supramolecular systems, including cyclic dimers, chemosensors, supramolecular polymers, and trans-membrane channels. Recently, the photoresponsive aggregation of the host–guest complex with the pillar[6]arene and the trans form of an azobenzene-containing guest has been utilized as a switch for the formation of either irregular aggregates or vesicle-like aggregates.59 This new pillar[6]arene-based photoresponsive host–guest recognition motif functions in organic solvents and reported as an alternative to the widely used cyclodextrin–azobenzene recognition motif.
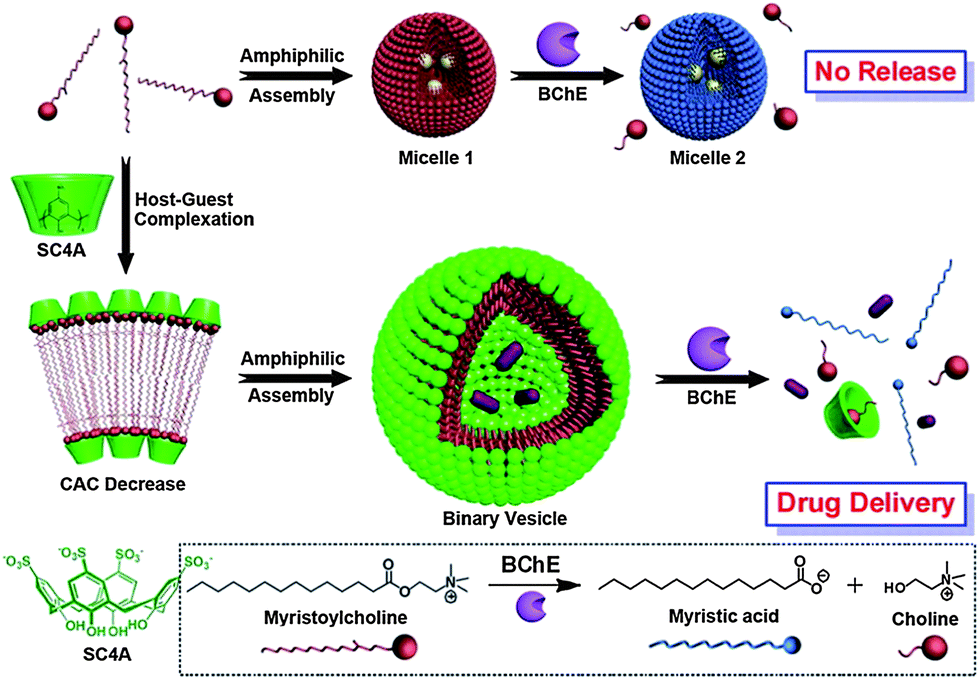 | ||
| Fig. 7 Schematic illustration of amphiphilic assemblies of myristoylcholine in the absence and presence of p-sulfonatocalix[4]arene (SC4Aa)58 Reprinted with permission from J. Am. Chem. Soc., 2012, 134, 10244–10250. Copyright (2012) American Chemical Society. | ||
Soft materials from calixarenes
Shinkai et al. reported p-acylcalix[n]arenes (n = 4, 6, 8) as unique gelators of various organic solvents such as toluene, carbon tetrachloride, carbon disulfide, hexane and isopropanol.60 The gels were temperature responsive and the sol–gel phase transition temperatures, Tgel, were determined. Below the Tgel, a fibrillar network was observed, whereas above the Tgel, it melts. This change is reversible. Xu et al. prepared a stable organogel that absorbs neutral organic molecules from the aqueous phase. This metallogel was immiscible with water and was stable over a wide range of pH, and temperatures (100 °C), and allows the efficient absorption of non-ionic organic molecules.61,62 The interaction of chiral calix[4]arene, functionalized with long tertiary alkyl groups at the upper rim, can form organogels with 2,3-dibenzoyltartaric acid.63 It is also able to gelatinize cyclohexane by the formation of vesicles. The size of the vesicles can be tuned by varying the alkyl chain length. At 20 °C, a mixture of calix[4]arene and (L)-2,3-dibenzoyltartaric acid in cyclohexane existed as a gel. When heated to 60 °C, this became a solution. However, for the corresponding mixture with the incorporation of (D)-2,3-dibenzoyltartaric acid being the only difference, at 20 °C, the mixture exists as a solution and when heated to 60 °C, the solution formed a gel (Fig. 8). In general, the long alkyl group attached onto calixarene drives the self-assembly of the amphiphile owing to hydrophobicity and van der Waals attraction between the long alkyl groups. Both mixtures can self-assemble into nanofibers. However, the fibrous assembles are not stable, due to the unmatched interaction between the chiral centers of calixarene and (D)-2,3-dibenzoyltartaric acid. Therefore, no gel formation occurs. The mismatched interaction decreases as the temperature increases, leading to gel formation. In the unstable state, the formation of vesicles reduces the repulsion between the chiral centers of calixarene and (D)-2,3-dibenzoyltartaric acid. Thus, at low temperature, the solution exists in a vesicular liquid form. | ||
| Fig. 8 Temperature-responsive calix[4]arene and 2,3-dibenzoyltartaric acid sol–gel behavior in cyclohexane. Reprinted with permission from Chem. Commun., 2007, 5200–5202. Copyright (2007) Royal Society of Chemistry. | ||
Liu et al. prepared a series of supramolecular binary hydrogels based on tetra-proline modified calix[4]arene (TPC) and basic amino acids (arginine, histidine and lysine).64 Binary gel systems can have tunable properties by varying the ratio of the components. The use of biocompatible amino acids as one of the components is advantageous for biomedical applications. In addition, this work looks towards the study of the influence of the amino acid structures on the properties of the resulting hydrogels, which can serve as a model for the development of other new functional binary gel-phase materials. The authors describe the use of aryl-extended calix[4]pyrrole as a responsive cavitand hydrogelator in combination with the tetramethylammonium cation (TMA).65 Calix[4]pyrrole possesses two aromatic cavities capable of including TMA. This compound was insoluble in neutral or acidic water, but readily dissolves at a basic pH. When TMA bromide (24 mM) was added to solutions of calix[4]pyrrole (6 mM) in aqueous NaOH, transparent hydrogels were formed. However, when tetraethylammonium or tetrabutylammonium bromide salt did not induce the same effect of gel formation. The hydrogelation process based on calix[4]pyrrole and TMA can be controlled by the concentration of the solution and responds to the presence of sodium cations or a change in pH. These gels formed in 100 mM NaOH solution, but not when LiOH or KOH were used as bases.
An electrolyte-triggered gelation of a proline-functionalised calix[4]arene was recently reported.66 The gelation process can be controlled by the electrolyte concentration and composition, as well as pH. In a follow-up report, the possible self-assembly mechanism that provides the structural information required to refine the properties of this unique hydrogelator was proposed.67 It was shown that the chirality of the molecule is a critical factor in the control of the bulk properties of the hydrogel formed.
Outlook and perspectives
Although the chemistry for the synthesis of macrocycles was discovered more than 100 years ago, the functionalization and application of these macrocycles is still in its infancy. In recent years, there has been intense research in this direction, including the establishment of standard protocols for the modification of these macrocycles. This has caught the interest of many researchers globally. The hydrophilic modifications of these macrocycles have led to these materials to be utilized in the formation of supramolecular structures in the aqueous medium. These are inherently useful for biomedical targeted applications, particularly for drug and protein delivery. Indeed, a variety of hierarchical structures, such as hydrogels, micelles and vesicles, have emerged from the self-assembly of polymer chains brought about by host–guest interactions. Some of the studies have even made it into the in vivo research setting, showing great promise for these materials. The development of biocompatible supramolecular materials remains a big challenge, as most of these materials have not been assessed for long term toxicity effects. For applications in the biomedical area, interactions of these supramolecular materials with bodily fluids, ionic buffers and protein-containing solutions have to be further evaluated. The wide use of these materials requires a larger quantity of materials to be prepared. The scale up production of these materials remains a major challenge, as most of the work is currently done at the intricate laboratory scale. Supramolecular host–guest polymeric materials have great potential to succeed on the biggest stage. Their ability to be manipulated by various stimuli, their ease of use by people of disparate technical backgrounds and potential wide applicability in different biomedical areas as sensors, drug delivery agents, cell supporting scaffolds and molecular switches make for exciting times for this unique class of materials in the coming few years.References
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- W. Zhao, Y. L. Su, C. Li, Q. Shi, X. Ning and Z. Y. Jiang, J. Membr. Sci., 2008, 318, 405–412 CrossRef CAS PubMed.
- A. Kawamura, A. Harada, K. Kono and K. Kataoka, Bioconjugate Chem., 2007, 18, 1555–1559 CrossRef CAS PubMed.
- C. Borde, V. Nardello, L. Wattebled, A. Laschewsky and J. M. Aubry, J. Phys. Org. Chem., 2008, 21, 652–658 CrossRef CAS.
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137–172 CrossRef CAS.
- Y. Wang, H. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849–2864 CrossRef CAS.
- X. Zhang and C. Wang, Chem. Soc. Rev., 2011, 40, 94–101 RSC.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- V. J. Stella and R. A. Rajewski, Pharm. Res., 1997, 14, 556–567 CrossRef CAS.
- V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037–2042 CAS.
- K. Wang, D.-S. Guo, H.-Q. Zhang, D. Li, X.-L. Zheng and Y. Liu, J. Med. Chem., 2009, 52, 6402–6412 CrossRef CAS PubMed.
- A. Harada, Acc. Chem. Res., 2001, 34, 456–464 CrossRef CAS PubMed.
- A. Harada and A. Hashidzume, Aust. J. Chem., 2010, 63, 599–610 CrossRef CAS.
- A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS PubMed.
- A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875–882 RSC.
- A. Hashidzume and A. Harada, Polym. Chem., 2011, 2, 2146–2154 RSC.
- Y. Liu, C. Y. Yu, H. B. Jin, B. B. Jiang, X. Y. Zhu, Y. F. Zhou, Z. Y. Lu and D. Y. Yan, J. Am. Chem. Soc., 2013, 135, 4765–4770 CrossRef CAS PubMed.
- A. Pourjavadi, M. Adeli and M. Yazdi, New J. Chem., 2013, 37, 295–298 RSC.
- Z. Zhang, J. X. Ding, X. F. Chen, C. S. Xiao, C. L. He, X. L. Zhuang, L. Chen and X. S. Chen, Polym. Chem., 2013, 4, 3265–3271 RSC.
- S. H. Wang, Q. X. Shen, M. H. Nawaz and W. A. Zhang, Polym. Chem., 2013, 4, 2151–2157 RSC.
- X. Y. Huan, D. L. Wang, R. J. Dong, C. L. Tu, B. S. Zhu, D. Y. Yan and X. Y. Zhu, Macromolecules, 2012, 45, 5941–5947 CrossRef CAS.
- T. Sun, Q. Guo, C. Zhang, J. C. Hao, P. Y. Xing, J. Su, S. Y. Li, A. Y. Hao and G. C. Liu, Langmuir, 2012, 28, 8625–8636 CrossRef CAS PubMed.
- A. Harada, R. Kobayashi, Y. Takashima, A. Hashidzume and H. Yamaguchi, Nat. Chem., 2011, 3, 34–37 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 2, 511 CrossRef PubMed.
- H. Yamaguchi, Y. Kobayashi, R. Kobayashi, Y. Takashima, A. Hashidzume and A. Harada, Nat. Commun., 2012, 3, 603 CrossRef PubMed.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Nakahata, T. Kakuta, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270 CrossRef PubMed.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS PubMed.
- S. Y. Jon, N. Selvapalam, D. H. Oh, J. K. Kang, S. Y. Kim, Y. J. Jeon, J. W. Lee and K. Kim, J. Am. Chem. Soc., 2003, 125, 10186–10187 CrossRef CAS PubMed.
- N. Zhao, G. O. Lloyd and O. A. Scherman, Chem. Commun., 2012, 48, 3070–3072 RSC.
- J. Kim, I. S. Jung, S. Y. Kim, E. Lee, J. K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- J. Z. Zhao, H. J. Kim, J. Oh, S. Y. Kim, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 4233–4235 CrossRef CAS.
- K. M. Park, K. Suh, H. Jung, D. W. Lee, Y. Ahn, J. Kim, K. Baek and K. Kim, Chem. Commun., 2009, 71–73 RSC.
- H. K. Lee, K. M. Park, Y. J. Jeon, D. Kim, D. H. Oh, H. S. Kim, C. K. Park and K. Kim, J. Am. Chem. Soc., 2005, 127, 5006–5007 CrossRef CAS PubMed.
- K. M. Park, D. W. Lee, B. Sarkar, H. Jung, J. Kim, Y. H. Ko, K. E. Lee, H. Jeon and K. Kim, Small, 2010, 6, 1430–1441 CrossRef CAS PubMed.
- E. A. Appel, J. Dyson, J. del Barrio, Z. Walsh and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 4185–4189 CrossRef CAS PubMed.
- U. Rauwald, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Commun., 2011, 47, 6000–6002 RSC.
- X. J. Loh, J. del Barrio, P. P. C. Toh, T. C. Lee, D. Z. Jiao, U. Rauwald, E. A. Appel and O. A. Scherman, Biomacromolecules, 2012, 13, 84–91 CrossRef CAS PubMed.
- X. J. Loh, M. H. Tsai, J. del Barrio, E. A. Appel, T. C. Lee and O. A. Scherman, Polym. Chem., 2012, 3, 3180–3188 RSC.
- D. Z. Jiao, J. Geng, X. J. Loh, D. Das, T. C. Lee and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 9633–9637 CrossRef CAS PubMed.
- Y. J. Jeon, P. K. Bharadwaj, S. W. Choi, J. W. Lee and K. Kim, Angew. Chem., Int. Ed., 2002, 41, 4474–4476 CrossRef CAS.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS PubMed.
- E. A. Appel, X. J. Loh, S. T. Jones, F. Biedermann, C. A. Dreiss and O. A. Scherman, J. Am. Chem. Soc., 2012, 134, 11767–11773 CrossRef CAS PubMed.
- E. A. Appel, X. J. Loh, S. T. Jones, C. A. Dreiss and O. A. Scherman, Biomaterials, 2012, 33, 4646–4652 CrossRef CAS PubMed.
- M. J. Rowland, E. A. Appel, R. J. Coulston and O. A. Scherman, J. Mater. Chem. B, 2013, 1, 2904–2910 RSC.
- H. Yang, H. Chen and Y. B. Tan, RSC Adv., 2013, 3, 3031–3037 RSC.
- Y. Lin, L. Li and G. Li, Carbohyd. Polym., 2013, 92, 429–434 CrossRef CAS PubMed.
- K. M. Park, J. A. Yang, H. Jung, J. Yeom, J. S. Park, K. H. Park, A. S. Hoffman, S. K. Hahn and K. Kim, ACS Nano, 2012, 6, 2960–2968 CrossRef CAS PubMed.
- H. Jung, K. M. Park, J. A. Yang, E. J. Oh, D. W. Lee, K. Park, S. H. Ryu, S. K. Hahn and K. Kim, Biomaterials, 2011, 32, 7687–7694 CrossRef CAS PubMed.
- H. Yang, Y. B. Tan and Y. X. Wang, Soft Matter, 2009, 5, 3511–3516 RSC.
- C. L. Tu, L. J. Zhu, P. P. Li, Y. Chen, Y. Su, D. Y. Yan, X. Y. Zhu and G. Y. Zhou, Chem. Commun., 2011, 47, 6063–6065 RSC.
- Y. X. Wang, D. S. Guo, Y. Cao and Y. Liu, RSC Adv., 2013, 3, 8058–8063 RSC.
- P. F. Gou, W. P. Zhu and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5643–5651 CrossRef CAS.
- V. Francisco, N. Basilio, L. Garcia-Rio, J. R. Leis, E. F. Marques and C. Vazquez-Vazquez, Chem. Commun., 2010, 46, 6551–6553 RSC.
- D. S. Guo, K. Wang, Y. X. Wang and Y. Liu, J. Am. Chem. Soc., 2012, 134, 10244–10250 CrossRef CAS PubMed.
- G. C. Yu, C. Y. Han, Z. B. Zhang, J. Z. Chen, X. Z. Yan, B. Zheng, S. Y. Liu and F. H. Huang, J. Am. Chem. Soc., 2012, 134, 8711–8717 CrossRef CAS PubMed.
- M. Aoki, K. Nakashima, H. Kawabata, S. Tsutsui and S. Shinkai, J. Chem. Soc., Perkin Trans. 2, 1993, 347–354 RSC.
- B. A. Xing, M. F. Choi, Z. Y. Zhou and B. Xu, Langmuir, 2002, 18, 9654–9658 CrossRef CAS.
- B. G. Xing, M. F. Choi and B. Xu, Chem. Commun., 2002, 362–363 RSC.
- J. L. Zhou, X. J. Chen and Y. S. Zheng, Chem. Commun., 2007, 5200–5202 RSC.
- J. Zhang, D. S. Guo, L. H. Wang, Z. Wang and Y. Liu, Soft Matter, 2011, 7, 1756–1762 RSC.
- B. Verdejo, F. Rodriguez-Llansola, B. Escuder, J. F. Miravet and P. Ballester, Chem. Commun., 2011, 47, 2017–2019 RSC.
- T. Becker, C. Y. Goh, F. Jones, M. J. McIldowie, M. Mocerino and M. I. Ogden, Chem. Commun., 2008, 3900–3902 RSC.
- C. Y. Goh, T. Becker, D. H. Brown, B. W. Skelton, F. Jones, M. Mocerino and M. I. Ogden, Chem. Commun., 2011, 47, 6057–6059 RSC.
| This journal is © The Royal Society of Chemistry 2014 |