Self-assembly of aligned rutile@anatase TiO2 nanorod@CdS quantum dots ternary core–shell heterostructure: cascade electron transfer by interfacial design†
Fang-Xing
Xiao
,
Jianwei
Miao
and
Bin
Liu
*
School of Chemical and Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, Singapore 637459, Singapore. E-mail: liubin@ntu.edu.sg; Fax: +65 6794-7553; Tel: +65 6513-7971
First published on 2nd October 2013
Abstract
A novel self-assembly approach based on electrostatic interactions has been developed for the synthesis of a rutile@anatase TiO2 nanorod (NR)@CdS quantum dots (QDs) ternary core–shell heterostructure, in which an in situ formed monodisperse anatase TiO2 layer was intimately sandwiched between rutile TiO2 NRs and CdS QDs. It has been demonstrated that the well-defined bilayer interface significantly improves the photocatalytic performance of the ternary heterostructure (i.e. rutile@anatase TiO2 NR@CdS QDs), owing predominantly to the appropriate band alignment of the constituent semiconductors, thus facilitating photogenerated electron–hole separation and charge collection under simulated solar light irradiation.
Introduction
One-dimensional (1D) nanostructured metal oxide arrays, such as nanotubes, nanorods and nanowires, have received growing interest in a wide range of application fields because of their unique geometrical structure providing large surface-to-volume ratios, direct pathways for charge transport, and low reflectance induced by light scattering and trapping.1–5 Particularly, TiO2 nanorod arrays (TiO2 NRs) grown vertically on transparent conductive substrates have received the most attention for the development of novel nanostructured electrodes as they have shown significant enhancements in charge separation, transport, and light absorption.6–8 In spite of these great efforts, TiO2, as a quintessential wide bandgap semiconductor, fails to absorb visible photons, which limits its potential in photocatalysis remarkably.To improve visible light absorption, a number of research efforts have been devoted to reducing the bandgap of TiO2 by doping with transition metal cations or non-metal anions,9,10 depositing noble metal nanoparticles,11 and coupling with secondary semiconductors.12 Coupling TiO2 with narrow bandgap semiconductors such as CdX (X = S, Se, Te),13–15 PbS,16 and InP,17 substantially extends the visible absorption of TiO2 and improves the quantum efficiency.18–20 Although many TiO2 based binary nanocomposites have been extensively investigated for this purpose by targeted modifications, nevertheless, higher excitonic efficiency of nanocomposites can be further achieved if a ternary hybrid system is well-designed via judicious integration of band alignment at the interface of different semiconductor components. For example, it has been well-established that an intermediate ZnS or ZnSe layer integrated between two different semiconductors contributes to enhanced charge separation by forming a built-in potential gradient band structure,21,22 thus resulting in remarkably enhanced photoelectrochemical performances.23–25 On the other hand, a number of works have previously demonstrated the advantage of the core–shell nanocable architecture, in which carrier separation takes place in the radial direction, with a carrier collection distance smaller or comparable to the minority of carrier diffusion lengths.26–30
Inspired by these ideas, herein we have designed a rutile@anatase TiO2 NRs (TN)@CdS QDs ternary core–shell heterostructure via a facile self-assembly approach, in which an intermediate anatase TiO2 layer was sandwiched in the interface between rutile TiO2 NRs and CdS QDs, and intimate integration between the nano-building blocks was achieved. It was found that the core–shell bilayer system plays an important role in light absorption and charge separation, therefore contributing to significantly improved photocatalytic performances.
Results and discussion
Scheme 1 depicts a flowchart for the self-assembly of the rutile@anatase TN@CdS QDs ternary core–shell heterostructure. Single-crystalline rutile TiO2 NRs were grown vertically on the fluorine-doped tin oxide (FTO) substrate via a hydrothermal approach. In order to obtain a positively charged surface, cationic poly(diallyldimethylammonium chloride) (PDDA) was used to modify the TiO2 NRs surface. In this way, negatively charged titanium(IV) bis(ammonium lactato)dihydroxide (TALH), a typical anatase TiO2 precursor,31–33 could be spontaneously adsorbed on the surface of the TiO2 NRs via pronounced electrostatic interactions, leading to a rutile@anatase TN core–shell heterostructure after facile refluxing.33 It should be emphasized that the as-prepared rutile@anatase TN nanostructure was subjected to calcination (450 °C, 1 h, in air) in order to improve its photoelectrochemical performance and simultaneously remove the PDDA layer. Finally, tailor-made negatively charged CdS QDs were self-assembled onto the 3-aminopropyltrimethoxysilane (APS) modified positively charged rutile@anatase TN scaffold, yielding a hierarchical rutile@anatase TN@CdS QDs ternary core–shell heterostructure, in which intermediate anatase TiO2 layer was sandwiched closely at the interface. | ||
| Scheme 1 Schematic illustration of the self-assembly of rutile@anatase TN@CdS QDs ternary core–shell heterostructure. | ||
As revealed by the field-emission scanning electron microscopy (FESEM) images shown in Fig. 1a and b and Fig. S2,† self-oriented TiO2 NRs with an average diameter of ca. 120 nm are uniformly distributed over the entire surface of the FTO and are nearly perpendicular to the substrate. The top surface of some large nanorods demonstrates a tetragonal shape, which contains many rough step edges (Fig. S2d†), while the side surface is very smooth (Fig. S2c†). Intriguingly, as displayed in Fig. 1c and S3,† the surface of the TiO2 NRs was evenly wrapped with a rough layer consisting of many monodisperse anatase TiO2 nanoparticles (NPs), resulting from the hydrolysis and condensation of TALH after refluxing; this is in stark contrast to the original morphology of the pure TiO2 NRs (Fig. S2†). Fig. 1d demonstrates a typical FESEM image of the rutile@anatase TN@CdS QDs ternary core–shell heterostructure, the morphology of which is distinct from that of pure TiO2 NRs and rutile@anatase TN. Note that the TiO2 NRs were covered by compact and monodisperse CdS QDs over the entire FTO surface (Fig. S4†), and no agglomeration of the CdS QDs was observed.
 | ||
| Fig. 1 Panoramic FESEM images of (a and b) rutile TNs, (c) rutile@anatase TN and (d) rutile@anatase TN@CdS QDs ternary core–shell heterostructure. | ||
The morphology and crystal structure of TiO2 NRs were characterized by transmission electron microscopy (TEM) and high-resolution TEM (HRTEM). The NRs consist of single crystalline rutile phase TiO2 with a [001] growth direction (Fig. 2a and b), which is consistent with TiO2 NRs reported previously.6,34 The image in Fig. 2c clearly shows that a thin anatase TiO2 layer was sandwiched at the interface between rutile TiO2 NRs and CdS QDs, and CdS QDs closely cover the surface with increased surface roughness. The HRTEM image in Fig. 2d demonstrates well-resolved lattice fringes of about 3.52 Å, which corresponds to the (101) crystal plane of anatase TiO2, suggesting the good crystallinity of the intermediate anatase TiO2 layer. The HRTEM image of the part shown in Fig. 2c reveals a distinct lattice fringe of ca. 3.36 Å, corresponding to the (220) crystal plane of CdS QDs.
 | ||
| Fig. 2 TEM images of (a) a single TiO2 NR and (c) the rutile@anatase TN@CdS QDs core–shell heterostructure. HRTEM images of (b)TiO2 NR and (d) the rutile@anatase TN@CdS QDs core–shell heterostructure. | ||
XRD patterns of the samples (Fig. S5†) reveal that the TiO2 NRs deposited on the FTO substrate are single-crystalline rutile TiO2, which is in good agreement with our previous results.6 Note that only weak diffraction peaks belonging to anatase TiO2 and CdS were observed (Fig. S5c†), which may be attributed to the relatively low deposition percentage of anatase TiO2 and CdS QDs ingredients. Moreover, the uniform deposition of the anatase TiO2 layer between the rutile TiO2 NRs and CdS QDs may also shield the characteristic diffraction peaks of the intermediate anatase TiO2 component.
The composition and chemical valence states of the elements in the ternary core–shell heterostructure were determined by X-ray photoelectron spectroscopy (XPS), which is displayed in Fig. S6.† As shown in Fig. S6f,† the Ti 2p3/2 and Ti 2p1/2 peaks located at ca. 458.35 and 464.10 eV, respectively, indicate that the Ti element is in the +4 oxidation state.35 In the high-resolution XPS spectrum of Cd 3d (Fig. S6e†), the peaks at ca. 405.03 and 411.76 eV for Cd 3d5/2 and Cd 3d3/2 are ascribed to the +2 oxidation state of Cd.36 Moreover, combined with high-resolution spectrum of S 2p (Fig. S6d†) which corresponds to chemical species of S2−,37 it can be concluded that CdS QDs were successfully deposited onto the rutile@anatase TN substrate, chemical valence states of which were retained during the self-assembly process. On the other hand, predominant peaks in the high-resolution spectrum of O 1s (Fig. S6c†) positioned at ca. 529.61 and 530.90 eV are assigned to lattice oxygen in TiO2 and Ti–OH groups on the TiO2 surface, respectively, suggesting that the synthesized TiO2 NRs are well crystallized. The detailed chemical bond species versus binding energy is tabulated in Table S1.†
UV-vis diffuse reflectance spectra (DRS) were used to determine the optical absorption properties of the samples. It can be seen from Fig. 3a that enhanced absorbance in the visible region ranging from ca. 400 to 800 nm is observed with the introduction of CdS QDs into the core–shell heterostructure. A plot obtained via transformation based on the Kubelka–Munk function versus the energy of light (Fig. 3b) reveals the estimated bandgaps of the samples to be ca. 3.02, 3.00, and 2.76 eV, corresponding to blank rutile TN, rutile@anatase TN, and the rutile@anatase TN@CdS QDs heterostructure, respectively. This suggests a narrowing of the bandgap of TiO2 NRs due to the integration of CdS QDs into the heterostructure. Meanwhile, we note that the light absorbance of rutile@anatase TN is significantly enhanced in comparison with that of pure rutile TN in the UV region, which can be ascribed primarily to the homogeneous passivation of rutile TiO2 NRs by the anatase TiO2 layer, thus contributing to the enhanced harvesting of UV light.
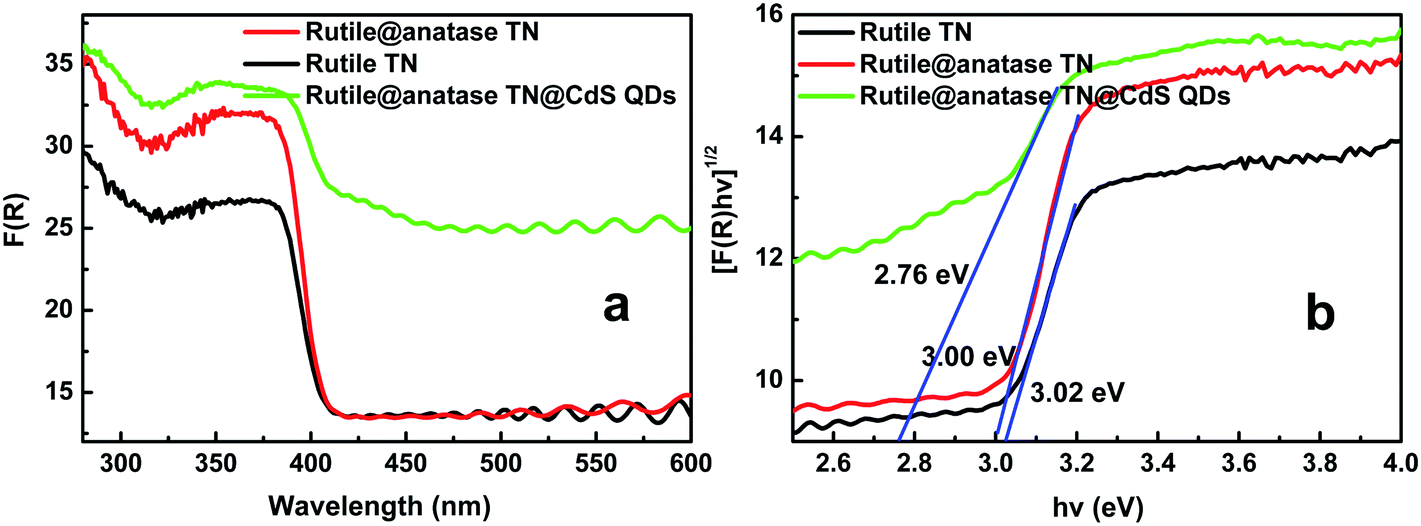 | ||
| Fig. 3 (a) UV-vis diffuse reflectance spectra (DRS) of rutile TiO2 NRs, rutile@anatase TN and the rutile@anatase TN@CdS QDs ternary core–shell heterostructure, and (b) plot of transformed Kubelka–Munk function versus energy of light. | ||
As displayed in Fig. S7,† Fourier transformed infrared (FTIR) spectra of the samples further corroborate the deposition of CdS QDs on the TiO2 NRs framework via the self-assembly strategy, since a substantial stretching vibration peak positioned at ca. 1578 cm−1, arising from various deprotonated carboxyl groups (–COO−) on the CdS QDs surface, was clearly observed. Besides, characteristic peaks corresponding to Ti–OH and Ti–O bonds of TiO2 could also be clearly identified.
Fig. 4a shows the transient photocurrent responses of the samples under intermittent simulated solar light irradiation. The introduction of CdS QDs significantly enhances the photocurrent of the nanocomposites, which rapidly decreases to zero once the light is switched off. It has been well-established that photocurrent is formed due to the diffusion of photogenerated electrons to the back contact and the simultaneous take up of the photogenerated holes by the hole acceptor in the electrolyte.38 As such, the superior photocurrent of the ternary core–shell heterostructure over its rutile TN@CdS QDs counterpart prepared via the direct self-assembly of CdS QDs onto APS-modified rutile TiO2 NRs using the same method indicates the more efficient separation and longer lifetime of the photoexcited electron–hole pairs. This result highlights the advantage of the anatase TiO2 layer between CdS QDs and rutile TiO2 NRs for light absorption and charge separation.
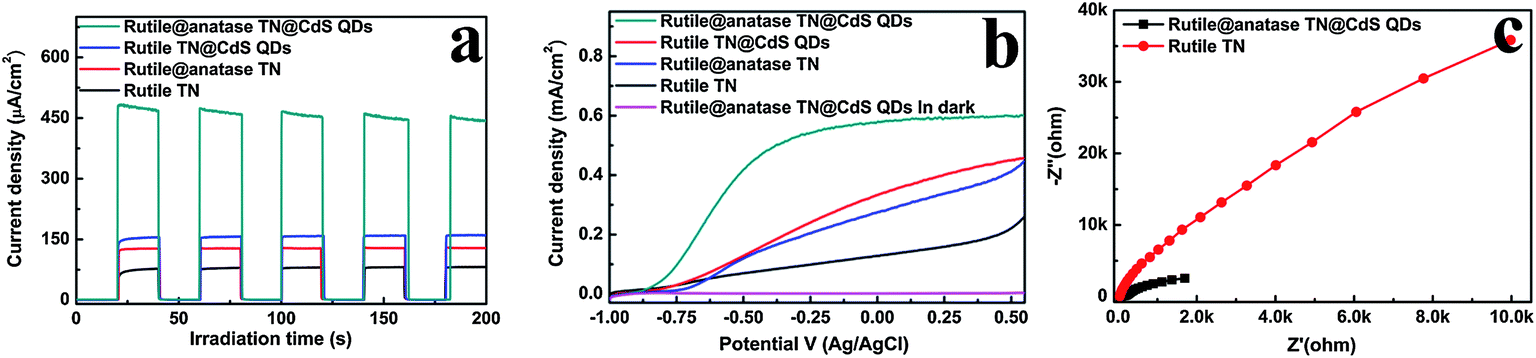 | ||
| Fig. 4 (a) Transient photocurrent responses and (b) current density versus potential curves of rutile TN, rutile@anatase TN, rutile@CdS QDs and the rutile@anatase TN@CdS QDs ternary core–shell heterostructure, (c) electrochemical impedance spectroscopy (EIS) Nyquist plots of samples in 0.1 M Na2S aqueous solution with zero bias under simulated solar light irradiation (100 mW cm−2), the amplitude of the sinusoidal wave was set at 10 mV and the frequency varied from 100 kHZ to 0.01 HZ. | ||
Fig. 4b demonstrates the photocurrent density versus the applied voltage plot recorded under irradiation with simulated solar light. It is found that the photocurrent density of the samples increases with the forward bias voltage, indicative of a typical n-type semiconductor.39 Among the samples, the ternary core–shell heterostructure demonstrates remarkably enhanced photocurrent compared to the other structures over the whole potential profile, which agrees well with the result in Fig. 4a. On the other hand, the electrochemical impedance spectroscopy (EIS) Nyquist plots in Fig. 4c show the dramatic decrease of the high-frequency arc for the ternary core–shell heterostructure as compared to that of pure rutile TN, suggesting that interfacial charge transfer over ternary core–shell heterostructure is quite efficient. Consequently, based on the above analysis, the imperative role of the intermediate anatase TiO2 layer in the ternary core–shell heterostructure was ascertained for prolonging the lifetime of the photogenerated charge carriers.
Photocatalytic performances of the samples was examined by the photodegradation of an organic pollutant under ambient conditions and simulated solar light irradiation. As shown in Fig. S8,† blank experiments (without light or photocatalyst) show negligible degradation, confirming that the reaction was driven by a photocatalytic process. It can be seen from Fig. 5a that the pure rutile TN exhibited almost no photoactivity, and in distinct contrast, the rutile@anatase TN binary nanostructure demonstrates improved photoactivity, which can be ascribed to the uniform deposition of a photoactive anatase TiO2 layer on the rutile TN surface. Moreover, when CdS QDs were additionally self-assembled on the rutile@anatase TN substrate, the photocatalytic performance of the ternary core–shell heterostructure was remarkably enhanced, along with favorable photostability (Fig. S9†). It should be noted that the photocatalytic performance of rutile TN@CdS QDs is inferior to that of the ternary heterostructure under the same experimental conditions, thus once again highlighting that the intermediate anatase TiO2 layer is of great importance for suppressing the recombination of photoexcited electron–hole charge carriers due to the intimate interfacial interaction between rutile TiO2 NRs and CdS QDs. On the other hand, the photocatalytic reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) over the different samples was also probed under ambient conditions (Fig. 5b and S10†). During the photocatalytic reduction process, photogenerated holes were completely quenched by a hole scavenger (HCO2NH4) in the presence of saturated argon flow, and thus the majority of the surviving photoelectrons could selectively reduce 4-NP to 4-AP. It was found that the photoreduction performances of the samples exhibited an analogous trend, among which the ternary core–shell heterostructure demonstrates the best activity among the samples.
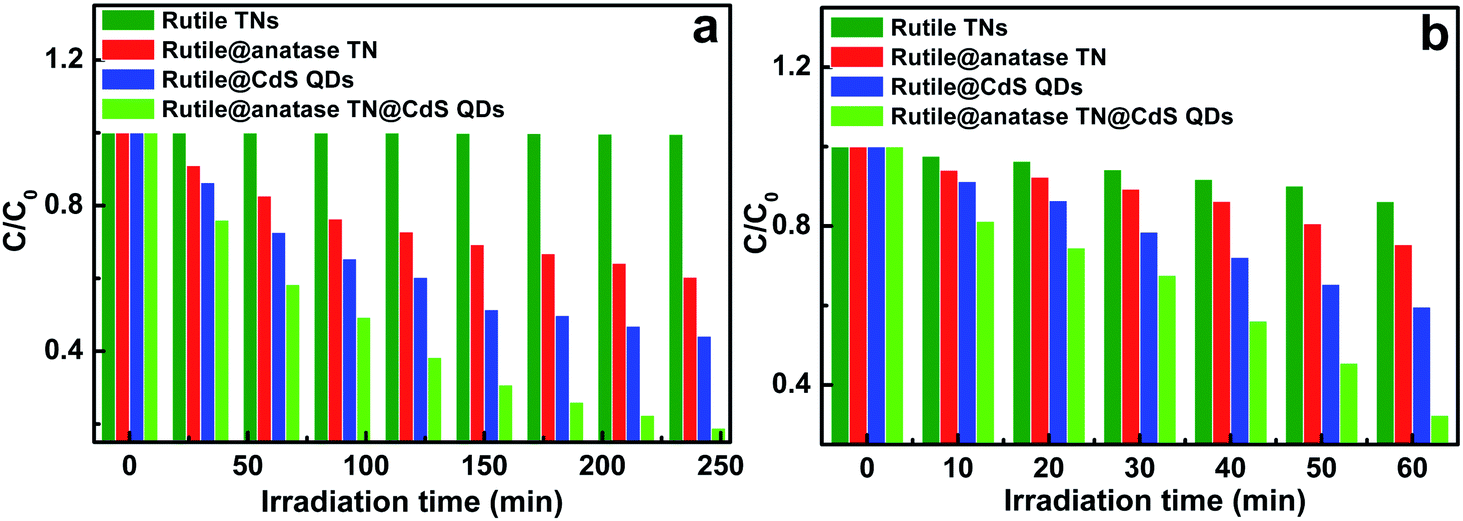 | ||
| Fig. 5 Photocatalytic performances of rutile TN, rutile@anatase TN, rutile TN@CdS QDs, and rutile@anatase TN@CdS QDs ternary core–shell heterostructure toward (a) the photooxidation of MO and (b) the selective photocatalytic reduction of 4-nitrophenol (4-NP) under simulated sunlight irradiation (AM 1.5, 100 mW cm−2) at ambient conditions. | ||
Accordingly, a possible photocatalytic mechanism for the rutile@anatase TN@CdS QDs ternary core–shell heterostructure is proposed, as illustrated in Fig. S11.† Under simulated solar light irradiation, the photogenerated electrons are excited from the valence band (VB) of the CdS QDs to the conduction band (CB) under visible light irradiation, along with a small portion of the photoelectrons originating from the CB of the anatase and rutile TiO2 phases in the near-UV photon region, thereby leaving holes in the VB. Notably, the CB of anatase TiO2 is located between the CBs of CdS QDs and rutile TiO2 (Fig. S11†), thus forming a unique band arrangement which efficiently facilitates the smooth and gradual transfer of photogenerated electrons from CdS QDs via the intermediate anatase TiO2 layer to rutile TiO2, and retards their back diffusion as well as recombination within the CdS QDs.7 Moreover, it is worth noting that the framework of rutile TiO2 NRs is a single-crystalline electron collector self-aligned on the FTO substrate with strong adhesion, which further promotes the shuttling of injected electrons from the CdS QDs to TiO2 and collection by the FTO substrate.7 In the meantime, photogenerated holes from the intermediate anatase TiO2 layer and rutile TiO2 NRs resulting from UV light excitation may also be effectively and gradually transferred to the VB of the CdS QDs, contributing to a more efficient separation and longer lifetime of the electron–hole pairs in the ternary core–shell heterostructure.
Conclusions
In summary, a facile and efficient self-assembly strategy for the preparation of a rutile@anatase TN@CdS QDs ternary core–shell heterostructure was developed, in which an intermediate anatase TiO2 layer was judiciously integrated at the interface between rutile TiO2 NRs and CdS QDs. The ternary core–shell heterostructure demonstrated remarkably enhanced photoelectrochemical and photocatalytic performances as a result of the monodisperse deposition of an anatase TiO2 layer and CdS QDs in an intimate fashion onto a framework of rutile TiO2 NRs, which provides the gradual transfer of photogenerated electrons for charge separation and visible light absorption. It is expected that our work could open the way for the design of various 1D semiconductor based core–shell heterostructures for promising photocatalytic applications.Acknowledgements
This work was supported by the Nanyang Technological University startup grant: M4080977.120, and the Singapore Ministry of Education Academic Research Fund (AcRF) Tier 1: M4011021.120. We thank Mr. Song Xiaohui, Mr. Fang Zhanxi, Prof. Zhang Hua and Prof. Chen Hongyu for their kind help for TEM measurements.References
- A. H. Hochbaum and P. D. Yang, Chem. Rev., 2010, 110, 527–546 CrossRef CAS PubMed.
- M. Law, L. E. Greene, J. C. Johnson, R. Saykally and P. Yang, Nat. Mater., 2005, 4, 455–459 CrossRef CAS PubMed.
- O. L. Muskens, J. G. Rivas, R. E. Algra, E. P. A. M. Bakkers and A. Lagendijk, Nano Lett., 2008, 8, 2638–2642 CrossRef CAS PubMed.
- F. Zhang, S. M. Niu, W. X. Guo, G. Zhu, Y. Liu, X. L. Zhang and Z. L. Wang, ACS Nano, 2013, 7, 4537–4544 CrossRef CAS PubMed.
- K. S. Leschkies, R. Divakar, J. Basu, E. Enache-Pommer, J. E. Boercker, C. B. Carter, U. R. Kortshagen, D. J. Norris and E. S. Aydil, Nano Lett., 2007, 7, 1793–1798 CrossRef CAS PubMed.
- B. Liu and E. S. Aydil, J. Am. Chem. Soc., 2009, 131, 3985–3990 CrossRef CAS PubMed.
- J. T Li, M. W. G. Hoffmann, H. Shen, C. Fabrega, J. D. Prades, T. Andreu, F. Hernandez-Ramirez and S. Mathur, J. Mater. Chem., 2012, 22, 20472–20476 RSC.
- J. Bang and P. V. Kamat, Adv. Funct. Mater., 2010, 20, 1970–1976 CrossRef CAS.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- A. Dawson and P. V. Kamat, J. Phys. Chem. B, 2001, 105, 960–966 CrossRef CAS.
- I. Robel, V. Subramanian, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2006, 128, 2385–2393 CrossRef CAS PubMed.
- W. T. Sun, Y. Yu, H. Y. Pan, X. F. Gao, Q. Chen and L. M. Peng, J. Am. Chem. Soc., 2008, 130, 1124–1125 CrossRef CAS PubMed.
- S. Banerjee, S. K. Mohapatra, P. P. Das and M. Misra, Chem. Mater., 2008, 20, 6784–6791 CrossRef CAS.
- A. Kongkanand, K. Tvrdy, K. Takechi, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2008, 130, 4007–4015 CrossRef CAS PubMed.
- R. Plass, S. Pelet, J. Krueger and M. Gratzel, J. Phys. Chem. B, 2002, 106, 7578–7580 CrossRef CAS.
- A. Zaban, O. I. Micic, B. A. Gregg and A. J. Nozik, Langmuir, 1998, 14, 3153–3156 CrossRef CAS.
- A. L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- L. M. Peter, D. J. Riley, E. J. Tull and K. G. Wijayantha, Chem. Commun., 2002, 1030–1031 RSC.
- Y. Tak, H. Kim, D. Lee and K. Yong, Chem. Commun., 2008, 4585–4587 RSC.
- I. Mora-Sero and J. Bisquert, J. Phys. Chem. Lett., 2010, 1, 3046–3052 CrossRef CAS.
- K. Y. Yan, L. X. Zhang, J. H. Qiu, Y. C. Qiu, Z. L. Zhu, J. N. Wang and S. H. Yang, J. Am. Chem. Soc., 2013, 135, 9531–9539 CrossRef CAS PubMed.
- M. Shalom, J. Albero, Z. Tachan, E. Martinez-Ferrero, A. Zaban and E. Palomares, J. Phys. Chem. Lett., 2010, 1, 1134–1138 CrossRef CAS.
- Y. Myung, D. M. Jang, T. K. Sung, Y. Sohn, G. Jung, Y. Cho, H. Kim and J. Park, ACS Nano, 2010, 4, 3789–3800 CrossRef CAS PubMed.
- H. Kim, J. Kim, W. Kim and W. Choi, J. Phys. Chem. C, 2011, 115, 9797–9805 CAS.
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- Y. Zhang and L.-W. Wang, Nano Lett., 2007, 7, 1264–1269 CrossRef CAS PubMed.
- B. Tian, X. Zheng, T. J. Kempa, Y. Fang, N. Yu, G. Yu, J. Huang and C. M. Lieber, Nature, 2007, 449, 885–889 CrossRef CAS PubMed.
- B. Tian, T. J. Kempa and C. M. Lieber, Chem. Soc. Rev., 2009, 38, 16–24 RSC.
- Y. J. Hwang, A. Boukai and P. Yang, Nano Lett., 2009, 9, 410–415 CrossRef CAS PubMed.
- F. Caruso, X. Y. Shi, R. A. Caruso and A. Susha, Adv. Mater., 2001, 13, 740–744 CrossRef CAS.
- A. Hanprasopwattana, T. Rieker, A. G. Sault and A. K. Dayte, Catal. Lett., 1997, 45, 165–175 CrossRef CAS.
- Y.-G. Guo, J.-S. Hu, H.-P. Liang, L.-J. Wan and C.-L. Bai, Adv. Funct. Mater., 2005, 15, 196–202 CrossRef CAS.
- I. S. Cho, Z. B. Chen, A. J. Forman, D. R. Kim, P. M. Rao, T. F. Jaramillo and X. L. Zheng, Nano Lett., 2011, 11, 4978–4984 CrossRef CAS PubMed.
- O. I. Micic, M. T. Nenadovic, M. W. Peterson and A. J. Nozik, J. Phys. Chem., 1987, 91, 1295–1297 CrossRef CAS.
- J. Zhang, J. G. Yu, M. Jaroniec and J. R. Gong, Nano Lett., 2012, 12, 4584–4589 CrossRef CAS PubMed.
- T. Nakanishi, B. Ohtani, K. Shimazu and K. Uosaki, Chem. Phys. Lett., 1997, 278, 233–237 CrossRef CAS.
- N. Zhang, Y. H. Zhang, X. Y. Pan, X. Z. Fu, S. Q. Liu and Y.-J. Xu, J. Phys. Chem. C, 2011, 115, 23501–23511 CAS.
- M. J. Natan, J. W. Thackeray and M. S. Wrighton, J. Phys. Chem., 1986, 90, 4089–4098 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mh00097d |
| This journal is © The Royal Society of Chemistry 2014 |