Growth of polythiophene nano-walls and their unique electrochemical and optical properties†
Bartlomiej
Kolodziejczyk
*,
Orawan
Winther-Jensen
,
Chun Hin
Ng
,
Shenghuang
Lin
,
Qiaoliang
Bao
and
Bjorn
Winther-Jensen
Department of Materials Engineering, Monash University, Clayton, 3800 Victoria, Australia. E-mail: bartlomiej.kolodziejczyk@monash.edu
First published on 29th April 2014
Abstract
It has been a persistent challenge to develop ordered conducting polymer nano- and micro-structures with a high active area. We herein report the method to produce and characterise a network of nanometre-sized walls (nano-walls) which forms on the surface of a conducting co-polymer composite film based on bithiophene and terthiophene prepared using vapour-phase polymerisation (VPP). The density and dimension of the walls are tunable in order to suit the application demands. The presence of nano-walls on the film surface increases the capacitance of the coating up to 3.4 times, making it a very promising candidate for energy and organic electronics applications. The nano-walls have also proved to be great candidates for photonic applications, efficiently guiding light. A two-step growth mechanism is proposed, which includes the reduction of Fe(III) and part of the tosylate ligands in the Fe(III) para-toluene sulphonate oxidant. This is the first report of a self-assembling conducting polymer material with nano-wall features induced by the conditions during VPP.
Conducting polymers are a rapidly developing field in materials science. They have found applications in organic electronics,1,2 energy applications,3–5 corrosion protection,6 tissue engineering7 and optical devices amongst many others. There are various approaches to the manufacturing of nano-structured materials in order to increase their available surface area. Previous attempts to create nano-structured materials of any kind required either complex manufacturing techniques (e.g. replica molding8), specialized equipment,9 high temperatures9,10 or expensive and rare catalysts.10a Vapour–liquid–solid growth10a,11 (VLS) and vapour–solid growth12,13 (VS) are the most common techniques applied today for producing one dimensional and two dimensional nano- and micro-structures. Most of the techniques mentioned above can be applied only to inorganic materials. Recently, self-assembly procedures have gained significant importance in the field of nanotechnology and materials science. Self-assembly is a process in which a disordered system of existing components or molecules forms an organized structure or pattern as a consequence of specific, local interactions between the components themselves, without external stimulus (molecular self-assembly). Electro-polymerization has been reported to be a viable way to create various spherical and circular conducting polymer nano-structures.14,15 However, electro-polymerization has not been reported to be suitable for creating rod-, wall- or wire-like nano-structures.
A particular oxidative (chemical) method for the polymerization of conducting polymers is vapor phase polymerization (VPP). VPP of conjugated polymers is by nature a VLS technique, but has not been reported to produce nano-structured materials till now. VPP has been used to form polymer materials with high conductivity and a high degree of order.16–18 VPP is a simple, fast and cheap way of creating good quality conducting thin films, and although the name may suggest otherwise, the polymerisation during VPP actually takes place in the condensed phase. In this procedure, a substrate is coated with an oxidant and placed in a heated chamber containing the appropriate monomer, and is held at a temperature above the monomer's melting point. Monomer vapour in the chamber then condenses on the substrate where it is oxidized to form polymer chains. Samples are later washed to remove excess oxidant and monomer. Recent work has shown that some sulphonic acid oxidants decompose during the oxidation process to highly volatile products, allowing for the washing and drying process to be avoided.19,20 The creation of a co-polymer material with enhanced properties through the use of VPP has been attempted previously.21 However, copolymerization using VPP has been shown to be difficult as there are many parameters to consider during the polymerization process – most important of which is the monomer vapour pressure. Monomer vapour pressure is directly related to temperature, and in many cases, the combination of monomers used to create copolymers has very different vapour pressures, and hence polymerization rates at a given reaction temperature. In this instance, the evaporation rates (i.e. amounts) of the monomers must be adjusted accordingly. The first copolymerized material using VPP was made of pyrrole and N-methylpyrrole using iron(III) chloride as an oxidant.22
We have previously reported VPP co-polymerisation of the two monomers bi- and ter-thiophene to produce films with enhanced (broader) absorption in the visible range.21 In the current paper, we report the surprise finding whereby under certain VPP conditions, polymer nano-walls were formed at longer polymerisation times sitting atop a polymer film. To examine the nature of the nano-wall formation, the progress of the nano-wall growth over time was studied. From the scanning electron microscopic (SEM) images in Fig. 1 it can be seen that the nano-walls only start to develop after ∼1.5 h of polymerisation and continue to grow higher with longer polymerisation times. The initial 30 minutes of polymerisation only results in a porous poly-thiophene layer and the thickness of this layer grows only slightly with longer polymerisation times. As the melting temperature of bi-thiophene is lower – and hence the vapour pressure is higher – than that of ter-thiophene, the rapid formation of this initial layer can be attributed to the polymerisation of bi-thiophene through the traditional oxidative route with the Fe(III)PTS oxidant. Similar polymerisation speeds for the VPP of bi-thiophene using Fe(III)PTS as an oxidant have been reported previously for lower temperatures than those used here.23
 | ||
| Fig. 1 SEM images of the surface (top six images) and cross-section (middle six images) of poly(bi–ter-thiophene) films polymerized for (a) 30 min, (b) 60 min, (c) 90 min, (d) 2 h, (e) 3 h and (f) 6 h. EDX analysis of the poly(bi–terthiophene) film performed at 15 kV; (g) SEM image, (h) sulphur distribution and (i) carbon distribution. | ||
It is expected that the formation of the initial polythiophene layer proceeds until all available Fe(III) is consumed and several experiments were therefore performed in order to determine whether the nano-walls are polymeric rather than an ordered combination of the bi- or ter-thiophene monomers. Samples were washed in ethanol, thiophene (Th) and acetonitrile (in which bithiophene and terthiophene monomers are highly soluble) without damaging the nano-walls. Samples were also left for around 120 hours in the oven at 110 °C; the melting point of bithiophene is 32–33 °C and the melting point of terthiophene is 93–95 °C. Leaving the polymer in the oven at this temperature for such an extended period of time would result in the evaporation of any remaining monomer. SEM images (Fig. S1, ESI†) of the samples after the treatments described above show no degradation in the nano-wall structure, thereby strongly indicating that the nano-walls are polymeric materials.
Elemental analysis using energy-dispersive X-ray spectroscopy (EDX) in Fig. 1g–i shows that the main components, carbon and sulphur, are evenly distributed throughout both the film and the nano-walls. No other elements (like iron or oxygen) were found in significant amounts. This indicates that the nano-walls are similar in composition to the underlying film, namely an organic thiophene-based material.
It then follows that a second oxidant source apart from Fe(III) must be present for the oxidative polymerisation of the nano-walls to proceed. We have recently shown that the para-toluene sulfonate ligand is able to act as an oxidant for VPP at very long polymerisation times.19,20 It is hereby proposed that this mechanism is primarily responsible for the formation of polythiophene nano-walls.
The proposed polymerisation mechanism is as follows. The initial bithiophene film is formed through oxidative polymerisation with Fe(III), resulting in a thin, spongy film:
| n/2(BTh) + nFe3+ + 3n(CH3C6H6SO3−) → (BTh)n/2 + nH+ + nFe2+ + 3n(CH3C6H6SO3−) |
After which, two PTS− molecules (per Fe3+) form the salt with Fe2+ while one PTS− can combine with a proton from the polymerisation reaction to give the acid-form of PTS, PTSa (CH3C6H4SO3H):
| nH+ + n(CH3C6H6SO3−) → n(HCH3C6H6SO3H) |
The melting point of PTSa is 38 °C and ∼103 °C for PTSa·H2O. PTSa can decompose at elevated temperatures (>100 °C) to produce volatile toluene and sulphur trioxide:19
| PTSa·H2O ⇄ C6H5CH3 + SO3 + H2O |
The equilibrium of this reaction strongly favours the production of PTSa (the reaction of SO3 with toluene is the commercial route to produce PTSa). However, SO3 is known to be a strong oxidant to thiophenes:19,20
| m/2(TTh) + m(HCH3C6H6SO3H) → (TTh)m/2 + mH+ + m(C6H5CH3 + SO2 + H2O) |
Note that the above equation assumes that TTh is the sole building-block for the nano-walls. This suggests that both monomers and the in situ produced SO3 oxidant are present as vapour inside the VPP chamber. The polymeric thiophene layer that is already formed is able to act as seeding for VLS growth,10a,11 thereby facilitating the slow growth of one- or two-dimensional nano-structures. Furthermore, it is hypothesised that the evaporated conjugated monomers present in the chamber slowly stack on top of each other, similar to the formation of Bechgaard salts, while being simultaneously polymerised.
In 1980, Bechgaard synthesized di-(tetramethyltetraselenafulvalene)-hexafluorophosphate – the first organic superconductor.24 This discovery led to the creation of the wide family of similar organic compounds known as Bechgaard salts.25 Bechgaard salts are simply crystals formed by conjugated organic molecules through the evaporation of the molecules and spontaneous stacking to form crystal structures. In addition to superconductivity at low temperatures, Bechgaard salts exhibit good conductivity at room-temperature in the π-stacking direction and a great number of other unique properties.25 A spontaneous stacking of traditional monomers for conducting polymer, such as thiophenes, during VPP or vacuum deposition has, to the best of our knowledge, not been reported previously.
From the SEM images in Fig. 1, it can be seen that while the underlying bithiophene film is porous and spongy, the nano-walls sitting atop are dense. It is reasoned that the terthiophene monomer is condensing on the top of the film to form crystals, which are being oxidized and polymerised at a later point. At first, it can be seen that the walls formed locally cross the surface of the film; with time the geometrical properties, namely the width, height and length, of the individual walls increase, thereby connecting single-standing nano-walls into bulk networks. In order to confirm this growing step, a simple control experiment was performed whereby a fixed amount of Fe(III)PTS oxidant was deposited on an identical substrate but instead of mixing the monomers inside the chamber, the oxidant-coated substrate was first exposed to (only) bithiophene for one hour and then transferred to a chamber with only terthiophene, where the polymerisation process was continued. The resulting material showed the same kind of “nano-wall on porous film” structure as seen with mixed monomers (Fig. S2, ESI†). This strongly indicates that bi- and ter-thiophene have separate roles in the process i.e. bithiophene being the main component of the porous film and terthiophene the main building-block of the nano-walls.
Fig. 2 below shows the effect of polymerisation time on several nano-wall properties. As expected, walls grow higher, wider and cover more area of the film as the polymerisation time increases. However, between 3 and 6 hours the further growth in wall height is very limited, which indicates that the process is running out of SO3 oxidant. This limitation makes sense as no further PTSa is produced after the formation of the initial polythiophene film.
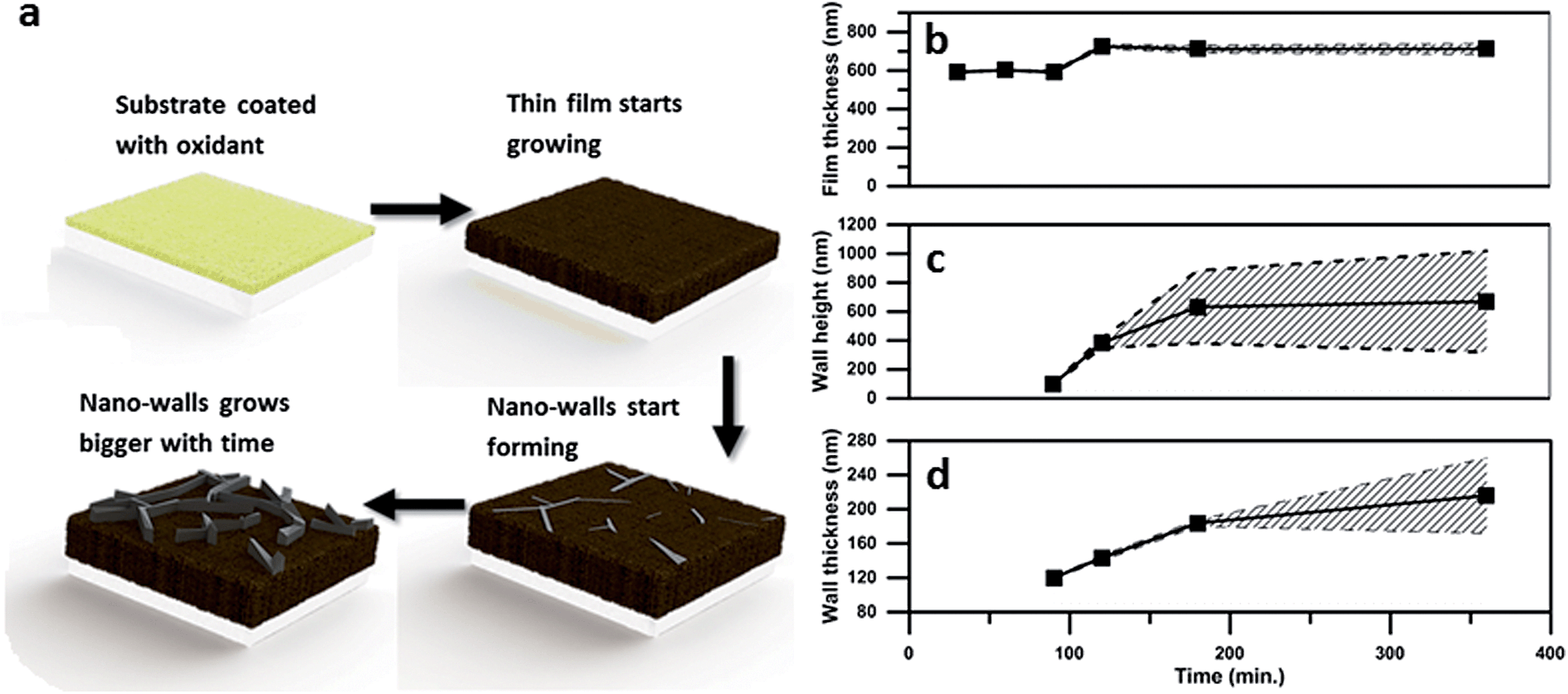 | ||
| Fig. 2 Geometrical properties of the film and nano-walls depending on the polymerization time. (a) Growth mechanism, (b) thickness of the underlying polymer film, (c) wall height, and (d) wall thickness. The average taken over several measurements is shown by the continuous black line. The shaded slashed background is the deviation calculated from the measurements (please see ESI† for the detailed calculations). | ||
The total surface area of the nano-walls was estimated by calculations based on SEM images and it was found that the surface area of the samples polymerized for 3 hours is 3.4 times higher than that of the flat film obtained after 1 h polymerisation.
The structural properties of nano-walls influence the contact angle between a water droplet and the polymerised thiophene material. The contact angle increases proportionally with the nano-wall geometry (width and height) and the coverage density of nano-walls on the polymer film, indicating the “lotus” effect well-known for nano-patterned hydrophobic materials. The increase in contact angle between polymer films synthesized for 30 min and polymer films with nano-features synthesized for 6 hours is almost doubled, reaching hydrophobicity with contact angle over 120°. A graph showing the increase in contact angle versus polymerization time is presented in the ESI (Fig. S6†).
Nano-wall growth was found to be substrate dependent, with the best quality nano-walls forming on oxidant-coated glass slides. Nano-walls can, however, be grown on other substrates such as gold-coated Mylar and silicon wafer. The difference in growth on various substrates may be linked to the thermal conductivity of the substrate materials, which can lead to local changes in growth conditions. Furthermore, ratios of BTh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TTh other than 2:3 (1:1 and 1:3) have also been shown to result in the production of nano-walls. The influence of polymerisation temperature and other polymerisation parameters on the wall growth is being investigated in detail and will be reported separately.
:TTh other than 2:3 (1:1 and 1:3) have also been shown to result in the production of nano-walls. The influence of polymerisation temperature and other polymerisation parameters on the wall growth is being investigated in detail and will be reported separately.
Raman spectroscopy was performed and the results revealed two polythiophene modes, called v1 and v2 as previously reported elsewhere.16 The 1500 cm−1 band can be identified as the v1 mode, associated with the end ring deformation on the polymer chain. The 1455 cm−1 band can be ascribed to the v2 mode, which is associated with the ring deformation in the central part of the polymer chain. The Effective Conjugation Coordinate (ECC) theory associates a shift in the v1 mode with a lower wavenumber when the polymer conjugation is increased. The ratio of v1/v2 intensity gives an estimation of the polymer chain length, where lower values are related to longer chain lengths. These two relations support the fact that relative chain lengths and conjugation lengths of polythiophene in nano-walls are slightly longer than those of the polymer film underneath. The plotted data are averages of several measurements taken at different spots for both the nano-walls and the polymer film. The difference in v1 shift is over 2 cm−1 and is considered a statistically significant shift. Longer conjugation and chain lengths for polythiophene are normally considered to produce a more conductive material.
FT-IR measurements performed on the powdered samples of the polymer film and the polymer film with nano-walls revealed similarities to previously reported data.26,27 Spectra for samples with and without nano-walls are almost identical with the only difference being the amplitude of the four peaks at around ∼1324, 1202, 1120 and 1029 cm−1,27 which are contributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1320 cm−1) and C–C (1202, 1120 and 1029 cm−1) ring stretching vibrations27 and associated with higher doping levels in the film.27 The increase in dopant level for thin films with nano-wall features would explain higher conductivity of the samples (Fig. 3).
C (1320 cm−1) and C–C (1202, 1120 and 1029 cm−1) ring stretching vibrations27 and associated with higher doping levels in the film.27 The increase in dopant level for thin films with nano-wall features would explain higher conductivity of the samples (Fig. 3).
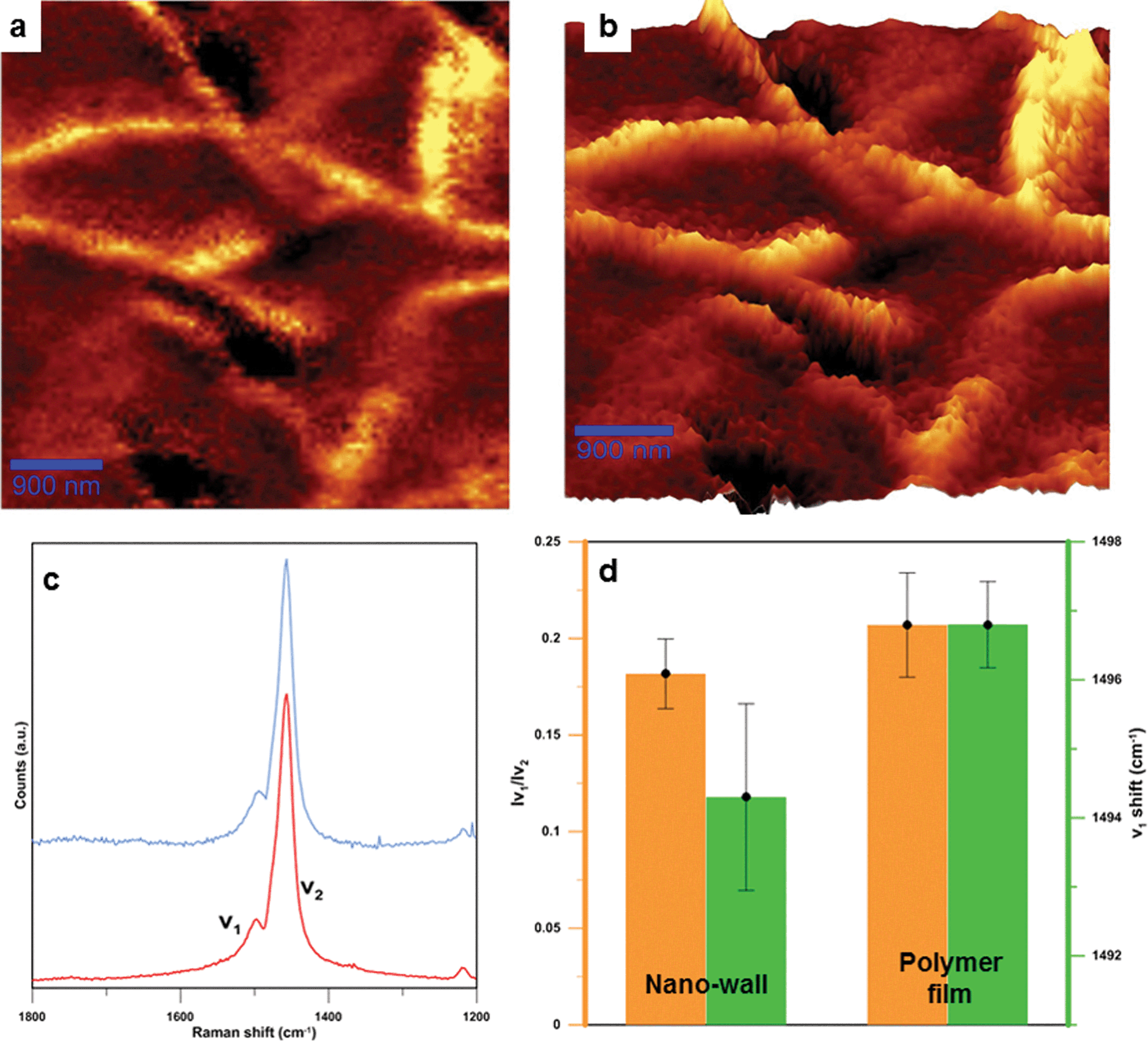 | ||
| Fig. 3 Raman spectroscopy characterizations of nano-walls. (a) Raman map of integrated v2 mode (ranging from 1450 to 1600 cm−1). (b) 3D plot of Raman map, in which brighter areas represent nano-walls while darker areas are polymer films. (c) Raman spectra for the nano-wall (blue) and the polymer film (red), with two visible modes named v1 and v2. The enlarged Raman graph can be found in Fig. S8 in the ESI.† (d) Average chain length (orange) and conjugation length (green) for the polymer film and the nano-wall. | ||
Cyclic voltammetry (CV) measurements were performed to determine the electrochemical properties and to compare the capacitance of samples with varying polymerisation times (Fig. 4a). The cyclic voltammograms of the samples with and without walls are consistent with previous reports for bi- and terthiophene.23,28 Peaks occurring during oxidation at ∼0.6 V vs. Ag/AgCl and during reduction at ∼−1.5 V vs. Ag/AgCl are attributed to “charge trapping” (conformational relaxation) oxidation and reduction peaks, respectively.29 The redox capacitance of the films has been estimated using the reduction peaks shown in Fig. 4b. As shown by the shaded areas, the presence of nano-walls greatly enhances the redox capacitance of the nano-wall poly(bi–terthiophene) films. This confirms the redox activity of the nano-walls and that there is no significant difference in the redox signature of the walls compared to other polythiophene materials. A peak area ratio of 3.4 was found when comparing samples with and without nano-walls, polymerised for 3 h and 0.5 h, respectively. This is in the same range as the calculated increase in surface area (see ESI Table S1†) indicating that the increased redox activity, under the measured conditions, is largely linked to the increase in electrolyte/polymer nano-wall area rather than the volume or height of the polymer nano-walls. The overall conductivity of the samples was measured and found to be in the range of 2.2 S cm−2, this is in agreement with previously reported values.21 The conductivity difference between the flat poly(thiophene) films and the nano-wall poly(thiophene) films is in the range of 0.2 S cm−2 and considered negligible. This is due to the nature of the experiment, the measurements were performed on dry samples where the nano-wall contribution to overall conductivity is minimal. Conductivity of individual nano-walls will be reported in a separate communication.
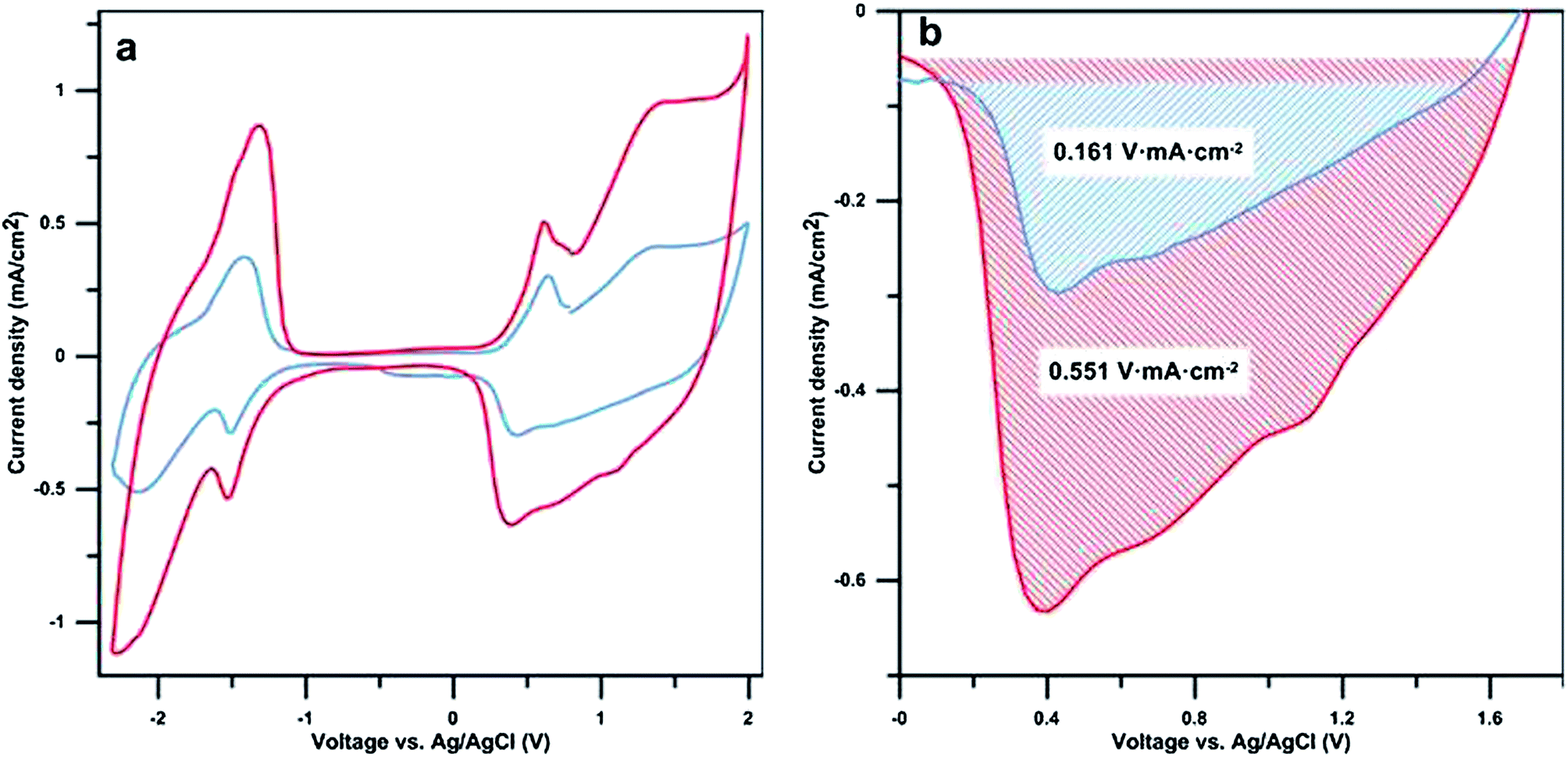 | ||
| Fig. 4 (a) Cyclic voltammetry measurements were performed under a nitrogen atmosphere on poly(bi–terthiophene) films polymerized for 3 hours (with nano-walls, red line) and 30 min (without nano-walls, blue line). (b) A close-up view of the reduction peak at 0.4 V from (a). The scan rate was 20 mV s−1 in an electrolyte consisting of 0.1 M tetrabutylammonium hexafluorophosphate in propylene carbonate. Both samples were polymerized at 105 °C. | ||
To demonstrate the potential use of nano-walls as a waveguide, an experiment using a point light source and an optical microscope in a dark field mode was performed. The result showed that some nano-walls can act as efficient nano-waveguides in the optical spectrum. In this type of single-mode strip waveguide, light travels through the polymer nano-wall material with negligible losses; the loss in light intensity has been calculated for nano-walls “A-B1” and “A-B2” and is equal to 0.7 and 4.7 μW μm−1 (1.4 and 9.4% μm−1), respectively. The higher loss from waveguide “A-B2” is due to the branched structure of the wall, where light is scattered while passing through the joints (Fig. 5a). Surprisingly, the same light is being absorbed by the polymer film beneath the nano-walls when light is shone directly on it. From these results, an obvious advantage to be considered is that the nano-walls can create a network of branched waveguides in which case light can travel not only along single nano-walls but also split and travel in multiple directions. In some cases, nano-walls show significant light leakage, we believe that this can be explained by the non-uniform geometry of the wall as well as different refractive indices between the walls which lead to a low coupling efficiency. This effect can be found in the ESI (Fig. S5†).
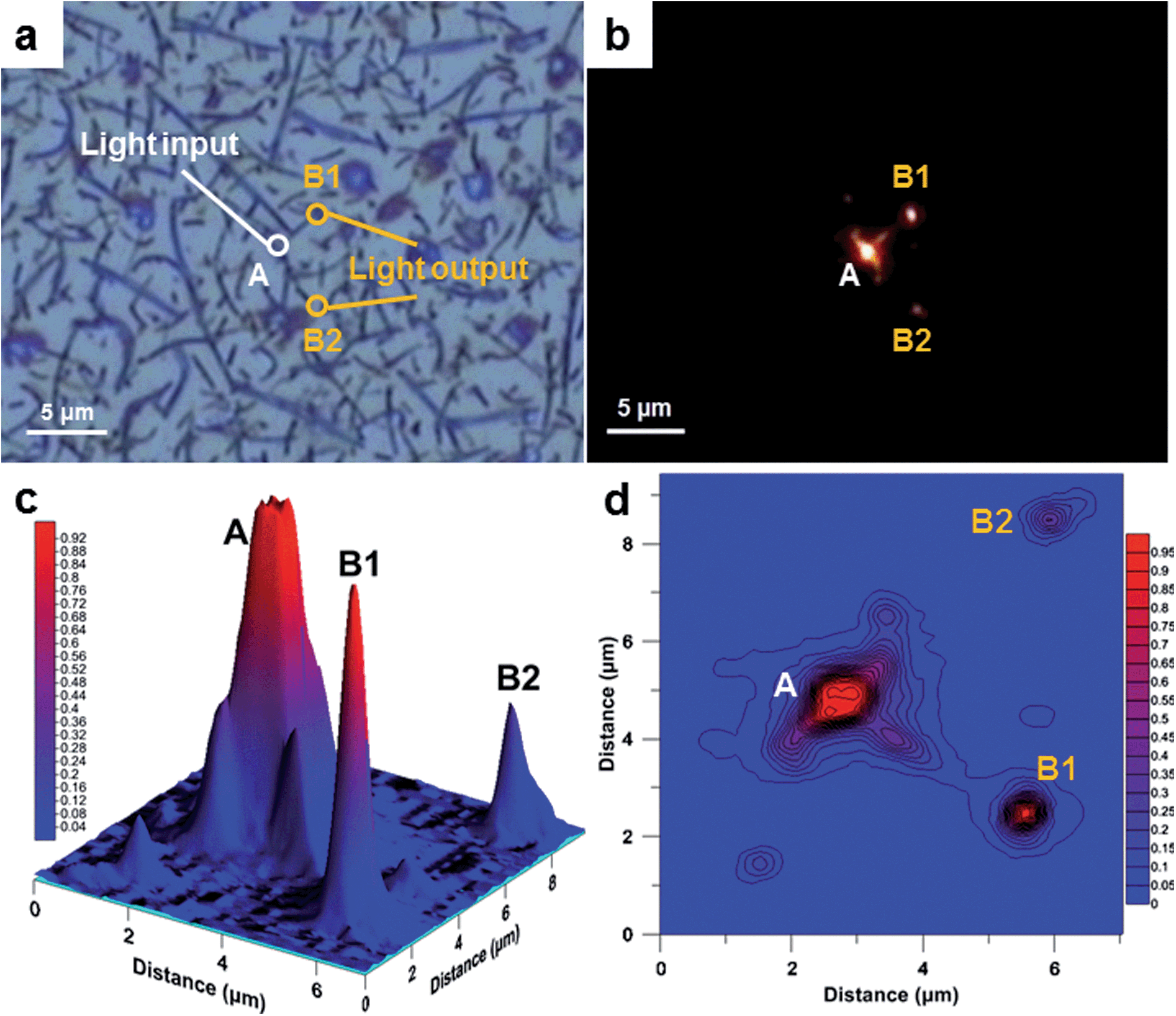 | ||
| Fig. 5 Waveguide experiment. (a) Optical image showing the excitation spot at the nano-wall. (b) A dark field image with the visible excitation spot and the two light outputs from branched nano-walls. (c) 3D map of the light intensity. (d) Contour map of the light intensity. | ||
We have reported the first conducting polymer nano-walls grown from the vapor phase at relatively low temperatures through a combination of monomer stacking and oxidative polymerisation without the use of expensive catalysts. The results from various experiments confirm that walls are made of a dense polymeric material and not from condensed or crystallized monomers. Samples from a fixed mass ratio of BTh:TTh monomers of 2:3 have been found to produce the best quality nano-walls. The presence of nano-walls drastically increases the redox capacity of the film in accordance with a three-fold increase in surface area. Nano-wall growth is found to be substrate-dependent with the best quality nano-walls produced on glass slides. The wall morphology can be easily tuned by varying polymerization parameters. This discovery opens up a new field of nanostructured polymer materials for a variety of applications including energy, medical and optical devices where high surface areas are desired. Finally, nano-walls are shown to be good waveguides. Due to their nano-size, polythiophene waveguides have the potential to find application in nano-photonics or as components in integrated optical nano-circuits.
Methods
2,2′-Bithiophene (BTh) and 2,2′:5′,2′′-terthiophene (TTh) were supplied by Sigma-Aldrich. Ferric p-toluenesulfonate (Fe(III)PTS) in 40% butanol was obtained from YACOO Chemical Reagent Co. Ltd. All materials were used without further purification. In short, Fe(III)PTS solution was spin-coated onto the desired substrate using a Laurell spin-coater at 1500 rpm for 30 seconds. A closed chamber was placed in a heated silicon oil bath, as shown in Fig. S4.† The temperature in the bath was controlled using a thermocouple. Monomer was placed inside the chamber once the desired temperature was reached. After 2–5 min, the oxidant-coated substrate was placed in the chamber. For poly(bi–terthiophene) films, both monomers were added to the chamber simultaneously at 105 °C. The mass ratio between bithiophene and terthiophene was fixed at 2:3 (12 mg:18 mg) as guided by the differences in vapour pressure. Poly(terthiophene) and poly(bi–terthiophene) were polymerized for 3 hours at 105 °C whereas poly(bithiophene) was polymerized for 1 hour at 70 °C. Polymerised films were left to cool down to room temperature, rinsed carefully with ethanol, and kept in ethanol for around 12 hours. Films were prepared on microscope glass slides for SEM and EDX analyses. Glass sputter-coated with a thin conductive gold layer was used as the substrate for electrochemical measurements.
Contact angle measurements were performed using a simple setup with a stage and a camera. An automatic pipette was used to produce distilled water droplets with the same volume in every case. The water droplet volume was 10 μl. Gathered images were processed in Matlab and the contact angle was calculated. Measurements have been reproduced several times for every sample to calculate the average and the standard deviation. The contact angle graph for samples polymerized at different times can be found in Fig. S6 in the ESI.†
Cyclic voltammetry and capacitance measurements were performed using a standard 3-electrode setup in propylene carbonate with a 0.1 M tetrabutylammonium hexafluorophosphate supporting electrolyte. Propylene carbonate and tetrabutylammonium hexafluorophosphate were dried using molecular sieves under vacuum. All electrochemical experiments were performed in a glove box under a nitrogen atmosphere. The reference electrode was a 0.01 M Ag/AgClO4 calibrated to the ferrocene/ferrocenium (0.1 M) redox couple. The counter electrode was a platinum wire. The experiment was performed on a Princeton Applied Research VMP2 multichannel potentiostat at 20 mV s−1.
SEM images were obtained using a JEOL 7100F Field Emission Gun Scanning Electron Microscope at 5 kV for the morphology study. EDX was performed at 15 kV for elemental analysis. Experiments were performed on gold sputter-coated samples.
Estimation of the area coverage of the walls and total area increase was done by processing the SEM images using Matlab software and Image Processing Toolbox. For more information please refer the ESI.†
Raman mapping has been performed at room temperature on a WITec Confocal Raman Microscope alpha300 R equipped with a WITec UHTS 300 Raman Spectrometer and a green laser source at 532 nm excitation wavelength. Raman intensity from 1450 to 1600 cm−1 has been integrated to build the maps.
Waveguide measurements were performed on the Raman setup described above. Instead of the laser point, light has been introduced to the walls. The experiment was performed in a dark field mode to capture the image of guided light.
Raman spectra were obtained on a Jobin Yvon T64000 Raman spectrometer equipped with a blue 487.9 nm laser. A baseline correction was conducted and the resulting spectra normalised by using the v2 mode intensity. Deconvolution was done using Spekwin32 software. Intensity ratio between v1 and v2 mode and v1 shift have been averaged from several data points for both the polymer films and nano-walls and the standard deviation was calculated. Nano-walls for Raman measurements were gently removed from the polymer film using a cotton swab and placed on a glass substrate.
Conductivity values were calculated using the film thickness and sheet resistance of the samples. Thickness measurements were performed on a Veeco DekTak 150 Surface Profiler and the sheet resistance was measured using a four point probe (Jandel Model RM3). The test was performed three times in different places of the sample and the average value was calculated.
The FT-IR experiments were performed on a Perkin Elmer Spectrum100 FTIR with diamond ATR on previously powdered samples.
Contributions
B.W.J. and B.K. initiated the study and designed the experiments. B.K. prepared samples and performed the electrochemical measurements. O.W.J. performed SEM and EDX imaging. B.K. developed the SEM image processing technique and prepared the manuscript. CH.H.N. and S.L. performed the Raman experiment. S.L. performed the waveguide experiment under Q.B.'s supervision. B.W.J. coordinated the project. All authors revised and approved the manuscript.Acknowledgements
B.W.J. gratefully acknowledges the Australian Research Council for fellowship. Prof. Douglas MacFarlane is acknowledged for valuable discussions. The microscopic facilities under the ARC's COE for Design in Light Metals funding scheme within Monash Centre for Electron Microscopy are gratefully acknowledged. Qiaoliang Bao acknowledges the support from ARC DECRA, Monash Larkins Fellowship and MCN Technology Fellowship.References
- L. H. Jimison, S. A. Tria, D. Khodagholy, M. Gurfinkel, E. Lanzarini, A. Hama, G. G. Malliaras and R. M. Owens, Measurement of Barrier Tissue Integrity with an Organic Electrochemical Transistor, Adv. Mater., 2012, 24(44), 5919–5923 CrossRef CAS PubMed.
- M. G. Harrison and R. H. Friend, Optical Applications, in Electronic Materials: The Oligomer Approach, Wiley-VCH Verlag GmbH, 2007, pp. 516–558 Search PubMed.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Conjugated Polymer-Based Organic Solar Cells, Chem. Rev., 2007, 107(4), 1324–1338 CrossRef PubMed.
- G. A. Snook, P. Kao and A. S. Best, Conducting-polymer-based supercapacitor devices and electrodes, J. Power Sources, 2011, 196(1), 1–12 CrossRef CAS PubMed.
- B. Kolodziejczyk, O. Winther-Jensen, D. R. MacFarlane and B. Winther-Jensen, Conducting polymer alloys for photo-enhanced electro-catalytic oxygen reduction, J. Mater. Chem., 2012, 22(21), 10821–10826 RSC.
- M. Rohwerder, Intelligent Corrosion Protection by Conducting Polymers, in Smart Coatings II, American Chemical Society, 2009, vol. 1002, pp. 274–287 Search PubMed.
- J. H. Collier, J. P. Camp, T. W. Hudson and C. E. Schmidt, Synthesis and characterization of polypyrrole–hyaluronic acid composite biomaterials for tissue engineering applications, J. Biomed. Mater. Res., 2000, 50(4), 574–584 CrossRef CAS.
- H. Yoon, A. Ghosh, J. Y. Han, S. H. Sung, W. B. Lee and K. Char, Nanowalls: Lateral Buckling of High Aspect Ratio Janus Nanowalls, Adv. Funct. Mater., 2012, 22(17), 3530 CrossRef CAS.
- Y. Wu, B. Yang, B. Zong, H. Sun, Z. Shen and Y. Feng, Carbon nanowalls and related materials, J. Mater. Chem., 2004, 14(4), 469–477 RSC.
- (a) H. T. Ng, J. Li, M. K. Smith, P. Nguyen, A. Cassell, J. Han and M. Meyyappan, Growth of Epitaxial Nanowires at the Junctions of Nanowalls, Science, 2003, 300(5623), 1249 CrossRef CAS PubMed; (b) V. Krivchenko, P. Shevnin, A. Pilevsky, A. Egorov, N. Suetin, V. Sen, S. Evlashin and A. Rakhimov, Influence of the growth temperature on structural and electron field emission properties of carbon nanowall/nanotube films synthesized by catalyst-free PECVD, J. Mater. Chem., 2012, 22(32), 16458–16464 RSC.
- A. M. Morales and C. M. Lieber, A Laser Ablation Method for the Synthesis of Crystalline Semiconductor Nanowires, Science, 1998, 279(5348), 208–211 CrossRef CAS.
- X. Jiang, T. Herricks and Y. Xia, CuO Nanowires Can Be Synthesized by Heating Copper Substrates in Air, Nano Lett., 2002, 2(12), 1333–1338 CrossRef CAS.
- S. S. Brenner and G. W. Sears, Mechanism of whisker growth—III nature of growth sites, Acta Metall., 1956, 4(3), 268–270 CrossRef CAS.
- H. Karami, M. G. Asadi and M. Mansoori, Pulse electropolymerization and the characterization of polyaniline nanofibers, Electrochim. Acta, 2012, 61, 154–164 CrossRef CAS PubMed.
- M. C. Turhan, A. Sezai Sarac, A. Gencturk, H.-D. Gilsing, H. Faltz and B. Schulz, Electrochemical impedance characterization and potential dependence of poly[3,4-(2,2-dibutylpropylenedioxy)thiophene] nanostructures on single carbon fiber microelectrode, Synth. Met., 2012, 162(5–6), 511–515 CrossRef CAS PubMed.
- P. M. Bayley, B. Winther-Jensen, D. R. MacFarlane, N. M. Rocher and M. Forsyth, Enhanced properties in chemically polymerized poly(terthiophene) using vapour phase techniques, React. Funct. Polym., 2008, 68(7), 1119–1126 CrossRef CAS PubMed.
- A. Mohammadi, M. A. Hasan, B. Liedberg, I. Lundström and W. R. Salaneck, Chemical vapour deposition (CVD) of conducting polymers: Polypyrrole, Synth. Met., 1986, 14(3), 189–197 CrossRef CAS.
- D. Bhattacharyya, R. M. Howden, D. C. Borrelli and K. K. Gleason, Vapor phase oxidative synthesis of conjugated polymers and applications, J. Polym. Sci., Part B: Polym. Phys., 2012, 50(19), 1329–1351 CrossRef CAS.
- J. Hadiono So, D. Mayevsky, O. Winther-Jensen and B. Winther-Jensen, A novel route for polymerisation of thiophene based conducting polymers using trace-free oxidants, Polym. Chem., 2014, 5, 361–364 RSC.
- C. Ong, P. M. Bayley, O. Winther-Jensen and B. Winther-Jensen, Toward a trace-free oxidant[mdash]insight into unexpected high yields of vapor phase polymerized polyterthiophene, Polym. J., 2013, 45, 391–395 CrossRef CAS.
- B. Kolodziejczyk, D. Mayevsky and B. Winther-Jensen, Enhanced absorption spectra of conducting polymers co-polymerised from thiophene derivatives, RSC Adv., 2013, 3, 4568–4573 RSC.
- Y. Fu, R. A. Weiss, P. P. Gan and M. D. Bessette, Conductive elastomeric foams prepared by in situ vapor phase polymerization of pyrrole and copolymerization of pyrrole and N-methylpyrrole, Polym. Eng. Sci., 1998, 38(5), 857–862 CAS.
- B. Winther-Jensen, J. Chen, K. West and G. Wallace, Vapor Phase Polymerization of Pyrrole and Thiophene Using Iron(III) Sulfonates as Oxidizing Agents, Macromolecules, 2004, 37(16), 5930–5935 CrossRef CAS.
- D. Jérome, A. Mazaud, M. Ribault and K. Bechgaard, Superconductivity in a synthetic organic conductor (TMTSF)2PF 6, J. Phys., Lett., 1980, 41(4), 95–98 CrossRef.
- K. Bechgaard, C. S. Jacobsen, K. Mortensen, H. J. Pedersen and N. Thorup, The properties of five highly conducting salts: (TMTSF)2X, X = PF6−, AsF6−, SbF6−, BF4− and NO3−, derived from tetramethyltetraselenafulvalene (TMTSF), Solid State Commun., 1980, 33(11), 1119–1125 CrossRef CAS.
- T. Matsuura and Y. Shimoyama, Growth kinetics of self-assembled monolayers of thiophene and terthiophene on Au(111): An infrared spectroscopic study, Eur. Phys. J. E: Soft Matter Biol. Phys., 2002, 7(3), 233–240 CrossRef CAS.
- C. Yong and Q. Renyuan, IR and Raman studies of polythiophene prepared by electrochemical polymerization, Solid State Commun., 1985, 54(3), 211–213 CrossRef.
- A. Sezai Sarac, U. Evans, M. Serantoni and V. J. Cunnane, Electrochemical and morphological study of the effect of polymerization conditions on poly(tetrathiophene) with emphasis on carbon fiber microelectrodes: A cyclic voltammetry and atomic force microscopy study, Carbon, 2003, 41(14), 2725–2730 CrossRef.
- L. Pigani, R. Seeber, F. Terzi, O. Cerri, M. Innocenti, R. Udisti and G. Sanna, Relaxation phenomena and structural modifications of substituted polythiophenes during the p-doping processes. An electrochemical and morphological study, Electrochim. Acta, 2006, 51(13), 2698–2705 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mh00016a |
| This journal is © The Royal Society of Chemistry 2014 |