 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward the ideal synthesis and molecular function through synthesis-informed design
Paul A.
Wender
*
Department of Chemistry, Department of Chemical and Systems Biology, Stanford University, CA 94305, USA. E-mail: wenderp@stanford.edu
First published on 3rd March 2014
Abstract
This Highlight describes factors that contribute to an ideal synthesis, including economies (step, time, atom, solvent, energy) and orientations (target, diversity, safety, function), and the role of synthesis-informed design directed at function in advancing synthesis and its impact on science.
 Paul A. Wender | Paul Wender is a product of Wilkes (Stine), Yale (Ziegler), and Columbia (Stork) Universities. He has served on the faculties of Harvard and Stanford and is currently the Bergstrom Professor of Chemistry and Professor (by courtesy) of Chemical and Systems Biology. His research interests are directed at solving problems in synthesis, chemistry, biology, medicine and materials research. His group's efforts have been recognized with several ACS Awards and recently the Tetrahedron Prize (2012) and Prelog Medal (2013). He is also a recipient of the Hoagland, Bing, and Dean's Distinguished Teaching Awards. He is a member of the National Academy of Sciences, Fellow of the American Academy of Arts and Sciences and the American Association of Arts and Sciences, and an elected member of the Royal Spanish Academy of Sciences. |
“The great book, always open and which we should make an effort to read, is that of Nature”. Antoni Gaudi
3.8 billion years of chemical evolution has produced a molecular library of unsurpassed size, structural diversity and functional value – our planet's chemome. Only in the last few decades have we started to develop the analytical tools needed to “access and read” this vast treasure trove of molecularly encoded information. The lessons learned thus far have been transformative, revolutionizing chemistry and most other scientific disciplines from molecular anthropology to zoology. New drugs (e.g., taxol), imaging agents (e.g., green fluorescent protein), materials (e.g., biopolymers), research tools (e.g., RNAi), catalysts (e.g., ribozymes), reactants (e.g., enediynes) and reaction processes (e.g., self-replication, bond activation, photosynthesis) have been uncovered, providing a wealth of fundamentally new knowledge and inspiring new innovations of great societal, health and economic benefit.
Notwithstanding the enormous potential value of Nature's library, tapping this vast body of information and inspiration has had and continues to have its challenges. Amongst these is our varied ability to not only make but also supply Nature's molecules.1 Clearly, synthesis has made remarkable strides forward in addressing this challenge by enabling access to many natural products once considered difficult if not impossible to make. Woodward's view of erythromycin as a “hopelessly complex” synthetic target in the 1950s, for example, gave way with the emergence of new methodology and analytical tools and the benefit of brilliant and dedicated coworkers, to his group's impressive synthesis of this very same target some 25 years later.2 Imaginative solutions to comparably, and even more complex targets have appeared with increasing frequency, collectively demonstrating the emerging firepower of contemporary synthesis and the impact of those who have creatively advanced its frontiers. Most natural product targets can now be made in the laboratory if suitable resources are provided. The challenge now, and for many targets it is formidable, is to do so in a green, step- and time-economical, if not ideal, way.3 Addressing this challenge will take more than improvements in reaction and synthesis efficiency as a long synthesis, even if it proceeds in 100% overall yield and thus 100% selectivity, is still a long synthesis with associated costs in time, effort, resources, and environmental impact.
There are three principal ways to more commonly achieve supply-impacting syntheses (Fig. 1: 1–3). While varying in stages of advancement, all involve a more holistic approach to the problem of synthesis design, i.e., one involving not only optimization of single reactions but also of the entire strategy, its implementation and its downstream consequences (e.g., waste and environmental impact). The commonly used and often effective approach to a supply-impacting synthesis is to optimally sequence reactions selected from the current reaction lexicon (Fig.1: 1). For a given synthetic problem, some sequences work, others work even better and some do not impact supply (for metrics read on). Given that most reactions, including the venerable Diels–Alder cycloaddition, allow for only two target bonds to be formed per synthetic operation and many targets require numerous bonds to be formed to connect with starting materials, this approach has its mathematical limitations as longer syntheses generally produce less final product due to the arithmetic of overall yield attrition. Other economies are similarly adversely impacted by increased step count. To address this problem, there is a growing interest in and use of processes that allow for greater target-relevant complexity increases per step, i.e., for more bonds to be made per synthetic operation. The use of multicomponent reactions, serialized sequences (a.k.a., domino or cascade processes) and strategies derived therefrom provide attractive ways to achieve greater per-operation complexity increases and therefore shorter synthetic sequences and better economies.
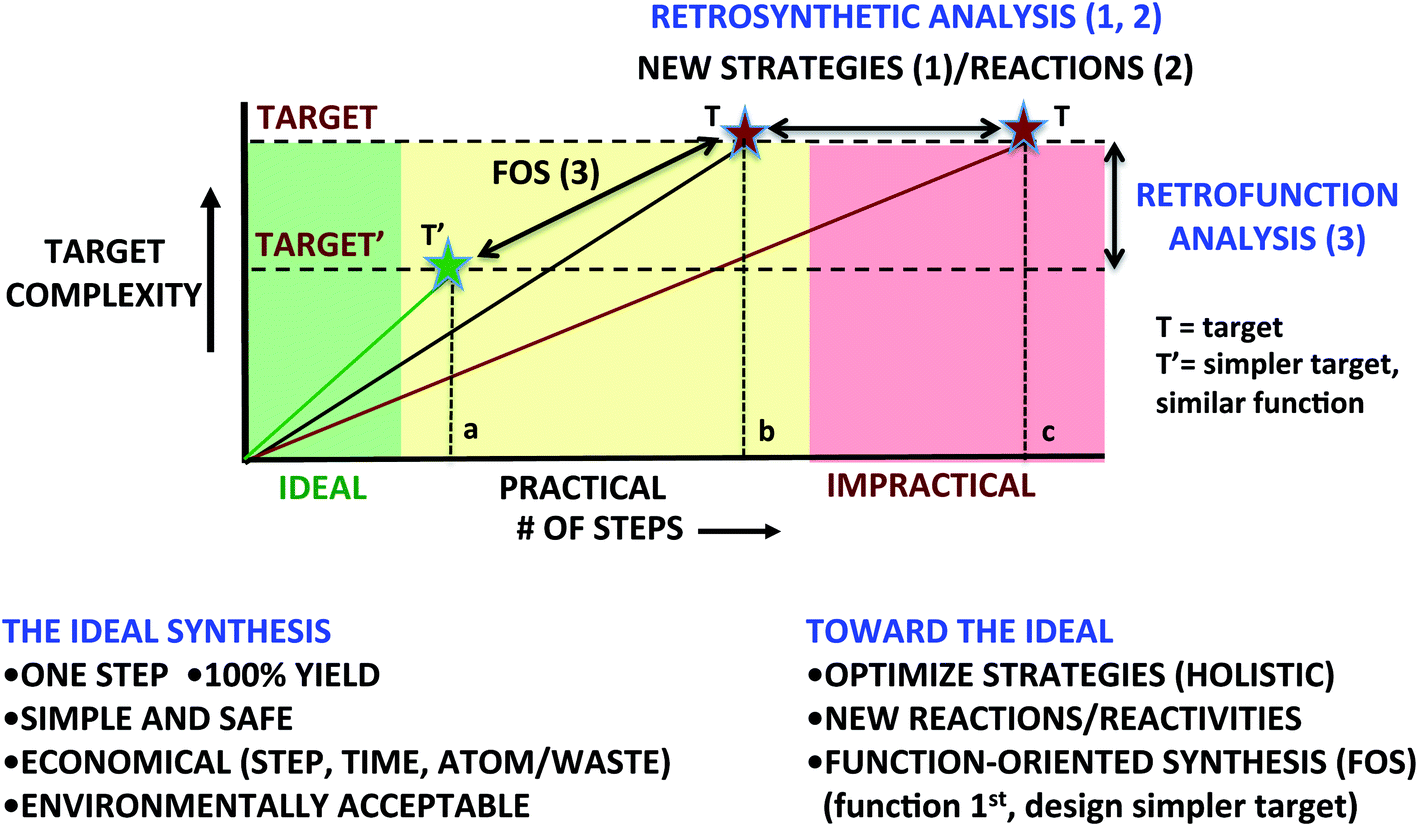 | ||
| Fig. 1 Toward the ideal synthesis. Optimized sequences, step-saving strategies (1): e.g., cascade, domino and multicomponent processes) and related new reactions (2) can reduce the number of steps needed to access a target of complexity “T” from “c” to “b”. Alternatively, if in addition to synthetic studies the goal is target function, synthesis-informed design of a simplified target “T′” with function equivalent to “T”, i.e. function-oriented synthesis (3), could lead to a greatly shortened (a) and thus practical synthesis. | ||
A related second approach, also based on achieving greater increases in target-relevant complexity, is obviously to expand the lexicon of reactions (Fig. 1(2)). New reactions and reactivities change the way one thinks about bond construction. They enable new and often greener synthetic strategies with more choices for route selection. Think about the synthesis of say 1-carbomethoxycyclohex-3-ene now and making the same target before the introduction of the Diels–Alder cycloaddition. The difference is transformative as the cycloaddition provides a new way to think about the problem and not just improvements over existing processes.
A third approach (Fig. 1(3)) to improved synthetic efficacy places an initial emphasis, not on target structure but on target function (e.g., imaging agents, medicinal agents, materials, diagnostics, catalysts, probes, etc.). This change in orientation from structure to function has a dramatic impact on how one defines the synthetic problem and thus on approaches to its solution. It is based on the view that most syntheses are directed at targets with some desired function. Indeed the first section of most publications and proposals related to synthesis often justify a synthesis effort on the basis of the target presenting both challenging synthetic problems and great potential or demonstrated value associated with its function. A central tenet of this third approach, which we have referred to as “function-oriented synthesis” (FOS),4 is that a given function can be derived from a variety of structures. It follows that one could design simpler and therefore synthetically more accessible targets with comparable or improved function. This does not eliminate the opportunity to address new synthetic challenges as one could also design targets that would serve that purpose as well. The FOS approach thus shifts the problem from an initial focus on structure and retrosynthetic analysis to an initial focus on function and “retrofunction” analysis. An especially attractive aspect of this approach is that one creates new targets – inspired by Nature or by de novo design, and then associated innovative strategies to address their supply. Synthetic chemists are superbly positioned to advance this field as it draws on translating modes of action, i.e., structure-driven mechanism, to new candidate structures that might exhibit that same function and selecting from the latter the ones that could be the most effective and most readily synthesized. It clearly creates many exciting opportunities for chemists to design new, synthetically accessible structures with improved or totally new functions.
Common to all three approaches (Fig. 1(1–3)) is the goal of moving synthesis to a more effective level marked by improvements in quality, speed and value. To do this, metrics are needed to quantify “improvements”. Comparisons with past achievements provide one means of measuring progress relative to the reaction and strategy lexicon of the past. Such comparisons generally favor newer work as newer science draws on the rich legacy of the past and new advances. Another approach is to start instead with a more aspirational measure of progress, comparing a plan against what might be an ideal solution to a problem. Such prospective comparisons do more to accelerate advancement of the field as they define and orient thinking toward ultimate goals and not just improvements over the past.
The “ideal synthesis” provides a template for measuring a synthetic plan against what might ultimately be possible. It has been defined in various ways and for various purposes over the years, often reflecting the intrinsic objectives of synthesis as well as the emerging priorities of the time. Jim Hendrickson – a friend and former collaborator, offered the following perspective drawn in part from his seminal contributions to computer-based synthesis design: “The ideal synthesis creates a complex skeleton from simpler starting materials and so must link several such synthon molecules via construction reactions. Ideally, the synthesis would start from available small molecules so functionalized as to allow constructions linking them together directly, in a sequence only of successive construction reactions involving no intermediary refunctionalizations, and leading directly to the structure of the target, not only its skeleton but also its correctly placed functionality. If available, such a synthesis would be the most economical, and it would contain only construction reactions.”5 There is great insight in this view especially as it relates to many aspects of computer-based design. There are also issues that are subject to interpretation and other unmentioned factors that go beyond connectivity analysis and are of major importance in contemporary synthesis design.
In the 1980s, sensitive to and emphasizing the importance of starting material availability (irrespective of its complexity), as well as operational ease, step economy, and most significantly safety – this last a matter of unique and pre-eminent importance in a comprehensive definition of an ideal synthesis, we offered the following: “ideal syntheses [are] those in which the target molecule is assembled from readily available starting materials in one simple, safe, economical, and efficient operation.”6 The motivation here was to chart progress by comparisons, numerical where possible, to what might ultimately be possible if not ideal and to integrate the importance of simple and safe operations into synthesis design plans.
There are points of significance in the above definition that merit emphasis. Safety, always a priority and indispensable determinant of a successful synthesis plan, clearly is a first priority in any comprehensive definition of an “ideal synthesis” as it increasingly influences if not drives decisions about synthesis design, route selection and execution. Indeed as we evolve the various “orientations” of synthesis (e.g., diversity-oriented synthesis (DOS), function-oriented synthesis (FOS), target-oriented synthesis (TOS)), “safety-oriented synthesis” (SOS), an emphasis introduced herein, will assume a position of unique importance and generality. Given the role of safe practices to the field, it is expected that SOS will receive even greater attention in future efforts to design, evaluate, and execute synthetic plans. It clearly addresses the interdependence of design and execution. In the final analysis, the exciting and demanding challenge of synthesis is that one must address all factors that are associated with the design and execution of a plan and safety is a consideration of pre-eminent importance and an essential component of any definition.
Starting material availability, whether simple or complex, is another obvious consideration in planning how best to address a synthetic need. While simple starting materials are often sought in a plan, it is more generally the availability of a starting material, irrespective of whether it is simple or complex, that determines whether the synthesis will indeed prove to be the most step economical and green if not ideal route. Indeed semi-synthesis, based generally on complex starting materials, has saved the day in many efforts to achieve supply-impacting syntheses including, for example, the semi-syntheses of the medicinal agent taxol7 and of prostratin,8 a preclinical lead for HIV/AIDS eradication. It will continue to be a major impact area for synthesis. As science moves forward and manufacturers and fine chemical producers provide more complex, commercially available building blocks, the pool of readily available starting materials will also increasingly include more complex components, thus leading to shorter syntheses. In essence, suppliers, exploiting the brilliant contributions from many laboratories to feedstock enhancements, are moving the starting line of simplicity closer to the finish line of complexity. Synthetic biology and engineered biosynthesis represent other sources of synthetic building blocks that are impacting synthesis now and will surely influence synthesis in the future including semi-synthesis. Hybrid efforts, combining, for example, biosynthesis and semi-synthesis, have already produced stunning practical syntheses in many fields (e.g., beta-lactam antibiotics).
Step economy is a major driver of synthetic success, a point we and others have emphasized for decades. It requires a holistic approach to design and execution rather than individual step optimizations. It serves to focus innovation not just on improvements but on seeking the best design (closest to the ideal) that accelerates the advancement and especially the broader impact of the field. While the ideal one-step synthesis is unlikely to be commonly achieved (although Wöhler and Reppe did so with urea and cyclooctatetraene, respectively, and many total syntheses have weighed in at under 5 steps), the importance of its inclusion in an “ideal” definition is that it provides a metric by which we could judge where we are and how close we come to an invariant ultimate goal. Such ideal goals drive innovation as they place emphasis on finding the best solution that our lexicon can deliver or defining what reactions might be needed to reach the ideal and not just better the past. Indeed it was this orientation that led many years ago, for example, to our search for more step economical ways to make triquinanes. That search for synthetic brevity approaching the ideal resulted in a three-step synthesis of silphinene, based on the arene-alkene metacycloaddition (Fig. 2).6 This remarkable process is still one of the most complexity-increasing reactions in our reaction lexicon, allowing for the formation of three new rings and up to six stereocenters in one synthetic operation from relatively simple starting materials.9
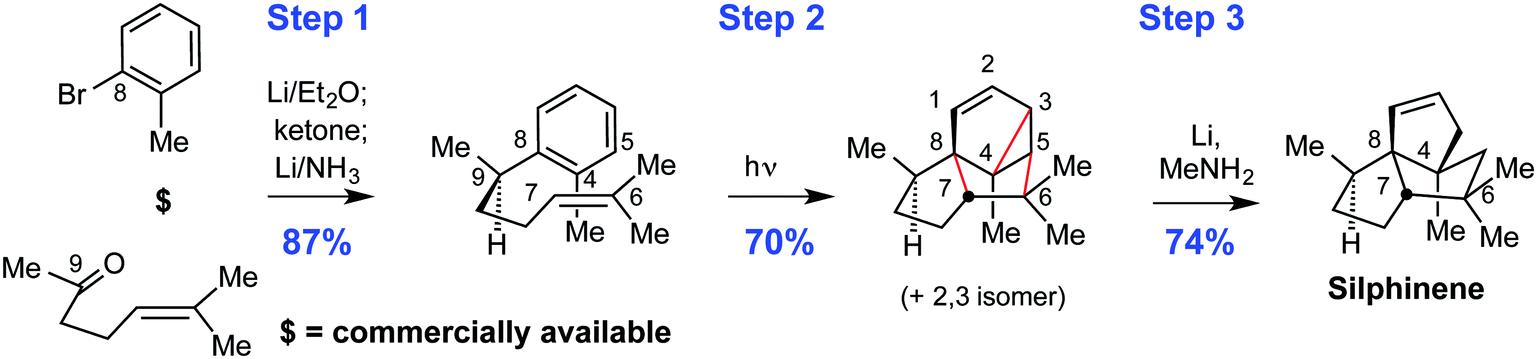 | ||
| Fig. 2 New reactions enable new strategies and more step- and time-economical access to natural and designed targets. | ||
In 1993, sensitive to the growing importance of time and environmental issues associated with synthesis design, we noted in a theoretical overview directed at the “ideal synthesis” and ways to achieve it that “while the best solution [to a synthetic problem] might be defined in various ways, most practitioners would agree that it will be the one that comes closest to the ‘ideal synthesis’…defined as one in which the target molecule is prepared from readily available starting materials in one simple, safe, environmentally acceptable, and resource-effective operation that proceeds quickly and in quantitative yield.”3 The terms “resource-effective” and “quickly” made their way into this definition as it is clear that even a short synthesis would be ineffective, i.e., far from the ideal, if it were to require too much effort and time to execute. Along with safety, a special priority is thus placed in this definition on conserving the time of those skilled in synthesis as their gift and time represent our most important resources. Most synthesis efforts whether directed at training and research in academia or synthesis campaigns in industry are heavily influenced by what we have referred to as time economy, thus necessitating its inclusion in an ideal synthesis.
Many of the economies of synthesis design are interdependent. For example, step economy favors time economy as steps require development and execution time. Similarly, step economy favors atom economy and more significantly waste minimization, as reaction solvents and purifications are the largest contributors to the waste stream of any process even though solvents are not included in the otherwise useful concept of atom economy.10 Mapping the fate of atoms in reactants into products is important but reaction and purification solvents often are the major contributors to atom loss in a synthesis because the solvent concentration in a reaction is generally significantly higher than that of reactants. Step reduction thus reduces solvent waste.11 Solvents can of course be recycled but that requires energy and time and incurs cost. While less emphasized, “energy economy” is a metric of importance in many processes and figures in recycling costs. Simply put, greater step economy correlates with greater “solvent/energy economy”. Finally, step economy is also logically at the core of redox economy and the importance of minimizing unnecessary protection steps as redox operations and protections and deprotections often add steps to a synthesis.12 Step economy more generally emphasizes eliminating unnecessary steps and using reactions and strategies that maximize increases in target relevant complexity.
While discussions about the ideal synthesis will and should continue, and new metrics (e.g., energy economy, solvent economy) will be added, the transition over the past 40 years from comparisons between current and previous syntheses to comparisons between current and ideal syntheses provides an important set of metrics (step count, atom loss, overall yield, overall time) to evaluate where we are and what more might ultimately be possible. A related aspirational set of metrics is what could be called minimally acceptable syntheses which in essence are those that supply what is minimally needed to solve the problem of material availability and route generality. The metrics here are hugely important to design and are context and problem dependent. Scalability, for example, plays a role. If one simply wants only to record a successful synthesis then one would only need to produce enough material to establish purity and to completely characterize what one has produced. If on the other hand one wishes to learn about the properties or activities of a target, then more material will be needed. Still more would be needed for more complex and compound demanding studies associated with, for example, in vivo work. Similarly, time economy will also influence design in varied ways. The difference in time constraints and thus synthesis design approaches between producing a creative synthesis of a molecule as part of a multi-year PhD program and producing a scale-up synthesis campaign of a compound cleared for first human dosing in say 6 months is huge and will dramatically influence what design is deemed acceptable. Metrics drive design and thus influence whether one seeks a plan that is minimally acceptable or closer to the ideal. Personnel, safety, development time, execution time, step count, scale, yield, cost, overall selectivity, purity, number of purifications, waste stream, environmental impact, energy requirements, equipment and toxicities are some of the measurable factors that figure in and influence synthesis design and thus the quality, timing and impact of a given plan and its execution.
The above analyses lead to an interesting question and problem and form the basis for the aforementioned third approach to design. What can one do when a target cannot be produced in a timely, safe, step-economical, and resource-effective manner? Obviously one approach would be to invent new reactions that would result in a practical synthesis. This is an important direction. However, reaction invention or discovery and development take time (even the metathesis reaction evolved over decades before reaching broad and practical use in the 1990s). If the target is needed now, a not uncommon situation for many molecules of say medicinal or similar importance, one has a problem principally dictated by time. Many synthetic targets could be produced in a practical fashion if given enough time and resources. That is often not the case. It was this problem (we need it now, but our reaction lexicon is not compatible with it being made now) that led to the genesis of what we have referred to as “function-oriented synthesis” (FOS). As we noted in our earlier overview of FOS: “the central principle of FOS is that the function of a biologically active lead structure can be recapitulated, tuned, or greatly enhanced with simpler scaffolds designed for ease of synthesis and also synthetic innovation.”4 In essence and as noted before, function is not the unique property of any one structure but could be exhibited in varying degrees by many structures. Simply put, one need not synthesize a bird to create a flying machine. Nature's library provides a lesson on flight (a bird) from which knowledge can be extracted (a wing's concavity provides lift) thereby enabling the design of many new structures (e.g., gliders, planes and jets) that function even better for intended human use (aviation).
Nature's library is rich with structures whose complexity is determined in part by their natural functions that generally would be irrelevant to many intended human uses of those structures. For example, if one needs a methylating agent, methyl iodide would be far less complex and easier to prepare than Nature's methylating agent, S-adenosyl methionine. Similarly, many natural products are derived from restricted biosynthetic pathways evolved to achieve distribution, target association, metabolism and clearance objectives peculiar to their ecosystem and mostly irrelevant to their potential use in say human therapy. It follows that only a subset of the structural features and therefore the complexity of a natural lead might be needed to recapitulate or even exceed its desirable function for human therapy. 25 years ago this FOS analysis provided the starting point for our phorbol ester tumor promotion and subsequent bryostatin studies. Computer modeling and structure-function studies led to a hypothesis about structural features of phorbol esters required for binding and modulation of protein kinase C (PKC) activity, a highly important protein family implicated in phorbol ester activity. This analysis produced a design blueprint that led to the first rationally designed PKC modulators (Fig. 3).13 The structural lead for this program, phorbol, was synthesized in 29 steps requiring years to execute.14 The first designed PKC modulators required 7 steps (step-economy) and two weeks (time-economy) to synthesize and exhibited function not unlike the endogenous PKC modulator.13
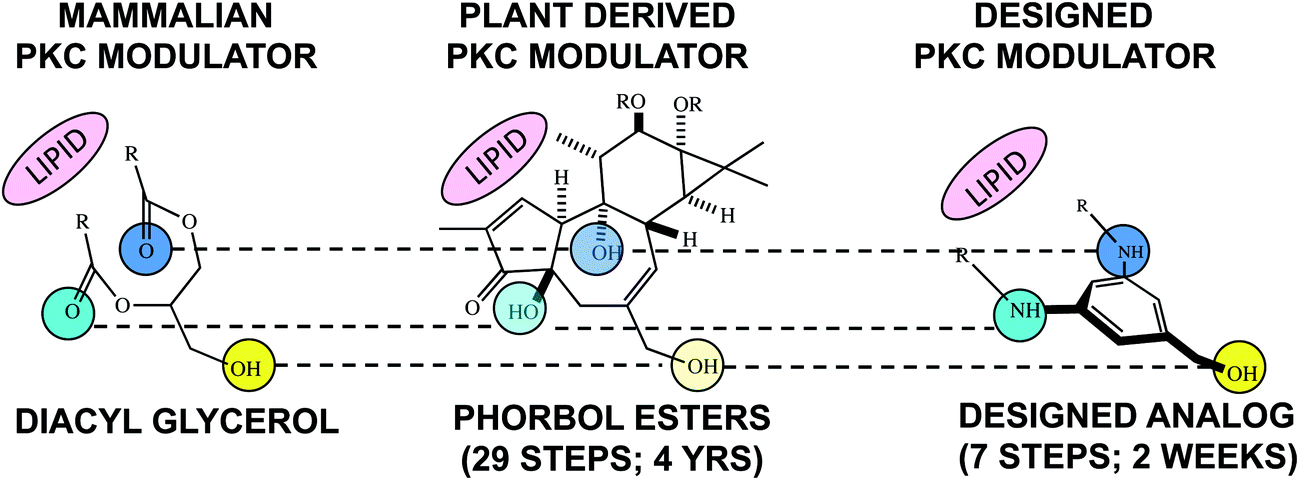 | ||
| Fig. 3 Step- and time-economy associated with the synthesis of structurally varied natural and designed compounds that exhibit a similar function, i.e., binding to and activation of PKC. | ||
In a subsequent FOS effort, bryostatin (Fig. 4), putatively produced in its ecosystem to deter predation of bryozoan larvae by fish, emerged as a significant medicinal lead and is now being studied in clinical trials for cancer and Alzheimer's disease. Our earlier FOS studies on phorbol suggested that bryostatin might associate with PKC through a similar array of H-bond donors and acceptors. This then gave way to similar computer based FOS studies on bryostatin. In 1998, the first designed bryostatin analog was produced.15 It weighed in at under 30 steps when otherwise impressive syntheses of the natural product at the time required over 70 steps. Significantly, the first bryostatin analogs, bryologs, were found to exhibit comparable or superior activity to bryostatin when screened by the National Cancer Institute for growth inhibition against a panel of human cancer cell lines. We and others have since produced additional bryologs based on this FOS approach. There are also now several impressive total syntheses of natural bryostatins, with three requiring only around 40 steps.16 In our case, the analog program actually preceded and paved the way for what eventually resulted in a concise total synthesis of bryostatin 9 using a then unexplored Prins macrolactonization strategy.17
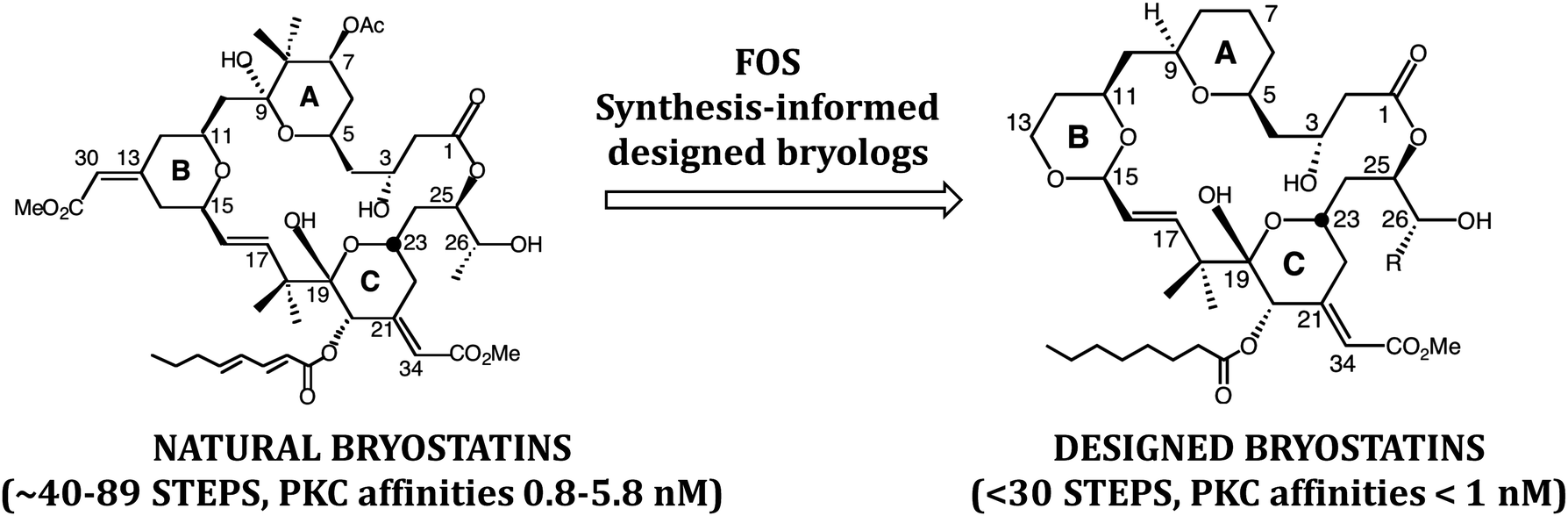 | ||
| Fig. 4 Simplified and more synthetically accessible bryologs inspired by the natural product bryostatin. | ||
Such FOS efforts are not restricted to small molecule design and synthesis in biology and medicine. Materials, polymers, aggregates and other chemical entities with sought after function are also addressable through synthesis-informed design. Indeed, the field of FOS is likely to be creatively led by those skilled in synthesis with an interest in functional targets. For example, new functional probes, e.g., molecular tweezers, molecular computers, molecular wires and on, representing the “molecularization” of functional materials, are possible through synthesis-informed design as exemplified by the rapidly growing list of amazing molecular devices.18
In another example of the interplay of function and synthesis design, 15 years ago we started a program on drug delivery based on the view that existing drugs and probes could be improved and others enabled through the introduction of better delivery technologies (function: molecules that breach biological barriers).19 Many molecules of therapeutic interest suffer from an inability to cross biological barriers and enter cells. Some of the most important new research tools, potential therapeutics, and globally recognized scientific targets of current interest, for example, such as siRNA, mRNA, DNA, and therapeutic proteins and peptides do not enter cells well if at all. As noted previously, Nature's library provides lessons on structure-function relationships and therein the basis to design biomimetics. With respect to drug delivery and barrier passage, one natural lead is found in the form of the protein HIV Tat (Fig. 5) that unlike most proteins enters cells. Research suggested that a sequence of 9 residues (RKKRRQRRR) might be responsible for cellular entry (function). Through a reverse-engineering effort involving systematic truncations, alanine scans and other modifications, we suggested for the first time that cellular entry of HIV Tat was a function of its arginine content and more specifically the number and spatial array of guanidinium groups in the so called Tat 9-mer.20
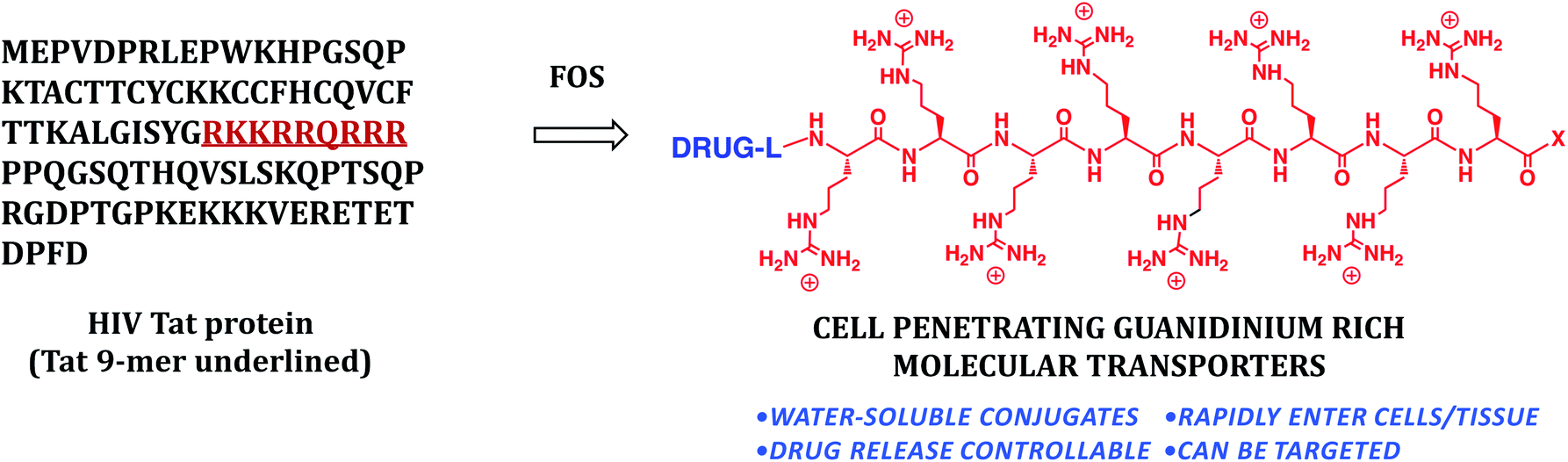 | ||
| Fig. 5 Cell penetrating guanidinium rich molecular transporters for general drug delivery inspired by the naturally occurring cell penetrating protein HIV Tat. | ||
The beauty of design is that once one understands how a function can be achieved one can create synthetically more accessible systems designed to function more effectively. We first found that octaarginine (R8: RRRRRRRRR) exhibits better cellular uptake than the Tat 9-mer. This “design” provided already a modest synthetic benefit as the Tat 9-mer requires 18 steps to synthesize while the octaarginine requires only 16 steps. More importantly, because R8 is a homooligomer of arginine, we proposed its assembly through a segment doubling strategy, reducing 18 steps to only 9 and showing how a focus on function could result in significant step economy with an actual gain in function. Unlike conventional solid phase synthesis for which two steps are required for each added subunit, in segment doubling three steps are needed for each doubling of length (starting monomers give in 3 steps a dimer which in 3 more steps give a 4-mer and 3 more give the 8-mer). Thus an 8-mer can be formed in three doubling events, i.e., 9 steps.21 This is a pure strategic reduction in steps arising from the doubling process and made possible by design of a more effective and more accessible homooligomer. This approach was enabled by knowledge of and focus on function (all arginines would work better) and synthesis-informed design with an eye on step- and time-economy.
It gets better. We have since used this information to make a variety of new guanidinium rich drug delivery systems that we refer to as cell penetrating, guanidinium rich molecular transporters (GRMoTrs). GRMoTrs have been shown to transport (function) small molecules, probes, imaging agents, metals, peptides, proteins, PNAs, RNAs and DNAs into cells in culture and across tissue barriers in animals.22 They have also been advanced into human clinical trials. We have also shown that attachment of transporters to drugs like taxol that are rendered ineffective due to Pgp export resistance, provides drug-conjugates that overcome the resistance of the drug itself (Fig. 6).23 In this case the step-economy is significant as one does not need to discover a new drug but rather use FOS to fix the functional problems of an existing drug. In all primary disease samples obtained from ovarian cancer patients, the taxol-transporter conjugate outperforms existing therapy presumably by evading Pgp export. The conjugates thus serve as molecular patches, rapidly getting into cells and releasing free drug only after cell entry at a rate determined by release design.
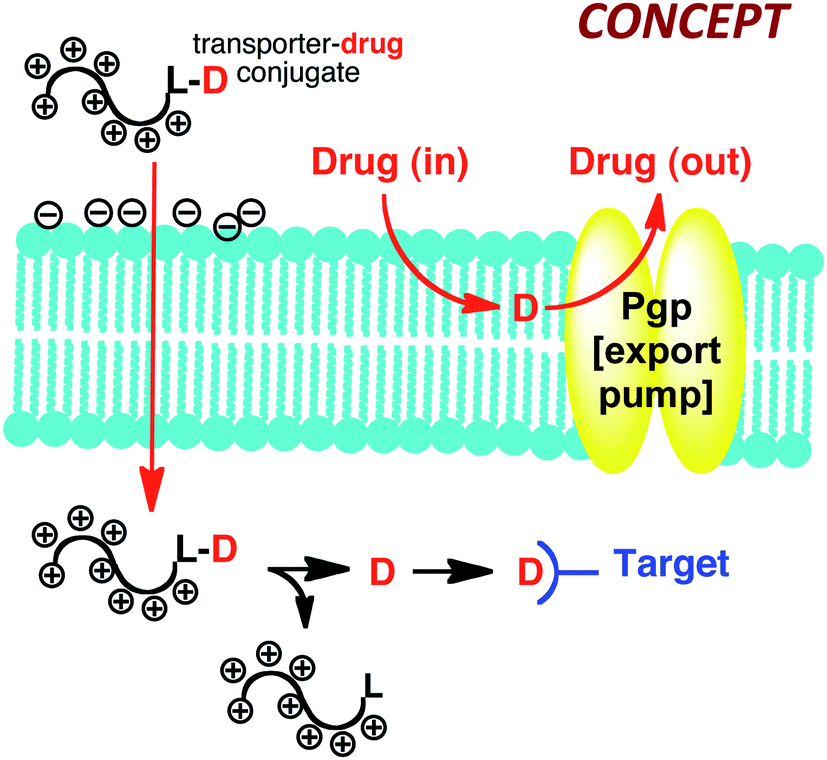 | ||
| Fig. 6 Free drug (D, e.g., taxol; L, releasable linker), as is often the case for membrane soluble (non-polar) drugs, is exported by the membrane bound export pump, rendering the cell drug resistant. Drug-MoTr conjugate is highly polar, enters cells through a different mechanism evading Pgp export and thus overcomes the very resistance that the free drug elicits. Once inside the cell, the drug-MoTr conjugate releases free drug to associate with its intracellular target. | ||
And it gets still better. Significantly, and related to the theme of synthesis-informed design directed at function, when we initiated efforts to study the use of GRMoTrs to transport RNAs, polyanions that do not cross the non-polar membrane of cells, we realized that access to even longer sequences of guanidinium rich (polycation) systems would be needed. To address the resultant need for a step economical strategy to make longer oligomers, we joined in a collaboration with Bob Waymouth, Jim Hedrick and their coworkers, and were able to show that one could produce guanidinium rich oligomers in one step (!) through an organocatalytic oligomerization process (Fig. 7).24 Simply put in terms of comparative metrics, making a 32-mer by solid phase synthesis would require 64 steps (2 steps/unit added). Even segment doubling would require 15 steps (3 steps/segment doubling). With the oligomerization process, one can produce 32-mers with good polydispersity in only one synthetic operation. Focus on the seemingly unattainable “ideal” in this case paid off. Indeed this approach theoretically allows one to make any length of oligomer in only one step. The value of this strategy is that it allows one to rapidly make many different types of oligomers, including di-, tri- and poly-block oligomers in one synthetic operation followed by Boc removal to liberate the free guanidinium groups. This near-ideal, time- and step-economical strategy allowed for a rapid study of factors that contribute to siRNA complexation and the subsequent use of these transporters to complex and deliver siRNA into cells.25 The amphipathic, guanidinium rich oligocarbonate-siRNA complexes were shown to enter HaCaT cells and selectively knocked down expression of a fluorescent protein (tdTomato) while not affecting expression of another fluorescent protein control (EGFP). This strategy is now driving development of new classes of biodegradable and non-biodegradable transporters for ferrying other oligonucleotides and a variety of other molecular cargos across biological barriers (e.g., membranes, skin, blood brain, ocular and most recently cell wall26 barriers).
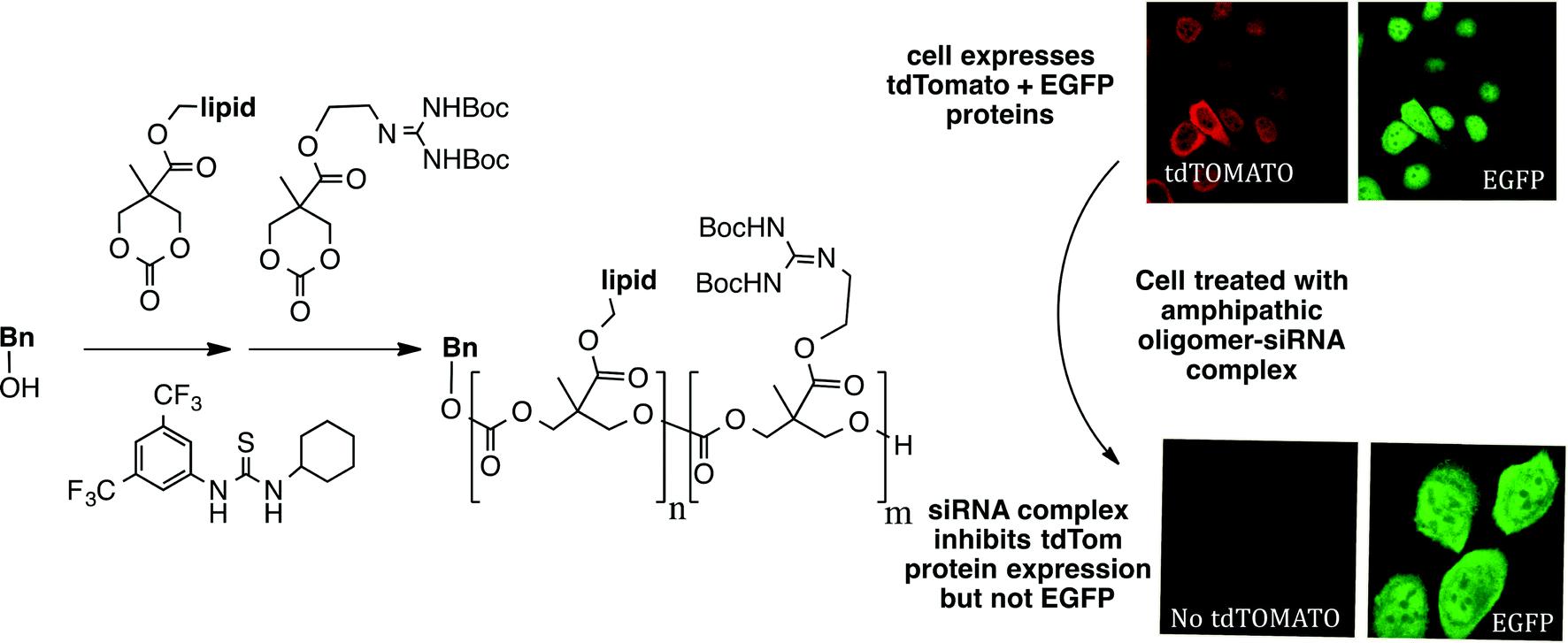 | ||
| Fig. 7 A one-step synthesis of amphipathic oligocarbonate molecular transporters that after Boc removal, complex, deliver and release siRNA in cells, suppressing the expression of only the target protein tdTomato and not expression of the control EGF protein. | ||
The powerful and general view that form follows function, the essence of function-oriented synthesis (FOS), can change how one thinks about synthesis especially the synthesis of compounds that exhibit sought after function (e.g., new medicinal agents; smart materials; devices; environmental sensors; imaging agents; energy collection, storage and conversion systems; and research tools) but whose structures are currently beyond the timely and practical reach of synthesis. Function-oriented synthesis is driven by design and it opens a field of opportunities for chemists to not only reproduce what Nature has made but to create new structures and associated functions, whether inspired by Nature or devised by de novo design, that might be better suited for their intended human use. FOS addresses the concern that some natural products are too complex and thus not likely to be supplied on scale in a timely fashion by focusing creative attention on the design of simpler and thus more accessible, often bio-inspired structures that can be accessed now through synthesis. It opens an exciting field of opportunities that synthetic chemists are uniquely able to lead and develop and with that to create new fields impacting all of molecular science. In short, through synthesis-informed design directed at function, one can achieve practical and creative syntheses in a time- and step-economical, simple, safe, resource effective, and environmentally acceptable fashion, of targets with improved or totally new functions that could transform science in ways that are of profound health, economic and societal benefit.
References
- P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef CAS PubMed.
- R. B. Woodward, E. Logusch, K. P. Nambiar, K. Sakan, D. E. Ward, P. Au-Yeung Balaram, L. J. Browne and P. J. Card, et al. , J. Am. Chem. Soc., 1981, 103, 3215–3217 CrossRef CAS.
- (a) P. A. Wender and B. L. Miller, Toward the ideal synthesis: connectivity analysis and multi-bond forming processes, in Organic synthesis: theory and applications, ed. T. Hudlicky, JAI, Greenwich, CT, 1993, vol. 2, pp. 27–66 Search PubMed; (b) P. A. Wender, D. Wright and S. Handy, Chem. Ind., 1997, 19, 765 Search PubMed.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed.
- J. B. Hendrickson, J. Am. Chem. Soc., 1975, 97, 5784–5800 CrossRef CAS.
- P. A. Wender and R. J. Ternansky, Tetrahedron Lett., 1985, 26, 2625–2628 CrossRef CAS.
- (a) J. N. Denis, A. E. Greene, D. Guénard, F. Guéritte-Voegelein and P. Potier, J. Am. Chem. Soc., 1988, 110, 5917 CrossRef CAS; (b) R. A. Holton; R. J. Biediger and P. D. Boatman, Semisynthesis of taxol and taoxtere, in Taxol: science and applications, ed. M. Suffness, CRC Press, Boc Raton, Fl, 1995, pp. 97–121 Search PubMed.
- P. A. Wender, J.-M. Kee and J. M. Warrington, Science, 2008, 320, 649–652 CrossRef CAS PubMed.
- D. Chappell and A. T. Russell, Org. Biomol. Chem., 2006, 4, 4409–4430 CAS.
- B. M. Trost, Science, 1991, 254, 1471–1477 CAS.
- (a) P. T. Anastas and J. C. Warner, Green chemistry: theory and practice, Oxford University Press, New York, NY, 1998, p. 30 Search PubMed; (b) R. A. Sheldon; I. Arends and U. Hanfeld, Green chemistry and catalysis, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (c) R. Sheldon, Introduction to green chemistry, organic synthesis and pharmaceuticals, in Green chemistry in the pharmaceutical industry, ed. P. J. Dunn, A. S. Wells and M. T. Williams, Wiley-VCH GmbH & KGaA, Weinheim, Germany, 2010 Search PubMed; (d) M. C. Bryan, B. Dillon, L. G. Hamann, G. J. Hughes, M. E. Kopach, E. A. Peterson, M. Pourashraf, I. Raheem, P. F. Richardson, D. T. Richter and H. F. Sneddon, J. Med. Chem., 2013, 56, 6007–6021 CrossRef CAS PubMed.
- Noah Z. Burns, Phil S. Baran and Reinhard W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed.
- P. A. Wender, K. F. Koehler, N. A. Sharkey, M. L. Dell'Aquila and P. Blumberg, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 4214–4218 CrossRef CAS.
- P. A. Wender, K. D. Rice and M. E. Schnute, J. Am. Chem. Soc., 1997, 119, 7897–7898 CrossRef CAS.
- P. A. Wender, J. De Brabander, P. G. Harran, J.-M. Jimenez, M. F. T. Koehler, B. Lippa, C.-M. Park, C. Siedenbiedel and G. R. Pettit, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6624 CrossRef CAS.
- For total syntheses of natural bryostatins, see: (a) M. Kageyama, T. Tamura, M. H. Nantz, J. C. Roberts, P. Somfai, D. C. Whritenour and S. Masamune, J. Am. Chem. Soc., 1990, 112, 7407–7408 CrossRef CAS; (b) D. A. Evans, P. H. Carter, E. M. Carreira, A. B. Charette, J. A. Prunet and M. Lautens, J. Am. Chem. Soc., 1999, 121, 7540–7552 CrossRef CAS; (c) K. Ohmori, Y. Ogawa, T. Obitsu, Y. Ishikawa, S. Nishiyama and S. Yamamura, Angew. Chem., Int. Ed., 2000, 39, 2290–2294 CrossRef CAS; (d) B. M. Trost and G. Dong, Nature, 2008, 456, 485–488 CrossRef CAS PubMed; (e) G. E. Keck, Y. B. Poudel, T. J. Cummins, A. Rudra and J. A. Covel, J. Am. Chem. Soc., 2011, 133, 744–747 CrossRef CAS PubMed; (f) P. A. Wender and A. J. Schrier, J. Am. Chem. Soc., 2011, 133, 9228–9231 CrossRef CAS PubMed; (g) Y. Lu, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 13876–13879 CrossRef CAS PubMed . For a formal synthesis, see: ; (h) S. Manaviazar, M. Frigerio, G. S. Bhatia, M. G. Hummersone, A. E. Aliev and K. J. Hale, Org. Lett., 2006, 8, 4477–4480 CrossRef CAS PubMed and references therein.
- P. A. Wender and A. J. Schrier, J. Am. Chem. Soc., 2011, 133, 9228–9231 CrossRef CAS PubMed.
- As an example relating to work of the guest editor on “molecular switches”, see: M. Stein, A. Breit, T. Fehrentz, T. Gudermann and D. Trauner, Angew. Chem., Int. Ed., 2013, 52, 9845–9848 CrossRef CAS PubMed.
- E. Geihe Stanzl, B. M. Trantow, J. R. Vargas and P. A. Wender, Acc. Chem. Res., 2013, 46, 2944–2954 CrossRef PubMed.
- P. A. Wender, D. J. Mitchell, K. Pattabiraman, E. Pelkey, L. Steinman and J. B. Rothbard, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13003–13008 CrossRef CAS PubMed.
- P. A. Wender, T. C. Jessop, K. Pattabiraman, E. T. Pelkey and C. L. VanDeusen, Org. Lett., 2001, 3, 3229–3232 CrossRef CAS PubMed.
- P. A. Wender, W. C. Galliher, E. A. Goun, L. R. Jones and T. H. Pillow, Adv. Drug Delivery Rev., 2008, 60, 452–472 CrossRef CAS PubMed.
- E. A. Dubikovskaya, S. H. Thorne, Th. H. Pillow, C. H. Contag and P. A. Wender, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12128–12133 CrossRef CAS PubMed; P. A. Wender, W. C. Galliher, N. M. Bhat, T. H. Pillow, M. M. Bieber and N. H. Teng, Gynecol. Oncol., 2012, 126, 118–123 CrossRef PubMed.
- C. B. Cooley, B. M. Trantow, F. Nederberg, M. K. Kiesewetter, J. L. Hedrick, R. M. Waymouth and P. A. Wender, J. Am. Chem. Soc., 2009, 131, 16401–16403 CrossRef CAS PubMed.
- E. I. Geihe, C. B. Cooley, J. R. Simon, M. K. Kiesewetter, J. A. Edward, R. P. Hickerson, R. L. Kaspar, J. L. Hedrick, R. M. Waymouth and P. A. Wender, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13171–13176 CrossRef CAS PubMed.
- J. M. Hyman, E. I. Geihe, B. M. Trantow, B. Parvin and P. A. Wender, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13225–13230 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |