 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acylthioureas as anion transporters: the effect of intramolecular hydrogen bonding†
Cally J. E.
Haynes
a,
Nathalie
Busschaert
a,
Isabelle L.
Kirby
a,
Julie
Herniman
a,
Mark E.
Light
a,
Neil J.
Wells
a,
Igor
Marques
b,
Vítor
Félix
b and
Philip A.
Gale
*a
aChemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: philip.gale@soton.ac.uk; Tel: +44 (0)23 8059 3332
bDepartamento de Química, CICECO and Secção Autónoma de Ciências da Saúde, Universidade de Aveiro, 3810-193 Aveiro, Portugal. E-mail: vitor.felix@ua.pt
First published on 12th September 2013
Abstract
Small molecule synthetic anion transporters may have potential application as therapeutic agents for the treatment of diseases including cystic fibrosis and cancer. Understanding the factors that can dictate the anion transport activity of such transporters is a crucial step towards their application in biological systems. In this study a series of acylthiourea anion transporters were synthesised and their anion binding and transport properties in POPC bilayers have been investigated. The transport activity of these receptors is dominated by their lipophilicity, which is in turn dependent on both substituent effects and the formation and strength of an intramolecular hydrogen bond as inferred from DFT calculations. This is in contrast to simpler thiourea systems, in which the lipophilicity depends predominantly on substituent effects alone.
The transport of inorganic anions such as chloride across cell membranes is an important biological process, and is regulated in the body by proteins embedded in the phospholipid bilayer. When naturally occurring anion transport processes are defective, a number of diseases (channelopathies) are known to result, including cystic fibrosis. There is a growing interest in the development of small molecular carriers for anions due to the hypothesis that they may serve as therapeutic replacements for defective transport proteins for the treatment of channelopathies or as tools for physiologists to study these conditions.1 Additionally, there is increasing evidence that anion carriers can induce apoptosis in various cancer cell lines, indicating a further possible use for these molecules as anticancer agents.1a,2
Research by our group and others has shown that small molecule anion receptors based on motifs including isophthalamides,3 ureas,2b,4 thioureas,2a,c,5 squaramides,6 cyanoguanidines7 and tambjamines2d,8 are effective anion antiporters. Schmitzer9 and Yang10 have reported small molecule anion channels that exhibit significant biological activity. In these previous studies it was observed that the activity of anion carriers is often strongly linked to the lipophilicity of the transporter. We have seen that substituent effects on a thiourea scaffold can dramatically modulate the transport activity. In a recent example, we reported a QSAR analysis of the anion transport by a series of 1-hexyl-3-phenylthioureas containing various substituents in the 4-phenyl position, including receptors 1–4, in which we correlated the transport activity to a range of molecular descriptors.5a We found that the variation in anion transport activity was mainly due to the varying lipophilicity of the receptors due to the substituent effects, with smaller contributions from the molecular size and hydrogen bond acidity (and hence anion binding capability) of the thiourea NH groups.
Controlling the lipophilicity of a scaffold is a key concept in drug design, as the lipophilicity of a potential drug molecule will greatly affect its membrane permeability and hence its absorption and distribution. Increasing the lipophilicity is often achieved through appending lipophilic functional groups onto the scaffold. Another strategy to achieve this when designing a compound is to introduce the possibility of forming intramolecular hydrogen bonds.11 Intramolecular hydrogen bonds decrease the ability of the molecule to interact with the environment (e.g. water), and hence the hydrophilicity is reduced. This effect has been previously reported as a strategy to improve brain penetration,12 bone tissue penetration,13 oral absorption14 and membrane permeability.15 A recent report by Shalaeva et al. considers methods to assess the propensity of drug molecules to form intramolecular hydrogen bonds16 and notes that the consequences of such interactions are of great significance to medicinal chemists but are often under-recognized and seldom predicted.
In this study, we examine the role that intramolecular hydrogen bonding has on the efficacy of a series of anion transporters. Given that simple thioureas are effective anion transporters, we decided to study related acylthiourea compounds as putative anion transporters. Acylthioureas may form six-membered intramolecular hydrogen-bonded rings that mask the hydrophilicity of the receptor. We found that acylthioureas are more lipophilic than analogous thioureas as a result of the intramolecular hydrogen bond. When comparing parent compounds of both series, the acylthiourea is a significantly more effective chloride/nitrate antiport agent than the thiourea. However, we found that if the transporters were delivered to a pre-formed lipid bilayer then the more lipophilic species resulted in less efficient transport, presumably due to the barrier to their delivery through the aqueous phase, whereas if these species were pre-incorporated into a lipid bilayer then the more lipophilic compounds were the best anion transporters.
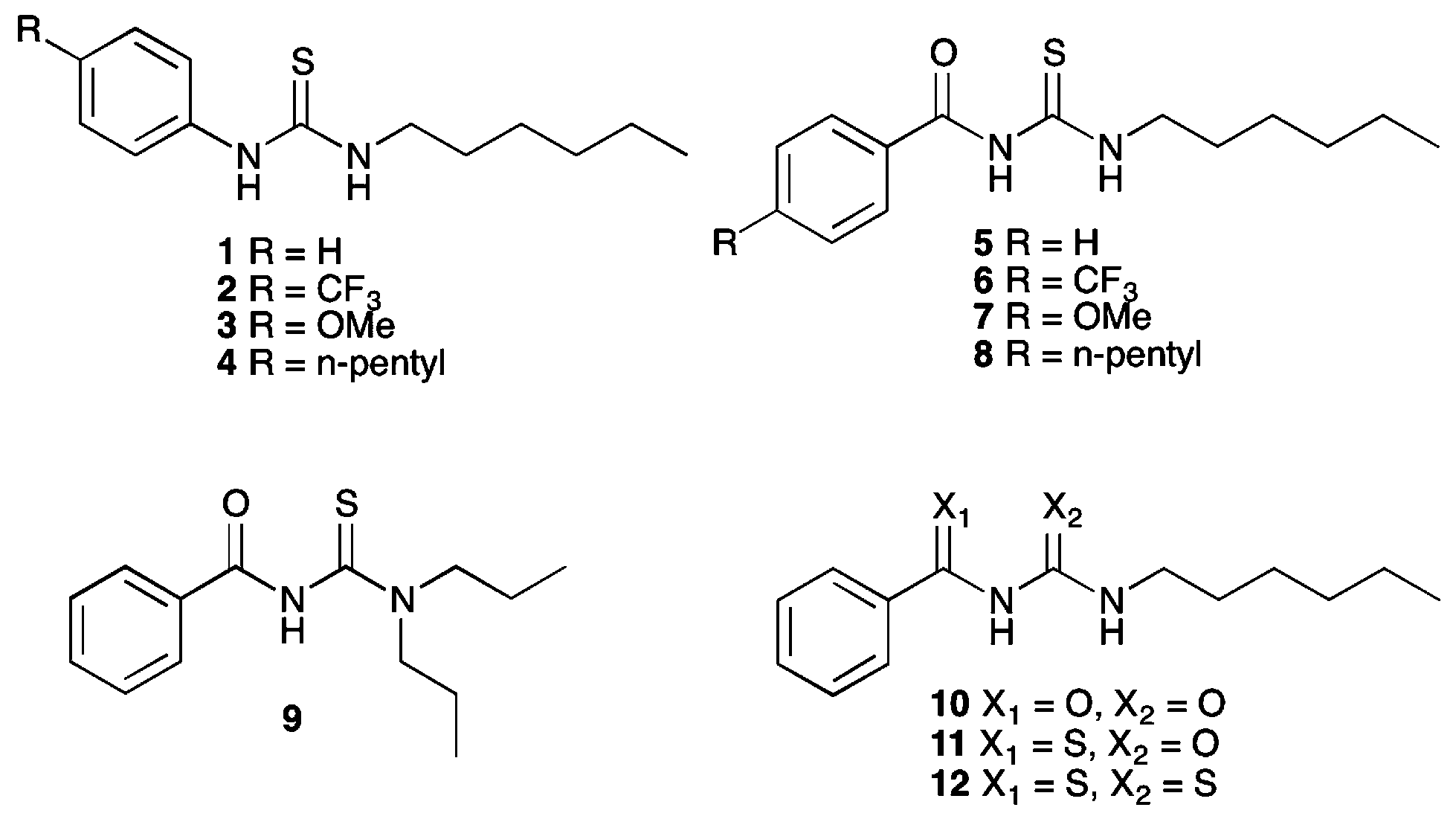
Previously, Wei and co-workers have reported that receptors based on an acylthiourea motif form an intramolecular hydrogen bond, as shown in Fig. 1, and this makes them highly selective receptors for basic anions such as F− and AcO−, as only these anions are strongly coordinating enough to disrupt this interaction.17 Acylthiourea based receptors have also been extensively studied for their affinity for soft transition metal ions, with the S and O atoms able to form a chelate ring.18 For example, Luckay et al., have reported the use of acylthioureas (including 9) for the extraction and transport of gold(III) ions.18d Acylthiourea derivatives have also been reported to have herbicidal,19 fungicidal,19 bactericidal,19 insecticidal20 and plant growth regulatory21 activity, and have been patented as anti-diabetic,22 anti-arthritic,23 anti-neoplastic24 and anti-coagulant25 agents for the treatment of cognitive problems26 and prostate disorder.27 Therefore their suitability for application to biological systems has been previously explored. Our group, in collaboration with J. T. Davis and Quesada, have previously used intramolecular hydrogen bonding to preorganise the anion-binding site of an anion receptor and transporter based on an isophthalamide structure.3
 | ||
| Fig. 1 Competition between intramolecular hydrogen bond formation and anion binding in acylthiourea receptors. | ||
Synthesis
Acylthiourea receptors 4–9 were synthesised by the reaction of the corresponding benzoyl isothiocyanate with 1-hexylamine or dipropylamine in ethyl acetate (Scheme 1).17b In the case of 6–8, the isothiocyanate was prepared in situ by the reaction of an acid chloride with ammonium thiocyanate, using PEG-400 as a phase transfer catalyst. Receptor 10 was similarly synthesised by the reaction of benzoyl isocyanate with 1-hexylamine in ethyl acetate, and was isolated in 30% yield. A number of these compounds have been previously reported.18 | ||
| Scheme 1 Synthesis of acylthiourea receptors 5–9. Reagents and conditions: (i) NH4SCN, PEG-400, EtOAc; (ii) 1-hexylamine (5–8) or dipropylamine (9). | ||
Receptors 11 and 12 were formed by the reaction of thiobenzamide with 1-hexyl isocyanate or 1-hexyl isothiocyanate in acetonitrile in the presence of potassium hydroxide pellets (Scheme 2). However, receptor 12 was found to be unstable. It appeared to decompose during purification by chromatographic methods, and as such was isolated with 15–20% impurities (see ESI†). After a period of 6 months, a sample of this receptor was found to have formed crystals of elemental sulfur (S8), and emitted a strong smell of hydrogen sulfide. Anion transport experiments were performed on a freshly prepared sample of this receptor, although it should be noted that the stability of this compound in buffered aqueous solutions is not known.
 | ||
| Scheme 2 Synthesis of thioacyl receptors 11 and 12. Reagents and conditions: (i) KOH, 1-hexyl isocyanate, yield = 30% (11) or 1-hexyl isothiocyanate (12), MeCN, yield 70%. | ||
X-ray crystallography
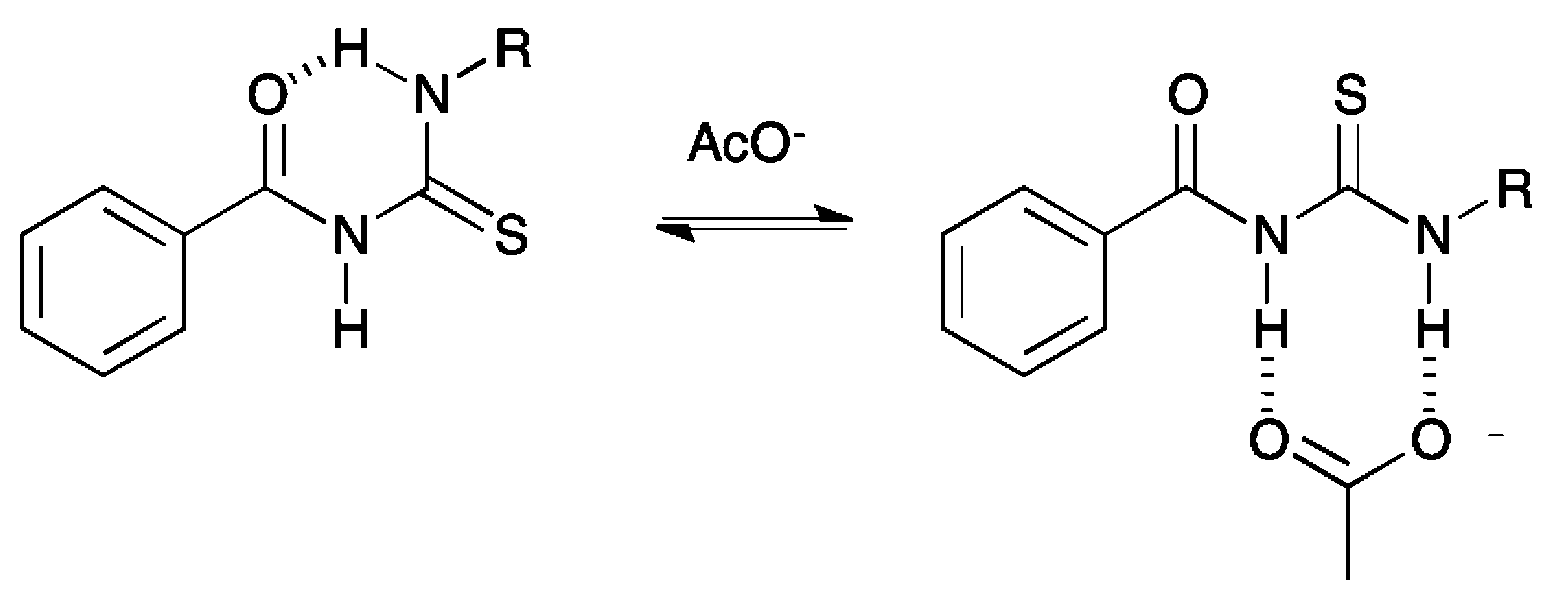
Compounds 6, 7, 10 and 11 were isolated as crystalline solids, and their crystal structures were elucidated by single crystal X-ray diffraction.‡ Full details of these structures can be found in the ESI.† The asymmetric unit of 6, composed by two independent molecules, is shown in Fig. 2. As with all of the structures elucidated here (and as previously observed for analogous systems17c,18a,28), this receptor forms a 6-membered hydrogen-bonded ring with the thiourea binding unit adopting an anti conformation. The donor–acceptor hydrogen bond distances (D⋯A) for this interaction is 2.619(2) and 2.620(2) Å, and the corresponding DH⋯A angles are 134 and 135°. By comparison, the analogous intramolecular hydrogen bonding lengths in receptor 7 and 10 are 2.678(3) Å and 2.941(3) Å respectively.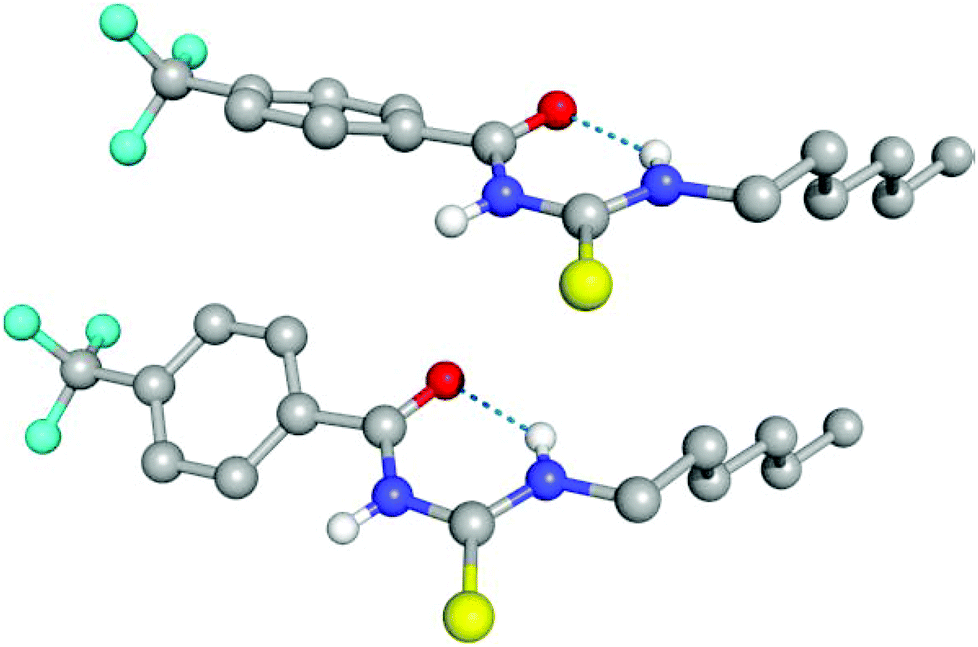 | ||
| Fig. 2 Asymmetric unit of the X-ray crystal structure of compound 6 showing the anti conformation with intramolecular hydrogen bonds drawn as dashed lines. Non-acidic hydrogen atoms have been omitted for clarity. | ||
Evidence for intramolecular hydrogen bonding in solution
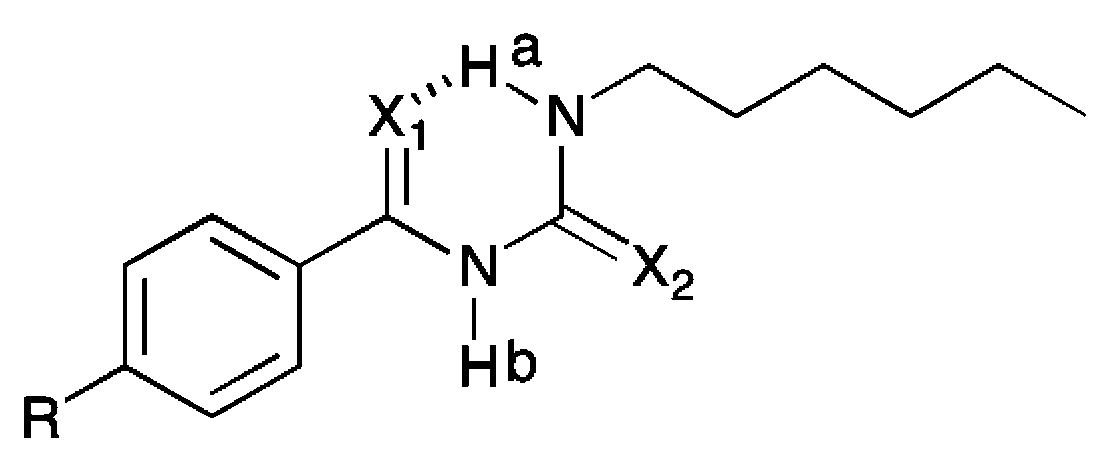
The X-ray crystal structures obtained indicated the formation of an intramolecular hydrogen bond in the solid state. Wei et al., have reported that acylthiourea receptors form an intramolecular hydrogen bond in DMSO–water solutions, and that this interaction competes with anion complexation.17b,c The 1H NMR spectra of 5–8 in DMSO-d6 show that the resonance of the NHa proton adjacent to the alkyl group, which is involved in the intramolecular hydrogen bonding, is shifted downfield compared to the NHa resonance of an analogous thiourea (Table 1). For example, the NHa resonance of thiourea 1 appears at 7.71 ppm, whilst the NHa resonance of analogous acyl thiourea 5 appears at 10.88 ppm, a difference of 3.17 ppm. The values for receptors 1–4 were previously reported and were therefore collected using a different batch of DMSO-d6 (with presumably a different water content), therefore the precise values are not directly comparable; however, the large magnitude of the difference in chemical shift of NHa between the thiourea and the acyl thiourea suggests that this comparison is still valid. The spectra for receptors 5–11 were all collected from samples prepared at the same concentration using the same batch of DMSO-d6. For all acyl thiourea receptors the NHb resonance also appears significantly downfield by comparison to the analogous thiourea, as a result of the extra acidity inferred by conjugation to both a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CS group.
O and CS group.
| Receptor | δ NHa (DMSO-d6) (ppm) | δ NHb (DMSO-d6) (ppm) | δ NHa (CDCl3) (ppm) | δ NHb (CDCl3) (ppm) |
|---|---|---|---|---|
| a Sample concentration 10 mM. b Previously reported.2c,7 | ||||
| 1 | 7.71b | 9.43 | — | — |
| 2 | 8.03b | 9.77 | — | — |
| 3 | 7.48b | 9.20 | — | — |
| 4 | 7.60b | 9.31 | — | — |
| 5 a | 10.88 | 11.27 | 10.73 | 8.95 |
| 6 a | 10.78 | 11.56 | 10.62 | 8.99 |
| 7 a | 10.95 | 11.07 | 10.79 | 8.89 |
| 8 a | 10.92 | 11.14 | 10.77 | 8.94 |
| 9 a | 8.66 | — | 8.20 | — |
| 10 a | 8.66 | 10.65 | 8.64 | 8.51 |
| 11 a | 9.26 | 11.74 | 9.93 | 9.18 |
According to Baell,29 and Llinas and Klein,30 comparing the chemical shift of this NH resonance in DMSO-d6 to the position of the resonance in CDCl3 can give an insight into how “solvent accessible” the NH group is. If the NH is strongly intramolecularly hydrogen bonded, it will have minimal interaction with solvent and thus a downfield shift should not be observed on moving to a more strongly coordinating system. We compared the chemical shift of NHa and NHb in both DMSO-d6 and CDCl3 for receptors 5–11. The NHa resonance was identified either by its multiplicity (triplet), or if the multiplicity of the NH peaks was not clear due to broadening, by COSY NMR (see ESI†). We found that, on moving from CDCl3 to DMSO-d6 as expected the NHb resonance of all of these receptors appeared significantly downfield in DMSO-d6 (for example from 8.95 ppm in CDCl3 to 11.27 ppm in DMSO-d6 for receptor 5). However, the resonance of NHa appeared at a similar chemical shift in both DMSO-d6 and CDCl3, which is evidence that leads us to suggest that this NH group is strongly intramolecularly hydrogen bonded and thus shielded from solvent effects.
A slow rate of deuterium exchange can also be used to indicate internal hydrogen bonding. We attempted a series of D2O shake experiments with receptors 5–8 and 10–11 in order to see if NHa is less readily exchanged than NHb. We prepared a 0.01 M solution of the receptors in DMSO-d6 (as supplied i.e. not dry), and monitored the 1H NMR spectrum on addition of 2 and 4 μL of D2O. A stack plot for the experiment with receptor 5 is shown in Fig. 3(a). We found that for every receptor, the resonance of NHa did not decrease in intensity as quickly as the resonance of NHb, indicating that the formation of the intramolecular hydrogen bond provides a barrier to its exchange. This is consistent with the comparison of the chemical shifts of NHa in DMSO-d6 and CDCl3, which indicated the presence of an intramolecular hydrogen bond in both solvent systems.
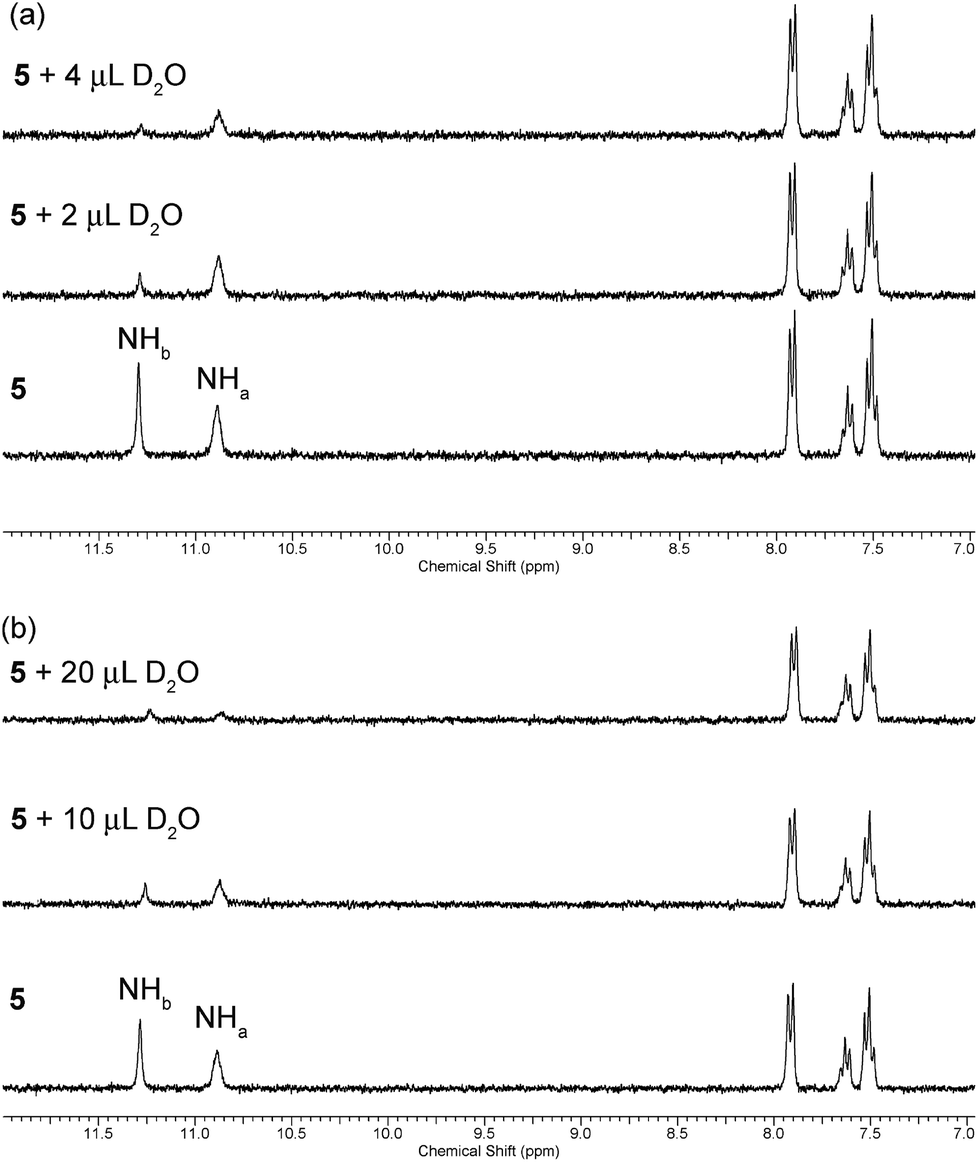 | ||
| Fig. 3 Changes to the downfield region of the 1H NMR spectrum of receptor 5 (5 mL of 0.01 M solution) in (a) DMSO-d6 and (b) DMSO-d6 with 1% H2O (v/v) upon the addition of D2O. | ||
However, it is reasonable to assume that the strength of an intramolecular hydrogen bond should be weakened in the presence of a more competitive solvent. Therefore, we repeated the D2O shake experiments using DMSO-d6 with 1% H2O added (v/v). A stack plot for this experiment with receptor 5 is shown in Fig. 3(b). In this case, a larger volume of D2O was required to cause the NH peaks to decrease in intensity, presumably because of competing deuterium exchange with the added H2O. We found that the resonance associated with NHa was less resistant to exchange in this case, which serves to reiterate that the formation and stability of a hydrogen bond is dynamic and highly dependent on the environment, and can be weakened in the presence of competitive solvent mixtures. Again, the same observation was made in the experiments with acylthioureas 6–8 (see ESI†). However, in the experiments with (thio)acyl ureas 10 and 11 the NHa resonance was found to retain its resistance to exchange in the more competitive solvent mixture. This indicates a difference in the behaviour of the acylthiourea receptors and their analogues.
Anion transport
The anion transport activity of receptors 5–12 was investigated using vesicle based methods.31 A sample of unilamellar POPC vesicles was prepared containing 489 mM NaCl buffered to pH 7.2 with 5 mM sodium phosphate salts. The vesicles were suspended at a lipid concentration of 1 mM in 489 mM NaNO3 buffered to pH 7.2 with 5 mM sodium phosphate salts. The experiment was initiated by the addition of a small amount of a DMSO solution of the receptor (2 mol% with respect to lipid), and the resulting chloride efflux was monitored using a chloride ion selective electrode (ISE). At the end of the experiment (300 s), the vesicles were lysed by adding a detergent, and the final electrode reading was used to calibrate 100% chloride release. Fig. 4 shows the chloride efflux mediated by receptor 5 in comparison to receptor 1, indicating that in this case the acylthiourea receptor is more active than its thiourea analogue.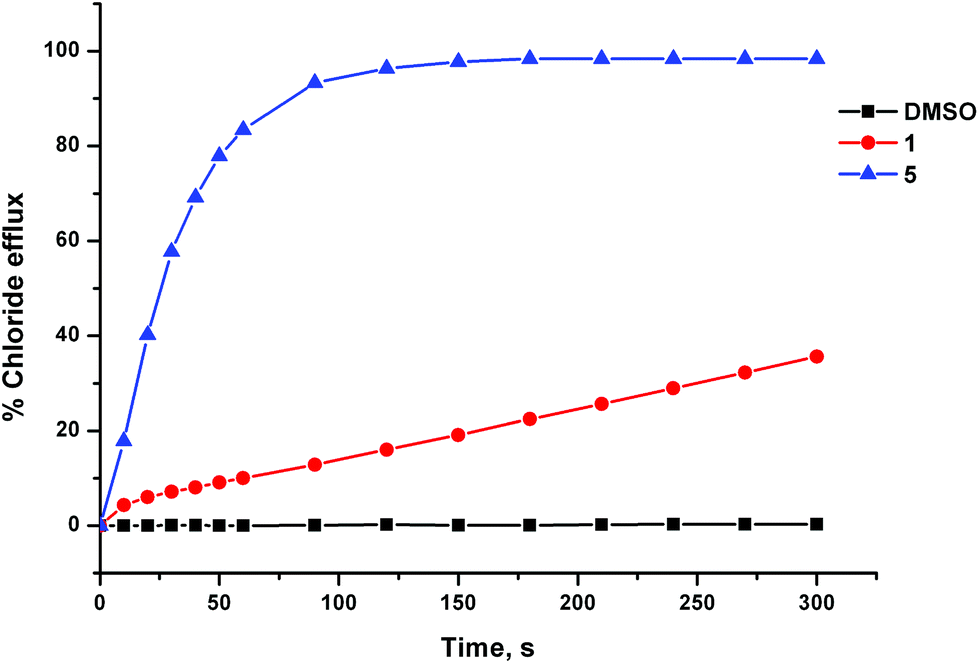 | ||
| Fig. 4 Chloride efflux mediated by unsubstituted receptors 1 and 5 (2 mol% with respect to lipid) from unilamellar POPC vesicles containing 489 mM NaCl buffered to pH 7.2 with 5 mM sodium phosphate salts. The vesicles were suspended in 489 mM NaNO3 buffered to pH 7.2 with 5 mM sodium phosphate salts. At the end of the experiment the vesicle were lysed with detergent in order to calibrate 100% chloride release. Each point represents the average of 3 trials. | ||
These studies showed that acylthioureas 5–8 are able to facilitate chloride transport under these conditions, and that each of these receptors could mediate 100% chloride efflux by the end of the experiment. However, analogous receptors 9–12 did not mediate any detectable chloride transport under these conditions.
In order to determine the mechanism of chloride release by receptors 5–8, we performed a second experiment using POPC vesicles containing 450 mM NaCl buffered to pH 7.2 with 20 mM sodium phosphate salts. This time, the vesicles were suspended in 162 mM Na2SO4 buffered to pH 7.2 with 20 mM sodium phosphate salts. On addition of a sample of receptors 5–8, there was negligible chloride release. This indicates that these receptors function by an anion exchange mechanism. In the first assay, the receptors were able to facilitate Cl−/NO3− exchange, but in this second experiment their activity was diminished due to the high hydrophilicity of the SO42− anion,32 which in most cases prevents its transport by synthetic transporters. After 120 s, we added a pulse of NaHCO3 solution, and found that the receptors were able to begin mediating chloride efflux, implying that these receptors are also able to facilitate a Cl−/HCO3− antiport mechanism. The lack of transport in the absence of a readily transportable counteranion was not perturbed by changing the encapsulated cation, thus implying that a M+/Cl− co-transport mechanism is not operating (see ESI†).
In order to quantify these anion transport processes, we performed a Hill analysis for Cl−/NO3− and Cl−/HCO3− antiport by receptors 5–8.33 These experiments allowed us to calculate the EC50 for these processes, a measure of activity defined as the effective receptor concentration required to mediate 50% chloride efflux after a specified time period. As is standard in our studies, this period is 270 s for Cl−/NO3− experiments and 390 s for Cl−/HCO3− experiments (corresponding to 270 s after the HCO3− spike). We were also able to determine the Hill coefficient (n), which can be used to give an indication of the number of monomers that form the active transport system.34 These values, and the comparable values for receptors 1–4 are shown in Table 1.
For each receptor, the Hill coefficient is relatively small (n < 3). This provides evidence that these receptors do not assemble into membrane spanning channels, as given the size of the receptors, this would require the aggregation of a large number of molecules. However, in each case for Cl−/NO3− antiport n > 1 therefore the transport process does not necessarily occur via a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 receptor:anion complex. Matile and co-workers have previously reported anion transport by a capsule system, in which more than one receptor molecule surrounds the anion, after which the complex is able to diffuse across the lipid bilayer,35 and we have observed high Hill coefficients for anion transport by tren based anion receptors.2a Transport by an aggregate species cannot be ruled out in this case, especially given the ambiguity as to how these receptors interact with anions.
:1 receptor:anion complex. Matile and co-workers have previously reported anion transport by a capsule system, in which more than one receptor molecule surrounds the anion, after which the complex is able to diffuse across the lipid bilayer,35 and we have observed high Hill coefficients for anion transport by tren based anion receptors.2a Transport by an aggregate species cannot be ruled out in this case, especially given the ambiguity as to how these receptors interact with anions.
In order to gather further evidence for a mobile carrier mechanism, we tested the Cl−/NO3− antiport activity of 5–8 in vesicles composed of POPC–cholesterol (7:3). Cholesterol is known to order the bilayer, and it is thought that the resulting increased viscosity should slow the diffusion of a mobile carrier. Compounds 5 and 7 did show a reduced rate of transport in the POPC–cholesterol system; however, compounds 6 and 8 showed an increased rate of transport in the presence of cholesterol. This has been previously observed in the study of receptors bearing lipophilic substituents including –CF3 and alkyl groups. We propose that the addition of cholesterol to the bilayer also affects the partitioning of receptors into the bilayer as well as the viscosity of the membrane, therefore interpreting the results of such assays is not as straightforward as previously believed. However, receptors 6 and 8 were found to have a limited but measurable ability to transport chloride through a nitrobenzene organic phase in a U-tube assay (see ESI†). This supports a mobile carrier mechanism, as the assembly of a multicomponent channel across a large organic phase is highly implausible.
We have previously reported that across the series of thioureas 1–4 there is large variation in the transport activity.5a In this case, the nature of the substituent is clearly dominating the transport properties of these molecules. In contrast, we found that the EC50 values for Cl−/NO3− antiport by 5–8 were similar. For this reason, each Hill plot was repeated 3 times in order to assess the reproducibility of this data and to determine whether these values were statistically distinct. These results indicate that the order of transport efficiency is 7 ≈ 5 < 6 < 8. However, for reasons discussed in more detail below, the EC50 value for receptor 8 should be treated with caution as the activity was found to be highly dependent on the stock receptor concentration. We also performed Hill plots for Cl−/HCO3− antiport by these molecules. The same approximate trend in EC50 values for this process was obtained as for Cl−/NO3− exchange, although receptor 8 performed significantly more poorly.
In some cases, the acylthiourea was significantly more active than the thiourea analogue. For example, receptor 7 is approximately 20 times more active than receptor 3 (i.e. the EC50 value of 7 is 20 times smaller than that of receptor 3). However, in the case of the –CF3 and –pentyl substituted receptors, the thioureas were more active than the acylthiourea (receptor 4 is 14 times more active than receptor 8).
The small variation in activity across the series 5–8 is evidence that substituent effects are not dominating the anion transport properties of these receptors. Adding lipophilic substituents (such as –CF3 or –pentyl) did not enhance the transport activity.
During the testing of compound 8 we observed some unusual behaviour, which implied that the delivery of the receptor through the aqueous phase was problematic. Firstly, the chloride efflux profiles we obtained appeared to be sigmoidal, as the receptor seemed to take more time to begin transporting chloride after being introduced to the system than we have observed previously for other transporter systems (this effect can be seen in Fig. 5). Secondly, the activity of compound 8 at a particular lipid:receptor ratio was highly dependent on the concentration of the DMSO stock solution it was added from. Fig. 5 shows chloride efflux mediated by receptor 8 at a loading of 2 mol% with respect to lipid, which was added to the experiment using different volumes of different concentration stock solutions. The more concentrated the stock solution is, the lower the anion transport activity observed, despite the overall quantity of receptor added being the same. This implies that the greater volume of DMSO present, when using the more dilute stock solution, was aiding the delivery of the receptor to the lipid bilayer. It has been previously reported that across a series of receptors, there will be an optimum lipophilicity for transport.4e,8 Less lipophilic compounds will partition less effectively with the lipid bilayer, while more lipophilic compounds will not be able to effectively pass the aqueous phase. We propose that compound 8 might therefore be too lipophilic. In contrast, compound 4 is the most efficient of the thiourea series, indicating that across that series, the optimum lipophilicity has not yet been exceeded. The Hill plot for compound 8 was performed using only a 5 mM stock solution of the compound (whereas commonly a number of serial dilutions of the stock solution are performed during a Hill plot). Clearly, the results would have been very different if a more concentrated stock solution was used.
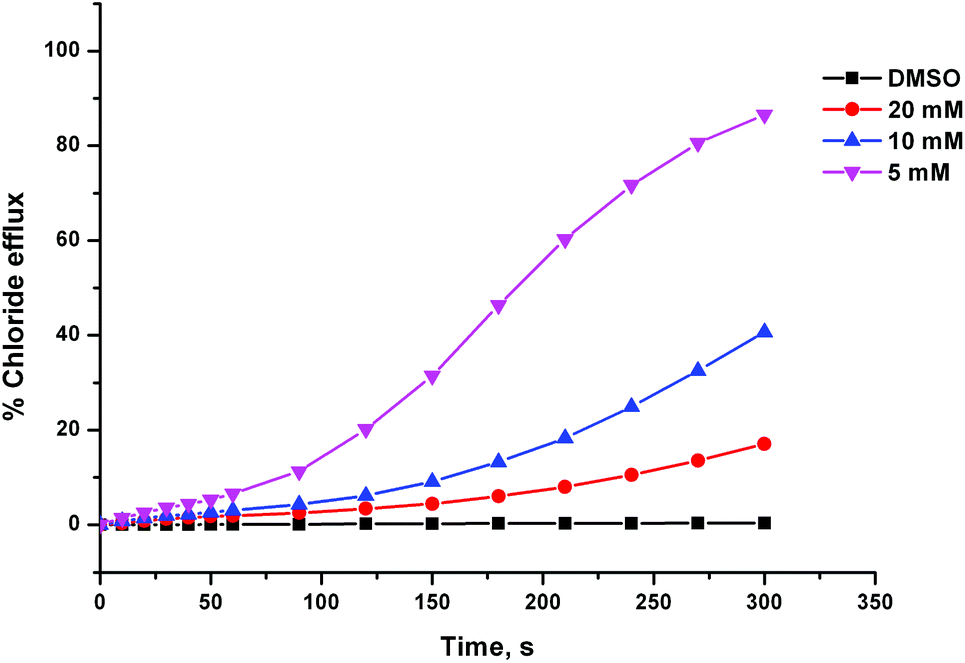 | ||
| Fig. 5 Chloride efflux mediated by compound 8 (2 mol% w.r.t. lipid), added from stock DMSO solutions of the receptor at varying concentration. The receptor was added to POPC vesicles containing 489 mM NaCl and suspended in 489 mM NaNO3, both buffered to pH 7.2 with 5 mM sodium phosphate salts. At the end of the experiment the vesicles were lysed with detergent in order to calibrate 100% chloride efflux. Each point represents the average of 3 trials. | ||
In order to test if the poor activity of receptor 8 was due to its problematic delivery to the bilayer, we adopted an anion transport assay based on the experiment reported by Davis and co-workers, in which the receptor is dispersed within the phospholipid prior to vesicle formation.5d As such, there is no aqueous barrier to delivering the receptor into the lipid bilayer, meaning highly lipophilic transporters can be tested.
We prepared a sample of POPC vesicles (1 mM lipid) with receptors 4–8 preincorporated at a concentration of 0.5 mol% with respect to lipid. The vesicles were prepared with internal and external NaNO3 (225 mM buffered to pH 7.2 with 5 mM sodium phosphate salts). The vesicles also contained 1 mM lucigenin, a fluorescent probe that is quenched in the presence of halide anions. On addition of a pulse of NaCl (25 mM) any resulting chloride influx was monitored via the corresponding decrease in fluorescence intensity of the lucigenin. The results are shown in Fig. 6. In this experiment, the trend in transport activity is reversed. The order of activity is 5 ≈ 7 < 6 < 8. This implies that although receptor 8 was the only receptor to show obvious signs of being too lipophilic to pass readily through the aqueous phase in the ISE experiments, perhaps this is a determining factor for all of the receptors, as when this barrier to transport is removed the expected trend in activity is observed.
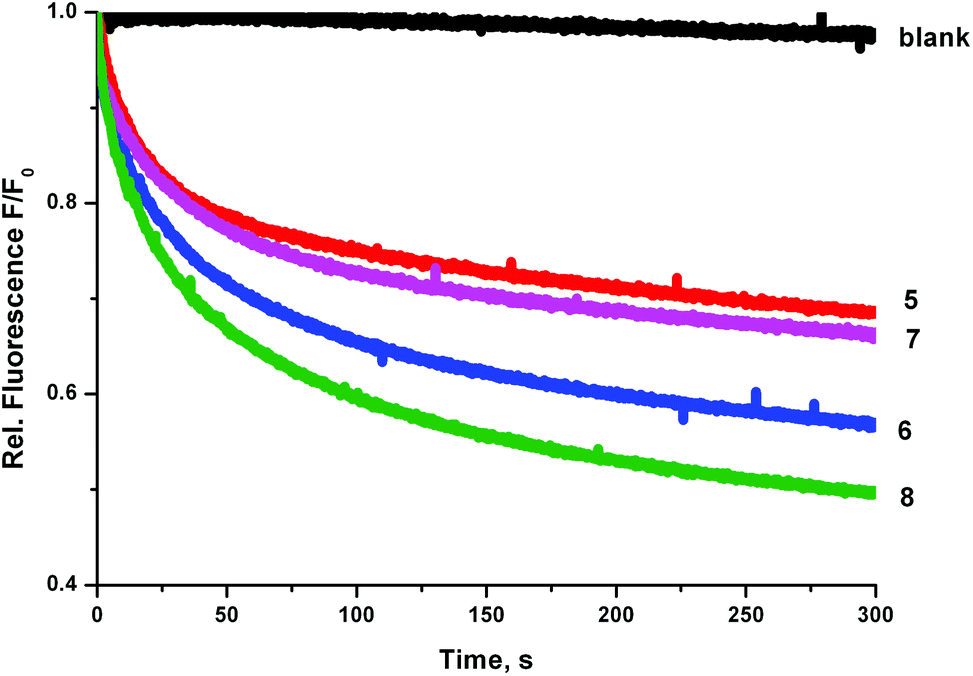 | ||
| Fig. 6 Chloride influx mediated by receptors 5–8 (preincorporated at 0.5 mol% with respect to lipid) into vesicles containing 225 mM NaNO3 buffered to pH 7.2 with sodium phosphate salts and 1 mM lucigenin. The vesicles were suspended in 225 mM NaNO3 and the experiment was initiated by adding 25 mM NaCl. Each experiment was repeated in triplicate. | ||
Receptor lipophilicity
A common technique to quantitatively assess the lipophilicity of a molecule is to determine a value of log P (the octanol–water partition coefficient). The log P values of a series of compounds have previously been shown to correlate to the retention time of the compounds on a reverse phase HPLC column.36 We performed an HPLC experiment using a reverse phase C18 column. The retention times of receptors 1–11 are shown in Table 2. The retention times of 1–4 correlate well with those previously reported (see ESI†).5a| Receptor | Retention time (min) | Cl−/NO3−a |
Cl−/HCO3−b |
||||
|---|---|---|---|---|---|---|---|
| EC50 | Error | n | Error | EC50 | n | ||
| a Transport data is the average of a minimum or 3 repeated Hill plot experiments. The error given is the standard deviation. b Transport data is the result of a single Hill plot analysis (therefore no errors are given). c Some transport data has been previously reported.2c,5a d Activity dependent on stock concentration of receptor solution – data gathered using only a 5 mM solution. e No anion transport activity detected. | |||||||
| 1 c | 4.9 | 2.7 | ±0.6 | 0.90 | ±0.10 | 3.1 | ±0.6 |
| 2 c | 8.3 | 0.44 | ±0.04 | 1.58 | ±0.17 | 0.89 | ±0.1 |
| 3 c | 5.1 | 5.5 | ±0.9 | 1.26 | ±0.12 | 5.8 | ±0.74 |
| 4 c | 10.8 | 0.08 | ±0.01 | 0.86 | ±0.17 | 0.10 | ±1.5 |
| 5 | 8.9 | 0.33 | ±0.06 | 2.07 | ±0.27 | 0.27 | ±0.78 |
| 6 | 10.5 | 0.69 | ±0.01 | 2.76 | ±0.24 | 8.1 | ±0.77 |
| 7 | 9.4 | 0.28 | ±0.02 | 2.03 | ±0.11 | 0.28 | ±0.91 |
| 8 | 12.9 | 1.1d | ±0.1d | 2.3d | ±0.21d | 6.5d | ±1.5d |
| 9 | 3.9 | —e | —e | —e | —e | —e | —e |
| 10 | 7.3 | —e | —e | —e | —e | —e | —e |
| 11 | 9.4 | —e | —e | —e | —e | —e | —e |
In general, all of the acylthiourea receptors are more lipophilic than the analogous thioureas despite containing an additional oxygen atom. However, tertiary acylthiourea 9, which cannot form an intramolecular hydrogen bond, is by far the least lipophilic compound studied. This indicates that the increased lipophilicity of 5–8 is not simply due to the functional groups present, implying that the formation of the intramolecular hydrogen bond is crucial. Further insights were obtained from DFT calculations on compounds 5 and 10–12 presented below.
Across the series 5–8, the varying substituent R-groups impose the same trend in lipophilicity as 1–4, i.e. –H < –OMe < –CF3 < –pentyl. However, the range of values is smaller across the acylthiourea series. The difference in retention time for receptors 1 and 4 is 5.9 min, while the difference between 5 and 8 is 4 min. Excluding receptors 4 and 8 (containing the most inherently lipophilic –pentyl substituent), the range is 3.4 min for the thioureas and 1.6 min for the acylthioureas. This implies that the overall lipophilicity of the acylthioureas is a balance between the lipophilicity of the substituents and the additional lipophilicity imparted by the intramolecular hydrogen bond. This balance is tipped in the favour of substituent effects in the case of the –pentyl substituted receptor, as increasing the length of an alkyl substituent results in an increase in lipophilicity without significant variation in electron donating character.
We examined the correlation between the activity of receptors 1–8 (represented by log(1/EC50)) and the retention time. The results are shown in Fig. 7. There is a negative correlation between the activity of the acylthioureas and their lipophilicity. Although the results for receptor 8 should be treated with caution due to the difficulties encountered in testing this compound, the negative correlation would still be evident excluding this data point.
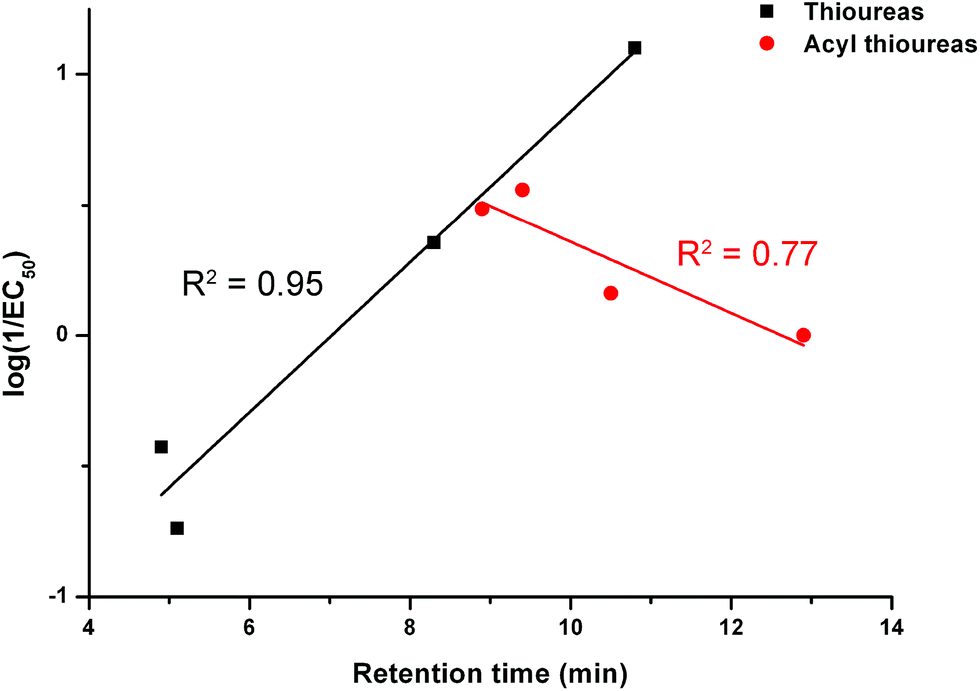 | ||
| Fig. 7 The relationship between retention time (determined by C18 HPLC) and transport activity of thioureas 1–4 and acylthioureas 5–8 as determined by an ISE assay. | ||
The trend in activity when the receptors were pre-incorporated is the opposite of the trend during the ISE assays. Therefore, the compounds with the longest retention times performed the best if they were pre-incorporated into the lipid bilayer. This provides further proof that within this series, the delivery of the receptors to the lipid bilayer is problematic (as reflected by the most lipophilic compounds being the poorest transporters in the ISE assay). However, if the receptors are pre-delivered to the lipid bilayer and this barrier is removed, the most lipophilic carriers are the most active, as might be expected. This rules out the possibility that the trends observed in the ISE assays were due to the more lipophilic carriers becoming trapped within the hydrophobic interior of the bilayer and thus unable to extract the anions from the aqueous interface.
Chloride binding in solution
Receptors 1–4 have been previously reported to interact weakly with tetrabutylammonium chloride in DMSO-d6–H2O 0.5%, with binding constants in the range of 10.6–27.9 M−1. The strength of binding was related to the Hammett constant of the R-group, with the strongest binding for compound 2 (electron withdrawing –CF3 group) and the weakest binding for compound 3 (electron donating –OMe group).5aIn order to assess the solution phase chloride binding properties of receptors 5–11, we performed 1H NMR titrations with tetrabutylammonium chloride in both a competitive solvent mixture (DMSO-d6–H2O 0.5%) and a non competitive solvent (CDCl3) at 298 K. Titrations were not performed with receptor 12 due to its instability and the presence of residual impurities.
Wei and co-workers have previously reported that acylthiourea receptors do not interact detectably with chloride anions under similar conditions as the intramolecular hydrogen bond effectively “locks” the receptor into a conformation that does not allow the anion to bind.17b No significant change in the 1H NMR spectrum was observed on the addition of up to ∼6 equivalents of chloride to receptor 5 in DMSO-d6–H2O 0.5%. In CDCl3, the NHa resonance was observed to broaden slightly, which may be indicative of a small change in the hydrogen bonding environment of this group due to interaction with the anion. Stack plots for all of the titrations performed can be found in the ESI.†
We reasoned that if the receptor is unable to interact with chloride due to the intramolecular hydrogen bond, it may be possible to observe a binding effect by first breaking the hydrogen bond by heating the system. We repeated the titration of receptor 5 with TBACl in DMSO-d6–H2O 0.5% at 323 K. In this experiment we observed a small change in chemical shift of the NH resonances on addition of the anion (see ESI†). This implies that chloride may be weakly coordinated by this receptor if the intramolecular hydrogen bonding interaction is first broken or weakened.
Across the series 6–8, the results of the titrations in both solvent systems were largely similar to those obtained for receptor 5, with negligible interaction observed in DMSO-d6–H2O 0.5% and peak broadening of the NHa resonance in CDCl3 (see ESI†). However, for receptors 9–11 a clearer interaction was observed in CDCl3. The only NH resonance of compound 9 and the NHa resonance in compound 11 were observed to undergo a small downfield shift indicative of anion binding, while the NHb resonance shifted slightly upfield. In compound 11 the NHa resonance shifted downfield and broadened, while NHb shifted upfield. All of these binding effects were too small to quantify; however, it is interesting that the compounds that were not able to mediate chloride transport again gave a different response to those that were effective transporters.
It is important to consider that, although the chloride binding by acylthioureas under the conditions of a 1H NMR titration experiment seems to be minimal, these conditions are not representative of a vesicle bilayer–aqueous phase interface. Additionally, the acylthiourea receptors only transport anions and not cations. Therefore it is reasonable to assume that there must be some interaction between the receptor and the anion under the conditions of the transport experiments, and that the lipophilicity of the scaffold cannot be the only determining factor towards transport, as this would impart no selectivity. The D2O shake experiments indicated that the strength of the intramolecular hydrogen bond is perturbed in the presence of increased aqueous content, which may imply binding is more likely to be possible at the bilayer–water interface.
Quantum calculations
Receptors 5–8 and 10–12 were optimised by DFT methods (B3LYP/6-311+G(d,p)) (see ESI† for computational details) to characterise the receptors’ binding affinity for anions, as well as the strength of the intramolecular hydrogen bonding interactions in the stabilization of the anti conformation of acylthioureas 6, 7, 10 and 11 found in the solid state (see Fig. 2). Therefore, the binding affinity was assessed through the most positive values mapped on the electrostatic potential surface, the VS,max, the importance of which in the characterization of anion transporters has previously been demonstrated,5a while the strength of the intramolecular NH⋯O/S bonds was described with second order interaction energies (E2).37 With the analysis of these parameters, along with anion efflux data, we attempted to establish how these quantum descriptors determine the transport ability of this series of molecules.The VS,max is related to the hydrogen bonding capacity and acidity38 and the values computed for compounds 1–8 and 10–12 are gathered in Table 3.
| Receptor | Substituent | syn a | anti a |
|---|---|---|---|
| a syn and anti refer to the two possible conformations adopted by the thiourea or urea binding units. | |||
| 1 | H | 59.8 | — |
| 2 | CF3 | 66.6 | — |
| 3 | OMe | 59.1 | — |
| 4 | n-Pentyl | 58.7 | — |
| 5 | H | 64.7 | 35.3 |
| 6 | CF3 | 70.6 | 42.0 |
| 7 | OMe | 64.0 | 33.2 |
| 8 | n-Pentyl | 62.9 | 34.4 |
| 10 | H | 64.0 | 39.3 |
| 11 | H | 63.2 | 36.7 |
| 12 | H | 62.6 | 31.6 |
In the subsequent analysis the compounds must be clustered in three sets: thioureas 1–4, acylthioureas 5–8 and compounds containing acylthiourea 5, acylurea 10, thioacylurea 11 and thioacylthiourea 12 motifs.
As would be expected, the VS,max values of the syn conformations of acylthioureas 5–8 are systematically higher when compared to the analogous thioureas due to the acyl motif. It is also evident that in both series the VS,max values increase, according to the electronic nature of the substituent, following the trend –pentyl < –OMe < –H < –CF3, which means that 2 and 6 with a para-electron withdrawing –CF3 group are the most acidic ones and, consequently, with higher binding affinity for the chloride anion. Furthermore, when the log(VS,max) are plotted against the corresponding EC50 values, a good correlation is obtained with both series of compounds (R2 = 0.77 for thioureas and R2 = 1.00 for acylthioureas, see Fig. SB3†), apart from the receptors with an n-pentyl substituent. In other words, in compounds 1–3 and 5–7 the transport activity appears to be related to the binding affinity, which is, in turn, mainly determined by the electronic nature of the different substituents. By contrast, the transport activity of 4 and 8, with a long n-pentyl alkyl chain is governed by the individual high lipophilicity (see Table 2), in agreement with the experimental transport studies reported above. It is also important to emphasise that the relationship between EC50 and VS,max parameters for thioureas and acylthioureas follow reverse trends, which seems to indicate that the intramolecular hydrogen bond in these molecules plays a role in the transport activity of these receptors, as apparent from the subsequent analysis.
The conformational change from syn to anti in acylthioureas leads to a significant decrease in VS,max values, with the –pentyl and –OMe substituents changing the order in the trend. The lower VS,max values for the anti structures of 5–8, which have an intramolecular hydrogen bond, indicate that these conformations are less acidic than the syn ones. Hence, the anti conformations are less able to bind the chloride anion, promoting its transport, in agreement with the absence of experimental transport activity found for 9, with a single NH binding group. However, it is noteworthy that the correlation found between EC50 and VS,max values of the anti structures is maintained with syn structures, when compound 8 is excluded from the data set, yielding a R2 = 0.98 (see Fig. SB3†). These correlations suggest that both conformations are relevant for the transmembrane transport of chloride. In the syn conformation of 5–8 the VS,max is positioned at the binding pocket defined by the two NH groups, while in the anti conformation it was found to be close of the free NHb group. The electrostatic potential mapped on the electron density surface of 6 together with the location VS,max is illustrated in Fig. 8 for syn (top) and anti (bottom) conformations. Equivalent graphical depictions are shown in Fig. SB1† for compounds 5, 7, 8 and 10–12.
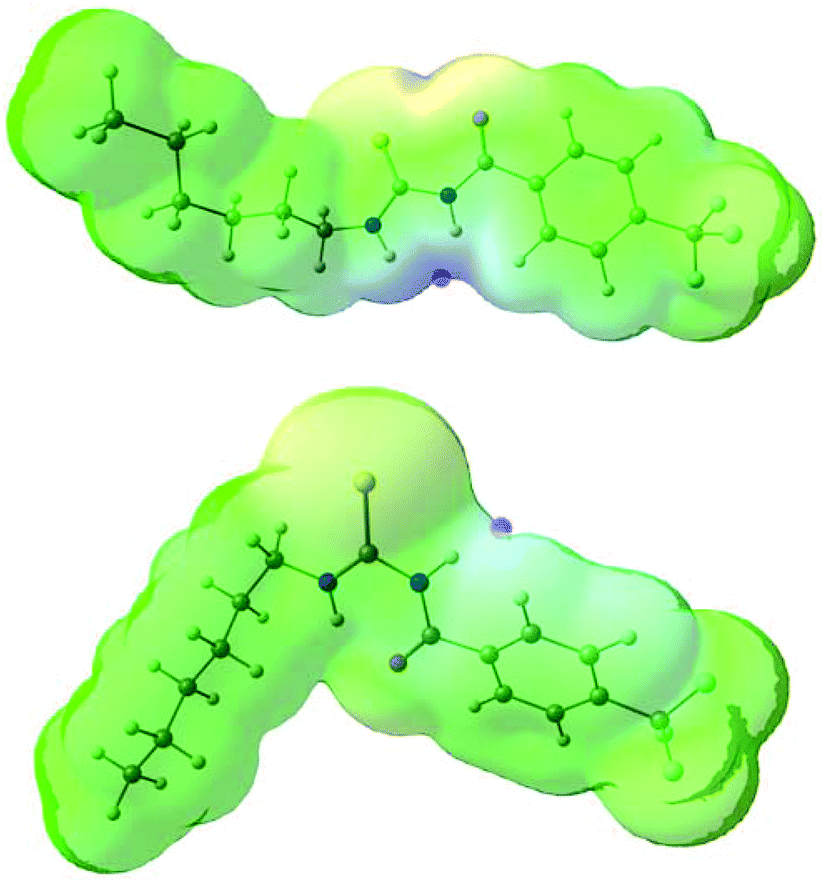 | ||
| Fig. 8 Computed electrostatic potential, mapped on the 0.001 electrons per Bohr3 surface of 6 for syn (top) and anti (bottom) conformations, with colours ranging between blue (+70.6 kcal mol−1) and red (−64.2 kcal mol−1) for the more positive and negative values, respectively. The location of VS,max for both conformations is emphasised as a pink dot. | ||
As observed for acylthioureas 5–8, the VS,max calculated for compounds 10–12 in both conformations indicate that the syn conformation is more acidic than the anti one. Furthermore, among the series composed of 5 and 10–12, the anti conformation in 10 with VS,max of 39.3 kcal mol−1 is slightly more acidic than in the remaining compounds with VS,max values ranging between 31.6 and 36.7 kcal mol−1.
The interaction energy associated with the formation of an intramolecular hydrogen bond in compounds 5–8 and 10–12 was estimated for the anti conformations via perturbation theory using the natural bond orbitals (NBO) analysis. The interaction energy values computed for NHa⋯O or NHa⋯S intramolecular bonding interactions are collected in Table 4. A graphical depiction of the interaction between the NBOs is given in Fig. SB2† for 5, which shows the interaction between the lone pair (LP) orbitals and the NHa antibonding (σ*) orbitals.
| Receptor |
|
|
|
|---|---|---|---|
| 5 | 2.97 | 7.87 | 10.84 |
| 6 | 2.80 | 7.47 | 10.27 |
| 7 | 3.09 | 8.18 | 11.27 |
| 8 | 3.04 | 8.05 | 11.09 |
| 10 | 2.19 | 6.31 | 8.50 |
| 11 | 2.04 | 14.41 | 16.45 |
| 12 | 2.78 | 18.49 | 21.27 |
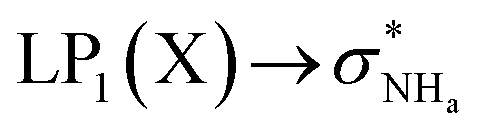
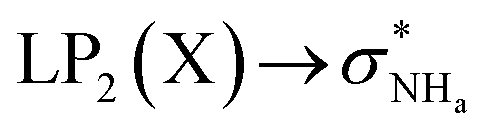

The active anion transporters 5–8 exhibit energies in a narrow range from 10.27 to 11.27 kcal mol−1. Across this series, as expected, receptor 7 has the strongest interaction as a result of the electron donating –OMe group, while the weakest interaction is found in receptor 6 with the electron withdrawing –CF3 group. In contrast, the strength of this interaction in receptor 10 (8.50 kcal mol−1) and receptors 11 and 12 (16.45 and 21.27 kcal mol−1) are outside of this energy range. These data suggest that chloride transport is dependent on an ideal interval of interaction energies, defined by the acylthiourea unit of 5–8. Thus, 10, with the lowest E2 energy, does not promote anion transport, while 11 and 12, although with higher E2 energy values, may be able to passively diffuse across the bilayer, but are almost unable to bind the anion, which is a necessary condition for the anion transmembrane transport. In other words, a significant amount of the energy should be required to break the NHa⋯S hydrogen bond in 11 or 12.
As previously found for the VS,max values, the E2 energies also seem to correlate well with the EC50 values (R2 = 0.90, see Fig. SB4†), thus suggesting the dependence between the strength of the intramolecular hydrogen bond and the transport ability. Also, the match between E2 and VS,max values for anti conformations of the transporters yields a good correlation (R2 = 0.97, Fig. SB5†), which indicates that a high VS,max value corresponds to a weak intramolecular hydrogen bond and vice-versa.
In summary, this quantum analysis shows that the interaction energies for this series of molecules are relatively low with exception of 11 and 12. Therefore, the intramolecular hydrogen bond is easily broken which leads to a conformational change. In this context, a delicate balance between the hydrogen bond strength, the anion binding affinity and the lipophilicity dictates the transport ability of the transports 5–8. Obviously, this analysis is limited by the number of studied molecules.
Conclusions
In conclusion, acylthioureas have been found to function as efficient anion antiporters. They are more lipophilic than analogous thioureas due to the formation of an intramolecular hydrogen bond, which shields the binding site from interactions with water. As such, the incorporation of lipophilic substituents results in the receptors becoming too lipophilic if passage through an aqueous phase is required.It is clear that the lipophilicity of simple anion transporters is crucial to their transport activity. Therefore, understanding the factors that dictate the lipophilicity and anion binding affinity of a structure is imperative. Intramolecular hydrogen bonding could be used as a design tool to enable the use of more hydrophilic scaffolds in transporter design. This would be advantageous in the design of new transporters for medicinal applications, in which absorption and distribution throughout the body would require a balance between solubility in the blood and in lipid bilayers.
Acknowledgements
We thank the EPSRC for funding (CJEH – EP/J009687/1) and for access to the crystallographic facilities at the University of Southampton. We thank the University of Southampton and A*STAR for a postgraduate scholarship (NB) and the University of Southampton for a teaching assistantship (ILK). PAG thanks the Royal Society and the Wolfson Foundation for a Royal Society Wolfson Research Merit Award. IM thanks the FCT (Fundação para a Ciência e a Tecnologia) for the PhD scholarship SFRH/BD/87520/2012. VF acknowledges the funding from QREN-FEDER, through the Operational Program Competitiveness Factors – COMPETE and National Funds through the FCT under project PTDC/QUI-QUI/101022/2008.Notes and references
- (a) N. Busschaert and P. A. Gale, Angew. Chem., Int. Ed., 2013, 52, 1374–1382 CrossRef CAS PubMed; (b) C. J. E. Haynes and P. A. Gale, Chem. Commun., 2011, 47, 8203–8209 RSC; (c) J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC; (d) A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC; (e) P. A. Gale, R. Peréz-Tomás and R. Quesada, Acc. Chem. Res., 2013 DOI:10.1021/ar400019p.
- (a) N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernandez, R. Perez-Tomas and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed; (b) S. J. Moore, C. J. E. Haynes, J. Gonzalez, J. L. Sutton, S. J. Brooks, M. E. Light, J. Herniman, G. J. Langley, V. Soto-Cerrato, R. Perez-Tomas, I. Marques, P. J. Costa, V. Felix and P. A. Gale, Chem. Sci., 2013, 4, 103–117 RSC; (c) S. J. Moore, M. Wenzel, M. E. Light, R. Morley, S. J. Bradberry, P. Gomez-Iglesias, V. Soto-Cerrato, R. Perez-Tomas and P. A. Gale, Chem. Sci., 2012, 3, 2501–2508 RSC; (d) P. I. Hernandez, D. Moreno, A. A. Javier, T. Torroba, R. Perez-Tomas and R. Quesada, Chem. Commun., 2012, 48, 1556–1558 RSC; (e) B. D. de Grenu, P. I. Hernandez, M. Espona, D. Quinonero, M. E. Light, T. Torroba, R. Perez-Tomas and R. Quesada, Chem.–Eur. J., 2011, 17, 14074–14083 CrossRef PubMed; (f) J. L. Sessler, L. R. Eller, W. S. Cho, S. Nicolaou, A. Aguilar, J. T. Lee, V. M. Lynch and D. J. Magda, Angew. Chem., Int. Ed., 2005, 44, 5989–5992 CrossRef CAS PubMed; (g) N. Busschaert, L. E. Karagiannidis, M. Wenzel, C. J. E. Haynes, N. J. Wells, P. G. Young, D. Makuc, J. Plavec, K. A. Jolliffe and P. A. Gale, Chem. Sci. 10.1039/C3SC52006D.
- (a) J. T. Davis, P. A. Gale, O. A. Okunola, P. Prados, J. C. Iglesias-Sanchez, T. Torroba and R. Quesada, Nat. Chem., 2009, 1, 138–144 CrossRef CAS PubMed; (b) P. V. Santacroce, J. T. Davis, M. E. Light, P. A. Gale, J. C. Iglesias-Sanchez, P. Prados and R. Quesada, J. Am. Chem. Soc., 2007, 129, 1886–1887 CrossRef CAS PubMed.
- (a) S. Hussain, P. R. Brotherhood, L. W. Judd and A. P. Davis, J. Am. Chem. Soc., 2011, 133, 1614–1617 CrossRef CAS PubMed; (b) L. W. Judd and A. P. Davis, Chem. Commun., 2010, 46, 2227–2229 RSC; (c) A. V. Koulov, T. N. Lambert, R. Shukla, M. Jain, J. M. Boon, B. D. Smith, H. Y. Li, D. N. Sheppard, J. B. Joos, J. P. Clare and A. P. Davis, Angew. Chem., Int. Ed., 2003, 42, 4931–4933 CrossRef CAS PubMed; (d) B. A. McNally, A. V. Koulov, T. N. Lambert, B. D. Smith, J. B. Joos, A. L. Sisson, J. P. Clare, V. Sgarlata, L. W. Judd, G. Magro and A. P. Davis, Chem.–Eur. J., 2008, 14, 9599–9606 CrossRef CAS PubMed; (e) C. J. E. Haynes, S. J. Moore, J. R. Hiscock, I. Marques, P. J. Costa, V. Felix and P. A. Gale, Chem. Sci., 2012, 3, 1436–1444 RSC.
- (a) N. Busschaert, S. J. Bradberry, M. Wenzel, C. J. E. Haynes, J. R. Hiscock, I. L. Kirby, L. E. Karagiannidis, S. J. Moore, N. J. Wells, J. Herniman, G. J. Langley, P. N. Horton, M. E. Light, I. Marques, P. J. Costa, V. Félix, J. G. Frey and P. A. Gale, Chem. Sci., 2013, 4, 3036–3045 RSC; (b) N. Busschaert, P. A. Gale, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis and W. A. Harrell, Chem. Commun., 2010, 46, 6252–6254 RSC; (c) N. J. Andrews, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis, W. A. Harrell and P. A. Gale, Chem. Sci., 2011, 2, 256–260 RSC; (d) B. A. McNally, A. V. Koulov, B. D. Smith, J. B. Joos and A. P. Davis, Chem. Commun., 2005, 1087–1089 RSC.
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef CAS PubMed.
- M. Wenzel, M. E. Light, A. P. Davis and P. A. Gale, Chem. Commun., 2011, 47, 7641–7643 RSC.
- V. Saggiomo, S. Otto, I. Marques, V. Felix, T. Torroba and R. Quesada, Chem. Commun., 2012, 48, 5274–5276 RSC.
- (a) C.-R. Elie, A. Hébert, M. Charbonneau, A. Haiun and A. R. Schmitzer, Org. Biomol. Chem., 2013, 11, 923–928 RSC; (b) C.-R. Elie, M. Charbonneau and A. R. Schmitzer, MedChemComm, 2012, 3, 1231–1234 RSC; (c) C. Chhun and A. R. Schmitzer, MedChemComm, 2011, 2, 987–990 RSC.
- (a) X. Li, B. Shen, X.-Q. Yao and D. Yang, J. Am. Chem. Soc., 2007, 129, 7264–7265 CrossRef CAS PubMed; (b) X. Li, B. Shen, x.-Q. Yao and D. Yang, J. Am. Chem. Soc., 2009, 131, 13676–13680 CrossRef CAS PubMed.
- B. Kuhn, P. Mohr and M. Stahl, J. Med. Chem., 2010, 53, 2601–2611 CrossRef CAS PubMed.
- V. A. Ashwood, M. J. Field, D. C. Horwell, C. Julien-Larose, R. A. Lewthwaite, S. McCleary, M. C. Pritchard, J. Raphy and L. Singh, J. Med. Chem., 2001, 44, 2276–2285 CrossRef CAS PubMed.
- A. A. Skjevik, B. E. Haug, H. Lygre and K. Teigen, Biophys. Chem., 2011, 154, 18–25 CrossRef CAS PubMed.
- S. Sasaki, N. Cho, Y. Nara, M. Harada, S. Endo, N. Suzuki, S. Furuya and M. Fujinov, J. Med. Chem., 2003, 46, 113–124 CrossRef CAS PubMed.
- T. Rezai, J. E. Bock, M. V. Zhou, C. Kalyanaraman, R. S. Lokey and M. P. Jacobson, J. Am. Chem. Soc., 2006, 128, 14073–14080 CrossRef CAS PubMed.
- M. Shalaeva, G. Caron, Y. A. Abramov, T. N. O'Connell, M. S. Plummer, G. Yalamanchi, K. A. Farley, G. H. Goetz, L. Phillipe and M. J. Shapiro, J. Med. Chem., 2013, 56, 4870–4879 CrossRef CAS PubMed.
- (a) Y. M. Zhang, J. D. Qin, Q. Lin and T. B. Wei, J. Fluorine Chem., 2006, 127, 1222–1227 CrossRef CAS PubMed; (b) T. B. Wei, W. Wei, C. Cao and Y. M. Zhang, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 1218–1228 CrossRef CAS; (c) J. H. Hu, L. Xu, J. Wang and T. B. Wei, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 1584–1591 CrossRef CAS.
- (a) M. Kodomari, M. Suzuki, K. Tanigawa and T. Aoyama, Tetrahedron Lett., 2005, 46, 5841–5843 CrossRef CAS PubMed; (b) P. Vest, M. Schuster and K. H. Konig, Fresenius’ J. Anal. Chem., 1991, 341, 566–568 CrossRef CAS; (c) P. Vest, M. Schuster and K. H. Konig, Fresenius’ Z. Anal. Chem., 1989, 335, 759–763 CrossRef CAS; (d) R. C. Luckay, F. Mebrahtu, C. Esterhuysen and K. R. Koch, Inorg. Chem. Commun., 2010, 13, 468–470 CrossRef CAS PubMed; (e) S. A. Bourne, O. Hallale and K. R. Koch, Cryst. Growth Des., 2005, 5, 307–312 CrossRef CAS; (f) K. R. Koch, Coord. Chem. Rev., 2001, 473–488 CrossRef CAS; (g) G. Binzet, B. Zeybek, E. Kilic, N. Kulcu and H. Arslan, J. Chem., 2013 DOI:10.1155/2013/201238; (h) V. Circu, D. Manaila-Maximean, C. Rosu, M. Ilis, Y. Molard and F. Dumitrascu, Liq. Cryst., 2009, 36, 123–132 CrossRef CAS; (i) M. Merdivan, F. Karipcin, N. Kulcu and R. S. Aygun, J. Therm. Anal. Calorim., 1999, 58, 551–557 CrossRef CAS; (j) A. Mohamadou, I. Dechampsolivier and J. P. Barbier, Polyhedron, 1994, 13, 1363–1370 CrossRef CAS; (k) N. Ozpozan, T. Ozpozan, H. Arslan, F. Karipcin and N. Kulcu, Thermochim. Acta, 1999, 336, 97–103 CrossRef CAS; (l) S. Ringmann and M. Schuster, Chem. Tech., 1997, 49, 217–226 CAS; (m) Y. M. Beasly, V. Petrow and O. Stephenson, J. Pharm. Pharmacol., 1961, 13, 694–697 CrossRef; (n) S. A. Bourne, O. Hallale and K. R. Koch, Cryst. Growth Des., 2005, 5, 307–312 CrossRef CAS; (o) K. R. Koch, Coord. Chem. Rev., 2001, 473–488 CrossRef CAS.
- J. T. Hackmann, Chem. Abstr., 1960, US Patent 2923656, 52028.
- A. Joos and W. Wirtz, Chem. Abstr., 1972, Ger. Patent 2059872, 88114.
- A. R. Katritzky, N. Kirichenko, B. V. Rogovoy, J. Kister and H. Tao, Synthesis, 2004, 1799–1805 CrossRef CAS PubMed.
- T. Sohda, H. Ikeda and Y. Momose, Chem. Abstr., 1990, Eur. Patent 369453, 191334.
- M. Missbach, Chem. Abstr., 1995, Int. Patent WO 9514685, 169640.
- K.-J. Hwang, K.-H. Park, C. O. Lee and S. U. Choi, Chem. Abstr., 1995, Int. Patent WO 9525096, 117305.
- G. S. Bisacchi, S. M. Seiler and R. M. Lawrence, Chem. Abstr., 2000, Int. Patent WO 0047563, 177117.
- W J Fanshewe and J. W. Epstein, Chem. Abstr., 1991, US Patent 5001157, 91862.
- (a) D A Holt, Chem. Abstr., 1995, Int. Patent WO 9521185, 314261; (b) A. Panzeri, E. Di Salle and M. Nesi, Chem. Abstr., 1991, Int. Patent WO 9112261, 256467.
- (a) A. Saeed, R. A. Khera, N. Abbas, M. Latif, I. Sajid and U. Florke, Turk. J. Chem., 2010, 34, 335–345 CAS; (b) D. C. Zhang, Y. Q. Zhang, Y. Cao and B. Zhao, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1716–1718 CrossRef; (c) Y. Cao, B. Zhao, Y. Q. Zhang and D. C. Zhang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1772–1774 CrossRef.
- G. Lessene, B. J. Smith, R. W. Gable and J. B. Baell, J. Org. Chem., 2009, 74, 6511–6525 CrossRef CAS PubMed.
- M. Llinas and M. P. Klein, J. Am. Chem. Soc., 1975, 97, 4731–4737 CrossRef CAS.
- B. D. Smith and T. N. Lambert, Chem. Commun., 2003, 2261–2268 RSC.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- A. V. Hill, Biochem. J., 1913, 7, 471–480 CAS.
- S. Bhosale and S. Matile, Chirality, 2006, 18, 849–856 CrossRef CAS PubMed.
- A. V. Jentzsch, D. Emery, J. Mareda, S. K. Nayak, P. Metrangolo, G. Resnati, N. Sakai and S. Matile, Nat. Commun., 2012, 3 Search PubMed.
- T. Braumann, J. Chromatogr., 1986, 373, 191–225 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- Y. G. Ma, K. C. Gross, C. A. Hollingsworth, P. G. Seybold and J. S. Murray, J. Mol. Model., 2004, 10, 235–239 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of the receptors, details of X-ray crystallography,39 stack plots for the 1H NMR titrations of the receptors with TBACl in DMSO–water and CDCl3, various vesicles assay methods, Hill plots and DFT calculations. CCDC 941594–941597. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob41522h |
| ‡ Structures were deposited with the Cambridge Crystallographic Database Centre (CCDC) and given the numbers CCDC 941594–941597. |
| This journal is © The Royal Society of Chemistry 2014 |