Hydrophilic tetracarboxy bacteriochlorins for photonics applications†
Jianbing
Jiang
,
Pothiappan
Vairaprakash
,
Kanumuri Ramesh
Reddy
,
Tuba
Sahin
,
M. Phani
Pavan
,
Elisa
Lubian
and
Jonathan S.
Lindsey
*
Department of Chemistry, North Carolina State University, Raleigh, NC 27695-8204, USA. E-mail: jlindsey@ncsu.edu
First published on 23rd October 2013
Abstract
Bacteriochlorins absorb strongly in the near-infrared (NIR, 700–900 nm) region and hence are well suited for photophysical studies and photomedical applications, yet such endeavors heretofore have been largely limited by the intrinsic lipophilicity of the bacteriochlorin macrocycle. Here, a new molecular design is investigated wherein 3,5-dicarboxyphenyl units are appended to the β-pyrrolic positions of the bacteriochlorin. Use of the 3,5-aryl substitution motif places the carboxylic acid groups, which are anionic at neutral pH, above and below the plane of the bacteriochlorin macrocycle. A de novo synthesis has been employed to create five such bacteriochlorins, which uses as intermediates two new 2,12-dibromobacteriochlorin building blocks and a known 3,13-dibromobacteriochlorin. The aryl groups with protected carboxylate moieties were introduced by Suzuki coupling; subsequent deprotection afforded the hydrophilic bacteriochlorins. The latter were characterized by absorption and fluorescence spectroscopy in DMF and in aqueous phosphate buffer (pH 7). In most cases, comparable sharp emission (FWHM of ∼25 nm) and modest fluorescence yields (0.060–0.11) were observed in aqueous phosphate buffer medium and in DMF. Aqueous solubility was examined by absorption spectral interrogation of samples over a 1000-fold concentration range with reciprocal change in pathlength (∼0.5, 5, 50, and 500 μM; 10, 1, 0.1, and 0.01 cm pathlength cuvettes). One hydrophilic bacteriochlorin was prepared that contains a single maleimido-terminated tether for bioconjugation; the tether was installed by the sequence of 15-bromination of the bacteriochlorin, Suzuki coupling, and DCC-mediated amide formation. The maleimido-bacteriochlorin was conjugated to a 48-residue cysteine-containing peptide analogue of a constituent from a bacterial photosynthetic light-harvesting complex. Taken together, the results show a new molecular design and facile de novo synthetic route for obtaining hydrophilic bacteriochlorins including a bioconjugatable group if desired.
Introduction
Bacteriochlorins (tetrahydroporphyrins) are attractive candidates in a wide variety of photochemical studies due to their strong absorption in the near-infrared (NIR) region (700–900 nm).1 Bacteriochlorophylls a, b and g contain the bacteriochlorin chromophore and provide the basis for light-harvesting processes and electron-transfer reactions in bacterial photosynthesis (Chart 1). Bacteriochlorophylls contain a full complement of substituents about the perimeter of the macrocycle and hence are only partially amenable toward semisynthetic tailoring2,3 as required for diverse studies such as cellular imaging, photodynamic therapy, clinical diagnostics, and artificial photosynthesis. The tailoring can include introduction of (i) auxochromes to tune the position of the long-wavelength absorption band, (ii) hydrophobic or hydrophilic groups to alter polarity, and (iii) one or more derivatizable groups for attachment to surfaces, macromolecules, or other entities.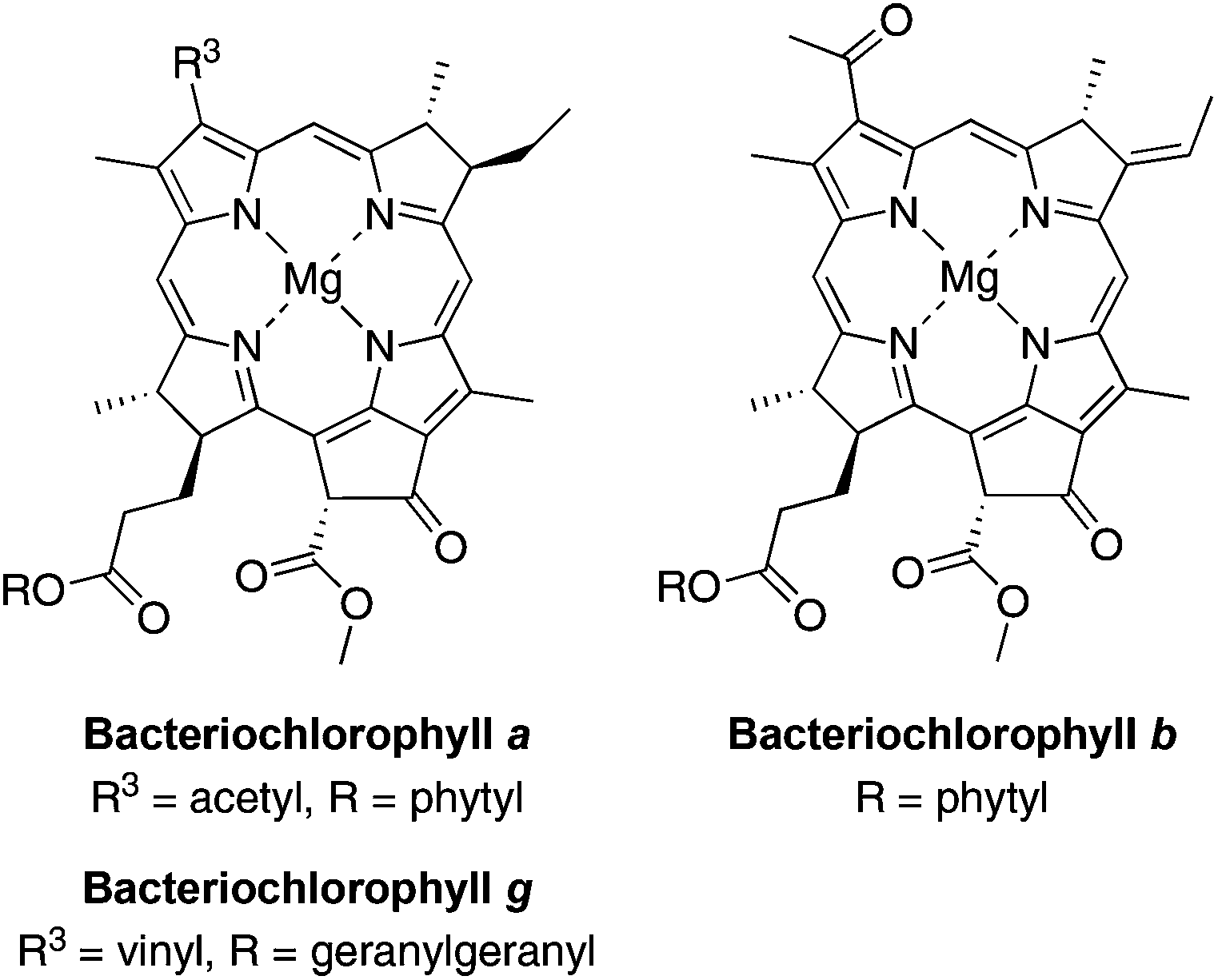 | ||
| Chart 1 Bacteriochlorophylls. | ||
Methods for the synthesis of bacteriochlorins are under active development by a number of groups.4–18 The methods range from the modification of native bacteriochlorophylls, hydrogenation or reductive addition to synthetic or native porphyrins or chlorins, and de novo synthesis.2,3,19,20 In the de novo synthesis of bacteriochlorins developed in our laboratory,21,22 the geminal dimethyl group in each reduced pyrroline ring increases stability by blocking adventitious oxidative pathways. The synthesis of bacteriochlorins relies on the self-condensation of a dihydrodipyrrin–acetal as shown in Scheme 1. The use of two identical dihydrodipyrrin–acetal molecules to form the bacteriochlorin affords synthetic expediency but has the drawback that the substituents at positions 2 and 12 are identical to each other, as are those at positions 3 and 13. A single group can be introduced at the 15-position of the bacteriochlorin by regioselective bromination of the 5-methoxybacteriochlorin.23 Wavelength tunability is achieved by installation of auxochromes at positions along the y-axis (e.g., 2, 3, 5, 12, 13, 15) as shown in Scheme 1.
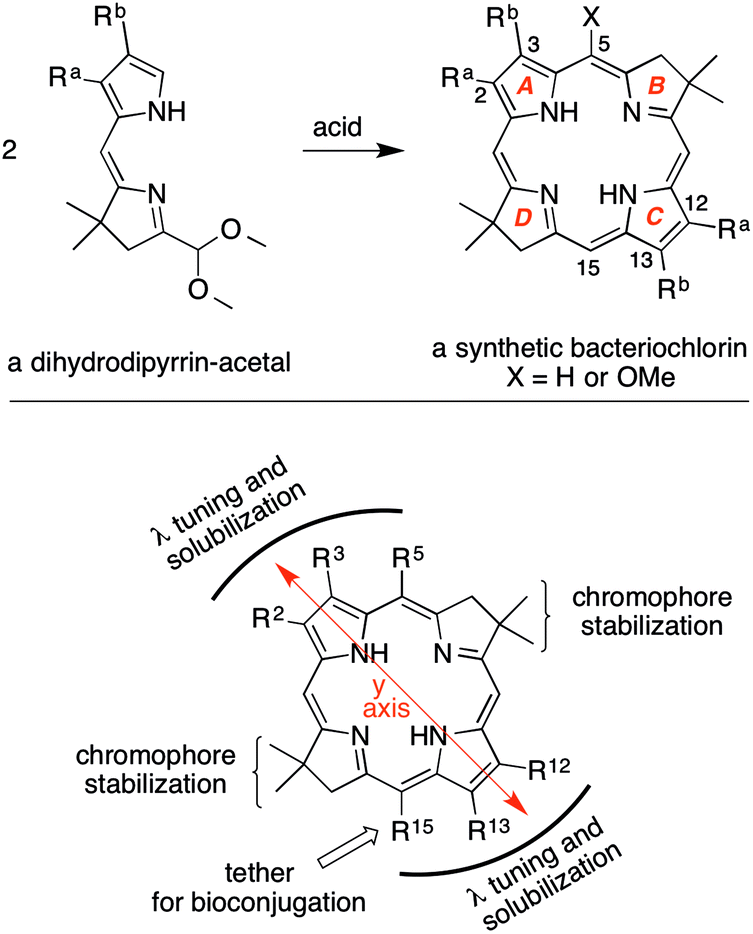 | ||
| Scheme 1 Synthesis of bacteriochlorins (top) and molecular design features (bottom). | ||
Using the approach in Scheme 1, families of bacteriochlorins have been created that are lipophilic,13,22,24,25 amphiphilic,26–28 or hydrophilic;25,29,30 lipophilic and wavelength-tunable;13,31 as well as lipophilic, bioconjugatable, and wavelength-tunable.13,14,32 The nexus of “hydrophilic and bioconjugatable” has heretofore not been attained with the approach shown in Scheme 1 although several hydrophilic, bioconjugatable bacteriochlorins based on other synthetic approaches have been prepared.4,5,7,9 Regardless, a general solution that opens the door to “hydrophilic, bioconjugatable, and wavelength-tunable” bacteriochlorins of de novo design has not been described. An excellent review of bioconjugatable tetrapyrrole macrocycles (chiefly porphyrins and chlorins) has been prepared by Boyle and coworkers.33 Recent reports describe semisynthetic routes to bacteriochlorins that are “lipophilic and wavelength-tunable,”34 “hydrophilic and wavelength-tunable,”35 or “lipophilic and bioconjugatable,”36 but again, routes to the nexus of all three features have not yet been developed.
The structures of representative hydrophilic bacteriochlorins that we prepared previously are shown in Chart 2. Compound BC-1 is compact and also was a precursor in the synthesis of BC-3 and analogues.25 Compounds BC-2 and BC-3 were prepared25 for studies in antimicrobial photodynamic therapy. Among several analogues prepared, BC-3 exhibited a negative log P value (−1.4) indicating preferential dissolution in water versus n-octanol.29 Compounds BC-4 and BC-5 represent an initial foray into hydrophilic bacteriochlorins that were thought to be compatible with subsequent elaboration with a bioconjugatable tether. Only limited photophysical studies have heretofore been carried out concerning the bacteriochlorins in Chart 2. Such studies employed the polar solvents DMF (BC-4, BC-5) or methanol (BC-1, BC-3), but only in one case examined water (BC-4). Regardless, none of the bacteriochlorins shown in Chart 2 is compatible with attachment of a bioconjugatable tether (vide infra).
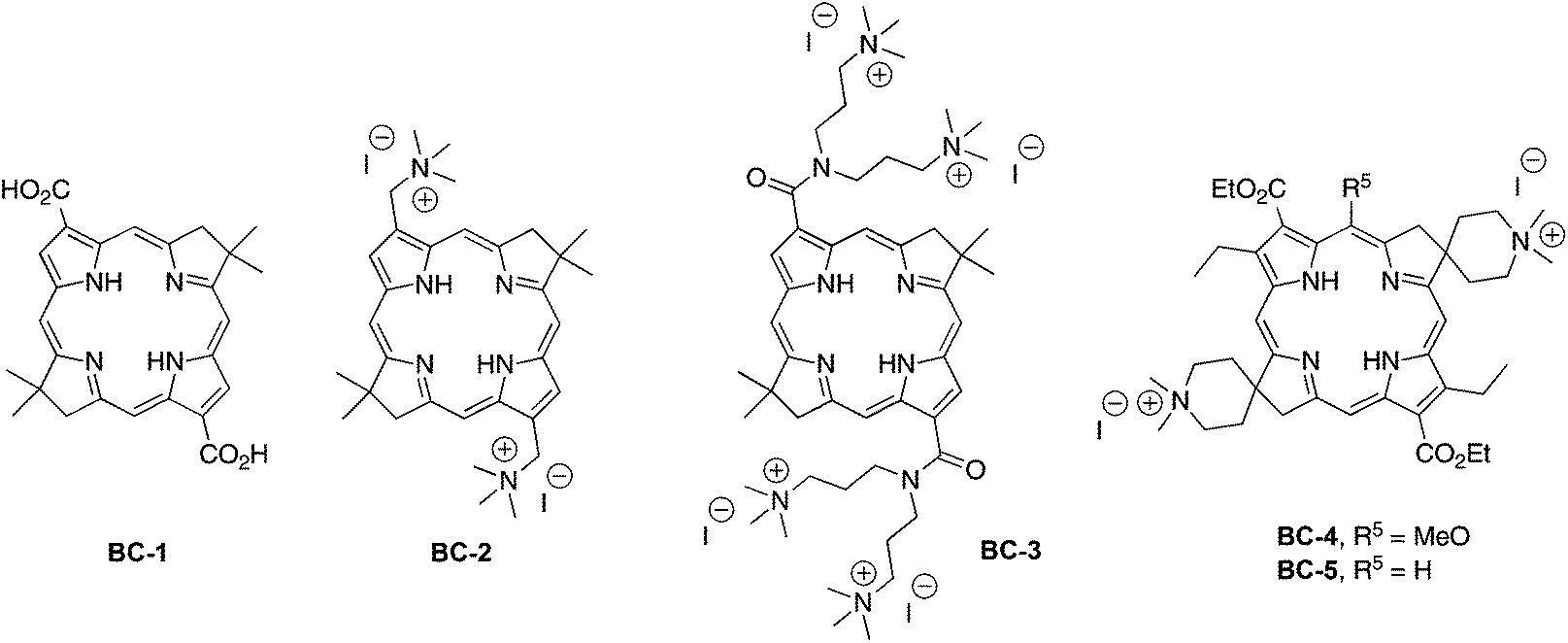 | ||
| Chart 2 Hydrophilic bacteriochlorins. | ||
A generic design of hydrophilic tetrapyrroles has been to employ ionized groups (ammonium,37–43 sulfonate,10,12,44,45 carboxylate46,47 or phosphonate48,49) or polar but nonionic groups (polyethylene glycol50–53 or glycoside54,55) at the synthetically accessible, meso-positions or at malleable groups at the perimeter of natural tetrapyrroles. A very attractive molecular design to impart hydrophilicity is to position ionizable groups (at neutral pH) above and below the plane of the macrocycle and thereby suppress aggregation by electrostatic repulsion. Pereira and coworkers synthesized water-soluble chlorin and bacteriochlorins (e.g., I) with arylsulfonic groups at the meso positions (although atropisomers are possible whereupon the two faces may be disproportionately “shielded”). Bacteriochlorin I (Chart 3) exhibits fluorescence quantum yield (Φf) of 0.012 in aqueous phosphate buffer (pH 7.4).10 A chlorin-diphosphonate (II) contains alkyl phosphonate groups projected above and below the plane of the macrocycle.56 Kobuke and coworkers prepared ethyne-linked porphyrin dyad (III), which bears carboxy-substituted swallowtail groups at the meso positions. The trans-AB-porphyrin IV incorporates a phosphono-substituted swallowtail group at one of the meso positions, is bioconjugatable due to the iodoacetamide unit, and exhibits high (>10 mM) aqueous solubility.49 A porphyrin (V) that bears a single tricarboxy-substituted aryl group is soluble at 3 mM in aqueous solution.57 All of these examples illustrate the use of facial encumbrance to impart higher solubility of tetrapyrrole macrocycles, an approach that stems from a very lengthy thread of research.57
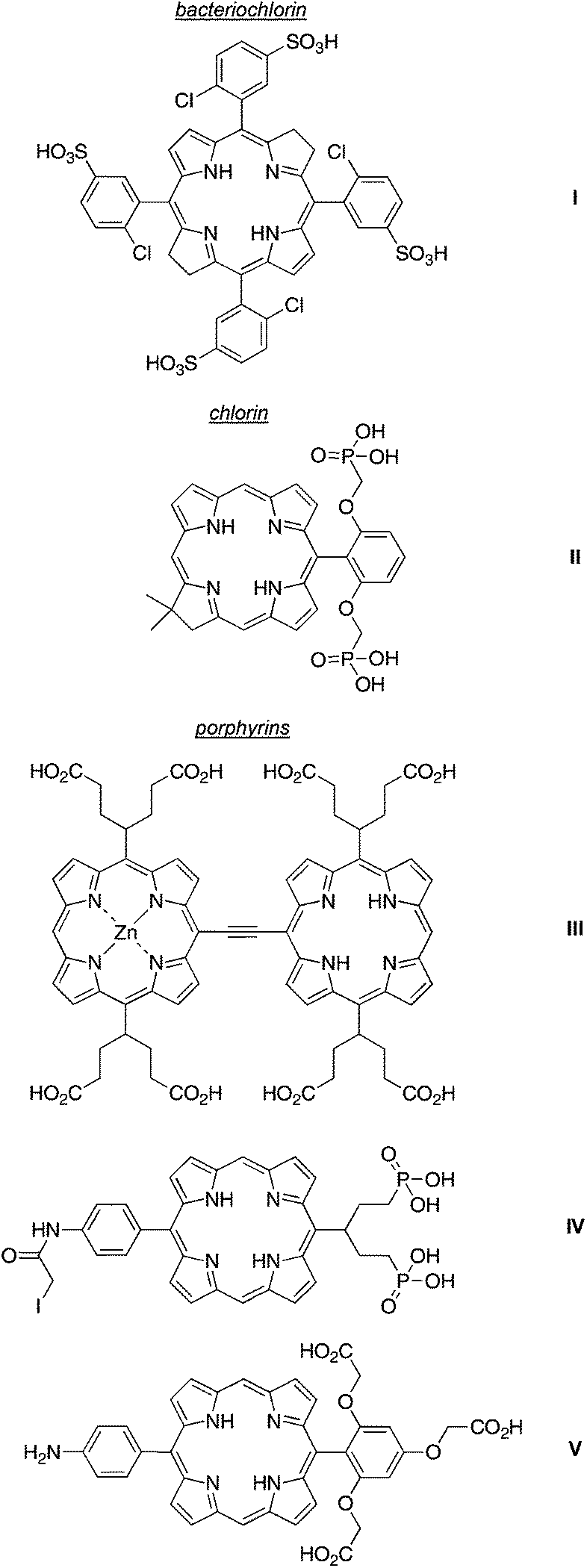 | ||
| Chart 3 Hydrophilic tetrapyrroles I–V. | ||
In this paper, we report the synthesis of three new bacteriochlorins that bear hydrophilic motifs. The hydrophilic motif is a 3,5-dicarboxyphenyl unit attached at two β-pyrrole positions of the bacteriochlorin (Chart 4), a design that was chosen to support (optional) installation of a bioconjugatable tether. Bacteriochlorin BC-6 contains 3,13-diaryl substituents, BC-7 contains 2,12-diaryl-3,13-dicarbomethoxy substituents, and BC-8 contains 2,12-diaryl substituents. The latter two architectures required synthesis of new dibromobacteriochlorin building blocks, which were then elaborated with the aryl groups via Suzuki coupling. One bioconjugatable, hydrophilic bacteriochlorin also was prepared and was reacted with a cysteine-containing 48-residue peptide analogous to a peptide from the light-harvesting antenna complex of a photosynthetic bacterium. The conjugation represents as an initial step in the preparation of a biohybrid light-harvesting antenna. The work also entails photophysical characterization of the four new hydrophilic bacteriochlorins as well as those prepared previously (BC-1–BC-5) that had not been examined thoroughly or at all for fluorescence properties.
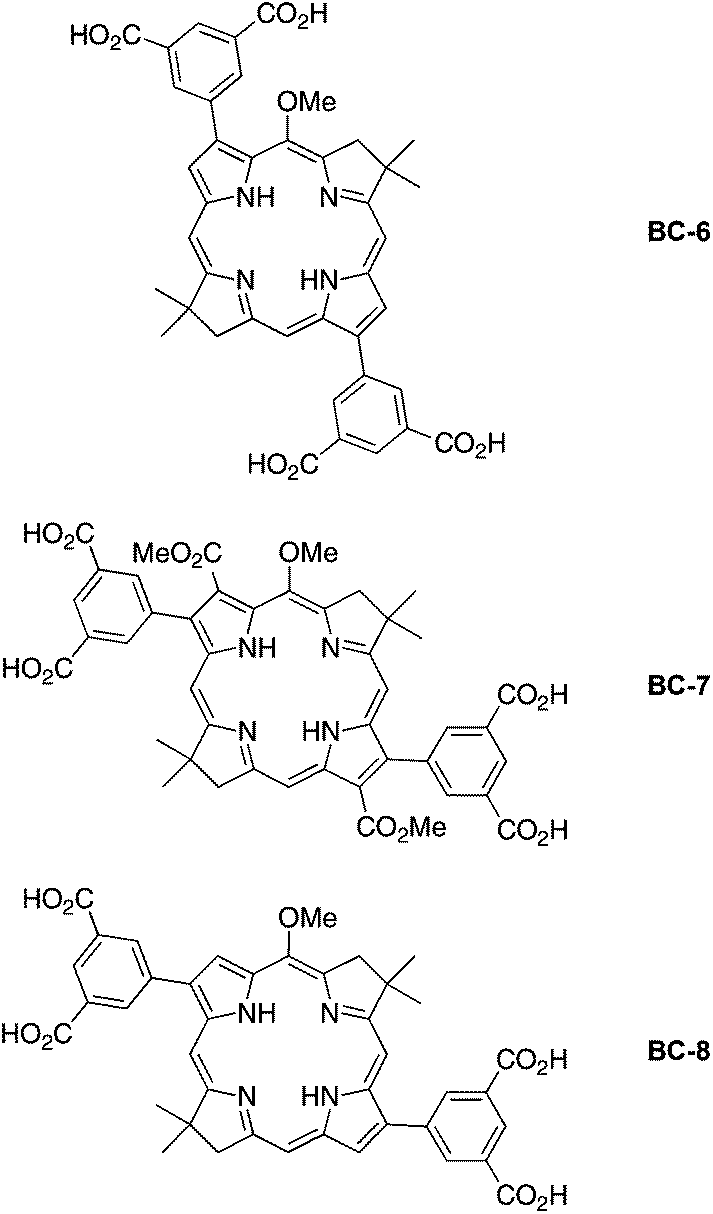 | ||
| Chart 4 New designs of candidate hydrophilic bacteriochlorins. | ||
Results and discussion
Reconnaissance
One strategy for introduction of a single group into a synthetic bacteriochlorin relies on bromination, which often (but not always) proceeds selectively at the 15-position for structures wherein the 5-methoxy group is present. Examples of successful 15-bromination include 5-methoxybacteriochlorins with no β-pyrrole substituents (entry 1, Table 1) or with electroneutral, p-tolyl substituents at the 2,12-positions (entry 2). On the other hand, strong electron-withdrawing groups (acetyl, carboethoxy) at the 3,13-positions led to failure of 15-bromination (entries 3 and 4), as did the protected acetyl (dioxolanyl) moiety (entry 5); by analogy, bacteriochlorins BC-1, the diformylbacteriochlorin precursor to BC-2, and BC-3 (Chart 2) are unsuitable for attachment of a bioconjugatable tether. The added presence of alkyl groups at the 2,12-positions results in effective 15-bromination, whether by counterbalancing the effect of the electron-withdrawing groups or merely by blocking otherwise open β-pyrrole sites (entries 6 and 7).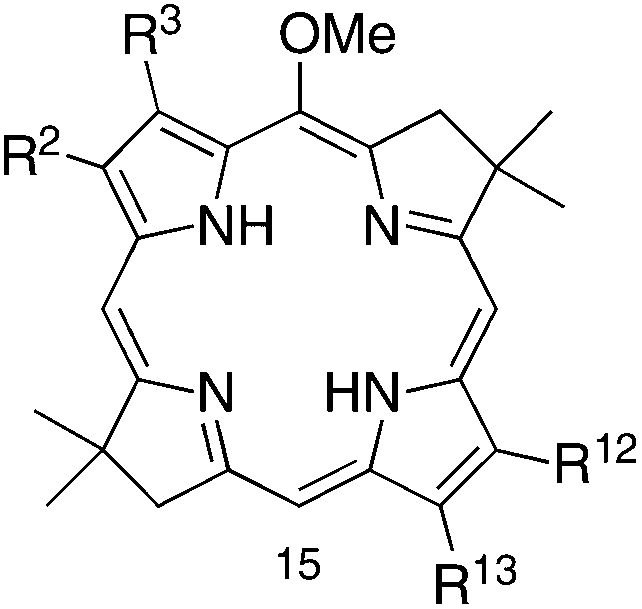
To address the question of whether electroneutral aryl groups at the 3,13-positions would be compatible with 15-bromination, 3,13-dibromobacteriochlorin BC-9 was treated to Suzuki coupling with 2-phenyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1) to give the corresponding 3,13-diphenylbacteriochlorin BC-10 (Scheme 2). The latter was subjected to standard conditions for 15-bromination (e.g., NBS in THF) whereupon the desired 15-bromobacteriochlorin BC-11 was smoothly formed. The result is included in Table 1 (entry 8) for comparison.
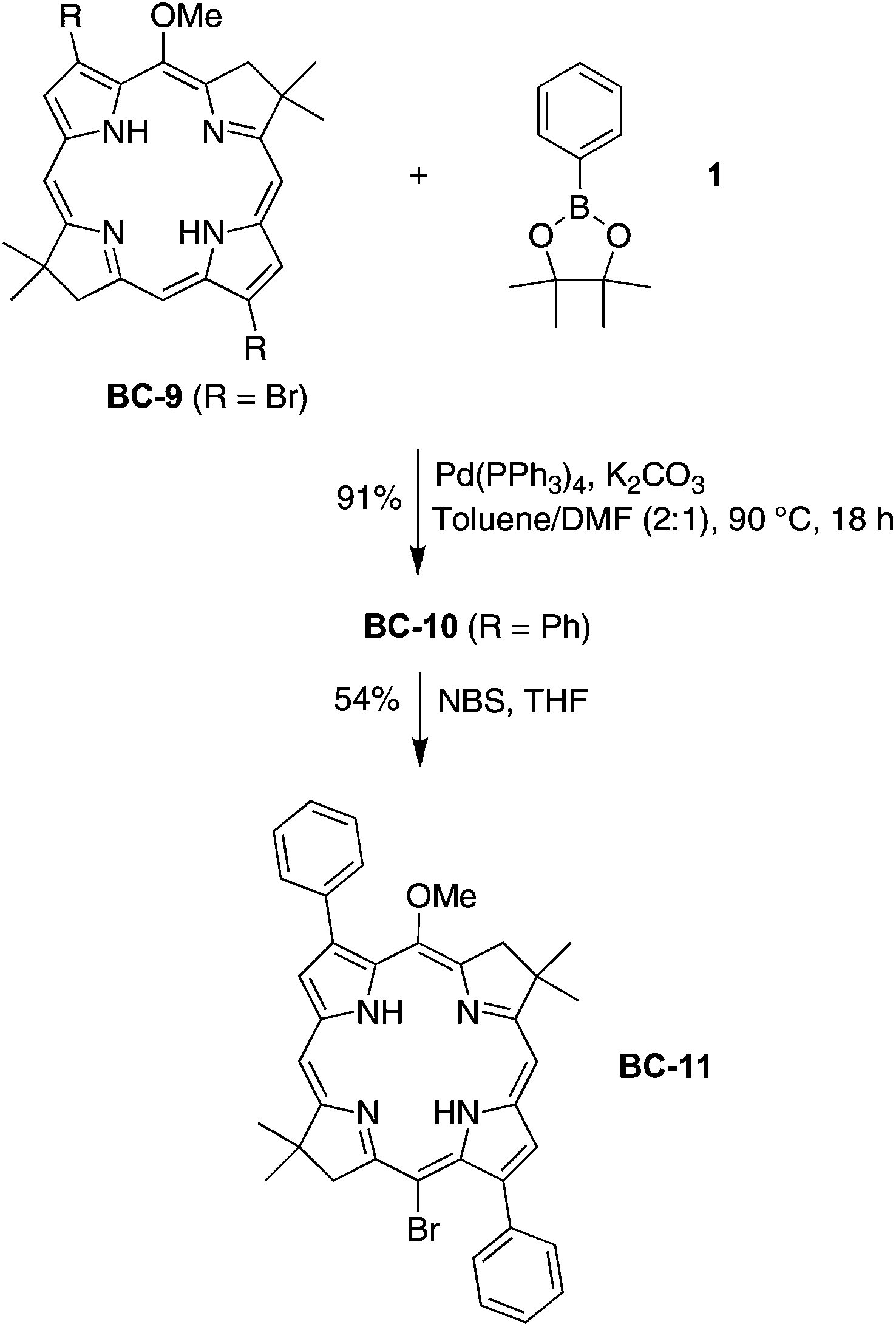 | ||
| Scheme 2 15-Bromination of a 3,13-diphenylbacteriochlorin. | ||
The strategy reported by Yu and Ptaszek13 wherein a 3,13-dibromo-5-methoxybacteriochlorin undergoes sequential coupling at the 13- and 3-sites was found to be successful, but of little advantage here because we sought to introduce two hydrophilic aryl groups and one bioconjugatable tether. Finally, the bacteriochlorins BC-4 and BC-5 are reasonably polar, yet N-protected (or unprotected) analogues thereof (inexplicably) do not undergo 15-bromination. All such considerations led to the following strategy: (1) prepare 2,12 or 3,13-dibromobacteriochlorins bearing a 5-methoxy group; (2) introduce hydrophilic aryl groups at the 2,12 or 3,13-positions by Suzuki coupling; and (3) if desired, install a bioconjugatable handle at the 15-position (via bromination/Suzuki coupling) as the last operation of the synthesis.
Synthesis
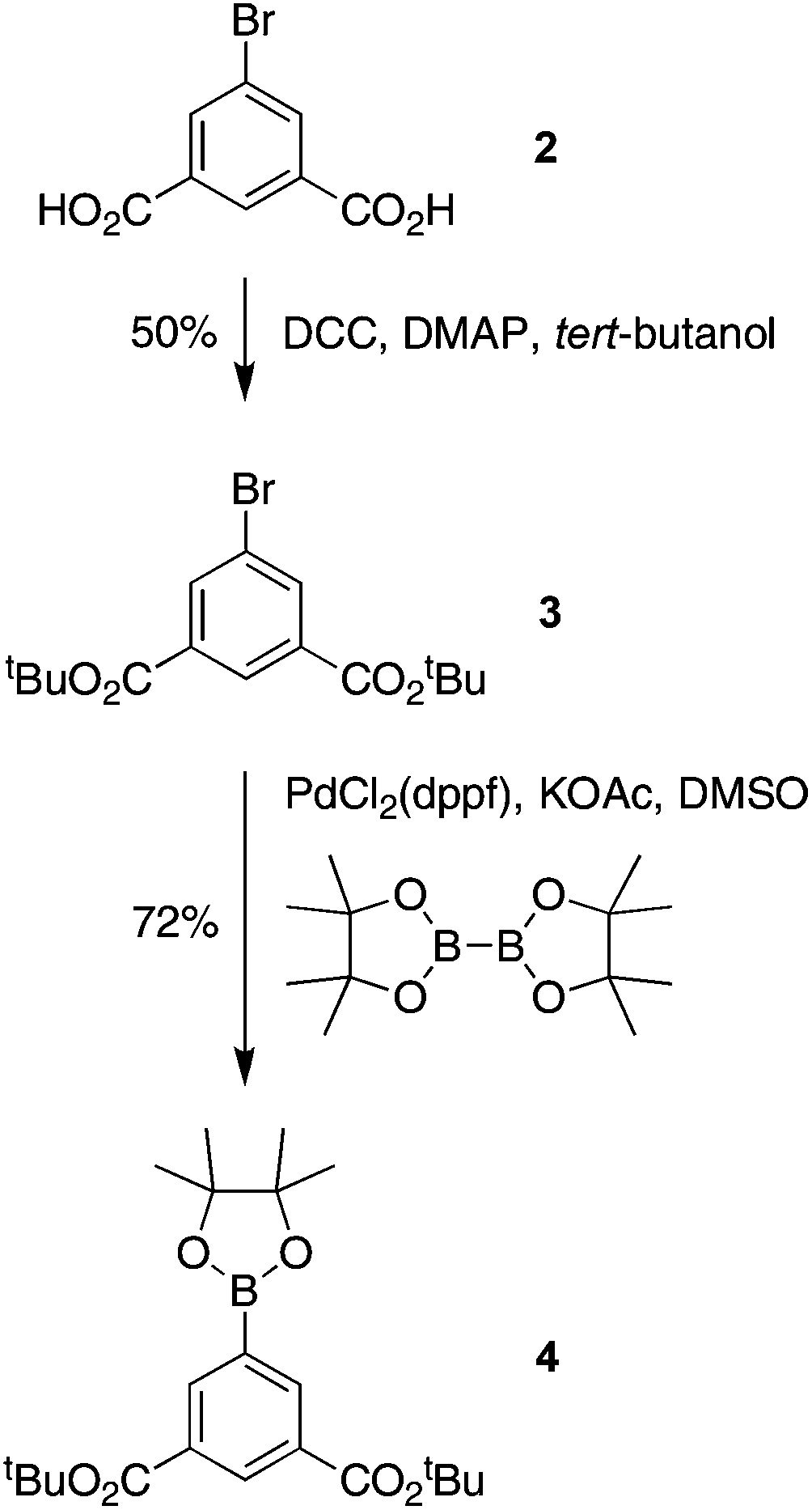 | ||
| Scheme 3 Synthesis of dicarboxy Suzuki coupling partner. | ||
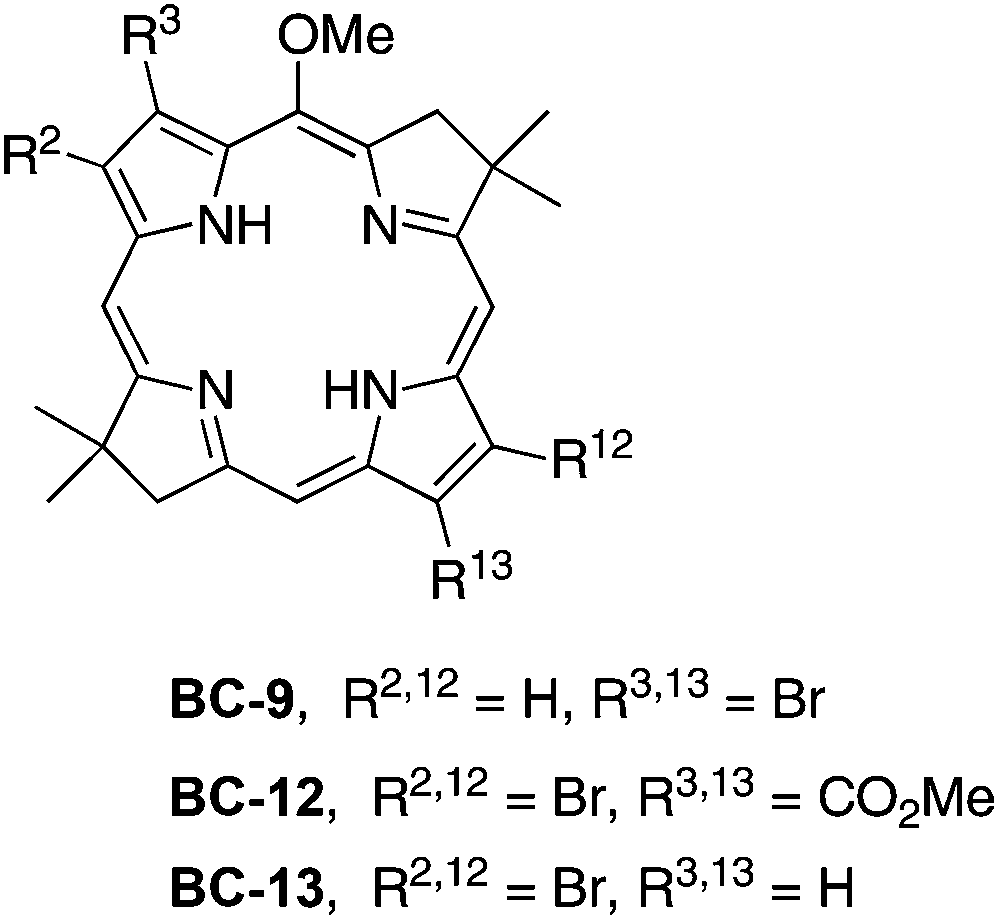 | ||
| Chart 5 Bacteriochlorin building blocks. | ||
The synthesis of the 2,12-dibromo-3,13-dicarbomethoxybacteriochlorin BC-12 began with formylation of 3-bromo-4-(methoxycarbonyl)pyrrole 6, available from 3,4-dibromo N-(triisopropylsilyl)pyrrole (5).60,61 Treatment of 6 with POCl3–DMF resulted in halogen exchange, affording 3-chloro-2-formyl-4-(methoxycarbonyl)pyrrole as the major product along with a trace amount of 3-bromo-2-formyl-4-(methoxycarbonyl)pyrrole 7. Attempts to separate the mixture failed. Although the mixture could be carried forward given that the halogen ultimately is to be displaced upon Pd-mediated coupling, we instead performed the Vilsmeier formylation using POBr3–DMF, which smoothly afforded the desired pyrrolecarboxaldehyde 7 in 72% yield (Scheme 4). The remainder of the synthesis followed standard methods:22,62 conversion of 7 to the nitroethylpyrrole 8, which upon Michael addition with 1,1-dimethoxy-4-methyl-3-penten-2-one (9)21,63 in the presence of DBU under solvent-free conditions yielded the nitrohexanone 10 in 75% yield. Reductive cyclization in the presence of buffered TiCl3 gave 11, which upon treatment with TMSOTf in the presence of the proton scavenger 2,6-di-tert-butylpyridine (DTBP)64 afforded the bacteriochlorin BC-12 in 25% yield. The pattern of substituents is established at the stage of formylation, and was confirmed by single-crystal X-ray analysis of 8 (see ESI, Fig. S1†).
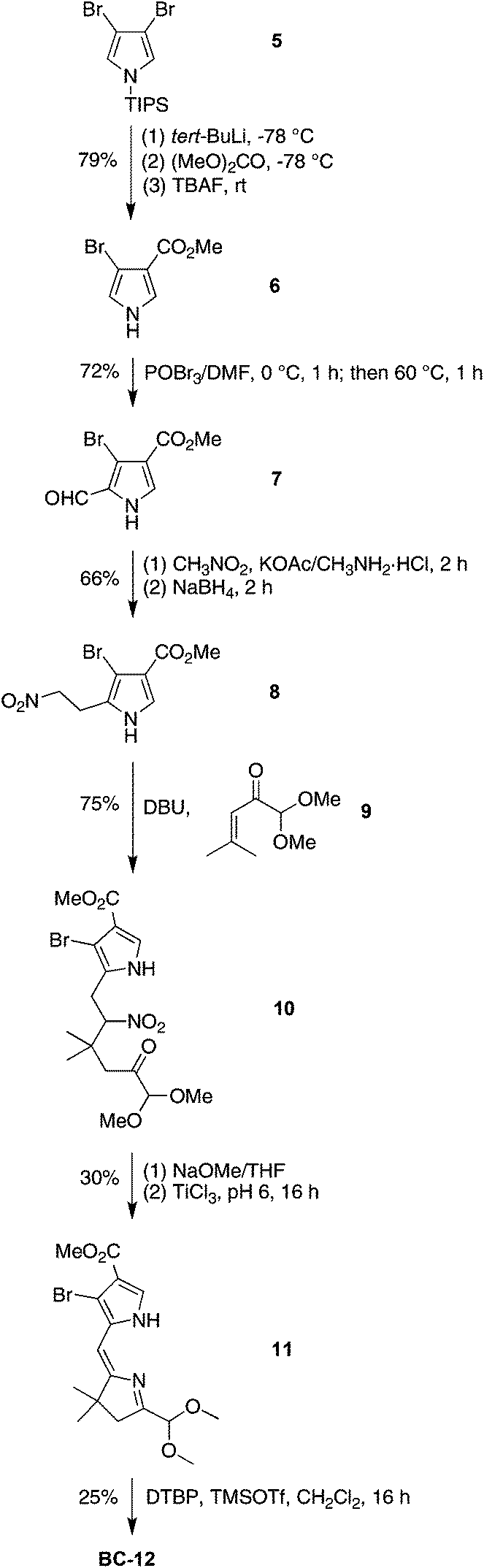 | ||
| Scheme 4 Synthesis of a dibromo-diester bacteriochlorin. | ||
To compare the ease of functionalization in the 15-position of bacteriochlorins having latent hydrophilic groups at the β-pyrrolic positions, we synthesized 2,12-dibromo-5-methoxybacteriochlorin BC-13. The 2,12-substitution pattern presents less hindrance toward further functionalization at the 15-position. The synthesis of BC-13 begins with 3-bromo-N-tosylpyrrole-2-carboxaldehyde (12),65,66 a known compound that has been prepared in 52 mg from a multistep synthesis starting with cinnamaldehyde, p-toluenesulfonamide and 3,3-diethoxyprop-1-yne. (The homologue 3-bromo-N-benzenesulfonylpyrrole-2-carboxaldehyde was recently prepared by Iwao and coworkers by direct lithiation of 3-bromo-N-benzenesulfonylpyrrole.67,68) We carried out the synthesis at larger scale and obtained 5.5 g of 12 (Scheme 5). Compound 12 was treated with nitromethane followed by reduction using LiBH4 at −10 °C to obtain the nitroethylpyrrole 13 in 20% yield. Treatment of 13 with 9 in the presence of DBU under solvent-free conditions yielded the Michael adduct 14 in 56% yield. The subsequent N-detosylation (5 equiv. of NaOMe for 2 h) and reductive cyclization (buffered TiCl3) were carried out in a one-flask process to give the dipyrrin 15 in 20% yield. Self-condensation of 15 under standard bacteriochlorin-forming conditions (DTBP and TMSOTf in CH2Cl2 at room temperature) gave the corresponding 2,12-dibromo-5-methoxybacteriochlorin BC-13 in 36% yield. A single-crystal X-ray structure of pyrrole 13 confirmed the substitution pattern of the adjacent 2-(2-nitroethyl) and 3-bromo substituents, and thereby substantiated the 2,12-dibromo substitution pattern in the corresponding bacteriochlorin target (Fig. S2†).
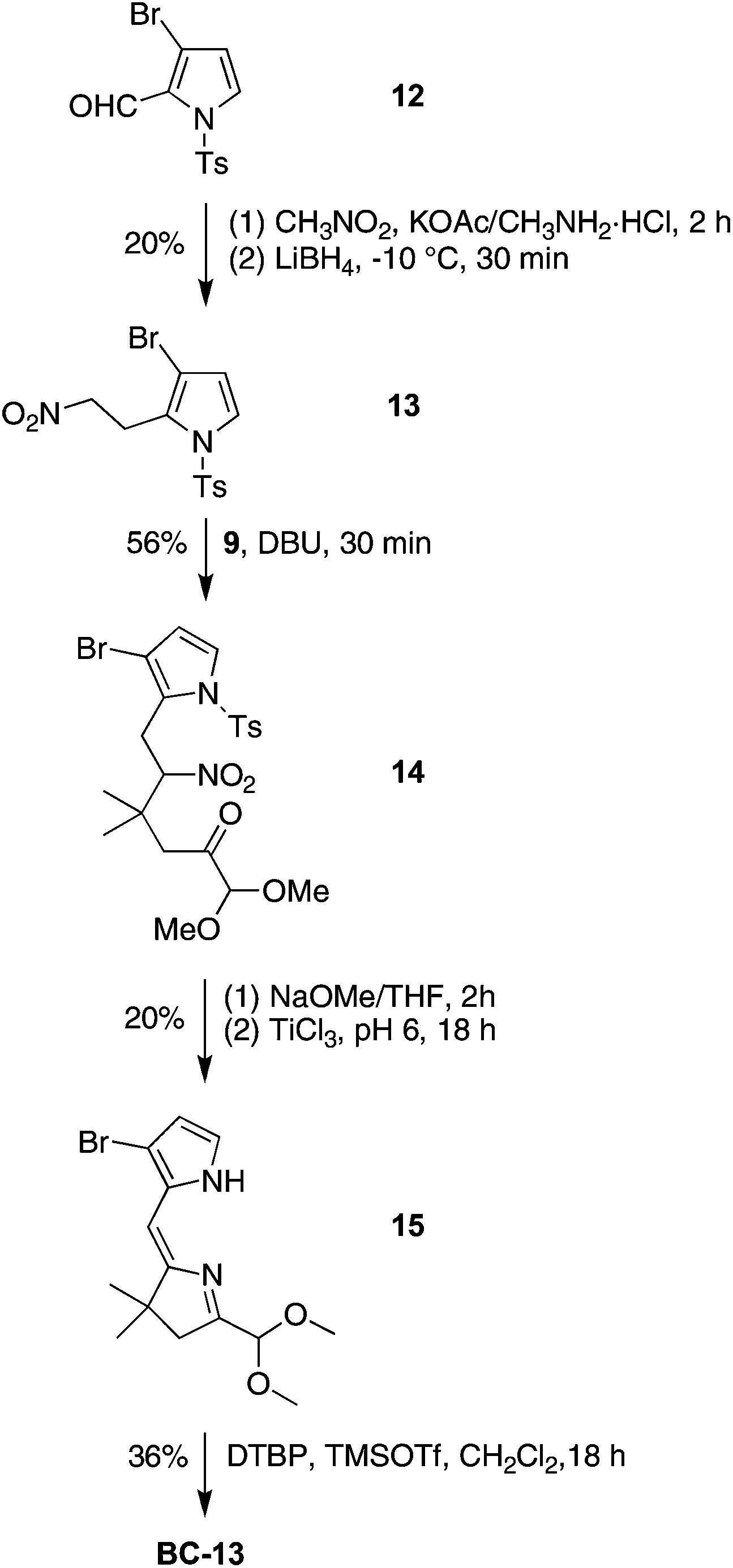 | ||
| Scheme 5 Synthesis of a 2,12-dibromobacteriochlorin. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) containing Pd(PPh3)4 and K2CO3 (or Cs2CO3) at 90 °C. The reactants and products are outlined in Scheme 6. Thus, the derivatization of 3,13-dibromobacteriochlorin BC-9 with the dicarboxy coupling partner 4 gave pro-BC-6 in 62% yield. When lesser quantities of reagents were employed, the mono-coupled mono-bromo bacteriochlorin intermediate was isolated, and could be subjected to a second round of Suzuki coupling (Fig. S3†). Yu and Ptaszek first reported selectivity in the Sonogashira coupling reaction with the same bacteriochlorin.13 In a similar manner, treatment of BC-12 with 4 afforded protected bacteriochlorin pro-BC-7 in 33% yield. Use of a lesser quantity of base (3 equiv. of Cs2CO3 instead of 12 equiv. of anhydrous K2CO3) afforded the protected bacteriochlorin pro-BC-7 in considerably increased yield (85%). Finally, reaction of 2,12-dibromobacteriochlorin BC-13 with 4 gave bacteriochlorin–tetraester pro-BC-8 in 30% yield.
:1) containing Pd(PPh3)4 and K2CO3 (or Cs2CO3) at 90 °C. The reactants and products are outlined in Scheme 6. Thus, the derivatization of 3,13-dibromobacteriochlorin BC-9 with the dicarboxy coupling partner 4 gave pro-BC-6 in 62% yield. When lesser quantities of reagents were employed, the mono-coupled mono-bromo bacteriochlorin intermediate was isolated, and could be subjected to a second round of Suzuki coupling (Fig. S3†). Yu and Ptaszek first reported selectivity in the Sonogashira coupling reaction with the same bacteriochlorin.13 In a similar manner, treatment of BC-12 with 4 afforded protected bacteriochlorin pro-BC-7 in 33% yield. Use of a lesser quantity of base (3 equiv. of Cs2CO3 instead of 12 equiv. of anhydrous K2CO3) afforded the protected bacteriochlorin pro-BC-7 in considerably increased yield (85%). Finally, reaction of 2,12-dibromobacteriochlorin BC-13 with 4 gave bacteriochlorin–tetraester pro-BC-8 in 30% yield.
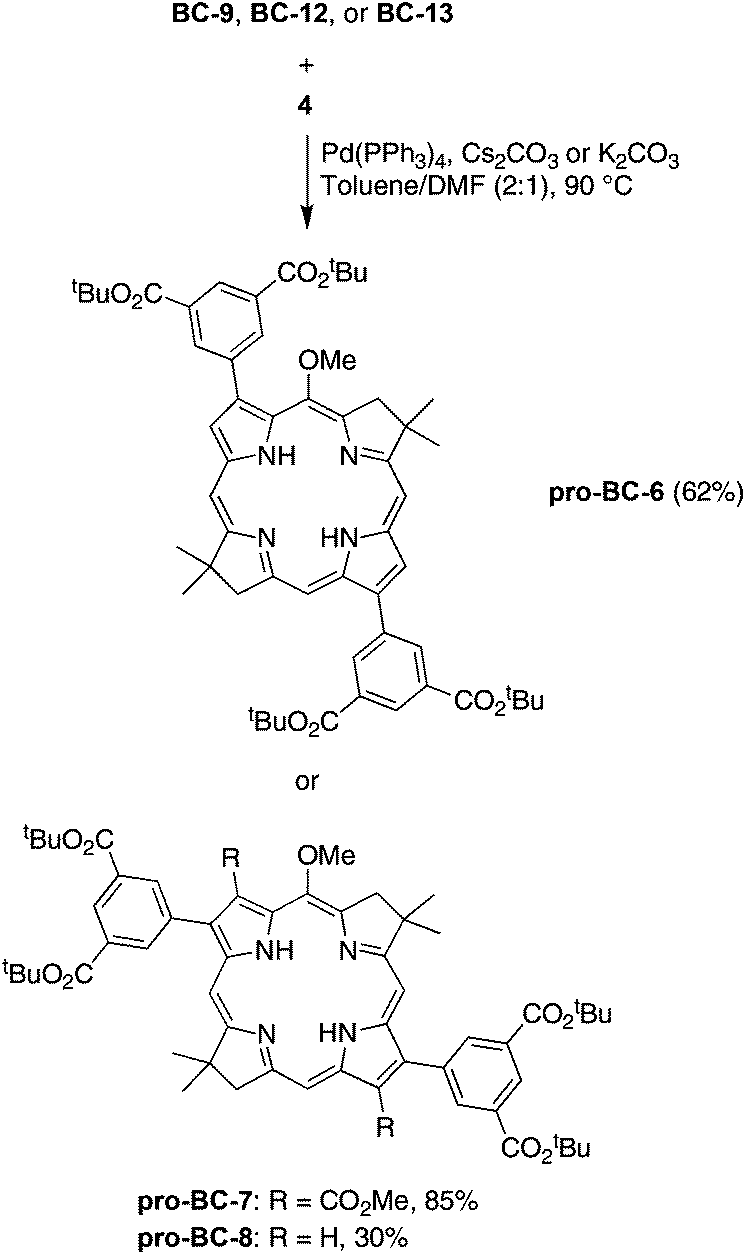 | ||
| Scheme 6 Suzuki coupling to give diarylbacteriochlorins. | ||
The tert-butyl esters were cleaved with 20% TFA in CH2Cl2 to give BC-6 (88% yield), BC-7 (89%), and BC-8 (90%). In the case of BC-7, there was no sign of hydrolysis of the carbomethoxy groups. The chemoselectivity and essentially quantitative nature of the protecting group cleavage reactions enabled the resulting bacteriochlorins BC-6–BC-8 to be characterized and used directly without purification.
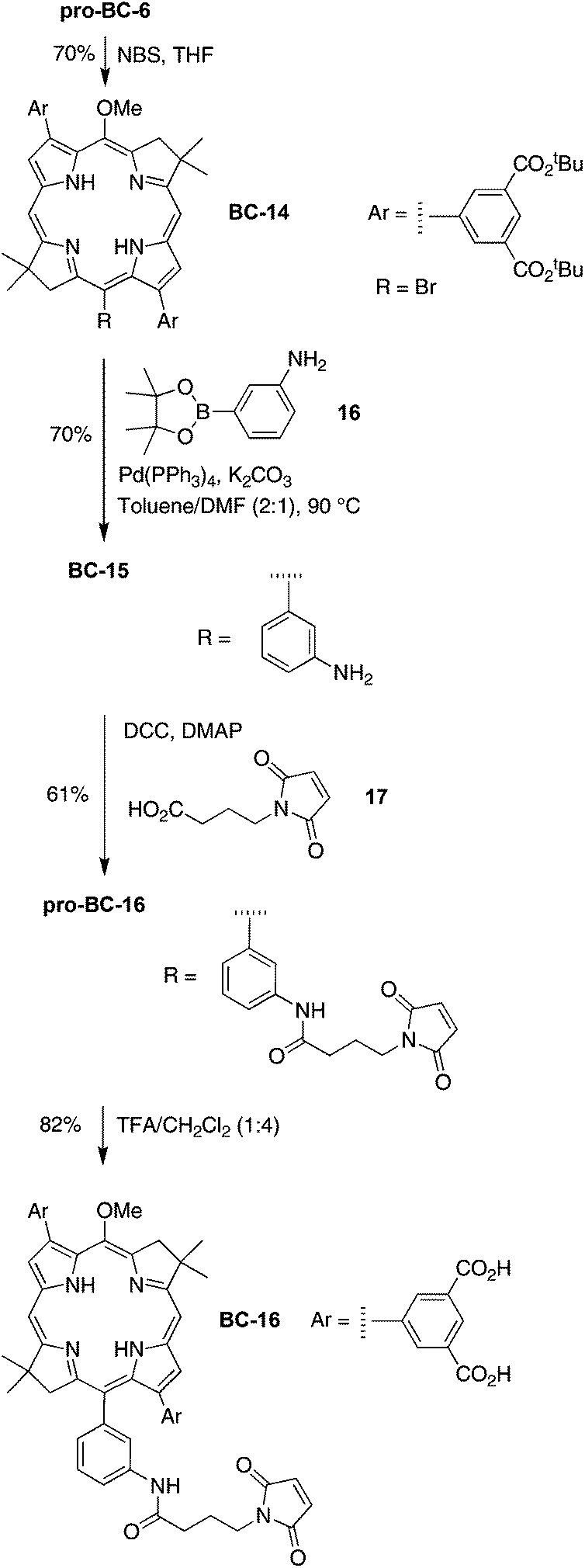 | ||
| Scheme 7 Hydrophilic bioconjugatable bacteriochlorin. | ||
The bacteriochlorins typically were characterized by absorption and fluorescence spectroscopy, 1H NMR spectroscopy, 13C NMR spectroscopy where quantity and solubility allowed, MALDI mass spectrometry, and ESI mass spectrometry. 13C NMR spectra were not collected for the compounds prepared to explore solubility features (BC-6–BC-8) owing to insufficient quantities; whereas BC-16 was characterized by 13C NMR spectroscopy.
Photophysical properties
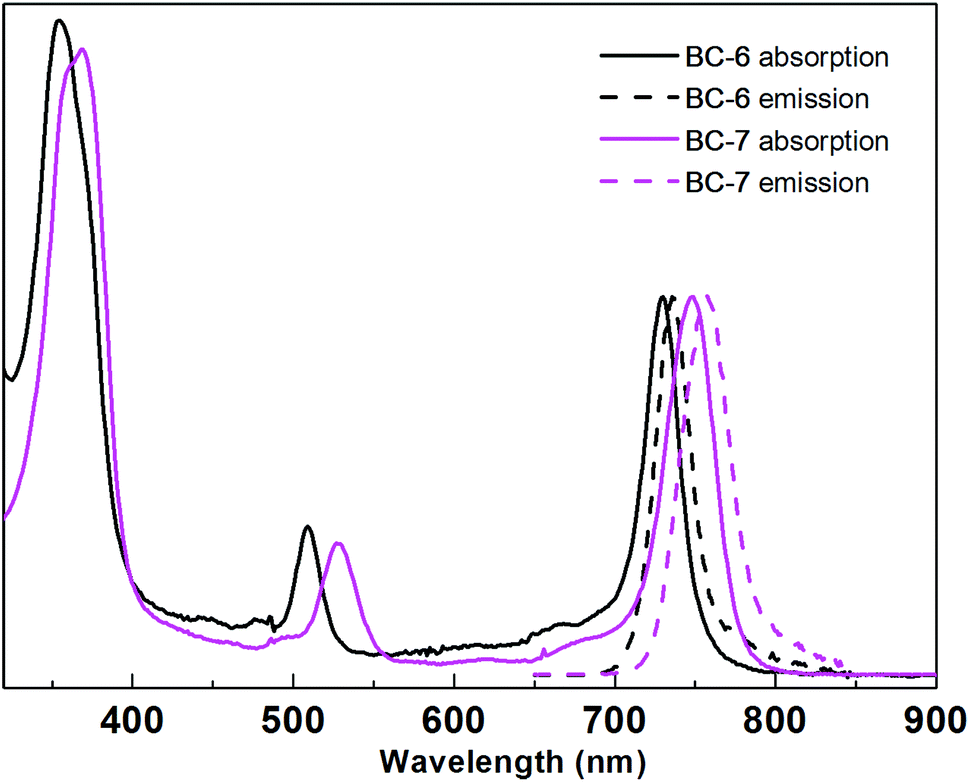 | ||
| Fig. 1 Normalized absorption spectra (solid) and emission spectra (dashed) of BC-6 and BC-7 in aqueous potassium phosphate buffer (0.5 M, pH 7.0) at room temperature. Spectral parameters are given in Table 2. | ||
| Reference | Compound | Solvent | λ abs/nm | FWHM/nm (Abs) | λ em/nm | FWHM/nm (Flu) | Φ f | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Each sample contains 1% DMF to facilitate initial dissolution. Pi buffer: 0.5 M potassium phosphate at pH 7.0. b Fluorescence yield data have not been reported previously. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-1 | DMF | 728 | 28 | 735 | 26 | 0.14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-1 | Pi buffera | 741 | 24 | 745 | 23 | 0.089 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-2 | DMF | 724 | 26 | 727 | 22 | 0.13 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-2 | Watera | 732 | 20 | 734 | 18 | 0.12 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-3 | DMF | 730 | 20 | 735 | 27 | 0.090 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-3 | Water | 733 | 20 | 739 | 22 | 0.11 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 30 b | BC-4 | DMF | 734 | 20 | 738 | 32 | 0.20 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 30 b | BC-4 | Watera | 732 | 23 | 738 | 22 | 0.096 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 30 b | BC-5 | DMF | 753 | 30 | 759 | 29 | 0.065 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 30 b | BC-5 | Watera | 752 | 31 | 758 | 33 | 0.010 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | pro-BC-6 | DMF | 731 | 24 | 738 | 25 | 0.18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-6 | DMF | 729 | 22 | 735 | 23 | 0.19 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-6 | Pi buffer | 730 | 26 | 736 | 26 | 0.078 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | pro-BC-7 | DMF | 753 | 25 | 759 | 26 | 0.18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-7 | DMF | 746 | 31 | 753 | 23 | 0.16 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-7 | Pi buffer | 749 | 35 | 758 | 37 | 0.11 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | pro-BC-8 | DMF | 736 | 22 | 742 | 23 | 0.20 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-8 | DMF | 732 | 25 | 739 | 23 | 0.14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-8 | Pi buffer | 734 | 29 | 740 | 27 | 0.077 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | pro-BC-16 | DMF | 728 | 22 | 735 | 25 | 0.21 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-16 | DMF | 726 | 21 | 732 | 24 | 0.16 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Here | BC-16 | Pi buffer | 729 | 23 | 735 | 25 | 0.074 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The key findings are as follows. (1) All bacteriochlorins regardless of medium or protecting group exhibited absorption and emission spectra characteristic of the bacteriochlorin chromophore, with a strong near-UV transition and a strong NIR (Qy) transition.69 (2) The position of the Qy band was little changed upon cleavage of the ester or upon change in environment from DMF to aqueous medium. The largest changes in DMF versus aqueous buffer were 13 or 8 nm for BC-1 or BC-2, respectively; the largest change upon deprotection was 7 nm in DMF for pro-BC-7versusBC-7. In all other cases, the position of the Qy band typically shifted by ≤3 nm. (3) The fwhm of the Qy absorption band was generally quite narrow and ranged from 20–35 nm; the emission band was similarly narrow (18–37 nm). (4) The Φf values generally range from 0.060–0.20 (with one outlier of 0.010 for BC-5 in water), with a typical value in aqueous medium diminished to approximately half that compared with DMF. Such a decrease in Φf value is precedented; chlorin II, for example, exhibits Φf = 0.11 in aqueous solution whereas that of an organic-soluble counterpart has Φf = 0.22 in toluene.56
In general, the spectroscopic properties in aqueous buffer of the new hydrophilic bacteriochlorins (BC-6–BC-8, BC-16) were quite similar to those of the prior hydrophilic bacteriochlorins (BC-1–BC-5). Bacteriochlorin BC-16 exhibited essentially identical properties to those of BC-6, indicating the absence of any adverse effects of the bioconjugatable tether. Moreover, in the one case examined (BC-16), the absorption spectrum was essentially unchanged when the sample was allowed to stand in aqueous buffer at room temperature over the course of 5 hours. The findings augur well for the use of the hydrophilic bacteriochlorins in photochemical applications wherein sharp absorption and fluorescence bands are required in an aqueous environment.
The spectral data of bacteriochlorin BC-6 measured at four concentrations (∼300, 30, 3 and 0.3 μM) were compared (Fig. 2). The concentrations reported here are based on the absorbance of the ∼30 μM solution measured at the Qy transition (assuming εQy = 100000 M−1 cm−1).21 The slight amount of broadening in the Qy band at higher concentration is consistent with some degree of aggregation, yet at all concentrations the general spectroscopic features indicative of bacteriochlorins were retained, and the sample remained visually clear. Similar results were observed with BC-7 and BC-8 (Fig. S5 and S6†).
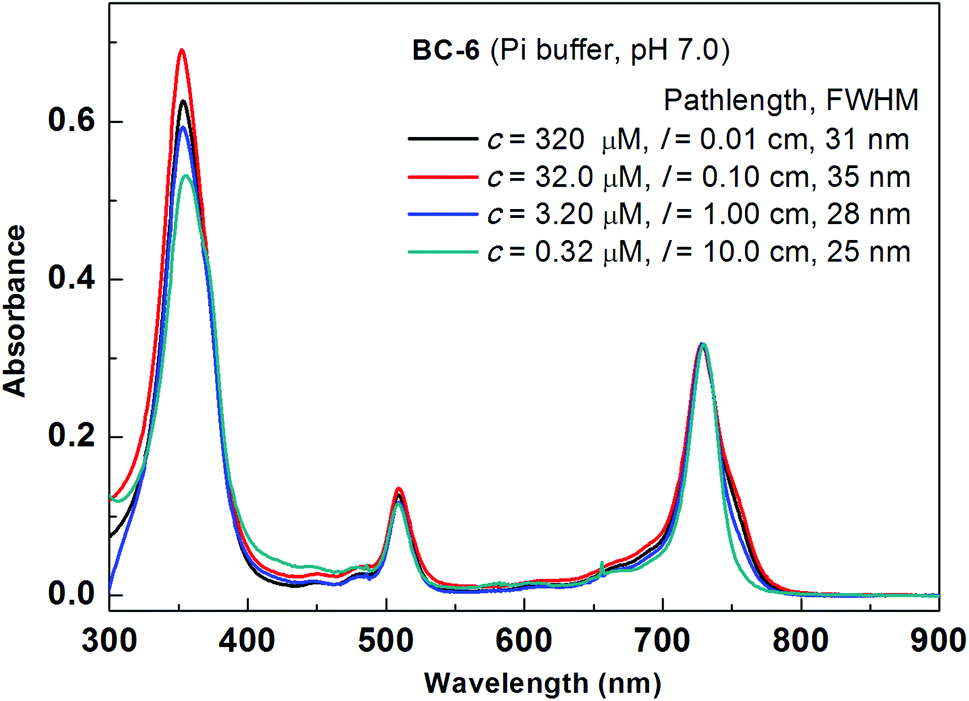 | ||
| Fig. 2 Absorption versus concentration of BC-6 over a range of 1000-fold. The spectra are normalized at the Qy band; the fwhm of the Qy band is indicated in the inset. | ||
Bioconjugation
One motivation for the development of bacteriochlorin chemistry concerns the development of biohybrid light-harvesting antennas wherein synthetic chromophores or other entities are integrated with constituents (or analogues) of the natural photosynthetic systems.32,71,72 Like the natural antennas, the biohybrid antennas form by a multi-tiered self-assembly process, exhibit structure of ∼10 nm dimensions, and incorporate up to several dozen chromophores in an integrated architecture.72 The design, preparation and characterization of biohybrid antennas thus touches diverse fields encompassing photosynthesis, biophysics, supramolecular chemistry, synthetic chemistry, and materials science.We have previously employed lipophilic maleimido–bacteriochlorins for attachment to peptides analogous to those found in the natural light-harvesting antennas of bacterial photosynthesis. The resulting conjugates were then examined in aqueous detergent (micellar) solutions. A biohybrid architecture also has been prepared in detergent solution by attachment of a lipophilic maleimido–terrylene dye to a recombinant photosynthetic antenna complex.73 The availability of a hydrophilic bioconjugatable bacteriochlorin (BC-16) holds out the possibility of examining analogous conjugates with lesser or no detergents in aqueous solution, ultimately broadening the scope of applications in materials science (such as attachment to diverse surfaces74). Thus, the helical β-peptide (Fig. 3) of Rhodobacter sphaeroides light harvesting complex I provided the inspiration for the choice of peptide to use herein. The native β-peptide has the sequence ADKSDLGYTGLTDEQAQELHSVYM−14SGLWLFSAVAIVAH0LAVYIWRPWF, where the site of histidine ligation to noncovalently bound bacteriochlorophyll a is indicated in bold (H0). The peptide employed herein is identical to the native β-peptide but contains a cysteine in lieu of the methionine residue (in bold at the −14 position). The resulting synthetic peptide, denoted β(-14Cys), thus contains a single cysteine for the ligation with BC-16.
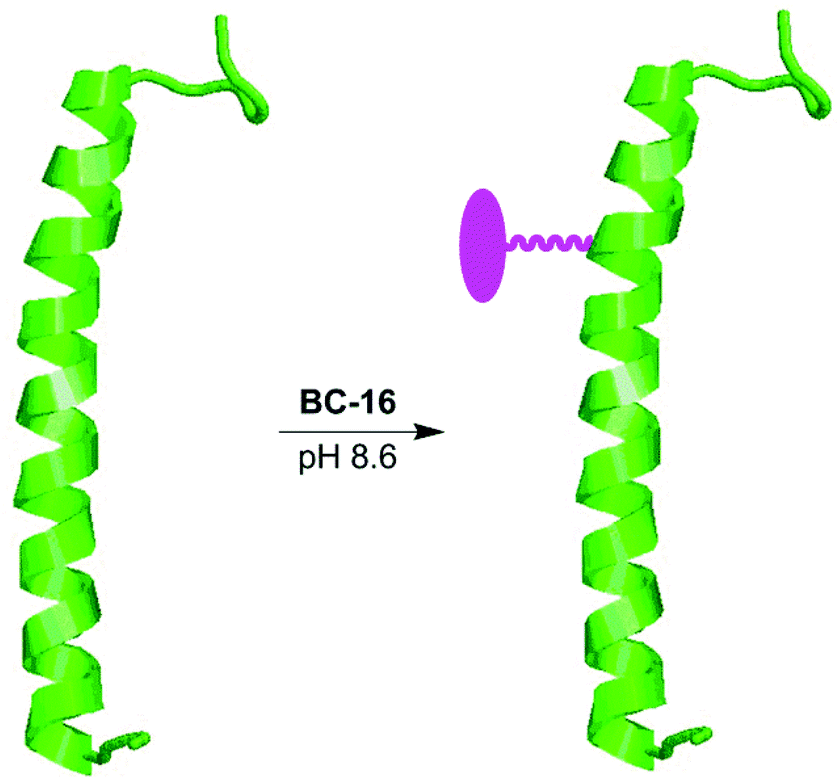 | ||
| Fig. 3 Preparation of β(-14Cys)BC-16 conjugate. The attached bacteriochlorin is illustrated in magenta. | ||
The reaction was carried out with β(-14Cys) at 2 mM and a 50% excess of BC-16 (3 mM) in a mixed aqueous-organic medium of Tris buffer (pH 8.6) and DMF (1:4) at room temperature for 3 hours. The starting peptide and the conjugate [denoted β(-14Cys)BC-16] closely chromatographed on reverse-phase HPLC using a C4 column. The desired conjugate was obtained in ∼50% yield by reverse-phase HPLC purification (Fig. S7†). It warrants mention that the conjugate is present as a mixture of diastereomers owing to the stereocenter created upon thio-maleimido conjugation. The absorption spectrum of the purified conjugate β(-14Cys)BC-16 clearly showed bands due to the peptide in the UV region (282 nm) and the bacteriochlorin in the near-UV (363 nm), visible (515 nm), and NIR (729 nm) region (Fig. S8†). Electrospray ionization mass spectrometry (ESI-MS) also gave a strong peak upon hypermass ion reconstruction of the intact bacteriochlorin–peptide conjugate at m/z = 6410.0550, to be compared with that for the peptide precursor at m/z = 5425.7052. The conjugate β(-14Cys)BC-16 will be employed in studies with bacteriochlorophyll a and the complementary native α-peptide to form self-assembled αβ-dyads and light-harvesting cyclic oligomeric antennas therefrom.32,71,72 The facile preparation, good yield, and ease of handling of the conjugate together augur well for the overall molecular design approach described herein.
Conclusions
The new molecular designs investigated herein entail introduction of polar aryl groups at the 2,12- or 3,13-positions of the bacteriochlorin macrocycle. The aryl groups investigated bear carboxylic acid entities at the 3,5-positions, thereby achieving a degree of facial encumbrance by ionized functional groups at neutral pH. The hydrophilic bacteriochlorins were readily prepared from the corresponding dibromobacteriochlorin building blocks. In one case examined, the presence of the hydrophilic aryl groups in protected form does not interfere with the attachment of a bioconjugatable tether. The resulting bacteriochlorins generally are soluble in polar media (aqueous or DMF) and exhibit spectroscopic properties – sharp absorption and emission bands, a small Stokes's shift, and modest fluorescence quantum yield – that are attractive for a wide range of photochemical applications. The molecular designs described herein also may prove instructive in guiding the development of bacteriochlorins that reside at the “hydrophilic, bioconjugatable, and wavelength-tunable” nexus.Experimental section
General methods
1H NMR (400 MHz) or 13C NMR (100 MHz) spectroscopy was performed at room temperature in CDCl3 unless noted otherwise. Silica gel (40 μm average particle size) was used for column chromatography. All solvents were reagent grade and were used as received unless noted otherwise. THF was freshly distilled from sodium/benzophenone ketyl. Anhydrous CH2Cl2 was used as received. Matrix-assisted laser-desorption mass spectrometry (MALDI-MS) was performed with the matrix 1,4-bis(5-phenyl-2-oxaxol-2-yl)benzene (POPOP)75 unless noted otherwise. ESI-MS data are reported for the molecular ion or cationized molecular ion. Noncommercial compounds 5,60,619,21,631265,66 and BC-922 were prepared following literature procedures. The peptide β(-14Cys) was purchased from Bio-Synthesis, Lewisville, TX in 90% purity. All other compounds were used as received from commercial sources.Recovery of 2,6-di-tert-butylpyridine (DTBP)
The condensation to form bacteriochlorins requires the use of substantial quantities of the hindered base DTBP, which is quite expensive. The following protocol enables recovery of DTBP: quench the crude bacteriochlorin-forming reaction mixture (carried out in CH2Cl2) with saturated aqueous NaHCO3. Separate the CH2Cl2 layer and dry over Na2SO4. Concentrate the CH2Cl2 extract by rotary evaporation to afford the crude product, which typically contains bacteriochlorin, byproducts derived from the dihydrodipyrrin–acetal reactants, and DTBP. Prepare a silica column with hexanes and apply the crude product to the top of the column. Elute with hexanes, whereupon DTBP elutes as a translucent band upon visualization via flashlight illumination from the backside of the column. Collect the translucent band and remove the solvent by rotary evaporation (≤40 °C, ∼50 mmHg). DTBP has bp ∼100 °C at 23 mmHg.64 DTBP is obtained as a colorless liquid after drying under high vacuum (oil pump, ∼0.05 mmHg, room temperature) for 5 min, whereas the typical commercial product is light yellow. In this manner, >95% of the DTBP can be recovered (at scales of 2–25 mL of DTBP). TLC analysis (silica, hexanes, Rf = 0.8) shows only one spot. The DTBP obtained in this manner is stable at 4 °C for at least 6 months, and can be used in bacteriochlorin-forming reactions with no adverse effects versus that obtained from commercial sources.Absorption versus concentration study
For each bacteriochlorin, four different solutions (A, B, C and D) in aqueous potassium phosphate buffer (0.5 M, pH = 7.0) were prepared. The concentration of solution A (∼500 μM) afforded absorbance (A) ∼0.5 for the Qy transition measured with a 0.01 cm pathlength cuvette. Successive serial dilution (10 times each) with buffer gave bacteriochlorin concentrations as follows: [solution A] = 10 × [solution B] = 100 × [solution C] = 1000 × [solution D].In a typical experiment for the aggregation studies (Fig. S1†), a stock solution was prepared in a small vial containing bacteriochlorin (∼0.1 mg; ∼0.1 μM) by adding a small amount of DMSO (12.5 μL, to facilitate dissolution) followed by aqueous potassium phosphate buffer (237.5 μL, 0.5 M, pH = 7.0). The resulting sample was sonicated for one minute and then filtered (poly-vinylidene difluoride high-volume low pressure filter, pore size 0.45 μm) to obtain solution A (∼500 μM). The absorbance of solution A was measured in a 0.01 cm pathlength cuvette. Solution B was obtained by mixing solution A (100 μL) with buffer (900 μL) in a small vial. The absorbance of solution B was measured in a 0.1 cm pathlength cuvette. For further dilution, solution B (300 μL) was mixed with buffer (2.70 mL) in a 1 cm pathlength cuvette to obtain solution C and the absorbance was measured. Solution C (2.50 mL) was transferred into a 25 mL standard measuring flask and made up to the mark with buffer. The absorbance of the resulting solution D was measured in a 10 cm pathlength cuvette. The procedure was followed for each bacteriochlorin BC-6–BC-8.
Fluorescence yield determinations
The Φf values (in DMF, water, or aqueous potassium phosphate buffer) were determined relative to that of 5-methoxy-8,8,18,18-tetramethyl-2,12-di-p-tolylbacteriochlorin in toluene (Φf = 0.18)21 without correction for refractive index differences.Di-tert-butyl 5-bromoisophthalate (3)
A mixture of 2 (2.45 g, 10.0 mmol), DCC (3.82 g, 18.5 mmol) and DMAP (0.244 g, 2.00 mmol) in CH2Cl2 (25 mL) and DMF (25 mL) was treated with tert-butyl alcohol (9.5 mL, 100 mmol). The mixture was stirred under argon at room temperature for 16 h. The resulting mixture was filtered to remove insoluble material. The filtrate was concentrated and chromatographed [silica, CH2Cl2–hexanes (1:1)] to yield a white solid (1.8 g, 50%): mp 95–96 °C; 1H NMR δ 1.60 (s, 18H), 8.25 (d, J = 1.6 Hz, 2H), 8.49 (t, J = 1.6 Hz, 1H); 13C NMR δ 28.3, 82.5, 122.4, 129.2, 134.2, 136.2, 163.9; ESI-MS obsd 379.0523, calcd 379.0515 [(M + Na)+, M = C16H21BrO4].
Di-tert-butyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isophthalate (4)
Following a general procedure,59 samples of 3 (250 mg, 0.700 mmol), bis(pinacolato)diboron (213 mg, 0.839 mmol), Pd(pddf)Cl2 (15.4 mg, 21.0 μmol), KOAc (206 mg, 2.10 mmol), and DMSO (4.2 mL, deaerated by bubbling with argon) was added to a Schlenk flask. The reaction mixture was deaerated by three freeze–pump–thaw cycles with argon. The reaction mixture was stirred at 80 °C for 2 h. The reaction mixture was cooled to room temperature, diluted with ethyl acetate and washed with brine. The organic layer was separated, dried (Na2SO4) and concentrated. Column chromatography [silica, CH2Cl2–ethyl acetate (47:3)] provided a white solid (203 mg, 72%): mp 195–196 °C; 1H NMR δ 1.36 (s, 12H), 1.62 (s, 18H), 8.54 (d, J = 1.6 Hz, 2H), 8.65 (t, J = 1.6 Hz, 1H); 13C NMR δ 25.1, 28.4, 81.7, 84.5, 131.9, 133.2, 139.5, 165.4; ESI-MS obsd 427.2277, calcd 427.2263 [(M + Na)+, M = C22H33BO6].
3-Bromo-4-(methoxycarbonyl)pyrrole (6)
A solution of tert-BuLi (25.0 mL, 1.7 M in pentane, 42.5 mmol) was slowly cannulated into a solution of 5 (10.7 g, 28.0 mmol) in THF (200 mL) at −78 °C under argon. After 30 min, dimethyl carbonate (7.58 mL, 90.0 mmol) was added and the stirring was continued for another 30 min. The reaction mixture was allowed to warm to 0 °C and treated with saturated aqueous NaHCO3. The reaction mixture was extracted with ethyl acetate. The organic extract was washed (water, brine), dried (Na2SO4) and concentrated by rotary evaporation. The resulting viscous material was mixed with excess hexanes and sonicated for a few minutes. A solid settled out, whereupon the supernatant was decanted. The solid was dried under reduced pressure, dissolved in THF (100 mL) and treated at room temperature with TBAF·3H2O (11.0 g, 35.0 mmol) for 30 min under argon. The reaction mixture was diluted with diethyl ether, washed (water, brine), dried (Na2SO4) and concentrated by rotary evaporation. The resulting residue was dissolved in a minimum amount of CH2Cl2, and then excess hexanes was added. The resulting precipitate was separated and dried under reduced pressure to afford a pale yellow solid (4.48 g, 79%): mp 96–98 °C; 1H NMR δ 3.83 (s, 3H), 6.83 (d, J = 2.4 Hz, 1H), 7.42 (dd, J1 = 2.4 Hz, J2 = 3.2 Hz, 1H), 8.58 (brs, 1H); 13C NMR (CDCl3–CD3OD) δ 51.1, 96.6, 113.5, 120.5, 125.1, 164.8; ESI-MS obsd 203.9655, calcd 203.9655 [(M + H)+, M = C6H6BrNO2].3-Bromo-2-formyl-4-(methoxycarbonyl)pyrrole (7)
A mixture of 6 (3.06 g, 15.0 mmol) and POBr3 (5.16 g, 18.0 mmol) under argon was treated with DMF (50.0 mL) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h and then at 60 °C for 1 h. The reaction mixture was treated with a mixture of saturated aqueous sodium acetate and CH2Cl2 [300 mL, 1:1 (v/v)] and stirred for 1 h. The aqueous phase was separated and extracted with CH2Cl2. The combined organic phase was washed with water (5 × 300 mL), dried (Na2SO4), and concentrated. The resulting solid was dissolved in a minimum amount of CH2Cl2, and then excess hexanes was added. The resulting precipitate was separated by filtration and dried under reduced pressure to afford a light brown solid (2.50 g, 72%): mp 187–189 °C; 1H NMR (CDCl3–CD3OD) δ 3.87 (s, 3H), 4.05 (brs, 1H), 7.69 (s, 1H), 9.67 (d, J = 0.8 Hz, 1H); 13C NMR (CDCl3–CD3OD) δ 51.7, 110.3, 116.5, 130.7, 131.0, 163.5, 180.1; ESI-MS obsd 231.9611, calcd 231.9604 [(M + H)+, M = C7H6BrNO3].
3-Bromo-4-(methoxycarbonyl)-2-(2-nitroethyl)pyrrole (8)
Following a general procedure,62 a stirred mixture of 7 (1.80 g, 7.75 mmol), potassium acetate (1.47 g, 15.0 mmol), and methylamine hydrochloride (1.01 g, 15.0 mmol) in absolute ethanol (10.0 mL) was treated with nitromethane (1.20 mL, 22.0 mmol). The resulting mixture was stirred at room temperature for 2 h, whereupon water was added. The resulting precipitate was separated by filtration, washed with water and dried under reduced pressure to afford a yellow solid, which was used directly in the next step. The crude solid material was dissolved in CHCl3–2-propanol (3:1, 100 mL). Silica (10 g) and NaBH4 (378 mg, 10.0 mmol) were added, and the mixture was stirred at room temperature under argon for 2 h. The reaction mixture was filtered, and the filtrate was concentrated. The resulting crude material was dissolved in CH2Cl2. The organic solution was washed with water, dried (Na2SO4) and concentrated to afford a pale brown solid (1.42 g, 66% yield): mp 148–150 °C; 1H NMR (CDCl3–CD3OD) δ 3.32 (t, J = 7.2 Hz, 2H), 3.81 (s, 3H), 4.63 (t, J = 7.2 Hz, 2H), 7.38 (s, 1H) (one pyrrolic proton was not observed); 13C NMR (CDCl3–CD3OD) δ 24.5, 51.3, 73.6, 96.8, 114.1, 125.0, 126.3, 164.8; ESI-MS obsd 276.9824, calcd 276.9818 [(M + H)+, M = C8H9BrN2O4].
6-(3-Bromo-4-(methoxycarbonyl)pyrrol-2-yl)-1,1-dimethoxy-4,4-dimethyl-5-nitrohexan-2-one (10)
Following a general procedure,62 a mixture of 8 (1.33 g, 4.80 mmol) and 9 (1.52 g, 9.60 mmol) was treated with DBU (3.60 mL, 24.0 mmol). The reaction mixture was stirred at room temperature for 16 h under argon. A saturated solution of cold aqueous NH4Cl was added. The mixture was extracted with ethyl acetate. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated. Column chromatography [silica, CH2Cl2–ethyl acetate (9:1)] afforded a pale yellow solid (1.57 g, 75% yield): mp 64–66 °C; 1H NMR δ 1.15 (s, 3H), 1.28 (s, 3H), 2.63, 2.74 (AB, 2J = 18.8 Hz, 2H), 3.15–3.35 (m, 2H), 3.42 (s, 3H), 3.43 (s, 3H), 3.81 (s, 3H), 4.38 (s, 1H), 5.19 (dd, 3J = 2.4 Hz, 3J = 11.2 Hz, 1H), 7.33 (d, J = 3.6 Hz, 1H), 8.82 (brs, 1H); 13C NMR δ 24.1, 24.4, 25.5, 36.7, 45.0, 51.4, 55.3, 55.4, 93.5, 97.6, 104.8, 114.9, 124.6, 126.3, 163.8, 203.6; ESI-MS obsd 435.0766, calcd 435.0761 [(M + H)+, M = C16H23BrN2O7].
7-Bromo-2,3-dihydro-8-(methoxycarbonyl)-1-(1,1-dimethoxymethyl)-3,3-dimethyldipyrrin (11)
Following a general procedure,62 in a first flask, a solution of 10 (1.30 g, 3.00 mmol) in freshly distilled THF (10.0 mL) at 0 °C was treated with NaOMe (486 mg, 9.00 mmol). The mixture was stirred and deaerated by bubbling argon through the solution for 45 min. In a second flask purged with argon, TiCl3 (12.0 mL, 20 wt% in 3% HCl solution, 19.0 mmol), 30.0 mL of THF and NH4OAc (12.0 g, 156 mmol) were combined under argon, and the mixture was deaerated by bubbling argon for 45 min. Then, the first flask mixture was transferred via cannula to the buffered TiCl3 mixture. The resulting mixture was stirred at room temperature for 16 h under argon. Then, the content was diluted with ethyl acetate and washed with saturated aqueous NaHCO3. The organic layer was separated, dried (Na2SO4) and concentrated. Column chromatography (silica, CH2Cl2) afforded a yellow solid (350 mg, 30% yield): mp 108–110 °C; 1H NMR (300 MHz) δ 1.25 (s, 6H), 2.65 (s, 2H), 3.44 (s, 3H), 3.45 (s, 3H), 3.83 (s, 3H), 5.04 (s, 1H), 5.98 (s, 1H), 7.48 (d, J = 3.6 Hz, 1H), 11.22 (brs, 1H); 13C NMR δ 26.9, 29.2, 40.7, 48.7, 51.3, 54.8, 97.2, 102.5, 104.4, 114.5, 125.4, 130.7, 162.9, 164.2, 176.9; ESI-MS obsd 385.0749, calcd 385.0757 [(M + H)+, M = C16H21BrN2O4].3-Bromo-2-(2-nitroethyl)-N-tosylpyrrole (13)
Following a general procedure,62 a mixture of 12 (5.25 g, 16.0 mmol), potassium acetate (1.57 g, 16.0 mmol), and methylamine hydrochloride (1.08 g, 16.0 mmol) in absolute ethanol (8.00 mL) was treated with nitromethane (2.76 mL, 52.0 mmol). The resulting mixture was stirred at room temperature for 3 h, whereupon water was added. The resulting precipitate was separated by filtration, washed with water and dried under reduced pressure to afford a yellow solid, which was used directly in the next step. The crude solid material was dissolved in THF (69.0 mL) and cooled to −10 °C (using an acetone bath with a few pieces of dry ice). The solution was treated with 95% LiBH4 (315 mg, 14.0 mmol) all-at-once under vigorous stirring. The reaction mixture was stirred for ∼30 min at −10 °C, whereupon the reaction mixture was quenched by slowly adding a cold saturated aqueous NH4Cl solution. The mixture was extracted with ethyl acetate. The organic solution was washed with brine, dried (Na2SO4) and concentrated. Column chromatography [silica, hexanes–CH2Cl2 (1:1)] afforded a white solid (1.20 g, 20% yield): mp 133–134 °C; 1H NMR δ 2.44 (s, 3H), 3.43 (t, J = 8.0 Hz, 2H), 4.50 (t, J = 8.0 Hz, 2H), 6.32 (d, J = 3.6 Hz, 1H), 7.33 (d, J = 3.6 Hz, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 8.4 Hz, 2H); 13C NMR δ 21.8, 24.2, 72.9, 105.4, 114.7, 123.2, 125.1, 127.0, 130.7, 135.2, 146.2; ESI-MS obsd 394.9663, calcd 394.9672 [(M + Na)+, M = C13H13BrN2O4S].
6-(3-Bromopyrrol-2-yl)-1,1-dimethoxy-4,4-dimethyl-5-nitrohexan-2-one (14)
Following a general procedure,62 a mixture of 13 (1.20 g, 3.20 mmol) and 9 (1.90 g, 16.0 mmol) was treated with DBU (3.60 mL, 24.0 mmol). The reaction mixture was stirred at room temperature for 30 min under argon. A saturated solution of cold aqueous NH4Cl was added at 0 °C. The mixture was extracted with ethyl acetate. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated. Column chromatography [silica, CH2Cl2–ethyl acetate (9:1)] afforded a pale yellow solid (950 mg, 56% yield): mp 92–93 °C; 1H NMR δ 1.13 (s, 3H), 1.29 (s, 3H), 2.43 (s, 3H), 2.66, 2.77 (AB, 2J = 18.8 Hz, 2H), 3.10 (dd, 3J = 3.2 Hz, 3J = 15.2 Hz, 1H), 3.42 (s, 3H), 3.43 (s, 3H), 3.56 (dd, 3J = 11.2 Hz, 3J = 15.2 Hz, 1H), 4.39 (s, 1H), 5.32 (dd, 3J = 2.8 Hz, 3J = 11.2 Hz, 1H), 6.26 (d, J = 3.6 Hz, 1H), 7.25 (d, J = 4.0 Hz, 1H), 7.33 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H); 13C NMR δ 21.9, 23.8, 24.0, 25.9, 36.8, 44.4, 55.2, 93.3, 104.7, 106.7, 116.0, 124.3, 126.5, 126.8, 130.5, 135.6, 145.9, 203.0; ESI-MS obsd 548.1060, calcd 548.1061 [(M + NH4)+, M = C21H27BrN2O7S].
7-Bromo-2,3-dihydro-1-(1,1-dimethoxymethyl)-3,3-dimethyldipyrrin (15)
Following a general procedure,62 in a first flask, a solution of 14 (530 mg, 1.00 mmol) in freshly distilled THF (5.0 mL) at 0 °C was treated with NaOMe (270 mg, 5.00 mmol). The mixture was stirred and deaerated by bubbling argon through the solution for 2 h. In a second flask purged with argon, TiCl3 (7.00 mL, 20 wt% in 3% HCl solution, 11.0 mmol), THF (20 mL) and NH4OAc (7.00 g, 90.0 mmol) were combined under argon, and the mixture was deaerated by bubbling with argon for 45 min. Then, the first flask mixture was transferred via cannula to the buffered TiCl3 mixture. The resulting mixture was stirred at room temperature for 18 h under argon. Then, the mixture was diluted with ethyl acetate and washed with saturated aqueous NaHCO3. The organic layer was separated, dried (Na2SO4) and concentrated. Column chromatography [silica, hexanes–CH2Cl2 (1:1)] afforded a yellow oil (75.0 mg, 20% yield): 1H NMR δ 1.24 (s, 6H), 2.63 (s, 2H), 3.45 (s, 6H), 5.02 (s, 1H), 5.93 (s, 1H), 6.19 (t, J = 2.8 Hz, 1H), 6.78 (t, J = 2.8 Hz, 1H), 10.75 (brs, 1H); 13C NMR δ 19.3, 29.2, 40.6, 48.5, 54.7, 97.7, 102.7, 104.8, 111.2, 119.4, 128.8, 161.0, 175.4; ESI-MS obsd 327.0706, calcd 327.0703 [(M + H)+, M = C14H19BrN2O2].
3,13-Bis[3,5-bis(tert-butoxycarbonyl)phenyl]-5-methoxy-8,8,18,18-tetramethylbacteriochlorin (pro-BC-6)
Following a general procedure,25 samples of BC-9 (17.3 mg, 31.0 μmol), 4 (75.2 mg, 186 μmol), Pd(PPh3)4 (21.5 mg, 18.6 μmol), K2CO3 (51.5 mg, 373 μmol) and toluene–DMF [3.1 mL (2:1), deaerated by bubbling with argon for 45 min] was added to a Schlenk flask and deaerated by three freeze–pump–thaw cycles. The reaction mixture was stirred at 90 °C for 20 h. The reaction mixture was cooled to room temperature, concentrated to dryness, diluted with CH2Cl2 and washed with saturated aqueous NaHCO3. The organic layer was separated, dried (Na2SO4) and concentrated. Column chromatography [silica, hexanes–CH2Cl2 (1:3)] afforded the title compound (18.4 mg, 62%): 1H NMR δ −1.85 (brs, 1H), −1.60 (brs, 1H), 1.68 (s, 18H), 1.71 (s, 18H), 1.96 (s, 6H), 1.98 (s, 6H), 3.64 (s, 3H), 4.38 (s, 2H), 4.40 (s, 2H), 8.64–8.66 (m, 2H), 8.67 (s, 1H), 8.72 (s, 1H), 8.75 (t, J = 2.4 Hz, 1H), 8.80 (t, J = 2.4 Hz, 1H), 8.84 (d, J = 2.4 Hz, 1H), 8.88 (d, J = 2.4 Hz, 2H), 8.94 (d, J = 2.4 Hz, 2H); 13C NMR δ 28.4, 28.5, 29.9, 31.3, 45.8, 45.9, 47.8, 52.2, 63.4, 81.8, 82.1, 96.7, 97.1, 97.3, 122.6, 122.7, 127.5, 129.0, 129.5, 130.1, 131.9, 132.0, 132.2, 133.2, 134.2, 134.7, 135.1, 135.5, 135.6, 136.0, 136.5, 137.0, 138.9, 154.5, 160.9, 165.5, 165.8, 169.6, 170.1; MALDI-MS obsd 952.5; ESI-MS obsd 952.4989, calcd 952.4981 (C57H68N4O9); λabs (CH2Cl2) 364, 511, 731 nm.
3,13-Bis(3,5-dicarboxyphenyl)-5-methoxy-8,8,18,18-tetramethylbacteriochlorin (BC-6)
A solution of pro-BC-6 (4.0 mg, 4.2 μmol) in CH2Cl2 (0.4 mL) was stirred at room temperature under argon for 2 min, followed by addition of TFA (0.1 mL). After 2 h, the reaction mixture was diluted with ethyl acetate and then washed with saturated aqueous NaHCO3, 2 N HCl solution, and water. The organic layer was separated, dried (Na2SO4) and concentrated. The resulting solid was treated with hexanes and methanol (49:1), sonicated in a benchtop sonication bath, and then centrifuged. The supernatant was discarded to afford a green solid (2.7 mg, 88%): 1H NMR (DMSO-d6) δ −1.93 (brs, 1H), −1.69 (brs, 1H), 1.89 (s, 6H), 1.93 (s, 6H), 3.54 (s, 3H), 4.33 (s, 2H), 4.39 (s, 2H), 8.67 (d, J = 1.2 Hz, 1H), 8.71 (d, J = 1.2 Hz, 1H), 8.74 (s, 1H), 8.81 (m, 2H), 8.86 (s, 1H), 8.90–8.92 (m, 3H), 8.93 (s, 1H), 9.19 (s, 1H), 13.4–13.6 (br, 4H); MALDI-MS obsd 728.7; ESI-MS obsd 729.2559, calcd 729.2555 [(M + H)+, M = C41H36N4O9]; λabs (CH2Cl2) 365, 511, 731 nm; λabs (100 mM aqueous potassium phosphate buffer, pH 7.6) 357, 508, 730 nm; λabs (DMF) 362, 509, 727 nm.
2,12-Bis[3,5-bis(tert-butoxycarbonyl)phenyl]-3,13-bis(methoxycarbonyl)-5-methoxy-8,8,18,18-tetramethylbacteriochlorin (pro-BC-7)
Following a general procedure,25 a mixture of BC-12 (25 mg, 0.037 mmol), 4 (90 mg, 0.22 mmol), Pd(PPh3)4 (25 mg, 0.022 mmol), and anhydrous K2CO3 (61 mg, 0.44 mmol) was deaerated under vacuum in a Schlenk flask for 1 h. Toluene–DMF [3.7 mL, (2:1), deaerated by bubbling argon] was added and the reaction mixture was deaerated by four freeze–pump–thaw cycles. The reaction mixture was heated at 90 °C for 18 h. After cooling to room temperature, the solvent was evaporated. The crude reaction mixture was diluted with CH2Cl2 and washed with saturated aqueous NaHCO3. The organic layer was separated, dried (Na2SO4) and concentrated. The residue was purified by column chromatography [silica, hexanes–ethyl acetate (9:1)] to afford a purple solid (13.4 mg, 33%): 1H NMR δ −1.44 (brs, 1H), −1.15 (brs, 1H), 1.66 (s, 18H), 1.67 (s, 18H), 1.81 (s, 6H), 1.87 (s, 6H), 4.04 (s, 3H), 4.20 (s, 3H), 4.27 (s, 3H), 4.36 (s, 2H), 4.43 (s, 2H), 8.36 (s, 1H), 8.52 (s, 1H), 8.73 (d, J = 1.6 Hz, 2H), 8.89 (d, J = 1.2 Hz, 4H), 9.65 (s, 1H); 13C NMR δ 28.2, 28.4, 29.9, 30.8, 30.9, 45.8, 46.3, 47.9, 51.6, 51.9, 53.1, 64.6, 81.8, 81.9, 95.9, 98.1, 98.6, 118.8, 124.9, 128.4, 129.9, 130.2, 131.8, 132.2, 132.6, 133.3, 134.2, 134.5, 135.0, 135.7, 136.0, 136.2, 136.4, 137.0, 157.2, 161.9, 165.0, 165.3, 166.4, 168.7, 169.5, 173.3; MALDI-MS obsd 1069.3; ESI-MS obsd 1069.5167, calcd 1069.5169 [(M + H)+, M = C61H72N4O13]; λabs (DMF) 373, 531, 753 nm.
The procedure was repeated at larger scale following the above procedure but with several changes. BC-12 (100 mg, 148 μmol), 4 (360 mg, 888 μmol), Pd(PPh3)4 (100 mg, 88.8 μmol) and Cs2CO3 (149 mg, 444 μmol) were reacted in toluene–DMF [10.0 mL, (2:1), deaerated by bubbling argon] for 22 h. After cooling to room temperature, the mixture was diluted with diethyl ether and washed (saturated aqueous NaHCO3, 0.5 N HCl, water, and brine), dried (Na2SO4) and concentrated. Chromatography afforded the title compound (135 mg, 85%) with characterization data identical with those listed above.
2,12-Bis(3,5-dicarboxyphenyl)-3,13-bis(methoxycarbonyl)-5-methoxy-8,8,18,18-tetramethylbacteriochlorin (BC-7)
A solution of pro-BC-7 (5.7 mg, 5.3 μmol) in anhydrous CH2Cl2 (1.4 mL) was treated with TFA (350 μL). The reaction mixture was stirred at room temperature under argon for 2 h. Ethyl acetate was added, and the mixture was washed with saturated aqueous NaHCO3. The aqueous phase was separated, acidified with 2 M HCl, and extracted with ethyl acetate. The organic layer was washed (brine, water), dried (NaSO4) and concentrated. The resulting solid was washed with hexanes to give the title compound as a purple solid (4.0 mg, 89% yield): 1H NMR (DMSO-d6) δ −1.44 (brs, 1H), −1.15 (brs, 1H), 1.82 (s, 6H), 1.76 (s, 6H), 3.99 (s, 3H), 4.10 (s, 3H), 4.22 (s, 3H), 4.35 (s, 2H), 4.43 (s, 2H), 8.38 (s, 1H), 8.57 (s, 1H), 8.71 (d, J = 1.2 Hz, 2H), 8.75 (t, J = 1.2 Hz, 1H), 8.78 (t, J = 1.6 Hz, 1H), 8.86 (d, J = 1.6 Hz, 2H), 9.52 (s, 1H), 13.56 (brs, 4H); MALDI-MS obsd 844.9; ESI-MS obsd 845.2648, calcd 845.2665 [(M + H)+, M = C45H40N4O13]; λabs (DMF) 360, 374, 529, 743 nm; λabs (100 mM aqueous potassium phosphate buffer, pH 7.6) 370, 527, 750 nm.2,12-Bis[3,5-bis(tert-butoxycarbonyl)phenyl]-5-methoxy-8,8,18,18-tetramethylbacteriochlorin (pro-BC-8)
Following a general procedure,25 a mixture of BC-13 (18.6 mg, 33.3 μmol), 4 (80.4 mg, 200 μmol), Pd(PPh3)4 (23.1 mg, 20.0 μmol), and K2CO3 (55.3 mg, 400 μmol) in toluene–DMF [3.33 mL (2:1)] in a Schlenk flask was deaerated by three freeze–pump–thaw cycles. The flask was placed in an oil bath at 90 °C and stirred for 20 h. The reaction mixture was allowed to cool to room temperature, concentrated, diluted with CH2Cl2 and washed with saturated aqueous NaHCO3. The organic layer was dried (Na2SO4) and concentrated. Column chromatography [silica, CH2Cl2–hexanes (2:1)] afforded a green solid (9.6 mg, 30% yield): 1H NMR δ −1.80 (s, 1H), −1.68 (s, 1H), 1.70 (s, 36H), 1.93 (s, 6H), 1.94 (s, 6H), 4.43 (s, 2H), 4.44 (s, 2H), 4.51 (s, 3H), 8.73 (s, 1H), 8.74 (s, 1H), 8.76 (s, 1H), 8.82 (s, 1H), 8.84 (t, J = 1.6 Hz, 1H), 8.85 (t, J = 1.6 Hz, 1H), 9.00 (t, J = 1.6 Hz, 2H), 9.03 (t, J = 1.6 Hz, 2H), 9.07 (d, J = 2.0 Hz, 1H); 13C NMR δ 25.1, 28.5, 29.9, 31.2, 31.3, 46.0, 46.3, 47.5, 51.9, 65.4, 82.0, 82.1, 95.4, 95.5, 98.4, 116.7, 121.7, 129.2, 129.7, 130.1, 132.3, 133.0, 133.2, 133.3, 134.7, 135.6, 135.7, 135.9, 136.8, 137.5, 153.6, 160.5, 165.4, 165.5, 170.2, 170.5; MALDI-MS obsd 953.8; ESI-MS 952.4982, calcd 952.4981 (C57H68N4O9); λabs (DMF) 368, 514, 735 nm.
2,12-Bis(3,5-dicarboxyphenyl)-5-methoxy-8,8,18,18-tetramethylbacteriochlorin (BC-8)
A solution of pro-BC-8 (3.1 mg, 3.2 μmol) in CH2Cl2 (300 μL) at room temperature was stirred under argon for 2 min followed by the addition of TFA (75 μL). The reaction mixture was stirred for 2 h. The mixture was diluted with ethyl acetate and treated with saturated aqueous NaHCO3. The ethyl acetate layer was then discarded. The aqueous phase (which contained the anionic bacteriochlorin) was separated, acidified with 2 N HCl, and extracted with ethyl acetate. The organic layer was washed (water, brine), dried (Na2SO4) and concentrated. The resulting solid was treated with hexanes and methanol (49:1), sonicated in a benchtop sonication bath, and then centrifuged. The supernatant was discarded. The residue was dried to afford a purple solid (2.1 mg, 90%): 1H NMR (DMSO-d6) δ −1.78 (s, 1H), −1.68 (s, 1H), 1.86 (s, 6H), 1.88 (s, 6H), 4.37 (s, 2H), 4.42 (s, 2H), 4.49 (s, 3H), 8.71 (s, 1H), 8.73 (s, 1H), 8.76 (s, 2H), 8.89 (s, 1H), 9.03 (s, 2H), 9.06 (s, 2H), 9.19 (s, 2H) (the four carboxylic acid protons were not observed); MALDI-MS obsd 729.2; ESI-MS obsd 729.2535, calcd 729.2555 [(M + H)+, M = C41H36N4O9]; λabs (DMF) 354, 512, 725 nm; λabs (100 mM aqueous potassium phosphate buffer, pH 7.6) 357, 511, 734 nm.
5-Methoxy-8,8,18,18-tetramethyl-3,13-diphenylbacteriochlorin (BC-10)
Following a general procedure,25 a mixture of BC-9 (20 mg, 0.036 mmol), 1 (22.0 mg, 0.108 mmol), Pd(PPh3)4 (12.5 mg, 0.0100 mmol), and anhydrous K2CO3 (60.0 mg, 0.432 mmol) was deaerated under vacuum in a Schlenk flask for 1 h. Toluene–DMF [3.6 mL, (2:1), deaerated by bubbling with argon] was added and the reaction mixture was deaerated by three freeze–pump–thaw cycles. The reaction mixture was heated at 90 °C for 18 h. After cooling to room temperature, the mixture was concentrated. The resulting residue was diluted with CH2Cl2 and washed with saturated aqueous NaHCO3. The organic layer was separated, dried (Na2SO4) and concentrated. Column chromatography [silica, CH2Cl2–hexanes (1:1)] afforded a green solid (18.0 mg, 91%): 1H NMR δ −1.91 (brs, 1H), −1.66 (brs, 1H), 1.94 (s, 6H), 1.97 (s, 6H), 3.64 (s, 3H), 4.38 (s, 4H), 7.51–7.57 (m, 1H), 7.58–7.68 (m, 3H), 7.72–7.79 (m, 2H), 8.09–8.13 (m, 2H), 8.16–8.20 (m, 2H), 8.61 (d, J = 2.4 Hz, 1H), 8.63 (s, 1H), 8.65 (s, 1H), 8.77 (d, J = 2.0 Hz, 1H), 8.79 (s, 1H); 13C NMR δ 31.2, 31.3, 45.7, 45.9, 47.7, 52.2, 63.4, 96.8, 96.9, 97.0, 122.3, 122.4, 127.0, 127.1, 127.8, 129.2, 131.3, 131.4, 133.9, 134.0, 135.4, 135.6, 136.3, 136.6, 136.7, 138.5, 153.9, 160.6, 169.0, 169.9; MALDI-MS obsd 552.2; ESI-MS obsd 553.2942, calcd 553.2962 [(M + H)+, M = C37H36N4O]; λabs (CH2Cl2) 361, 373, 508, 728 nm.
15-Bromo-5-methoxy-8,8,18,18-tetramethyl-3,13-diphenylbacteriochlorin (BC-11)
Following a general procedure,23 a solution of BC-10 (15.0 mg, 0.0271 mmol, 2.0 mM) in dry THF (13.5 mL) was treated dropwise (10 min) with a solution of NBS (4.82 mg, 0.0271 mmol, 4.0 mM) in THF (6.75 mL) and stirred at room temperature under argon for 1 h. The reaction mixture was diluted with CH2Cl2 and washed with saturated aqueous NaHCO3. The organic layer was separated, dried (Na2SO4) and concentrated. Column chromatography [silica, CH2Cl2–hexanes (1:1)] afforded a green solid (9.3 mg, 54%): 1H NMR δ −1.74 (brs, 1H), −1.45 (brs, 1H), 1.95 (s, 6H), 1.96 (s, 6H), 3.65 (s, 3H), 4.36 (s, 2H), 4.41 (s, 2H), 7.52–7.70 (m, 6H), 7.83–7.89 (m, 2H), 8.08–8.14 (m, 2H), 8.59 (s, 1H), 8.62–8.68 (m, 3H); 13C NMR δ 31.4, 31.6, 45.5, 45.7, 48.2, 54.8, 63.7, 96.9, 98.5, 124.7, 125.8, 127.4, 127.8, 128.0, 129.6, 130.9, 131.2, 131.6, 132.4, 135.3, 136.3, 136.3, 137.7, 139.4, 157.2, 159.7, 168.0, 171.3; MALDI-MS obsd 631.9; ESI-MS obsd 631.2046, calcd 631.2067 [(M + H)+, M = C37H35BrN4O]; λabs (CH2Cl2) 365, 520, 725 nm.
2,12-Dibromo-5-methoxy-3,13-bis(methoxycarbonyl)-8,8,18,18-tetramethylbacterio-chlorin (BC-12)
Following a general procedure,22 a solution of 11 (289 mg, 750 μmol) in anhydrous CH2Cl2 (40.0 mL) was treated first with DTBP (2.87 g, 15.0 mmol) and second with TMSOTf (680 μL, 3.75 mmol). The reaction mixture was stirred at room temperature for 16 h. The mixture was washed with saturated aqueous NaHCO3, dried (Na2SO4) and concentrated. Column chromatography [silica, CH2Cl2–hexanes (2:1)] afforded a dark purple solid (120 mg, 25% yield): 1H NMR δ −1.54 (s, 1H), −1.26 (s, 1H), 1.93 (s, 6H), 1.94 (s, 6H), 4.23 (s, 3H), 4.31 (s, 3H), 4.32 (s, 3H), 4.33 (s, 2H), 4.37 (s, 2H), 8.66 (s, 1H), 8.86 (s, 1H), 9.53 (s, 1H); 13C NMR δ 26.8, 29.9, 31.0, 31.2, 45.9, 46.2, 48.0, 51.7, 52.5, 53.6, 64.6, 95.7, 97.9, 98.6, 110.4, 115.4, 120.2, 126.6, 128.5, 131.9, 133.5, 135.1, 135.3, 157.5, 162.1, 165.3, 167.4, 170.0, 173.5; ESI-MS obsd 673.0636, calcd 673.0656 [(M + H)+, M = C29H30Br2N4O5]; λabs (CH2Cl2) 356, 367, 527, 749 nm.
2,12-Dibromo-5-methoxy-8,8,18,18-tetramethylbacteriochlorin (BC-13)
Following a general procedure,22 a solution of 15 (75.0 mg, 230 μmol) in anhydrous CH2Cl2 (13.0 mL) was treated first with DTBP (1.02 mL, 4.60 mmol) and second with TMSOTf (210 μL, 1.15 mmol). The reaction mixture was stirred under argon at room temperature for 18 h. The mixture was washed with saturated aqueous NaHCO3, dried (Na2SO4) and concentrated. Column chromatography [silica, CH2Cl2–hexanes (2:1)] afforded a green solid (23.2 mg, 36% yield): 1H NMR δ −2.06 (s, 1H), −1.95 (s, 1H), 1.94 (s, 6H), 1.96 (s, 6H), 4.33 (s, 2H), 4.35 (s, 2H), 4.43 (s, 3H), 8.54 (s, 1H), 8.67 (d, J = 2.0 Hz, 1H), 8.70 (s, 1H), 8.71 (s, 1H), 8.93 (d, J = 2.0 Hz, 1H); 13C NMR δ 31.2, 31.4, 45.9, 46.2, 47.5, 51.9, 65.3, 95.3, 98.1, 109.6, 113.3, 118.9, 123.8, 130.0, 132.9, 134.3, 135.2, 135.7, 153.9, 160.8, 170.2, 170.6; MALDI-MS obsd 556.5; ESI-MS obsd 556.0468, calcd 556.0468 (C25H26Br2N4O); λabs (CH2Cl2) 348, 358, 369, 504, 724 nm.
15-Bromo-3,13-bis[3,5-bis(tert-butoxycarbonyl)phenyl]-5-methoxy-8,8,18,18-tetramethylbacteriochlorin (BC-14)
Following a general procedure23 in more dilute solution, a solution of pro-BC-6 (27 mg, 28 μmol) in THF (58 mL) was treated with a solution of NBS (5.2 mg, 29 μmol) in THF (0.29 mL) at room temperature for 2 h. The reaction mixture was diluted with CH2Cl2 and washed with saturated aqueous NaHCO3. The organic layer was dried (Na2SO4), concentrated and chromatographed [silica, CH2Cl2–ethyl acetate (7:3)] to afford a reddish solid (21 mg, 70%): 1H NMR (300 MHz) δ −1.68 (s, 1H), −1.38 (s, 1H), 1.65 (s, 18H), 1.69 (s, 18H), 1.96 (s, 12H), 3.68 (s, 3H), 4.37 (s, 2H), 4.41 (s, 2H), 8.60 (s, 1H), 8.63 (d, J = 1.2 Hz, 2H), 8.65 (s, 1H), 8.66 (d, J = 2.7 Hz, 1H), 8.69 (d, J = 2.7 Hz, 1H), 8.76–8.78 (m, 2H), 8.88 (d, J = 1.2 Hz, 2H); 13C NMR δ 25.1, 28.5, 31.3, 31.4, 45.2, 46.0, 47.7, 52.0, 63.5, 81.8, 97.2, 97.5, 114.0, 114.2, 118.3, 121.0, 121.4, 123.1, 124.1, 125.2, 126.5, 128.1, 128.4, 129.1, 131.9, 132.3, 133.9, 134.0, 134.1, 135.8, 136.0, 136.2, 138.7, 139.0, 141.5, 145.9, 155.0, 161.1, 165.8, 169.2, 169.4; ESI-MS obsd 1030.4089, calcd 1030.4086 (C57H67BrN4O9); MALDI-MS obsd 1031.7; λabs (CH2Cl2) 367, 522, 727 nm.
15-(3-Aminophenyl)-3,13-bis[3,5-bis(tert-butoxycarbonyl)phenyl]-5-methoxy-8,8,18,18-tetramethylbacteriochlorin (BC-15)
Following a general procedure,23 samples of BC-14 (21 mg, 20 μmol), 16 (22 mg, 0.10 mmol), Pd(PPh3)4 (9.3 mg, 8.0 μmol), and K2CO3 (33 mg, 0.24 mmol) were placed in a Schlenk flask, and deaerated under vacuum for 30 min. Toluene–DMF [2.0 mL, (2:1), deaerated by bubbling with argon] was added under argon and deaerated by three freeze–pump–thaw cycles. The reaction mixture was stirred at 90 °C for 18 h. The reaction mixture was cooled to room temperature, concentrated to dryness and diluted with CH2Cl2. The solution was washed with saturated aqueous NaHCO3. The organic layer was separated, dried (Na2SO4) and concentrated. Column chromatography [silica, CH2Cl2–ethyl acetate (99:1)] provided a reddish solid (15 mg, 70%): 1H NMR δ −1.57 (s, 1H), −1.23 (s, 1H), 1.64 (s, 18H), 1.69 (s, 18H), 1.80 (s, 3H), 1.90 (s, 3H), 1.97 (s, 3H), 1.99 (s, 3H), 3.66 (s, 3H), 3.92 (d, J = 17.6 Hz, 1H), 4.08 (d, J = 17.6 Hz, 1H), 4.38 (s, 2H), 6.39–6.42 (m, 1H), 6.72 (s, 1H), 6.93–6.99 (m, 2H), 8.02 (br, 1H), 8.25 (br, 1H), 8.42 (t, J = 1.6 Hz, 1H), 8.63 (d, J = 2.4 Hz, 1H), 8.65 (s, 2H), 8.67 (d, J = 2.4 Hz, 1H), 8.76 (t, J = 1.6 Hz, 1H), 8.91 (d, J = 1.2 Hz, 2H), the NH2 protons were not observed); 13C NMR δ 28.5, 28.5, 31.4, 31.6, 45.6, 45.8, 48.2, 45.6, 45.8, 54.8, 63.7, 81.2, 81.9, 97.2, 97.3, 98.8, 125.0, 126.0, 129.4, 129.5, 130.2, 130.8, 131.9, 132.1, 132.5, 133.2, 134.3, 134.4, 135.4, 135.6, 135.8, 136.0, 136.1, 136.3, 138.1, 139.8, 157.7, 160.0, 165.7, 168.3, 171.8; MALDI-MS obsd 1044.6; ESI-MS obsd 1043.5406, calcd 1043.5408 (C63H73N5O9); λabs (CH2Cl2) 365, 517, 728 nm.
3,13-Bis[3,5-bis(tert-butoxycarbonyl)phenyl]-15-[(3-(4-maleimidobutyramido)phenyl)]-5-methoxy-8,8,18,18-tetramethylbacteriochlorin (pro-BC-16)
A mixture of 17 (54 mg, 0.29 mmol), DCC (61 mg, 0.29 mmol) and DMAP (0.80 mg, 5.9 μmol) in CH2Cl2 (2.5 mL) was treated with BC-15 (31 mg, 29 μmol) in CH2Cl2 (0.5 mL) and stirred at room temperature for 2.5 h. The resulting mixture was filtered to remove insoluble material. The filtrate was concentrated and chromatographed [silica, CH2Cl2–ethyl acetate (24:1)] to afford a reddish solid (22 mg, 61%): 1H NMR (300 MHz) δ −1.59 (s, 1H), −1.22 (s, 1H), 1.63 (s, 18H), 1.69 (s, 18H), 1.81 (s, 3H), 1.89 (s, 3H), 1.98–2.02 (m, 8H), 2.29 (t, J = 6.9 Hz, 2H), 3.63 (t, J = 6.3 Hz, 2H), 3.68 (s, 3H), 3.91 (d, J = 17.4 Hz, 1H), 4.03 (d, J = 17.4 Hz, 1H), 4.38 (s, 2H), 6.69, (s, 2H), 7.15 (t, J = 7.8 Hz, 1H), 7.34 (d, J = 7.2 Hz, 1H), 7.44–7.47 (m, 2H), 7.66 (s, 1H), 7.96 (s, 1H), 8.20 (s, 1H), 8.34 (t, J = 1.5 Hz, 1H), 8.63 (d, J = 2.7 Hz, 1H), 8.65 (t, J = 7.5 Hz, 2H), 8.68 (d, J = 2.1 Hz, 1H), 8.76 (t, J = 1.5 Hz, 1H), 8.91 (d, J = 1.8 Hz, 2H); 13C NMR δ 28.5, 28.5, 31.3, 31.4, 34.8, 37.3, 45.4, 46.0, 47.8, 52.2, 63.6, 81.6, 81.9, 97.2, 97.8, 113.3, 119.2, 123.3, 125.2, 126.6, 128.2, 128.3, 129.2, 129.4, 131.0, 132.0, 132.6, 133.8, 133.8, 134.3, 134.4, 134.9, 135.3, 136.0, 136.3, 137.4, 138.6, 139.0, 141.6, 155.3, 160.9, 165.8, 169.1, 169.6, 170.1, 171.3; MALDI-MS obsd 1210.9; ESI-MS obsd 1208.5848, calcd 1208.5829 (C71H80N6O12); λabs (CH2Cl2) 366, 517, 730 nm.
3,13-Bis(3,5-dicarboxyphenyl)-15-[3-(4-maleimidobutyramido)phenyl]-5-methoxy-8,8,18,18-tetramethylbacteriochlorin (BC-16)
A solution of pro-BC-16 (26 mg, 21 μmol) in CH2Cl2 (2.0 mL) at room temperature was stirred under argon for 2 min, followed by addition of TFA (0.5 mL). After 2 h, the reaction mixture was diluted with ethyl acetate and washed with saturated aqueous NaHCO3, 2 N HCl solution and water. The organic layer was separated, dried (Na2SO4) and concentrated. The resulting solid was treated with a mixture of hexanes and CH2Cl2 (3:2), sonicated in a benchtop sonication bath, and centrifuged. The supernatant was discarded to afford a green solid (17 mg, 82%): 1H NMR (DMSO-d6) δ −1.66 (s, 1H), −1.31 (s, 1H), 1.75 (s, 6H), 1.87 (s, 6H), 1.92 (s, 3H), 1.94 (s, 3H), 2.23 (t, J = 7.2 Hz, 2H), 3.43 (t, J = 6.8 Hz, 2H), 3.58 (s, 3H), 3.78 (d, J = 16.8 Hz, 1H), 4.09 (d, J = 16.8 Hz, 1H), 4.34 (s, 2H), 6.97–7.07 (m, 2H), 7.25–7.31 (m, 1H), 7.72 (s, 1H), 7.84 (br, 1H), 8.27 (s, 1H), 8.69 (s, 1H), 8.85–8.97 (m, 5H), 9.97 (s, 1H), 13.24 (br, 4H); 13C NMR (DMSO-d6) δ 24.5, 31.3, 31.4, 31.6, 34.2, 37.6, 45.3, 46.2, 47.5, 52.1, 63.7, 98.0, 98.4, 114.1, 117.5, 124.2, 127.6, 127.8, 127.9, 128.2, 128.5, 129.4, 130.5, 131.6, 132.1, 133.8, 134.1, 134.2, 135.2, 135.8, 135.9, 136.2, 138.5, 138.7, 139.5, 140.5, 155.5, 161.2, 164.2, 167.6, 169.1, 169.6, 169.8, 170.7, 171.8; MALDI-MS obsd 982.8; ESI-MS obsd 985.3403, calcd 985.3403 [(M + H)+, M = C55H48N6O12]; λabs (CH3OH) 363, 514, 725 nm; λabs (100 mM aqueous potassium phosphate buffer, pH 7.6) 360, 516, 729 nm; λabs (DMF) 367, 516, 727 nm.
Preparation of β(-14Cys)BC-16
Stock solutions of DMF and Tris buffer (0.1 M, pH 8.6) were sonicated for 5 min followed by bubbling with argon for 30 min to remove oxygen. A sample of β(-14Cys) (2.5 mg, 0.46 μmol) was dissolved in 92 μL of DMF, and 23 μL of Tris buffer (pH 8.6) was then added while the mixture was stirred under argon. The protein appeared to readily dissolve in DMF and stay in solution when Tris buffer was added. A sample of BC-16 (0.7 mg, 0.71 μmol) was dissolved in 92 μL of DMF and 23 μL of Tris buffer. The resulting BC-16 solution was then added dropwise to the β(-14Cys) solution under argon. The reaction mixture was stirred in the dark at room temperature for 3 h. The reaction mixture was treated with diethyl ether to cause precipitation. The resulting suspension was sonicated (benchtop sonication bath) and centrifuged (3800 rpm). The supernatant was discarded. The procedure (ether addition, sonication and centrifugation) was performed two additional times. The resulting greenish residue was dried under vacuum for 30 min. Addition of hexafluoroacetone trihydrate (50 μL) to the greenish solid followed by sonication for 3 min completely dissolved the solid, affording a blue solution. Addition of 150 μL of a solvent mixture that represented the initial HPLC eluant [composed of H2O (50%), isopropanol (33.3%), acetonitrile (17.6%) and TFA (0.1%)] caused a change from blue back to green. This mixture was centrifuged to remove (visibly evident) insoluble particles. An aliquot (30 μL) of the sample was injected into the HPLC instrument and a fraction at 15.5 min was collected. The fraction contained the title conjugate as well as unreacted peptide as observed upon ESI-MS analysis. The HPLC conditions are described in the Electronic Supplementary Information.† An aliquot of the HPLC fraction was reinjected to assess homogeneity (Fig. S7†), which showed the fraction to be essentially free of unreacted bacteriochlorin. The total (isolated) yield was determined to be 52% on the basis of absorption spectroscopy of the conjugate (bacteriochlorin Qy band; Fig. S8†). The fraction from the analytical HPLC was concentrated under vacuum to dryness and subjected to ESI-MS, which gave m/z = 6410.0550 for the monoisotopic ion (calcd 6410.0241 for M+, M = C309H417N67O82S) and m/z = 6414.0708 for the most intense peak. Such data can be compared with those for the peptide β(-14Cys), which gave m/z = 5425.7052 for the monoisotopic ion (calcd 5425.6905 for M+, M = C254H369N61O70S) and m/z = 5428.7056 for the most intense peak.Acknowledgements
This research was carried out as part of the Photosynthetic Antenna Research Center (PARC), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award no. DE-SC0001035. Mass spectra were obtained at the Mass Spectrometry Laboratory for Biotechnology at North Carolina State University. Partial funding for the facility was obtained from the North Carolina Biotechnology Center and the National Science Foundation.Notes and references
- H. Scheer, in Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Springer, Dordrecht, The Netherlands, 2006, pp. 1–26 Search PubMed.
- Y. Chen, G. Li and R. K. Pandey, Curr. Org. Chem., 2004, 8, 1105–1134 CrossRef CAS.
- M. A. Grin, A. F. Mironov and A. A. Shtil, Anti-Cancer Agents Med. Chem., 2008, 8, 683–697 CAS.
- J. M. Sutton, N. Fernandez and R. W. Boyle, J. Porphyrins Phthalocyanines, 2000, 4, 655–658 CrossRef CAS.
- J. M. Sutton, O. J. Clarke, N. Fernandez and R. W. Boyle, Bioconjugate Chem., 2002, 13, 249–263 CrossRef CAS PubMed.
- A. M. G. Silva, A. C. Tomé, M. G. P. M. S. Neves, A. M. S. Silva and J. A. S. Cavaleiro, J. Org. Chem., 2005, 70, 2306–2314 CrossRef CAS PubMed.
- J. R. McCarthy, J. Bhaumik, N. Merbouh and R. Weissleder, Org. Biomol. Chem., 2009, 7, 3430–3436 CAS.
- A. C. Tomé, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Porphyrins Phthalocyanines, 2009, 13, 408–414 CrossRef.
- S. Singh, A. Aggarwal, S. Thompson, J. P. C. Tomé, X. Zhu, D. Samaroo, M. Vinodu, R. Gao and C. M. Drain, Bioconjugate Chem., 2010, 21, 2136–2146 CrossRef CAS PubMed.
- J. M. Dabrowski, L. G. Arnaut, M. M. Pereira, C. J. P. Monteiro, K. Urbanska, S. Simoes and G. Stochel, ChemMedChem, 2010, 5, 1770–1780 CrossRef CAS PubMed.
- N. A. M. Pereira, S. M. Fonseca, A. C. Serra, T. M. V. D. Pinho e Melo and H. D. Burrows, Eur. J. Org. Chem., 2011, 3970–3979 CrossRef CAS.
- J. M. Dąbrowski, K. Urbanska, L. G. Arnaut, M. M. Pereira, A. R. Abreu, S. Simões and G. Stochel, ChemMedChem, 2011, 6, 465–475 CrossRef PubMed.
- Z. Yu and M. Ptaszek, Org. Lett., 2012, 14, 3708–3711 CrossRef CAS PubMed.
- V. M. Alexander, K. Sano, Z. Yu, T. Nakajima, P. L. Choyke, M. Ptaszek and H. Kobayashi, Bioconjugate Chem., 2012, 23, 1671–1679 CrossRef CAS PubMed.
- A. Kozyrev, M. Ethirajan, P. Chen, K. Ohkubo, B. C. Robinson, K. M. Barkigia, S. Fukuzumi, K. M. Kadish and R. K. Pandey, J. Org. Chem., 2012, 77, 10260–10271 CrossRef CAS PubMed.
- L. P. Samankumara, S. Wells, M. Zeller, A. M. Acuña, B. Röder and C. Brückner, Angew. Chem., Int. Ed., 2012, 51, 5757–5760 CrossRef CAS PubMed.
- M. M. Pereira, A. R. Abreu, N. P. F. Goncalves, M. J. F. Calvete, A. V. C. Simoes, C. J. P. Monteiro, L. G. Arnaut, M. E. Eusébio and J. Canotilho, Green Chem., 2012, 14, 1666–1672 RSC.
- J. Ogikubo, E. Meehan, J. T. Engle, C. J. Ziegler and C. Brückner, J. Org. Chem., 2013, 78, 2840–2852 CrossRef CAS PubMed.
- M. Galezowski and D. T. Gryko, Curr. Org. Chem., 2007, 11, 1310–1338 CrossRef CAS.
- C. Brückner, L. Samankumara and J. Ogikubo, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing Co., Singapore, 2012, vol. 17, pp. 1–112 Search PubMed.
- H.-J. Kim and J. S. Lindsey, J. Org. Chem., 2005, 70, 5475–5486 CrossRef CAS PubMed.
- M. Krayer, M. Ptaszek, H.-J. Kim, K. R. Meneely, D. Fan, K. Secor and J. S. Lindsey, J. Org. Chem., 2010, 75, 1016–1039 CrossRef CAS PubMed.
- D. Fan, M. Taniguchi and J. S. Lindsey, J. Org. Chem., 2007, 72, 5350–5357 CrossRef CAS PubMed.
- K. E. Borbas, C. Ruzié and J. S. Lindsey, Org. Lett., 2008, 10, 1931–1934 CrossRef CAS PubMed.
- C. Ruzié, M. Krayer, T. Balasubramanian and J. S. Lindsey, J. Org. Chem., 2008, 73, 5806–5820 CrossRef PubMed.
- C. Muthiah, M. Taniguchi, H.-J. Kim, I. Schmidt, H. L. Kee, D. Holten, D. F. Bocian and J. S. Lindsey, Photochem. Photobiol., 2007, 83, 1513–1528 CrossRef CAS PubMed.
- S. Sharma, M. Krayer, F. F. Sperandio, L. Huang, Y.-Y. Huang, D. Holten, J. S. Lindsey and M. R. Hamblin, J. Porphyrins Phthalocyanines, 2013, 17, 73–85 CrossRef CAS PubMed.
- K. Aravindu, O. Mass, P. Vairaprakash, J. W. Springer, E. Yang, D. M. Niedzwiedzki, D. F. Bocian, D. Holten and J. S. Lindsey, Chem. Sci., 2013, 4, 3459–3477 RSC.
- L. Huang, Y.-Y. Huang, P. Mroz, G. P. Tegos, T. Zhiyentayev, S. K. Sharma, Z. Lu, T. Balasubramanian, M. Krayer, C. Ruzié, E. Yang, H. L. Kee, C. Kirmaier, J. R. Diers, D. F. Bocian, D. Holten, J. S. Lindsey and M. R. Hamblin, Antimicrob. Agents Chemother., 2010, 54, 3834–3841 CrossRef CAS PubMed.
- K. R. Reddy, E. Lubian, M. P. Pavan, H.-J. Kim, E. Yang, D. Holten and J. S. Lindsey, New J. Chem., 2013, 37, 1157–1173 RSC.
- E. Yang, C. Kirmaier, M. Krayer, M. Taniguchi, H.-J. Kim, J. R. Diers, D. F. Bocian, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2011, 115, 10801–10816 CrossRef CAS PubMed.
- K. R. Reddy, J. Jiang, M. Krayer, M. A. Harris, J. W. Springer, E. Yang, J. Jiao, D. M. Niedzwiedzki, D. Pandithavidana, P. S. Parkes-Loach, C. Kirmaier, P. A. Loach, D. F. Bocian, D. Holten and J. S. Lindsey, Chem. Sci., 2013, 4, 2036–2053 RSC.
- F. Giuntini, C. M. A. Alonso and R. W. Boyle, Photochem. Photobiol. Sci., 2011, 10, 759–791 CAS.
- H. Tamiaki, M. Xu, T. Tanaka and T. Mizoguchi, Bioorg. Med. Chem. Lett., 2013, 23, 2377–2379 CrossRef CAS PubMed.
- N. Drogat, C. Gady, R. Granet and V. Sol, Dyes Pigm., 2013, 98, 609–614 CrossRef CAS PubMed.
- M. A. Grin, I. S. Lonin, L. M. Likhosherstov, O. S. Novikova, A. D. Plyutinskaya, E. A. Plotnikova, V. V. Kachala, R. I. Yakubovskaya and A. F. Mironov, J. Porphyrins Phthalocyanines, 2012, 16, 1094–1109 CrossRef CAS.
- T. Maisch, C. Bosl, R.-M. Szeimies, N. Lehn and C. Abels, Antimicrob. Agents Chemother., 2005, 49, 1542–1552 CrossRef CAS PubMed.
- H. Taima, A. Okubo, N. Yoshioka and H. Inoue, Tetrahedron Lett., 2005, 46, 4161–4164 CrossRef CAS PubMed.
- H. Taima, A. Okubo, N. Yoshioka and H. Inoue, Chem.–Eur. J., 2006, 12, 6331–6340 CrossRef CAS PubMed.
- G. Jori, C. Fabris, M. Soncin, S. Ferro, O. Coppellotti, D. Dei, L. Fantetti, G. Chiti and G. Roncucci, Lasers Surg. Med., 2006, 38, 468–481 CrossRef PubMed.
- T. Maisch, C. Bosl, R.-M. Szeimies, B. Love and C. Abels, Photochem. Photobiol. Sci., 2007, 6, 545–551 CAS.
- G. Simonneaux, P. Le Maux, S. Chevance and H. Srour, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing Co., Singapore, 2012, vol. 21, pp. 377–410 Search PubMed.
- C. Spagnul, R. Alberto, G. Gasser, S. Ferrari, V. Pierroz, A. Bergamo, T. Gianferrara and E. Alessio, J. Inorg. Biochem., 2013, 122, 57–65 CrossRef CAS PubMed.
- H. Garcia-Ortega and J. M. Ribo, J. Porphyrins Phthalocyanines, 2000, 4, 564–568 CrossRef CAS.
- I. Batinic-Haberle, J. S. Rebouças, L. Benov and I. Spasojevic, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing Co., Singapore, 2011, vol. 11, pp. 291–393 Search PubMed.
- S. J. Griffiths, P. F. Heelis, A. K. Haylett and J. V. Moore, Cancer Lett., 1998, 125, 177–184 CrossRef CAS.
- Y. Inaba, K. Ogawa and Y. Kobuke, J. Porphyrins Phthalocyanines, 2007, 11, 406–417 CrossRef CAS.
- C. M. Nixon, K. Le Claire, F. Odobel, B. Bujoli and D. R. Talham, Chem. Mater., 1999, 11, 965–976 CrossRef CAS.
- K. E. Borbas, H. L. Kee, D. Holten and J. S. Lindsey, Org. Biomol. Chem., 2008, 6, 187–194 CAS.
- M. F. Grahn, A. Giger, A. McGuinness, M. L. de Jode, J. C. M. Stewart, H.-B. Ris, H. J. Altermatt and N. S. Williams, Lasers Med. Sci., 1999, 14, 40–46 CrossRef.
- R. Hornung, M. K. Fehr, H. Walt, P. Wyss, M. W. Berns and Y. Tadir, Photochem. Photobiol., 2000, 72, 696–700 CrossRef CAS.
- C.-L. Peng, M.-J. Shieh, M.-H. Tsai, C.-C. Chang and P.-S. Lai, Biomaterials, 2008, 29, 3599–3608 CrossRef CAS PubMed.
- W. J. Kim, M. S. Kang, H. K. Kim, Y. Kim, T. Chang, T. Ohulchanskyy, P. N. Prasad and K. S. Lee, J. Nanosci. Nanotechnol., 2009, 9, 7130–7135 CAS.
- G. Zheng, A. Graham, M. Shibata, J. R. Missert, A. R. Oseroff, T. J. Dougherty and R. K. Pandey, J. Org. Chem., 2001, 66, 8709–8716 CrossRef CAS PubMed.
- S. K. Pandey, X. Zheng, J. Morgan, J. R. Missert, T.-H. Liu, M. Shibata, D. A. Bellnier, A. R. Oseroff, B. W. Henderson, T. J. Dougherty and R. K. Pandey, Mol. Pharm., 2007, 4, 448–464 CrossRef CAS PubMed.
- K. E. Borbas, V. Chandrashaker, C. Muthiah, H. L. Kee, D. Holten and J. S. Lindsey, J. Org. Chem., 2008, 73, 3145–3158 CrossRef CAS PubMed.
- A. Z. Muresan and J. S. Lindsey, Tetrahedron, 2008, 64, 11440–11448 CrossRef CAS PubMed.
- M. Krayer, E. Yang, J. R. Diers, D. F. Bocian, D. Holten and J. S. Lindsey, New J. Chem., 2011, 35, 587–601 RSC.
- T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508–7510 CrossRef CAS.
- P. W. Shum and A. P. Kozikowski, Tetrahedron Lett., 1990, 31, 6785–6788 CrossRef CAS.
- B. L. Bray, P. H. Mathies, R. Naef, D. R. Solas, T. T. Tidwell, D. R. Artis and J. M. Muchowski, J. Org. Chem., 1990, 55, 6317–6328 CrossRef CAS.
- M. Krayer, T. Balasubramanian, C. Ruzié, M. Ptaszek, D. L. Cramer, M. Taniguchi and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2009, 13, 1098–1110 CrossRef CAS.
- O. Mass and J. S. Lindsey, J. Org. Chem., 2011, 76, 9478–9487 CrossRef CAS PubMed.
- H. C. Brown and B. Kanner, J. Am. Chem. Soc., 1953, 75, 3865 CrossRef CAS.
- T. Masquelin and D. Obrecht, Synthesis, 1995, 276–284 CrossRef CAS PubMed.
- L. Ghosez, C. Franc, F. Denonne, C. Cuisinier and R. Touillaux, Can. J. Chem., 2001, 79, 1827–1839 CrossRef CAS.
- T. Fukuda, T. Ohta, E.-I. Sudo and M. Iwao, Org. Lett., 2010, 12, 2734–2737 CrossRef CAS PubMed.
- T. Fukuda and M. Iwao, Heterocycles, 2012, 86, 1261–1273 CrossRef CAS PubMed.
- M. Kobayashi, M. Akiyama, H. Kano and H. Kise, in Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Springer, Dordrecht, The Netherlands, 2006, pp. 79–94 Search PubMed.
- The examination of absorption upon reciprocal change of concentration and pathlength is time-honored, dating 150 years ago to the development of Beer's law.a,b Such studies have been carried out since to identify deviations from Beer's law particularly due to analyte association phenomena such as aggregation, oligomerization, or complexation.c,d Early experiments relied on variable-pathlength cuvettes (e.g., Baly tubese,f) whereas a series of fixed-pathlength cuvettes provides more exacting examination across a larger concentration range. Representative chromophores examined in the latter manner include thionine,g,h xanthene,i uroporphyrin,j and synthetic trans-AB-porphyrins.57 (a) A. Beer, Ann. Phys. Chem., 1852, 86, 78–88 Search PubMed; (b) E. I. Stearns, The Practice of Absorption Spectrophotometry, Wiley-Interscience, New York, 1969, pp. 70–72 Search PubMed; (c) W. C. Holmes, Ind. Eng. Chem., 1924, 16, 35–40 CrossRef CAS; (d) W. E. Speas, Phys. Rev., 1928, 31, 569–578 CrossRef CAS; (e) E. C. C. Baly and C. H. Desch, Astrophys. J., 1906, 23, 110–127 CrossRef PubMed; (f) G. R. Harrison, R. C. Lord and J. R. Loofbourow, Practical Spectroscopy, Prentice-Hall, Inc., New York, 1948, pp. 371–373 Search PubMed; (g) L. F. Epstein, F. Karush and E. Rabinowitch, J. Opt. Soc. Am., 1941, 31, 77–84 CrossRef CAS; (h) E. Rabinowitch and L. F. Epstein, J. Am. Chem. Soc., 1941, 63, 69–78 CrossRef CAS; (i) J. E. Selwyn and J. I. Steinfeld, J. Phys. Chem., 1972, 76, 762–774 CrossRef CAS; (j) D. Mauzerall, Biochemistry, 1965, 4, 1801–1810 CrossRef CAS.
- J. W. Springer, P. S. Parkes-Loach, K. R. Reddy, M. Krayer, J. Jiao, G. M. Lee, D. M. Niedzwiedzki, M. A. Harris, C. Kirmaier, D. F. Bocian, J. S. Lindsey, D. Holten and P. A. Loach, J. Am. Chem. Soc., 2012, 134, 4589–4599 Search PubMed.
- M. A. Harris, P. S. Parkes-Loach, J. W. Springer, J. Jiang, E. C. Martin, P. Qian, J. Jiao, D. M. Niedzwiedzki, C. Kirmaier, J. D. Olsen, D. F. Bocian, D. Holten, C. N. Hunter, J. S. Lindsey and P. A. Loach, Chem. Sci., 2013, 4, 3924–3933 Search PubMed.
- H. Wolf-Klein, C. Kohl, K. Müllen and H. Paulsen, Angew. Chem., Int. Ed., 2002, 41, 3378–3380 Search PubMed.
- M. Escalante, A. Lenferink, Y. Zhao, N. Tas, J. Huskens, C. N. Hunter, V. Subramaniam and C. Otto, Nano Lett., 2010, 10, 1450–1457 Search PubMed.
- N. Srinivasan, C. A. Haney, J. S. Lindsey, W. Zhang and B. T. Chait, J. Porphyrins Phthalocyanines, 1999, 3, 283–291 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray data for compounds 8 and 13; additional procedures for the stepwise conversion of BC-9 to pro-BC-6; complete procedural and experimental data for the absorption versus concentration studies of BC-6–BC-8; HPLC data for the preparation of β(-14Cys)BC-16. CCDC 958396 and 958397. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob41791c |
| This journal is © The Royal Society of Chemistry 2014 |