Precursor-directed biosynthesis of micacocidin derivatives with activity against Mycoplasma pneumoniae†
Martin F.
Kreutzer
a,
Hirokazu
Kage
a,
Jennifer
Herrmann
b,
Julia
Pauly
c,
Ron
Hermenau
a,
Rolf
Müller
b,
Dirk
Hoffmeister
c and
Markus
Nett
*a
aLeibniz Institute for Natural Product Research and Infection Biology, Hans-Knöll-Institute, Adolf-Reichwein-Str. 23, D-07745 Jena, Germany. E-mail: markus.nett@hki-jena.de; Fax: +49 3641 5320811; Tel: +49 3641 5321297
bDepartment of Microbial Natural Products, Helmholtz-Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research (HZI) and Pharmaceutical Biotechnology, Saarland University, Campus C2 3, D-66123 Saarbrücken, Germany
cDepartment of Pharmaceutical Biology at the Hans-Knöll-Institute, Friedrich Schiller Universität Jena, D-07745 Jena, Germany
First published on 31st October 2013
Abstract
Micacocidin is a promising natural product for the treatment of Mycoplasma pneumoniae infections. In the biosynthesis of this antibiotic, a fatty acid-AMP ligase (FAAL) activates the starter unit hexanoic acid as acyl-adenylate and forwards it to an iteratively acting polyketide synthase. Biochemical analysis of the FAAL revealed an extended substrate tolerance, thereby opening the door for the modification of a micacocidin residue that is barely accessible via semisynthesis. A total of six new analogues were generated by precursor-directed biosynthesis in this study and profiled against M. pneumoniae.
Introduction
Exploiting the promiscuity of secondary metabolite biosynthesis enzymes has been a successful tool for customising and improving the bioactivity of natural products for many years.1 Precursor-directed biosynthesis (PDB) aims at the derivatisation of microbial compounds by feeding analogues of their natural building blocks, which are likely to be tolerated by the native biosynthetic enzymes, to the producing organism.2 The incorporation of cognate precursors into secondary metabolites expands their natural diversity and can even allow for further semisynthetic modifications, e.g. through the introduction of clickable groups.3 PDB was shown to be particularly effective in polyketide natural products when their biosynthetic starter units were replaced.4 Examples include many medicinally significant compounds, such as avermectin, erythromycin and rapamycin.5 While the virtue of PDB clearly lies in its simplicity and versatility, this method suffers from its inherent empiricism. At the beginning of a study, it is usually not clear whether the fed precursors can be enzymatically activated and eventually further processed by the downstream biosynthetic machinery. By determining the substrate tolerance of known gatekeeping enzymes, it should be possible, however, to identify the most promising precursors for an incorporation and hence to minimise the failure rate of PDB. To evaluate the feasibility of this strategy, we chose the natural product micacocidin as an example. Micacocidin is a thiazoline-containing metabolite from the plant pathogenic bacterium Ralstonia solanacearum that is generated on a hybrid polyketide/nonribosomal peptide enzyme assembly line.6 The molecule is of pharmaceutical interest due to its pronounced activity against Mycoplasma pneumoniae,7 which is a common causative agent of community-acquired pneumonia.8 Notwithstanding its close relatedness to the siderophore yersiniabactin,9 micacocidin was shown to involve several unique features in its biosynthesis.10 Of particular note is an iterative type I polyketide synthase (iPKS) module, which promotes the conversion of acyl carrier protein (ACP)-tethered hexanoic acid into the distinctive pentylphenol moiety of micacocidin. The activation of hexanoic acid and its loading require a FAAL that is located on the same biosynthesis enzyme (MicC) as the aforementioned iPKS (Fig. 1).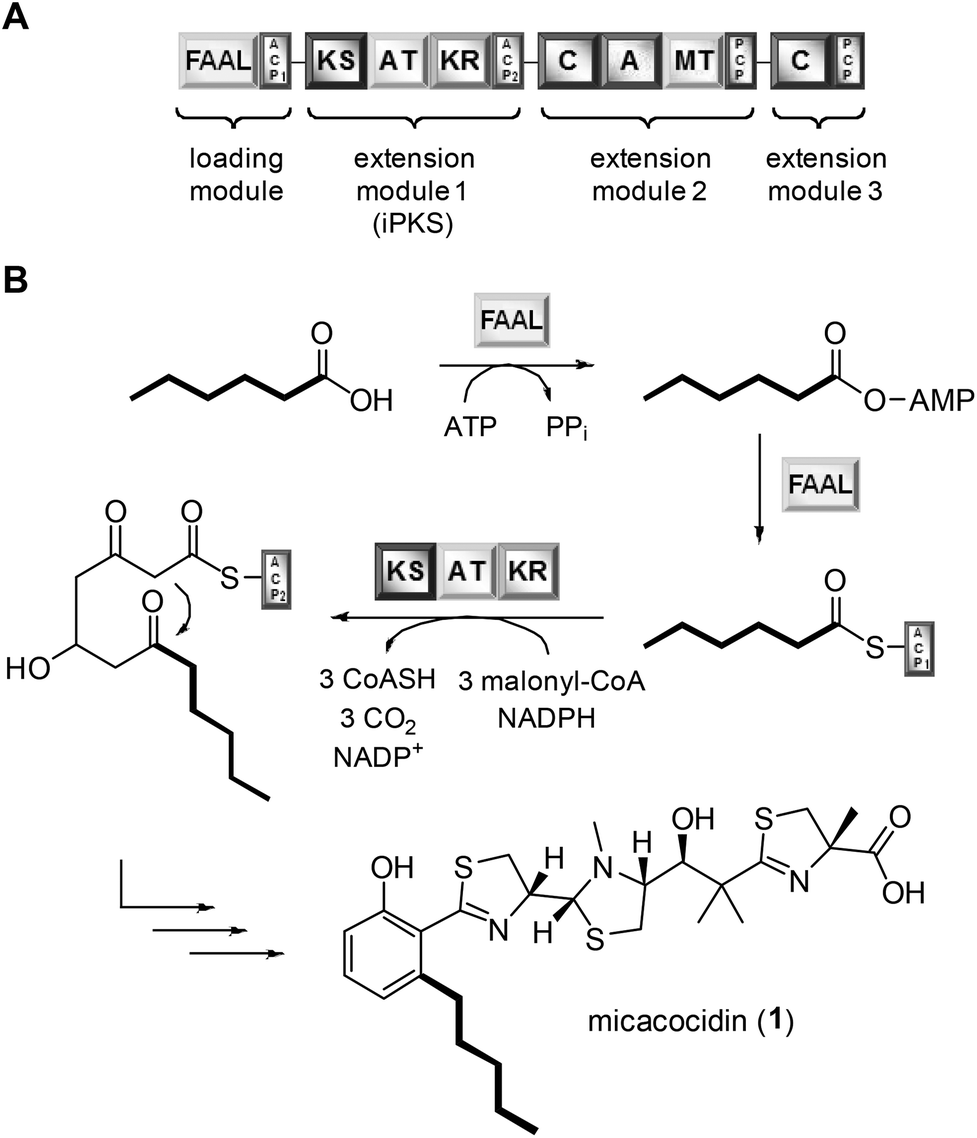 | ||
| Fig. 1 Domain architecture of the micacocidin biosynthesis initiating enzyme MicC (A). The thiotemplate-based assembly of 1 (B). Domain notation: ACP, acyl carrier protein; KS, ketosynthase; AT, acyl transferase; KR, ketoreductase; C, condensation; A, adenylation; MT, SAM-dependent methyltransferase; PCP, peptidyl carrier protein. | ||
A preliminary biochemical characterisation of the FAAL already indicated the possibility to recruit starter units other than hexanoic acid, albeit the detected conversion rates of the positively tested pentanoic acid and heptanoic acid were relatively low in comparison to the natural substrate.10 Motivated by these findings, we were poised to further explore this enzymatic plasticity. Here, we report the outcome of this study as well as the antibiotic activities of the micacocidin derivatives that were generated by PDB.
Results and discussion
Substrate specificity of the MicC-FAAL domain
To determine which compounds can enter micacocidin biosynthesis, the substrate tolerance of the gatekeeping MicC-FAAL domain was interrogated in the ATP-[32P]-pyrophosphate exchange assay.11 For this purpose, a gene fragment of micC encoding the FAAL and its adjacent ACP domain was overexpressed in E. coli KRX as an N-terminal hexahistidine fusion via the pET28a-derived vector pHiK007. The resulting protein was purified via metal-affinity and fast protein liquid chromatography. Subsequently, a total of 29 compounds were evaluated as potential substrates of the recombinant protein, along with the natural substrate hexanoic acid as a positive reference and water as a negative control (Fig. S1†). The tested compounds included a diverse set of saturated and unsaturated carboxylic acids, which consisted of up to ten carbon atoms. In terms of in vitro substrate activation, the substitution of the native starter unit was found to be quite permissive, provided that the cognate precursor included no amino or hydroxyl group (Fig. 2). On the other hand, halogen substitutions were surprisingly well tolerated. With the radiolabel exchange of hexanoic acid set to 100%, 5-bromo- and 5-chloropentanoic acid possessed almost identical turnover rates of 100% and 105%, respectively. Relevant activation was also observed for 6-heptenoic acid (73%), 4-pentenoic acid (65%), and 5-hexenoic acid (57%). Consistent with our previous study,10 the recombinant protein again showed only modest affinity toward pentanoic acid (35%) and heptanoic acid (31%). 2,2,3,3-Tetrafluorobutanedioic acid, hexanedioic acid and hex-2,4-dienoic acid displayed turnover rates comparable to the negative control, i.e., these compounds were not accepted as substrates. It is thus evident that the FAAL opposes any substrates with highly polar functional groups, whereas moderate changes in terms of carbon chain length and degree of unsaturation are tolerated.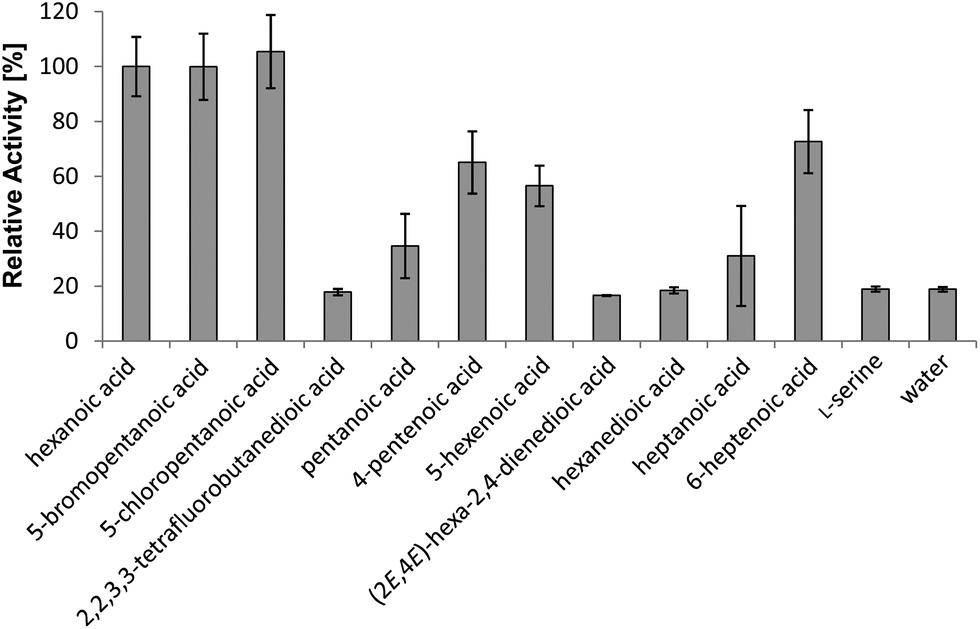 | ||
| Fig. 2 In vitro specificity of the MicC-FAAL domain towards natural and unnatural substrates, as determined by the ATP-[32P]-pyrophosphate exchange assay. Bars indicate the standard deviation. | ||
Precursor feeding and isolation of micacocidin derivatives
The seven carboxylic acids that had been positively tested in the ATP-[32P]-pyrophosphate exchange assay were selected for in vivo feeding studies in R. solanacearum GMI1000. The precursor surrogates were added aseptically to 50 mL of the cultivation medium prior to inoculation of the bacterium. Their incorporation was analysed in 3-day-old cultures by HPLC-MS after extraction of the fermentation broth with ethyl acetate. All metabolic profiles indicated the presence of micacocidin (1), confirming that the fed precursor concentrations (100 mg L−1) had not been toxic to the bacterium and that the unnatural building blocks did not interfere with micacocidin biosynthesis. In addition to the native natural product, six out of seven chromatograms included peaks, the masses of which corresponded to the anticipated derivatives (Fig. 3). As expected from its conversion rate in the ATP-[32P]-pyrophosphate exchange assay, 5-chloropentanoic acid was readily incorporated into micacocidin. Surprisingly, however, significant product yields were also achieved with the less preferred substrates pentanoic acid, 4-pentenoic acid, 5-hexenoic acid and 6-heptenoic acid. According to the HPLC-MS data, only the heptanoic acid-derived micacocidin analogue 5 was produced in minute amounts (<5% of 1), while a derivative incorporating 5-bromopentanoic acid could not be detected at all. The weak or missing in vivo turnover of the latter two carboxylic acids suggests steric constraints of downstream biosynthetic enzymes. Since the 6-heptenoic acid analogue 6 was produced in fairly good yields compared to 5, it is reasonable to assume that the chain length and spatial dimension of the substrate are limiting factors for a successful incorporation. In order to procure sufficient material for NMR-based structure verification and subsequent biological testing, the production of the derivatives 2–7 was scaled up in 3 to 15 L cultivations. All fermentations were run for 4 days, after which no increase in cell number could be observed. Following the extraction of the cultivation broths with ethyl acetate, micacocidin and its analogues were purified by HPLC. A detailed description of the chromatographic separation of all compounds is given in the Experimental section.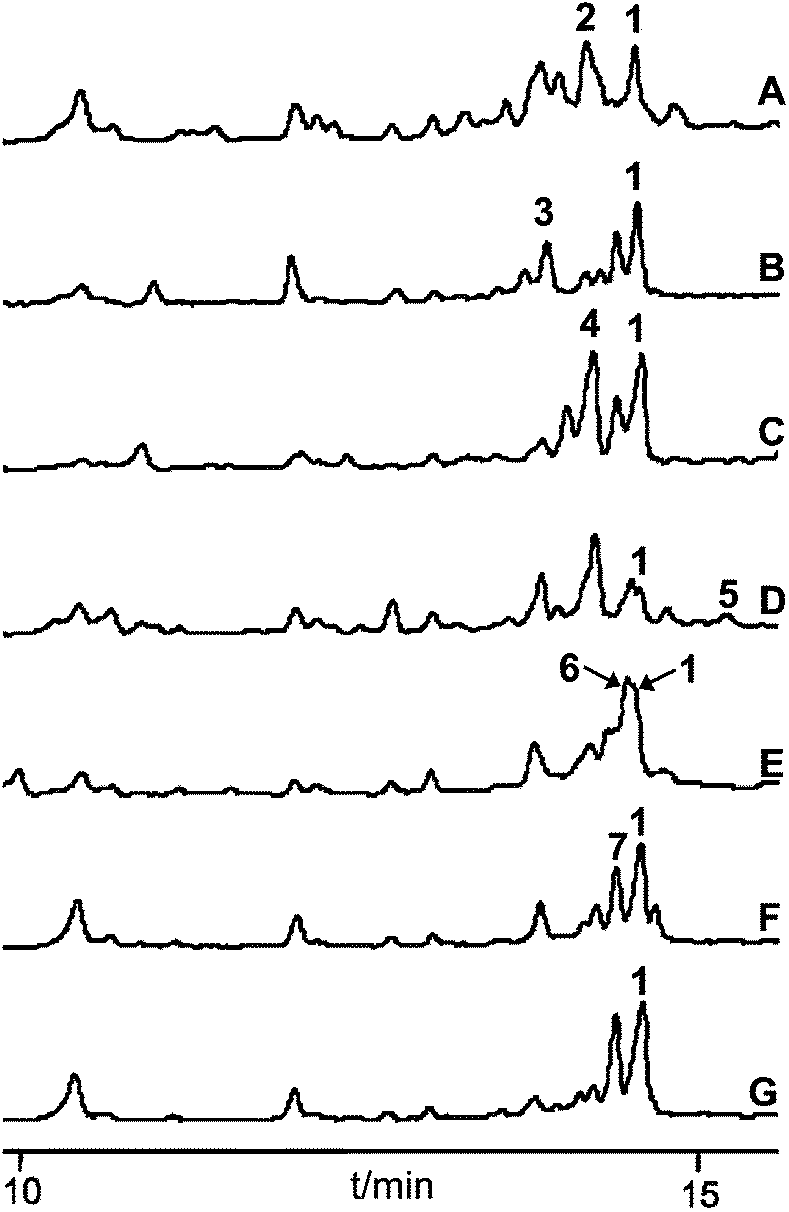 | ||
| Fig. 3 UV metabolic profiles of crude extracts from R. solanacearum GMI1000 cultures after feeding pentanoic acid (A), 4-pentenoic acid (B), 5-hexenoic acid (C), heptanoic acid (D), 6-heptenoic acid (E), 5-chloropentanoic acid (F) and 5-bromopentanoic acid (G). All chromatograms were recorded at 254 nm. The peaks of 1–7 were assigned by ESI-MS measurements. | ||
Structure elucidation of micacocidin derivatives
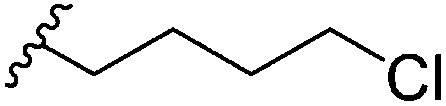
All generated analogues were fully characterised by NMR and high resolution (HR)-MS analyses after their conversion into gallium complexes.6 The 1H and 13C NMR spectra of 2 were almost identical with those of the parental molecule 1 except for the absence of resonances corresponding to a methylene group. This observation was consistent with MS data, which indicated a molecular formula of C26H37N3O4S3 for 2 (1: C27H39N3O4S3). An interpretation of 2D NMR spectra eventually confirmed the presence of a butyl side chain on the benzene ring, thus establishing the gross structure of 2. The HR-ESI-MS spectrum of 3 implied the loss of two hydrogen atoms in comparison to 2. Its 1H NMR spectrum included three characteristic signals in the olefinic range. Their homonuclear couplings were in accordance with the presence of an allyl group. COSY data revealed that the latter was part of a but-3-en-1-yl moiety that replaced the pentyl side chain of 1. Likewise, the 1H NMR spectrum of 4 showed three olefinic signals, but the molecular formula of this compound (C27H37N3O4S3) rather suggested a pent-4-en-1-yl substituent attached to the benzene ring. This assumption was confirmed by 2D NMR measurements. Compound 5, which was obtained after feeding heptanoic acid, possessed a molecular formula of C28H41N3O4S3. Although its 1H NMR spectrum did not allow a clear differentiation from 1, COSY and HMBC data unequivocally revealed the presence of a straight-chain hexyl unit on the benzene ring. The successful incorporation of 6-heptenoic acid, giving rise to 6, could be easily deduced from a comparison of the NMR and MS spectra with those of 3 and 4, respectively. Similar to these compounds, the alkyl side chain did not include a terminal methyl function, but exhibited an allyl group. The chlorinated micacocidin analogue 7 was identified by its characteristic MS isotopic pattern with a distinctively enlarged M + 2 peak as well as by NMR data. A COSY experiment established the connectivities in the alkyl side chain, in which the protons of the chlorine-bound methylene group were readily identified by their distinctive chemical shift at δH 3.62 ppm.Biological activity of micacocidin derivatives
The antimycoplasma activities of micacocidin and its derivatives were determined using a broth microdilution assay according to the recommendations of the Clinical and Laboratory Standards Subcommittee on Antimicrobial Susceptibility Testing of Human Mycoplasma.12 The respective minimal inhibitory concentrations (MICs) of the thiazoline antibiotics are listed in Table 1 in comparison to tetracycline, which served as a positive reference. All tested micacocidin gallium complexes showed promising activities against M. pneumoniae ATCC 29342, with MIC50 values ranging from 3 to 7 μM. The only exception was the pentenoic acid derivative 3 whose chelate complex exhibited significantly reduced activity (MIC50 26 μM). Since 3 was also the compound featuring the shortest alkyl side chain in the test series, it is possible that another truncation of this moiety may further increase the detrimental effects on the bioactivity. Future studies will address whether the small change from a butyl to a but-3-enyl side chain is really critical in this respect. On the other hand, the remaining modifications of the side chain, including its chlorination, were surprisingly well tolerated. 1 is actually less active than its analogues 2, 4, 6, and 7, but the observed differences in the MIC50 values do not appear to be significant. Noteworthy, the uncomplexed 1 showed a ∼4-fold increase in activity when compared to the corresponding Ga3+ complex. This observation might be explained by the fact that the non-chelated 1 is able to withdraw metal ions from its surrounding,6 thereby putting M. pneumoniae under the pressure of coping not only with a specific antibiotic effect, but also with a limitation of possibly essential micronutrients.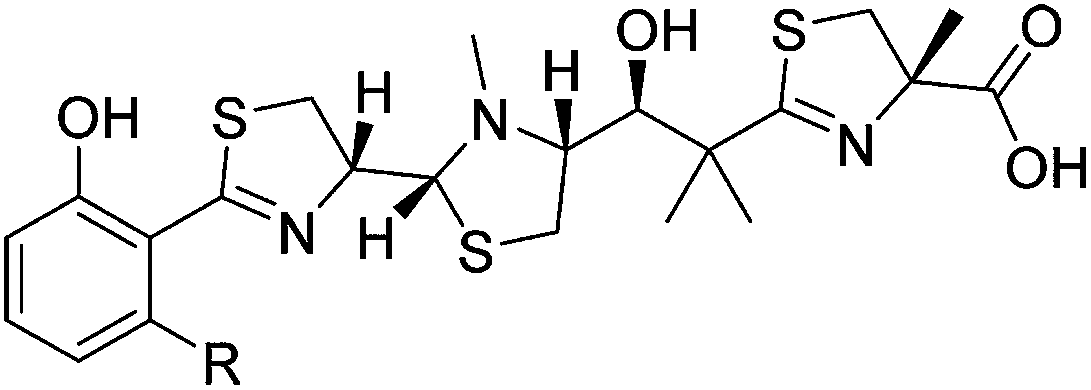


Conclusions
In this study, we have demonstrated that the micacocidin biosynthetic pathway provides sufficient enzymatic flexibility to allow the generation of unnatural analogues by PDB. Instead of carrying out a large number of empiric feeding experiments, we pursued a more targeted strategy to obtain new micacocidin derivatives. By analysing the substrate tolerance of the enzyme, which controls the entry to the biosynthetic assembly line, we were able to identify those cognate precursors that possessed a good chance for incorporation. The advantage of this approach was obvious. Six out of seven conducted feeding experiments led to the expected products, all of which were isolated and subjected to biological testing. While the described procedure is more time-consuming than the classical PDB, it allows a reduction of the number of in vivo studies by focusing on a few promising supplementation experiments. In the present case, this was highly desirable, as the micacocidin producing bacterium R. solanacearum is itself a pathogen.PDB was an efficient way to alter a moiety in micacocidin, which is not amenable to semisynthesis. Although the novel derivatives described in this paper could also be accessed by total synthesis, such an approach appears less attractive, as it would require the repeated use of identical linear transformations from the synthetic branching point at which the respective phenyl side chain is introduced. The work presented here, by contrast, allows for the convenient generation of micacocidin derivatives by feeding easily available short-chain carboxylic acids. For the first time, it was hence possible to determine the impact of modifications in the benzene side chain on the activity against M. pneumoniae. Previous structure–activity relationship (SAR) studies focused on the two hydroxy groups and the carboxylate function in 1. As a result of these investigations, it was proposed that the antimycoplasma activity of 1 largely depends on its ability to adopt a folded conformation.13 The alkyl side chain variations introduced in the present study are not likely to induce significant changes in the spatial folding. In accordance with this assumption, almost all tested derivatives exhibited MICs in the same range as 1. As evidenced by the decreased activity of 3, however, a minimum alkyl chain length might be important for the interaction with the biological target. The mode of action of micacocidin is currently under investigation in our laboratories.
Experimental
Biochemical methods
The ATP-[32P]-pyrophosphate exchange assay was carried out as described previously.11General experimental procedures
Production of micacocidin analogues was monitored on an HPLC-DAD system (Agilent, 1100 series) equipped with a C8 column (Zorbax Eclipse XDB C8, 150 × 4.6 mm, 5 μm; Agilent) coupled to an MSD trap operating in alternating ionisation mode. Peak separation was achieved using a methanol (B) and water + 0.1% formic acid (A) gradient elution running from 10% B to 90% B within 15 min with a flow rate of 1 mL min−1. HR mass analyses were carried out on an Exactive Mass Spectrometer (Thermo-Scientific). IR spectra were recorded using a benchtop FT/IR 4100 spectrometer (JASCO). One- and two-dimensional NMR measurements were conducted at 300 K on Bruker Avance III 500 and 600 MHz spectrometers, respectively. Samples were dissolved in methanol-d4 as an internal standard. The solvent signal was referenced to δH 3.31 ppm and δC 49.0 ppm, respectively.Production and isolation of micacocidin analogues
Ralstonia solanacearum GMI1000 was cultivated in iron-limited, modified 1/4 × M63 minimal medium supplemented with 0.2% glucose and 0.02% sodium acetate, as described earlier.6 For the production of micacocidin analogues, 3 L cultures were shaken in Erlenmeyer flasks at 30 °C and 120 rpm for 4 days. In the case of 5, the fermentation had to be repeated (5 × 3 L) in order to isolate sufficient material for a spectroscopic characterisation. At the end of cultivation, all R. solanacearum cultures were extracted three times with equal amounts of ethyl acetate. The organic layers were pooled and dried under vacuum. After resuspension in 1 mL of methanol, the extracts were filtered through a 0.22 μm pore size syringe filter to remove insoluble remnants. 10 mg of Ga(NO3)3 hydrate was added to the filtrate and left for complexation overnight at room temperature. Subsequently, the samples were purified by semipreparative HPLC using a Shimadzu UFLC system. Initial purification was achieved using a Nucleodur HTec column (250 × 10 mm, 5 μm; Macherey Nagel) or on an Eclipse-XDB C8 column (250 × 9.4 mm, 5 μm; Agilent) in the case of 5, using a linear gradient elution with water + 0.1% trifluoroacetic acid (A) and methanol (B). The gradient was conducted from 10% B to 90% B within 20 min at a flow rate of 2.5 mL min−1. While 2, 3 and 5 were already obtained in adequate purity, the fractions containing 4, 6 and 7 were subjected to a second HPLC step. 4 was separated on a Nucleodur Sphinx column (250 × 10 mm, 5 μm; Macherey Nagel) with acetonitrile as solvent B, using otherwise the same conditions as described for the first HPLC step. 6 was purified using a Nucleodur Pyramid column (250 × 10 mm, 5 μm; Macherey Nagel) and an isocratic elution of 45% methanol in water + 0.1% trifluoroacetic acid at a flow rate of 2.5 mL min−1. 7 was cleaned up using the same column with the gradient conditions described in the first HPLC step, but with acetonitrile as solvent B. All chromatograms were monitored with 254 nm detection wavelength. The fractionated compounds were dried under vacuum, redissolved in 1 mL methanol, and 5 μL were subjected to HR-MS analyses.Spectroscopic data of micacocidin analogues
The clean Ga-micacocidin analogues were freeze-dried to remove residual water and prepared for NMR analyses by dissolving in 120 μL of methanol-d4 using 2.5 mm NMR tubes.Broth microdilution assay
The assay was carried out according to an established protocol,12 except that M. pneumoniae ATCC 29342 (syn. M. pneumoniae DSM 23979) was cultured in PPLO (pleuropneumonia-like organisms) broth base (BBL 211458), supplemented with 30% (v/v) PPLO enrichment (Difco 283610) and 0.5% glucose. In order to monitor the growth of the bacterium, the medium also contained the pH indicator phenol red. For the determination of MICs, mycoplasmas were cultured in 96-well microtitre plates in the presence of twofold serial dilutions of the different test compounds (128 to 0.06 μg mL−1). The microdilution plates were incubated at 37 °C and 5% CO2 for 4 days, after which phenol red in the growth control well changed its color due to medium acidification. All experiments were run in duplicate. MIC50 values were calculated from the mean phenol red absorption at 545 nm by sigmoidal fitting of the data.Acknowledgements
Financial support was provided by the Leibniz Association and the Jena School for Microbial Communication (JSMC). We thank Andrea Perner and Heike Heinecke (Hans-Knöll-Institute Jena, Department of Biomolecular Chemistry) for recording HR-ESI-MS and IR spectra, respectively.Notes and references
-
(a) R. J. M. Goss, S. Shankar and A. A. Fayad, Nat. Prod. Rep., 2012, 29, 870 RSC
; (b) A. Kirschning, F. Taft and T. Knobloch, Org. Biomol. Chem., 2007, 5, 3245 RSC
-
(a) H. Steinmetz, W. Zander, M. A. M. Shushni, R. Jansen, K. Gerth, R. Dehn, G. Dräger, A. Kirschning and R. Müller, ChemBioChem, 2012, 13, 1813 CrossRef CAS PubMed
-
(a) A. Kirschning and F. Hahn, Angew. Chem., Int. Ed., 2012, 51, 4012 CrossRef CAS PubMed
- B. S. Moore and C. Hertweck, Nat. Prod. Rep., 2002, 19, 70 RSC
-
(a) T. S. Chen, E. S. Inamine, O. D. Hensens, D. Zink and D. A. Ostlind, Arch. Biochem. Biophys., 1989, 269, 544 CrossRef CAS
- M. F. Kreutzer, H. Kage, P. Gebhardt, B. Wackler, H. P. Saluz, D. Hoffmeister and M. Nett, Appl. Environ. Microbiol., 2011, 77, 6117 CrossRef CAS PubMed
- S. Kobayashi, Y. Ikenishi, Y. Takinami, M. Takema, W.-Y. Sun, A. Ino and Y. Hayase, J. Antibiot., 2000, 53, 532 CrossRef CAS
- K. B. Waites and D. F. Talkington, Clin. Microbiol. Rev., 2004, 17, 697 CrossRef CAS PubMed
- D. A. Miller, L. S. Luo, N. Hillson, T. A. Keating and C. T. Walsh, Chem. Biol., 2002, 9, 333 CrossRef CAS
- H. Kage, M. Kreutzer, B. Wackler, D. Hoffmeister and M. Nett, Chem. Biol., 2013, 20, 764 CrossRef CAS PubMed
- B. Wackler, P. Schneider, J. M. Jacobs, J. Pauly, C. Allen, M. Nett and D. Hoffmeister, Chem. Biol., 2011, 18, 354 CrossRef CAS PubMed
- K. B. Waites, L. B. Duffy, C. M. Bébéar, A. Matlow, D. F. Talkington, G. E. Kenny, P. A. Totten, D. J. Bade, X. Zheng, M. K. Davidson, V. D. Shortridge, J. L. Watts and S. D. Brown, J. Clin. Microbiol., 2012, 50, 3542 CrossRef CAS PubMed
- A. Ino, S. Kobayashi, K. Ueda, S. Hidaka and Y. Hayase, J. Antibiot., 2001, 54, 753 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ob41839a |
| This journal is © The Royal Society of Chemistry 2014 |