 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modular synthesis of cyclic cis- and trans-1,2-diamine derivatives†
Anna K.
Weber
a,
Josef
Schachtner
b,
Robert
Fichtler
a,
Timo M.
Leermann
a,
Jörg M.
Neudörfl
a and
Axel
Jacobi von Wangelin
*ab
aDepartment of Chemistry, University of Cologne, Germany
bInstitute of Organic Chemistry, University of Regensburg, Germany. E-mail: axel.jacobi@ur.de; Fax: +49 (0)941 943 4617; Tel: +49 (0)941 943 4802
First published on 9th June 2014
Abstract
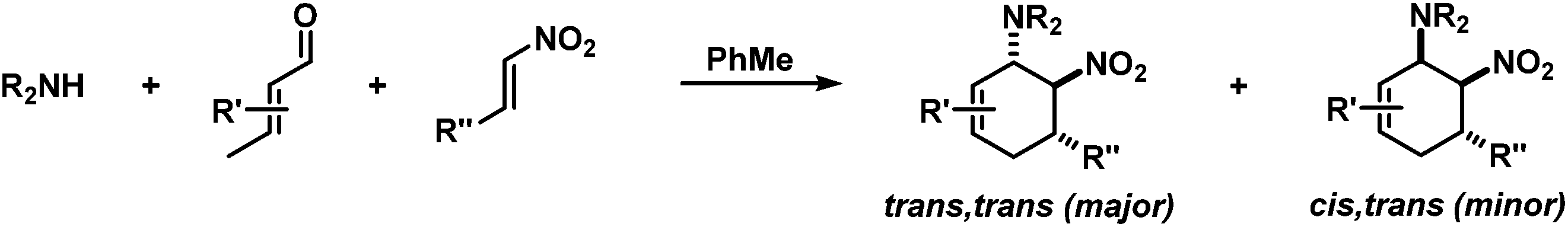
Structurally diverse carbocycles with two vicinal nitrogen-substituents were prepared in expedient three-component reactions from simple amines, aldehydes, and nitroalkenes. trans,trans-6-Nitrocyclohex-2-enyl amines were obtained in a one-pot domino reaction involving condensation, tautomerisation, conjugate addition, and nitro-Mannich cyclisation. Upon employment of less nucleophilic carboxamides, a concerted Diels–Alder cycloaddition mechanism operated to give the corresponding cis,trans-nitrocyclohexenyl amides. Both types of substituted carbocycles offer ample opportunities for chemical manipulations at the core and periphery. Ring oxidation with MnO2 affords substituted nitroarenes. Reduction with Zn/HCl provides access to various trans- and cis-diaminocyclohexenes, respectively, in a straight-forward manner. With enantiopure secondary amines, a two-step synthesis of chiral nitrocyclohexadienes was developed (82–94% ee).
Introduction
Functionalised cyclohexanes are one of the most prevalent molecular architectures in nature and synthesis. Today, a plethora of synthetic procedures is available to provide access to various saturation states and peripheral substitution patterns. The majority of syntheses involve functionalisation of already cyclic precursors whereas de-novo-syntheses of functionalised cyclohexanes from two or more acyclic starting materials exhibit much higher modularity and provide access to a large chemical space.1 The most prominent examples of such intermolecular cyclisation reactions include cyclo-additions2 and many variants of sequential condensation-addition reactions with carbonyl compounds.3 Heteroatom(X)-substituted cyclohexanes are arguably the most interesting structures for active pharmaceutical ingredients and fine chemical building blocks due to the distinct stereoelectronic properties and chemical reactivities of polar C–X bonds and the available lone pairs at X.4 Aminodienes were widely used in method developments, syntheses of pharmaceutically active molecules, natural products and materials (Scheme 1).5 | ||
| Scheme 1 1-Amino-1,3-butadienes in organic synthesis. | ||
Aminodienes can be easily prepared from condensations of unsaturated carbonyl compounds and amines and exhibit high reactivity in normal-electron demand cycloadditions and nucleophilic additions due to their high-lying HOMO (highest occupied molecular orbital).6 Further N-based substituents can be incorporated into the product structure by employment of α-electrophilic nitrogen compounds. Nitroolefins are a readily available class of vinylogous nitrogenous electrophiles with widespread applications in Michael-type additions and cycloadditions.7 We envisioned a domino process involving initial condensation of unsaturated aldehydes with amines followed by selective cyclisation of the resultant 1-amino-1,3-dienes with nitroolefins.8 Such strategy would provide an expedient access to highly functionalised cyclohexenes containing two chemically orthogonal N-based substituents in vicinal positions (NR2, NO2). 1,2-Diaminocyclohexane motifs constitute important building blocks of pharmacologically active molecules, fine chemicals and catalysts (Scheme 2).9
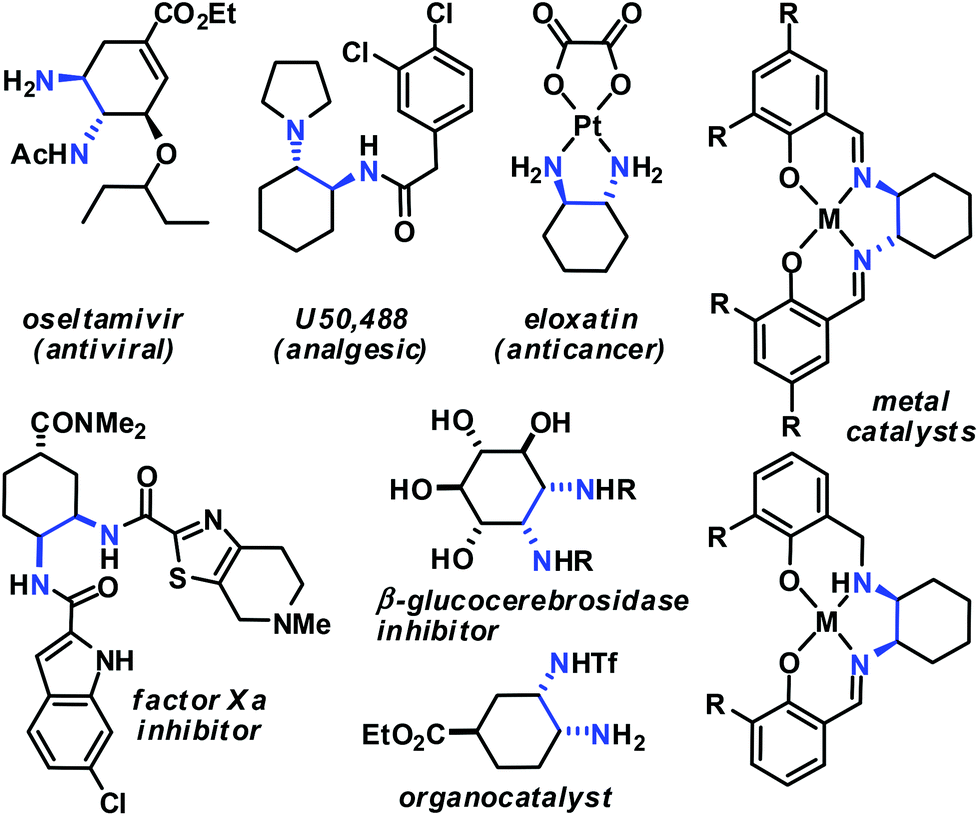 | ||
| Scheme 2 Applications of trans- (top) and cis-1,2-diaminocyclohexanes. | ||
The electronic nature of the intermediate aminodienes can be easily tuned by the introduction of various N-substituents. It has been demonstrated that highly nucleophilic aminodienes bearing electron-donating (EDG) alkyl substituents engage in rapid Michael-type additions to electrophiles.10 However, a change of mechanism can be effected by electron-withdrawing (EWG) N-substituents. 1-N-Acyl-amino-1,3-butadienes undergo concerted [4 + 2]-cycloadditions with electron-deficient dienophiles.6 Here, we report the viability of controlling the reaction mechanism and stereoselectivity by the employment of secondary amines (R1 = EDG) or less nucleophilic acylamines (R1 = EWG) in three-component cyclisations with α,β-unsaturated aldehydes and nitroalkenes to give trans- or cis-1,2-diaminocyclohexenes (Scheme 3).
 | ||
| Scheme 3 The stereoselectivity switch: amines vs. N-acyl amines. | ||
Results and discussion
Synthesis of trans-nitrocyclohexenyl amines
As an extension of earlier work on functionalised aminocyclo-hexenes,8 we optimised reaction conditions for the three-component cyclisation of secondary amines, α,β-unsaturated aldehydes and nitroalkenes. We chose pyrrolidine, crotonaldehyde and β-nitrostyrene as model substrates (Table 1). High selectivities were observed in toluene as solvent and upon slow addition of a slight excess of the aldehyde.10|
|
||
|---|---|---|
| Entry | Conditions | Yieldb [%] |
| a Optimised conditions: Pyrrolidine (2 mmol), β-nitrostyrene (2 mmol), PhMe (3 mL), slow addition of crotonaldehyde (2.4 mmol in 1 mL PhMe) over 2 h, 30 °C, 20 h. b GC yields vs. internal hexadecane. c 60 °C. d Diastereomeric ratio (dr). e 83% isolated yield. | ||
| 1 | CH3CN | 27 (40)c |
| 2 | CH2Cl2 | 58 |
| 3 | DMF | 72 |
| 4 | PhMe | 76 |
| 5 | PhMe, 2 equiv. aldehyde | 68 |
| 6 | PhMe, 2 equiv. amine | 29 |
| 7 | PhMe, slow addition (∼2 h) of 1.2 equiv. aldehyde | 90 (19/1)d,e |
| 8 | As entry 8, but with pyrrolidine/benzoic acid (1/1) | 64 (18/1)d |

Rapid aldehyde addition resulted in oligomer formation. The three-component cycloadduct 1-(6-nitro-5-phenylcyclohex-2-enyl)pyrrolidine (1) was obtained with high thermodynamic stereocontrol (trans,trans/cis,trans = 19/1). The minor diastereomer exhibited a cis-relation of the amino and nitro groups. The preferential formation of the all-trans stereoisomer and the observation of an identical outcome from reactions with (Z)-β-nitrostyrene suggest the operation of a stepwise mechanism (Scheme 4).10 Initial condensation between the amine and aldehyde gives rise to the formation of an equilibrating mixture of imine, enamine and aminal derivatives, of which the aminodiene undergoes reversible conjugate addition to the nitroolefin.11 Subsequent nitro-Mannich ring-closure affords the thermodynamic product containing three vicinal equatorial substituents.12
 | ||
| Scheme 4 Nitro-Mannich mechanism with secondary amines. | ||
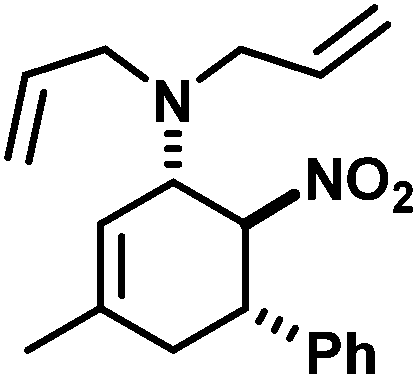
We extended the reaction conditions to various other amines, aldehydes and nitroolefins (Table 2).13 The trans,trans-isomers were preferentially formed in diastereomeric ratios of >10/1.10 Diastereomeric ratios (dr) were assigned based on high-resolution 1H-NMR spectra. The trans-configurations of the vicinal amino/nitro and nitro/aryl groups, respectively, at the cyclohexene result in dihedral angles of 170–180° and 3JHH coupling constants of 10–12 Hz. Employment of morpholine gave lower dr values (13, 14). Products with substituents in the 2-position formed rather slowly (15, 18, 19), possibly due to steric congestion with the amine substituent in the planar aminodiene species. Similarly, crotonaldehydes bearing γ-substituents reacted very slow due to steric inhibition of the Michael-type attack onto β-nitrostyrenes (14, 19). The latter trend allowed selective conversion of citral to the 3-alkyl cycloadduct via the terminal aminodiene isomer (20, kinetic control). Cyclisation with diallylamine proceeded highly effective to give 21 which allows facile access to the free amine. Highly electrophilic methyl 2-methyl-4-oxobut-2-enoate dimerised upon rapid aldehyde addition (Scheme 5).14
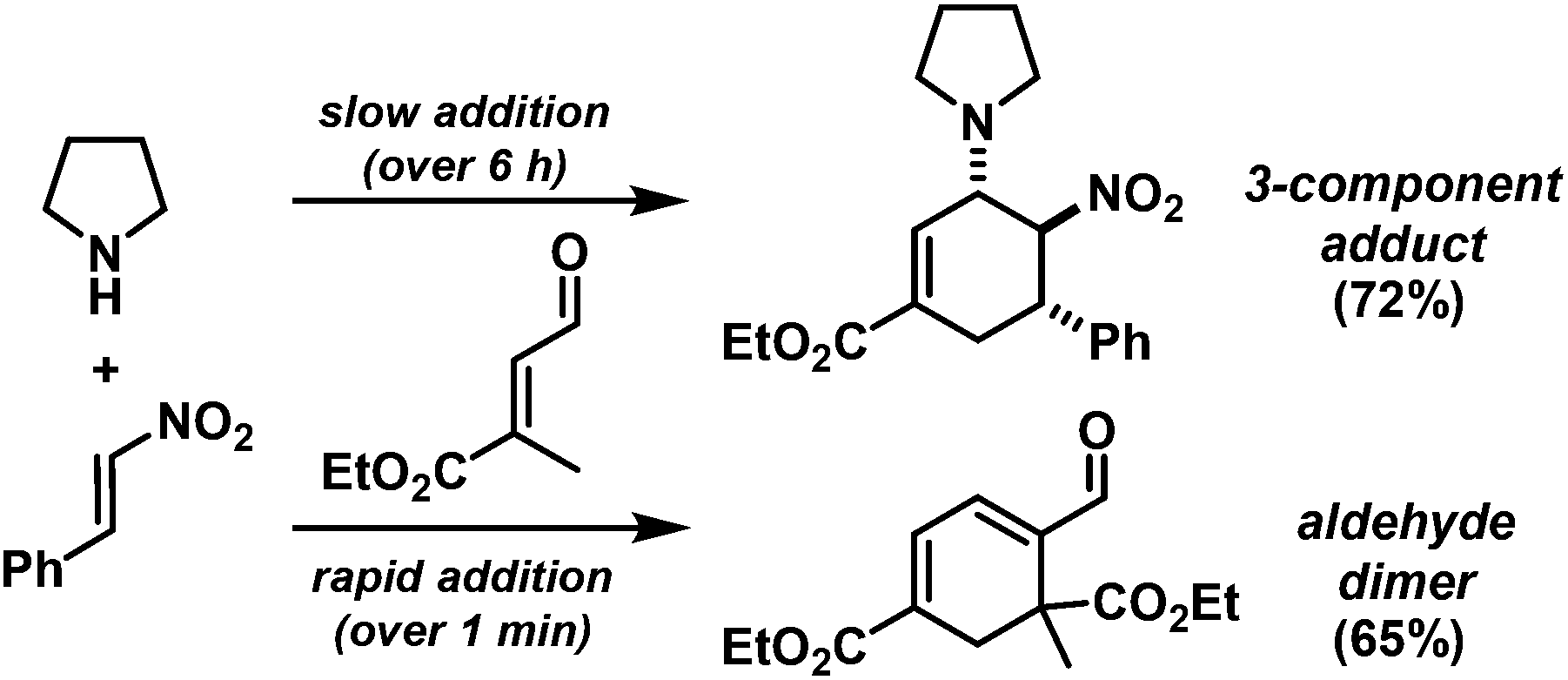 | ||
| Scheme 5 Rate of aldehyde addition governs selectivity. | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Major isomer | Substituents | dr | Yieldb [%] | Major isomer | Substituents | dr | Yieldb [%] | ||
| a Amine (4 mmol), aldehyde (1.2 equiv.) and nitroalkene in PhMe were stirred for 20 h at 30 °C. b Isolated yields of isomer mixtures. c After treatment with SiO2 or wet CDCl3. d Major diastereomer shown, other isomers not assigned. e From citral. f Ar = 3,5-bis(trifluoromethyl)phenyl. | |||||||||

|
R′′ = Ph | 1 | 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
83 |

|
20 | 6:1 |
82e | |
| 2,3-(MeO)2–C6H3 | 2 | 15:1 |
69 | ||||||
| 3,4-(OCH2O)–C6H3 | 3 | 34:1 |
59 | ||||||
| 2,6-Cl2–C6H3 | 4 | 30:1c |
66 | ||||||

|
R′′ = Ph | 5 | 28:1 |
94 |
|
21 | 35:1 |
93 | |
| 2,3-(MeO)2–C6H3 | 6 | 14:1 |
62 | ||||||
| 3,4-(OCH2O)–C6H3 | 7 | 34:1 |
78 | ||||||
| 2-F–C6H4 | 8 | 18:1 |
91 | ||||||
| 2,4-Cl2–C6H3 | 9 | 15:1 |
90 | ||||||
| 2-NO2–C6H4 | 10 | 5:1 |
90 | ||||||
| 2,6-Cl2–C6H3 | 11 | 10:1c |
91 | ||||||
| 2-Furyl | 12 | 12:1 |
90 | ||||||

|
R′ = 3-Me | 13 | 13:1 |
70 |
|
22 | 10:1 |
46 | |
| R′ = 4-Me | 14 | 7:2:1d |
53 | ||||||

|
15 | 2:1 |
38 |

|
R′′ = Ph | 23 | 28:1:1 |
86 | |
| 2,4-Cl2–C6H3 | 24 | 19:1 |
82 | ||||||
| 3,4-(OCH2O)–C6H3 | 25 | 27:1 |
75 | ||||||
| 2-Furyl | 26 | 11:1 |
69 | ||||||
| 2-NO2–C6H4 | 27 | 2:1 |
74 | ||||||
| 2-F–C6H4 | 28 | 11:1 |
96 | ||||||

|
16 | 6:1 |
72 |

|
29 | 15:1 |
68 | ||

|
17 | 50:1 |
90 |

|
R′′ = Ph | 30 | 40:1 |
82 | |
| 2,4-Cl2–C6H3 | 31 | 6:1 |
82 | ||||||

|
R′ = H | 18 | 3:1d |
16 |

|
R′′ = Ph | 32 | 14:9:1 |
63 |
| Me | 19 | 28:6:1d |
39 | 2-F–C6H4 | 33 | 1.5:1 |
69 | ||
| 2,4-Cl2–C6H3 | 34 | 4:3:1 |
62 | ||||||
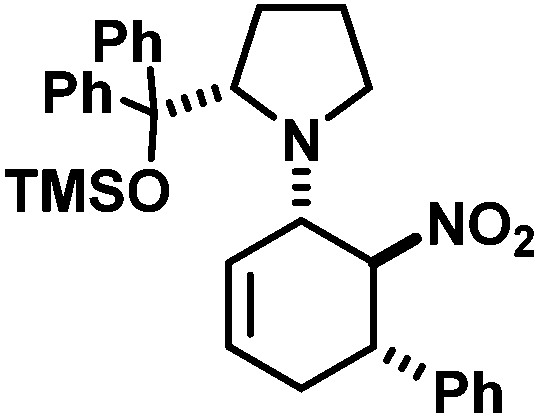
The three contiguous stereocenters evolve under thermo-dynamic control. We have employed prolinol derivatives (22–34) among which bulky diarylprolinols exhibited the most effective facial discrimination of the intermediate iminium ion. Consistently, the trans,trans-isomers were formed preferentially. Crystal structure analysis confirmed the absolute and relative configuration of 24 (Fig. 1). However, bulky 1-nitro-2-(2′,6′-dichlorophenyl)ethylene preferentially gave cis,trans-35 (cis,trans:trans,trans 5:1). Slow epimerisation at C-1 occurred during work-up (SiO2) or in the presence of acid (wet CDCl3, HCl) to give the thermodynamic trans,trans-35 (cis,trans:trans,trans ∼1:1) possibly via elimination of the axial amine group (Scheme 6, Fig. 2).13
 | ||
| Fig. 1 Crystal structure of trans,trans-24. | ||
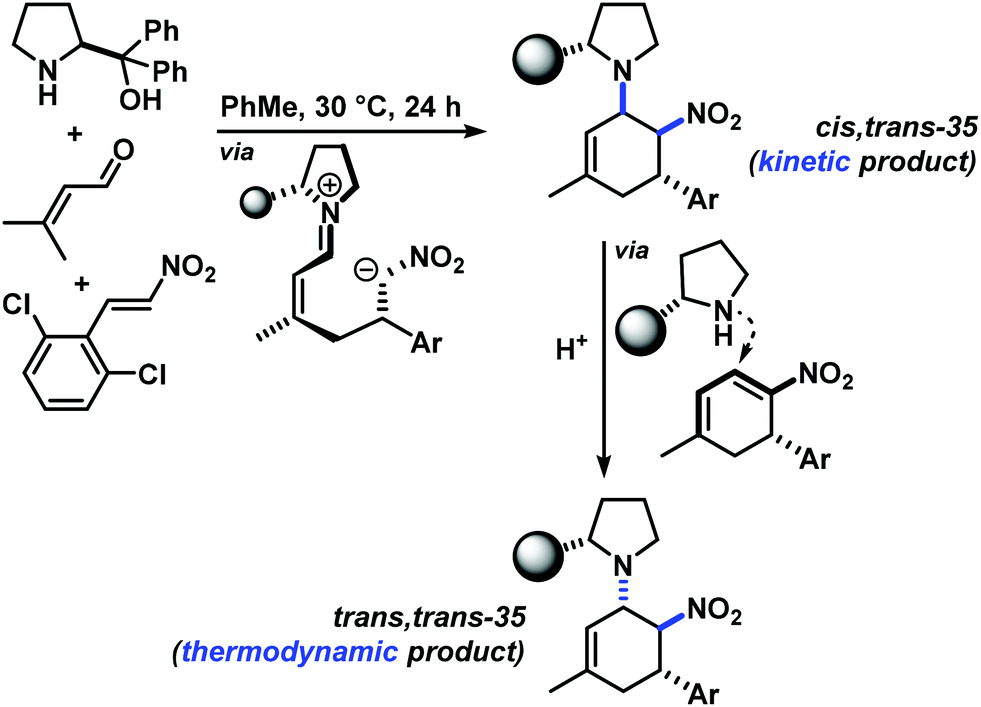 | ||
| Scheme 6 Kinetic cis,trans-35 and thermodynamic trans,trans-35. | ||
 | ||
| Fig. 2 Crystal structure of the kinetic product cis,trans-35. | ||
Reactions of trans-nitrocyclohexenyl amines
We have studied various structural manipulations at the core and periphery of the synthesised cycloadducts. Eliminations, oxidations, and reductions were realised to give access to diene, arene and diamine derivatives (Scheme 7).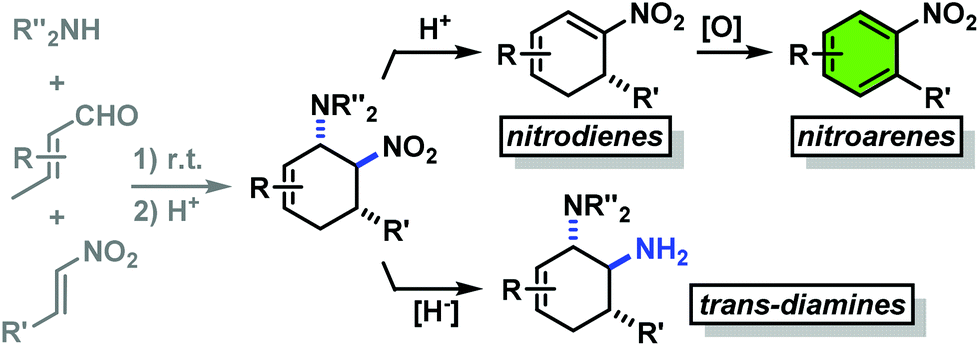 | ||
| Scheme 7 Core and peripheral reactions to dienes, arenes and diamines. | ||
Oxidation of nitrocyclohexenyl amines with manganese dioxide (MnO2) resulted in unselective decomposition involving dehydrogenation and amine or nitrite elimination.15 Similarly, various Nef conditions (base (NaH/n-BuLi), then acid (HCl); TiCl3; KMnO4)16 afforded complex mixtures. In no case was the desired ketone detected (GC-MS, IR). The solid cycloadducts appeared to be stable toward air and water; their solutions in dichloromethane or toluene withstood exposure to weak acids and bases. The equatorial amino group inhibits base-mediated elimination (E1cb pathway). However, benzoic acid effected slow elimination of pyrrolidine to nitrocyclohexadienes (Scheme 8). Stronger acids (HCl, TFA) allowed elimination of prolinols to give the nitrocyclohexadienes 36, 37, 39 and 41 in good enantiomeric purity (82–94% ee, Table 3).13 Amine/acid-co-catalyzed reactions of aldehydes and nitroalkenes gave low selectivities.13,17
 | ||
| Scheme 8 Acid-mediated elimination of pyrrolidine. | ||



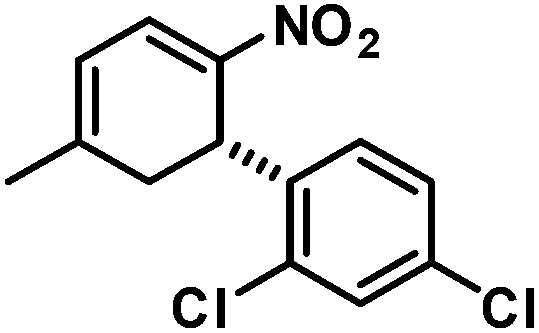
The nitrocyclohexadienes were prone to oxidation under aerobic conditions but could be stored under nitrogen at 0 °C for days. However, selective dehydrogenation would render a straight-forward access to substituted nitroarenes. Several methods for the aromatisation of carbocycles were reported (Pt, Pd, Ni catalysts, elemental sulfur or selenium, quinones, oxygen, MnO2, SeO2etc.).18 We obtained good yields of 2-aryl nitrobenzenes with 5 equiv. MnO2 at 80 °C (Scheme 9). This strategy allows the three-step synthesis of 2-nitrobiaryls.
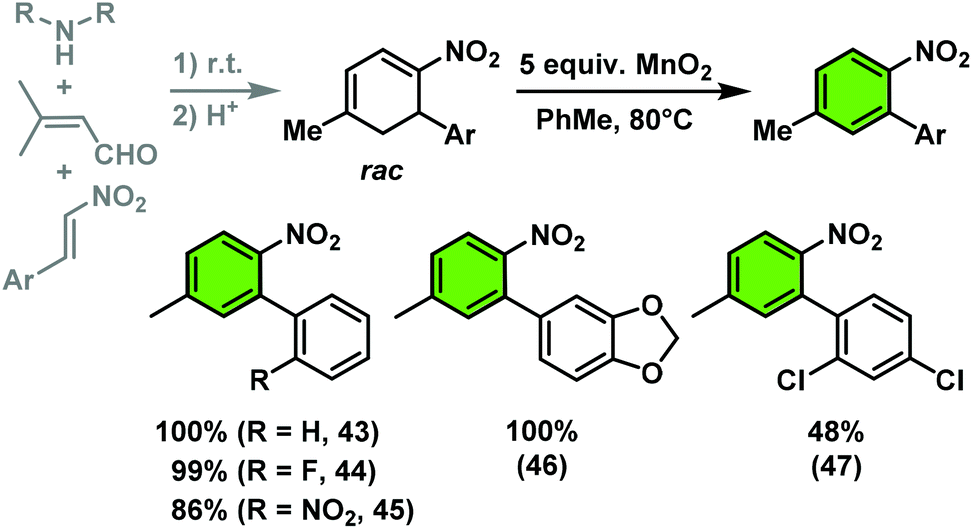 | ||
| Scheme 9 Three-step synthesis of 2-nitrobiaryls. | ||
The reduction of a nitro function to a primary amine is an especially useful manipulation.19 Application of such selective manipulation of one substituent of our three-component cycloadducts renders an expedient access to carbocyclic 1,2-trans-diamines (Scheme 10). The overall two-step sequence thus involves stereoselective three-component cyclisation with nitroolefins followed by reduction of the nitro substituents in with Zn/HCl in ethanol. The nitrocyclohexenyl amines 1, 11 and 21 were cleanly converted to the corresponding diamine derivatives 48–50 in excellent yields with complete retention of stereochemistry.13
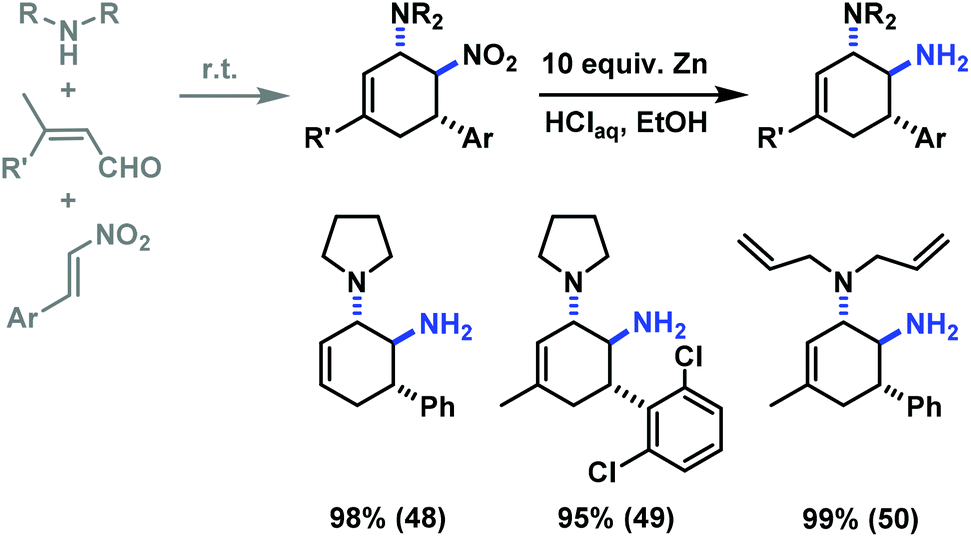 | ||
| Scheme 10 Two-step synthesis of trans-1,2-diaminocyclohexenes. | ||
The stereoselectivity switch: synthesis of cis-nitrocyclohexenyl amides
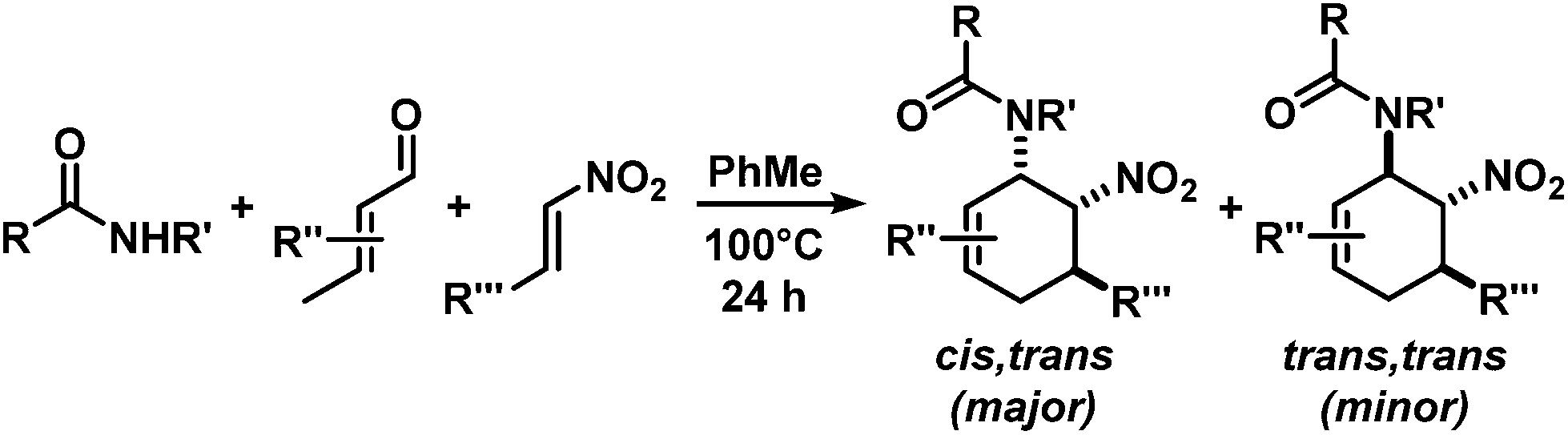
The nature of the N-substituents controls all three elemental steps in the Michael/Mannich-type mechanism with amines. The nucleophilicity of the amine directly affects the rate of condensation with the α,β-unsaturated aldehyde and the nucleophilic reactivity of the aminodiene intermediate at the terminal δ-position (Scheme 4). Stereoelectronics also govern the formation of the 1-amino-2-nitroethylene moiety via a formal nitro-Mannich reaction. It is therefore obvious that fine-tuning of the N-substituents directly effects the cyclisation mechanism. Literature reports on Diels–Alder mechanisms of cyclisations of 1-N-acylaminodienes with electron-deficient olefins are in full accord with this notion.5,6 The significantly lower nucleophilicity of carboxamides (vs. amines) favours an orbital-controlled, more or less concerted, cycloaddition pathway (over a stepwise charge-controlled Michael-Mannich pathway). In order to provide a stereo-chemical complement of the three-component synthesis of trans-nitrocyclohexenyl amines, we have replaced the secondary amines with simple carboxamides. Indeed, formation of the cis-nitrocyclohexenyl amides was observed in high diastereoselectivities (Scheme 11). The major product results from an endo-selective [4 + 2]-cycloaddition and bears the carboxamide in axial position. Despite the presence of an axial hydrogen atom at the CH–NO2 moiety, the low propensity of carboxamide to act as leaving group under the reaction conditions prevents E2-type elimination (cf. Scheme 6). Table 4 shows a selection of cycloadducts prepared from three-component reactions of carboxamides, unsaturated aldehydes and nitrostyrenes.13 The initial amide/aldehyde condensation required elevated temperature (100 °C). The trans,trans-N-acyl-6-nitrocyclohex-2-enyl amines were formed as minor stereoisomers. | ||
| Scheme 11 Endo-selective Diels–Alder mechanism with carboxamides. | ||
|
|
||||
|---|---|---|---|---|
| Major isomer | Substituents | dr | Yield [%] | |
| a Amide, aldehyde, β-nitrostyrene (each 3 mmol) in PhMe, 100 °C, 24 h. | ||||

|
R′′′ = Ph | 51 | 18:1 |
66 |
| 2,6-Cl2–C6H3 | 52 | 50:1 |
67 | |
| 2,4-Cl2–C6H3 | 53 | 28:1 |
57 | |
| 2-F–C6H4 | 54 | 15:1 |
44 | |

|
R′′ = H | 55 | 8:1 |
75 |
| 4-Me | 56 | 50:1 |
39 | |
| 2,4-Me2 | 57 | 40:1 |
74 | |

|
R′′′ = H | 58 | 26:1 |
56 |
| 3-Me | 59 | 32:1 |
79 | |
| 2,4-Me2 | 60 | 13:1 |
18 | |
The relative configurations were assigned based on the 3JHH coupling constants from 1H-NMR spectra. Crystal structure analyses of cis,trans-51 and trans,trans-51 confirmed the relative configurations of the substituents (Fig. 3).
 | ||
| Fig. 3 Crystal structures of cis,trans-51 (left) and trans,trans-51 (right). | ||
Reactions of cis-nitrocyclohexenyl amides
Similar post-synthesis modifications as for the amine series were evaluated with the synthesised cis-nitrocyclohexenyl amides. We were delighted to observe that direct oxidation of 57 with MnO2 proceeded with good selectivity (Scheme 12).15 However, the related C3-substituted cyclohexenyl amines 51, 53 and 54 exhibited poor dehydrogenation selectivity. Competing NO2-elimination was observed which resulted in mixtures of nitro-acetanilides (62–64) and benzoxazoles (65–67, Scheme 13).20 | ||
| Scheme 12 Two-step synthesis of substituted 2-nitro acetanilide. | ||
 | ||
| Scheme 13 Oxidative formation of acetanilides and benzoxazoles. | ||
We have also applied the Zn/HCl reduction protocol to the cycloaddition products.19b Clean reduction of the nitro substituents gave the corresponding cis-diamines in excellent yields with retention of stereochemistry (Scheme 14). The overall synthetic strategy allows the straight-forward preparation of carbocyclic 1,2-diamine derivatives with high cis-stereocontrol in a modular manner starting from simple amide, aldehyde, and nitroethylene precursors.
 | ||
| Scheme 14 Synthesis of cis-1,2-diaminocyclohexenes. | ||
Conclusions
We have applied an operationally simple one-pot protocol to the synthesis of multi-substituted carbocycles upon three-component reaction of amines, aldehydes, and nitroalkenes. The synthesis is highly modular, operates under practical reaction conditions with cheap starting materials (20–100 °C, toluene as solvent), and tolerates ester, ether, chloro, fluoro, thioether, and carbamate substituents. Upon wide variations of the starting materials, the synthesis allows the access to diverse cycloadducts in combinatorial fashion. The electronic properties of the employed amine component dictate the reaction mechanism and product stereochemistry. Reactions with secondary amines proceed via reversible Michael/nitro-Mannich reactions12 to give trans,trans-6-nitrocyclohex-2-enyl amines. The less nucleophilic carboxamides trigger a Diels–Alder-type mechanism toward N-acyl cis,trans-6-nitrocyclo-hex-2-enyl amines.6 Reduction with Zn/HCl afforded the corresponding 1,2-diamine derivatives. The tri-, tetra-, and penta-substituted carbocycles constitute derivatives of cis- and trans-1,2-diaminocyclo-hexanes which are key building blocks of various natural products, fine chemicals, drugs and catalysts.9 In the presence of enantiopure prolinols, one enantiomerically pure set of diastereomers was formed.21 Acid-mediated amine eliminations resulted in the formation of chiral cyclohexadienes. Oxidative aromatisations proceeded with MnO2 to give 2-nitrobiaryls. It is important to note that the presence of two chemically orthogonal N-based substituents (NHR, NO2) allows various selective manipulations of the general structure. Likewise, the electronic polarisation (push–pull substitution) is of high relevance to photoactive materials.Experimental section
General
Unless otherwise noted, all synthesised cycloadducts are racemic mixtures of one diastereomer. For reasons of clarity, only one enantiomer is depicted in the schemes. For atom numbering used in the spectroscopic data assignment, see chemical structures in schemes and tables above. All chemicals were purchased from commercial suppliers and used without further purification unless otherwise noted. Solvents were dried and distilled before use. 1H and 13C NMR spectra were recorded on a Bruker Avance II 600 (600.20 and 150.94 MHz), a Bruker DRX 500 (500.13 and 125.77 MHz) and a Bruker Avance 300 (300.13 and 75.48 MHz) at 298 K. Chemical shifts (δ in ppm) are referenced to tetramethylsilane (TMS). Abbreviations for 1H-NMR data: s (singlet), d (doublet), t (triplet), q (quartet), sept (septet), m (multiplet), q* (apparent quartet), m* (apparent multiplet). Peaks were assigned based on H,H-COSY, H,C-HMQC, and H,C-HMBC. IR-ATR spectroscopy was performed on a Perkin-Elmer 100 Paragon FT-IR. ESI-MS were measured with a Finnigan MAT 900S and an Agilent LC/MSD VL G1956A, respectively. EI-MS were measured with a Finnigan Incos 50 Galaxy and a Finnigan MAT 95, respectively (ionisation 70 eV). Exact masses (HR-MS) were determined by peak matching method. GC-MS analyses were performed on a 6890N Agilent system with 5975 MS detector on an HP-5MS column (30 m × 0.25 mm i.d. × 0.25 μm film thickness) with 5% phenylmethyl siloxane from Macherey-Nagel. H2 was the mobile phase. Standard method 50–300 M: 50 °C (2 min hold), +25 °C min−1→300 °C (5 min hold). Determination of enantiomeric excesses (ee) were performed on a HP6890 GC-FID with chiral BGB 176 column (30 m, 0.25 mm i.d., 0.25 μm film thickness). Helium was the mobile phase. Standard method: 50 °C (2 min hold), 3 °C min−1→150 °C, 1 °C min−1→180 °C. Chiral HPLC spectra were recorded on a Merck Hitachi HPLC with D-6000 interface, L-4000A UV-detector, L-6200A intelligent pump, and a Merck differential refractometer RI-71. Mixtures of n-hexane and 2-propanol were used as mobile phase. Crystal structure data were collected on a Nonius Kappa CCD diffractometer using monochromated Mo-Kα radiation, structures refined by shelxs97 and shelxl97 (Table 5).22| Compound | 24 | 35 | syn-51 | trans-51 |
|---|---|---|---|---|
| CCDC number | 973119 | 973120 | 973121 | 973122 |
| Formula | C37H34Cl2F12N2O3Si | C30H30Cl2N2O3 | C15H18N2O3 | C15H18N2O3 |
| Formula mass | 881.65 | 537.46 | 274.31 | 274.31 |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic | Monoclinic |
| a/Å | 10.8393(5) | 8.9169(13) | 19.3984(10) | 19.0719(12) |
| b/Å | 16.7856(3) | 17.351(3) | 9.6352(5) | 5.2738(3) |
| c/Å | 42.689(2) | 17.602(3) | 16.2688(9) | 27.2994(14) |
| α/° | 90.00 | 90.00 | 90.00 | 90.00 |
| β/° | 90.00 | 90.00 | 108.737(2) | 90.632(4)° |
| γ/° | 90.00 | 90.00 | 90.00 | 90.00 |
| Unit cell vol./Å3 | 7767.0(5) | 2723.2(8) | 2879.6(3) | 2745.6(3) |
| Temperature/K | 100(2) | 100(2) | 100(2) | 100(2) |
| Space group | C2221 | P212121 | P21/c | C2/c |
| Formula units/cell, Z | 8 | 4 | 8 | 8 |
| Reflections measured | 13598 |
12769 |
13657 |
7864 |
| Independent refl. | 6278 | 5633 | 5758 | 3000 |
| R int | 0.0651 | 0.0783 | 0.0667 | 0.0540 |
| R 1 (I > 2σ(I)) | 0.0658 | 0.0630 | 0.1537 | 0.0540 |
| wR(F2) (I > 2σ(I)) | 0.1413 | 0.1281 | 0.0713 | 0.1300 |
| R 1 (all data) | 0.1379 | 0.1520 | 0.1482 | 0.0948 |
| wR(F2) (all data) | 0.1657 | 0.1519 | 0.1851 | 0.1456 |
General procedures (selected examples)13
1-(5-(2,4-Dichlorophenyl)-3-methyl-6-nitrocyclohex-2-enyl) pyrrolidine (9). TLC (ch–ea 5/1): Rf 0.25. Yield: 90%; dr (trans,trans/cis,trans) 15/1. Mp. 137 °C. 1H NMR (300 MHz, CDCl3): δ 7.39 (s, 1H), 7.21 (m, 2H), 5.50 (s, 1H), 4.95 (t, 10.7 Hz, 1H), 4.26 (d, 8.1 Hz, 1H), 4.21–4.11 (m, 1H), 3.14–2.90 (m, 1H), 2.81 (d, 6.5 Hz, 3H), 2.75–2.61 (m, 2H), 1.84–1.69 (m, 7H). 13C NMR (75 MHz, CDCl3): δ 136.3 (2C), 135.6, 129.8, 129.0, 128.2, 127.7, 118.8, 89.2, 61.3, 47.6 (2C), 40.3, 38.0, 24.7 (2C), 24.1. FT-IR (ATR):
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) [cm−1] 2963 (m), 2912 (m), 2847 (m), 1668 (w), 1551 (s), 1474 (s), 1437 (w), 1368 (m), 1326 (w), 1277 (w), 1172 (w), 1125 (w), 1103 (m), 1046 (w), 910 (w), 865 (m), 823 (s), 764 (w), 744 (m), 732 (m). LR-MS (EI, 70 eV): m/z 354, 308, 272, 256, 152, 137, 122. HR-MS (ESI): calcd [M + H]+ 355.0974, found 355.0977.
[cm−1] 2963 (m), 2912 (m), 2847 (m), 1668 (w), 1551 (s), 1474 (s), 1437 (w), 1368 (m), 1326 (w), 1277 (w), 1172 (w), 1125 (w), 1103 (m), 1046 (w), 910 (w), 865 (m), 823 (s), 764 (w), 744 (m), 732 (m). LR-MS (EI, 70 eV): m/z 354, 308, 272, 256, 152, 137, 122. HR-MS (ESI): calcd [M + H]+ 355.0974, found 355.0977.
N,N-Diallyl-3-methyl-6-nitro-5-phenylcyclohex-2-enyl-1-amine (21). TLC (ch–ea 5/1): Rf 0.3. Yield: 93%; dr (trans,trans/cis,trans) 35/1. Pale yellow solid. Mp. 128 °C. 1H NMR (300 MHz, CDCl3): δ 7.43–6.97 (m, 5H), 5.83–5.74 (m, 1H), 5.74–5.66 (m, 1H), 5.43 (s, 1H), 5.19 (m, 1H), 5.13 (s, 2H), 5.10 (s, 1H), 4.86 (dd, 11.8, 9.8 Hz, 1H), 4.25–4.13 (m, 1H), 3.45 (ddd, 11.8, 9.4, 7.7 Hz, 1H), 3.33–3.28 (m, 1H), 3.28–3.22 (m, 1H), 3.04 (d, 7.7 Hz, 1H), 2.99 (d, 7.7 Hz, 1H), 2.32 (d, 7.7 Hz, 2H), 1.74 (m, 3H). 13C NMR (75 MHz, CDCl3): δ 132.4, 131.3, 130.8, 94.9, 66.2, 64.4, 56.8, 48.8, 41.9, 26.2. FT-IR (ATR):
[cm−1] 2968, 2920, 2831, 2806, 1643, 1548, 1495, 1418, 1373, 1315, 1261, 1176, 1109, 1086, 996, 936, 913, 860, 816, 763, 698, 655, 621, 596, 541, 460, 438. HR-MS (EI): calcd [M]+ 312.1838, found 312.1843.
1-[5-(2,4-Dichlorophenyl)-3-methyl-6-nitrocyclohex-2-enyl]-2-(S)-[di-(3,5-bis(trifluoromethyl)phenyl) (trimethylsilyloxy)-methyl]pyrrolidine (24). TLC (ch–ea 20/1): Rf 0.4. Yield: 82%; dr (trans,trans/cis,trans) 19/1. 1H NMR (300 MHz, CDCl3): δ 7.94 (s, 4H), 7.87 (s, 2H), 7.40 (s, 1H), 7.19 (d, 13.9 Hz, 2H), 5.17 (s, 1H), 5.09 (s, 1H), 4.60 (s, 1H), 4.10 (d, 8.9 Hz, 1H), 3.49 (s, 1H), 2.83 (dd, 16.2, 8.5 Hz, 1H), 2.45–2.25 (m, 2H), 2.08–1.82 (m, 3H), 1.69 (s, 3H), 1.65–1.55 (m, 1H), 0.25–0.10 (m, 1H), −0.15 (d, 3.1 Hz, 9H). 13C NMR (75 MHz, CDCl3): δ 145.1, 143.7, 135.7, 135.2, 135.0, 133.8, 131.4, 131.2, 131.0, 130.8, 130.1, 129.5, 128.7, 127.7, 127.2, 125.0, 123.8, 122.0, 121.4, 117.8, 84.8, 83.7, 67.5, 62.3, 40.0, 37.4, 29.1, 23.3, 22.3, 1.5. FT-IR (ATR):
[cm−1] 3094 (w), 2956 (m), 2917 (m), 2855 (w), 1676 (w), 1620 (w), 1588 (w), 1547 (s), 1474 (s), 1446 (w), 1368 (s), 1337 (m), 1316 (m), 1275 (s), 1170 (s), 1125 (s), 1047 (m), 1014 (w), 983 (w), 934 (m), 905 (s), 870 (s), 840 (s), 761 (m), 753 (m), 709 (s), 681 (s). LR-MS (EI, 70 eV): m/z 881, 875, 873, 871. HR-MS (ESI): [M + H]+ calcd 881.1596, found 881.1592.
2,4-Dichloro-1-(5-methyl-2-nitrocyclohexa-2,4-dienyl)-benzene (39). From 24 (with TFA): TLC (ch–ea 12/1): Rf = 0.25. Yield: 60%; 93% ee (HPLC). 1H NMR (300 MHz, CDCl3): δ 7.72 (d, 6.3 Hz, 1H), 7.44 (d, 2.3 Hz, 1H), 7.11 (d, 8.4 Hz, 1H), 6.99 (d, 8.4 Hz, 1H), 5.97 (d, 7.2 Hz, 1H), 4.81 (d, 11.1 Hz, 1H), 3.05 (dd, 18.5, 11.3 Hz, 1H), 2.46 (d, 18.0 Hz, 1H), 1.83 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 147.4, 145.0, 135.8, 133.5, 133.5, 131.9, 130.1, 128.0, 127.3, 117.3, 37.2, 33.4, 23.9. FT-IR (ATR):
[cm−1] 3067 (w), 2870 (w), 2666 (w), 2544 (w), 1688 (s), 1602 (m), 1583 (s), 1558 (w), 1503 (s), 1466 (m), 1450 (w), 1426 8 m), 1316 (s), 1289 (m), 1269 (w), 1103 (w), 1085 (w), 1050 (w), 1025 (w), 938 (w), 866 (w), 841 (m), 812 (m), 711 (s), 663 (m). LR-MS (EI, 70 eV): m/z 283, 266, 148, 236, 216, 202, 183, 165. HR-MS (EI, 70 eV): calcd 283.0167, found 283.018.
2,4-Dichloro-5′-methyl-2′-nitrobiphenyl (47). TLC (ch–ea 10/1): Rf 0.2. Yield: 48%. 1H NMR (300 MHz, CDCl3): δ 8.05 (d, 8.4 Hz, 1H), 7.46 (s, 1H), 7.40–7.30 (m, 2H), 7.19 (d, 8.0 Hz, 1H), 7.11 (s, 1H), 2.48 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 146.1, 144.6, 136.2, 134.5, 133.6, 133.4, 132.7, 131.0, 130.5, 130.0, 129.8, 129.2, 128.2, 127.5, 127.3, 124.8, 115.8, 21.4. FT-IR (ATR):
[cm−1] 2922 (w), 1609 (w), 1586 (m), 1519 (s), 1466 (m), 1377 (w), 1342 (s), 1100 (m), 1066 (w), 1025 (w), 888 (w), 866 (w), 837 (s), 790 (s), 759 (m), 688 (w). LR-MS (EI, 70 eV): m/z 248, 246 [M − Cl]+, 216, 165, 154, 127, 97, 81, 69, 57, 55.
N2,N2-Diallyl-3-methyl-5-phenylcyclohex-2-ene-1,2-diamine (50). Yield: 99%. Mp. 100 °C. 1H NMR (300 MHz, CDCl3): δ 7.39–7.24 (m, 5H), 6.27–6.07 (m, 2H), 5.64–5.40 (m, 1H), 5.32–5.20 (m, 4H), 3.73 (d, 10.5 Hz, 1H), 3.58–3.51 (m, 2H), 3.47–3.24 (m, 3H), 3.13–2.98 (m, 2H), 2.46–2.14 (m, 2H), 1.77 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 139.8, 139.6, 129.7, 128.1, 121.9, 116.1, 77.4, 54.5, 38.6, 23.3. FT-IR (ATR):
[cm−1] 2912, 1600, 1454, 1150, 1098, 1065, 998, 965, 814, 760, 701, 645, 604, 574, 526, 461. HR-MS (EI): calcd [M]+ 282.2096; found 282.20976.
N-3-Methyl-(6-nitro-5-phenylcyclohex-2-enyl) acetamide (51). TLC (ch–ea 1/3): Rf 0.3. Yield: 66%; dr (cis,trans/trans,trans) 18/1. Mp. 205 °C. 1H NMR (300 MHz, CDCl3): δ 7.41–7.09 (m, 5H), 5.93 (d, 9.0 Hz, 1H), 5.47 (d, 3.3 Hz, 1H), 5.34–5.21 (m, 1H), 5.13 (dd, 11.1, 4.9 Hz, 1H), 3.53 (td, 10.3, 6.1 Hz, 1H), 2.45 (dd, 18.4, 6.0 Hz, 1H), 2.24 (dd, 18.5, 10.0 Hz, 1H), 1.98 (s, 3H), 1.77 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 169.7, 140.0, 138.1, 129.0 (2C), 127.6, 127.1 (2C), 118.8, 88.0, 45.6, 39.9, 37.6, 23.1, 22.8. FT-IR (ATR):
[cm−1] 3271 (w), 3030 (w), 2970 (w), 2912 (w), 1732 (w), 1654 (s), 1549 (s), 1455 (w), 1438 (w), 1369 (m), 1281 (w), 1246 (w), 1055 (w), 1029 (w), 975 (w), 832 (w), 802 (w), 785 (w), 758 (m), 699 (m). LR-MS (EI, 70 eV): m/z 274 [M]+, 227 [M − NO2]+. HR-MS (ESI): calcd [M + Na]+ 297.1215, found 297.121.
N-(2,4-Dimethyl-6-nitro-5-phenylcyclohex-2-enyl) acetamide (57). TLC (ch–ea 1/1): Rf 0.2. Yield: 74%; dr (cis,trans/trans,trans) 40/1. Mp 203 °C. 1H NMR (300 MHz, CDCl3): δ 7.26–7.19 (m, 5H), 5.64 (d, 1H, 9.6 Hz), 5.52 (s, 1H), 5.26 (m, 1H), 5.22 (dd, 1H, 12.3/4.9 Hz), 2.81 (dd, 1H, 12.3/10.3 Hz), 2.36 (m, 1H), 2.02 (s, 3H), 1.80 (s, 3H), 0.91 (d, 3H, 6.9 Hz). 13C NMR (75 MHz, APT, CDCl3): δ 170.3, 138.9, 131.5, 130.3, 128.9, 128.0, 127.7 (5C), 89.0, 49.3, 46.7, 38.1, 23.2, 20.6, 19.5. IR (ATR):
[cm−1] 3271 (w), 2970 (w), 1733 (m), 1653 (s), 1550 (s), 1454 (m), 1371 (s), 1241 (s), 1042 (m), 913 (w), 725 (m). LR-MS (ESI, MeOH/CH2Cl2): m/z 289 [M + H]+, 242 [M − NO2]+, 200, 183, 157. HR-MS (ESI): calcd [M + H]+ 289.1552, found 289.154.
N-(4,6-Dimethyl-2-nitrobiphen-3-yl) acetamide (61). TLC (ch–ea 1/1): Rf 0.6. Yield: 71%. 1H NMR (500 MHz, MeOH-d4): δ 7.46–7.35 (m, 5H), 7.08 (s, 1H), 2.54 (s, 3H), 2.52 and 2.27 (2s, 6H), NH resonance obscured. 13C NMR (125 MHz, APT, MeOH-d4): δ 165.1, 150.5, 138.9, 136.4, 133.5, 131.0, 129.4, 128.7, 128.6, 128.5, 123.7, 19.8, 16.3, 14.1. IR (ATR):
[cm−1] 2920 (m), 2860 (w), 1720 (w), 1607 (m), 1486 (m), 1441 (m), 1383 (m), 1250 (m), 1223 (s), 1061 (s), 926 (m), 759 (s), 700 (s). LR-MS (ESI, MeOH): m/z 238 [M − NO2]+, 223 [M − NO2 − Me]+, 213, 186, 137, 129, 105.
[cm−1] 3256 (w), 3027, (w), 2963 (w), 1643 (s), 1536 (s), 1493 (m), 1452 (m), 1370 (m), 1285 (w), 1092 (w), 1092 (w), 1036 (w), 907 (m), 755 (s), 727 (s), 700 (s). LR-MS (EI, 70 eV): m/z 258 [M]+, 199 [M − AcNH]+, 184, 167, 139, 119, 108. HR-MS (ESI): calcd [M + Na]+ 281.1624, found 281.162.
Acknowledgements
This work was financed by the 7th European framework program (Cataflu.Or).Notes and references
- (a) M. J. Palframan and A. F. Parsons, in Science of Synthesis: Houben-Weyl Methods of Molecular Transformations, ed. H. Hiemstra, Thieme, Stuttgart, 2009, vol. 48, pp. 647–693 Search PubMed; (b) L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed.
- (a) Cycloaddition Reactions in Organic Synthesis, ed. S. Kobayashi and K. A. Jorgensen, Wiley-VCH, Weinheim, 2002 Search PubMed; (b) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS; (c) W. Oppolzer, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 5, pp. 315–399 Search PubMed.
- C. Thebtaranonth and Y. Thebtaranonth, Cyclization Reactions, CRC Press, Boca Raton, 1994 Search PubMed.
- Selected examples: (a) S. Danishefsky, Acc. Chem. Res., 1981, 14, 400–406 CrossRef CAS; (b) V. K. Das, Synlett, 2011, 430–431 CrossRef CAS PubMed; (c) M. C. Aversa, A. Barattuci, P. Bonaccorsi, P. Giannetto and D. N. Jones, J. Org. Chem., 1997, 62, 4376–4384 CrossRef CAS PubMed; (d) S. A. Kozmin and V. H. Rawal, J. Am. Chem. Soc., 1999, 121, 9562–9573 CrossRef CAS; (e) R. C. Cookson, M. C. Cramp and P. J. Parsons, J. Chem. Soc., Chem. Commun., 1980, 197–198 RSC; (f) J. Becher, H. C. Nielsen, J. P. Jacobsen, O. Simonsen and H. Clausen, J. Org. Chem., 1988, 53, 1862–1871 CrossRef CAS; (g) C. J. Suckling, M. C. Tedford, L. M. Bence, J. I. Irvine and W. H. Stimson, Bioorg. Med. Chem. Lett., 1992, 2, 49–52 CrossRef CAS.
- (a) Dienamine catalysis: D. B. Ramachary and Y. V. Reddy, Eur. J. Org. Chem., 2012, 865–887 CrossRef CAS; (b) Somatostatin analogues: M. Sukopp, R. Schwab, L. Marinelli, E. Biron, M. Heller, E. Varkondi, A. Pap, E. Novellino, G. Keri and H. Kessler, J. Med. Chem., 2005, 48, 2916–2926 CrossRef CAS PubMed; (c) Photoactive luminols: H. Neumann, S. Klaus, M. Klawonn, D. Strübing, S. Hübner, D. Gördes, A. Jacobi von Wangelin, M. Lalk and M. Beller, Z. Naturforsch., B: Chem. Sci., 2004, 59, 431–438 CAS; (d) Aza-Corollosporins: H. Neumann, D. Strübing, M. Lalk, S. Klaus, S. Hübner, A. Spannenberg, U. Lindequist and M. Beller, Org. Biomol. Chem., 2006, 4, 1365–1375 RSC; (e) Gephyrotoxin: L. E. Overman, D. Lesuisse and M. Hashimoto, J. Am. Chem. Soc., 1983, 105, 5373–5379 CrossRef CAS; (f) Dendrobine: S. F. Martin and W. Li, J. Org. Chem., 1991, 56, 642–650 CrossRef CAS; (g) Aspidosperma alkaloids: S. A. Kozmin, T. Iwama, Y. Huang and V. H. Rawal, J. Am. Chem. Soc., 2002, 124, 4628–4641 CrossRef CAS PubMed; (h) Azasteroidal building blocks: A. Jacobi von Wangelin, H. Neumann, D. Gördes, S. Hübner, C. Wendler, S. Klaus, D. Strübing, A. Spannenberg, H. Jiao, L. El Firdoussi, K. Thurow, N. Stoll and M. Beller, Synthesis, 2005, 2029–2038 Search PubMed; (i) Automated phthalate synthesis: A. Jacobi von Wangelin, H. Neumann, D. Gördes, S. Klaus, H. Jiao, A. Spannenberg, T. Krüger, C. Wendler, K. Thurow, N. Stoll and M. Beller, Chem. – Eur. J., 2003, 9, 2273–2281 CrossRef PubMed.
- (a) Review: D. Enders and O. Meyer, Liebigs Ann., 1996, 1023–1035 CAS. Selected examples: (b) W. Oppolzer, Angew. Chem., Int. Ed. Engl., 1977, 16, 10–24 CrossRef; (c) W. Oppolzer, L. Bieber and E. Francotte, Tetrahedron Lett., 1979, 16, 981–984 CrossRef; (d) W. Oppolzer, L. Bieber and E. Francotte, Tetrahedron Lett., 1979, 16, 4537–4540 CrossRef; (e) L. E. Overman, G. F. Taylor, C. B. Petty and P. J. Jessup, J. Org. Chem., 1978, 43, 2164–2167 CrossRef CAS; (f) L. E. Overman, L. A. Clizbe, R. L. Freerks and C. K. Marlowe, J. Am. Chem. Soc., 1981, 103, 2807–2815 CrossRef CAS; (g) L. E. Overman, R. L. Freerks, C. B. Petty, L. A. Clizbe, R. K. Ono, G. F. Taylor and P. J. Jessup, J. Am. Chem. Soc., 1981, 103, 2816–2822 CrossRef CAS; (h) C. A. Zezza and M. B. Smith, J. Org. Chem., 1988, 53, 1161–1167 CrossRef CAS; (i) M. B. Smith, Org. Prep. Proced. Int., 1990, 22, 315–335 CrossRef CAS; (j) R. Sustmann, M. Rogge, U. Nüchter and H. Bandmann, Chem. Ber., 1992, 125, 1647–1656 CrossRef CAS; (k) A. R. Katritzky, A. V. Ignatchenko and H. Lang, J. Org. Chem., 1995, 60, 4002–4005 CrossRef CAS; (l) D. A. Alonso, E. Alonso, C. Najera and M. Yus, Synlett, 1997, 491–492 CrossRef CAS PubMed; (m) J. Barluenga, F. Aznar, C. Ribas and C. Valdés, J. Org. Chem., 1997, 62, 6746–6753 CrossRef CAS; (n) J. M. Janey, T. Iwama, S. A. Kozmin and V. H. Rawal, J. Org. Chem., 2000, 65, 9059–9068 CrossRef CAS PubMed; (o) A. N. Thadani, A. R. Stankovic and V. H. Rawal, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5846–5850 CrossRef CAS PubMed; (p) M. R. Tremblay, T. J. Dickerson and K. D. Janda, Adv. Synth. Catal., 2001, 343, 577–585 CrossRef CAS; (q) S. Hübner, H. Neumann, A. Jacobi von Wangelin, S. Klaus, D. Strübing, H. Klein and M. Beller, Synthesis, 2005, 2084–2089 Search PubMedS. Hübner, H. Neumann, A. Jacobi von Wangelin, S. Klaus, D. Strübing, H. Klein and M. Beller, Synthesis, 2005, 2270 Search PubMed; (r) A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 2458–2459 CrossRef CAS PubMed; (s) N. Momiyama, T. Konno, Y. Furiya, T. Iwamoto and M. Terada, J. Am. Chem. Soc., 2011, 133, 19294–19297 CrossRef CAS PubMed.
- (a) A. G. M. Barrett and G. G. Graboski, Chem. Rev., 1986, 86, 751–762 CrossRef CAS; (b) N. Ono, H. Miyake, A. Kamimura and A. Kaji, J. Chem. Soc., Perkin Trans. 1, 1987, 1929–1935 RSC; (c) R. Ballini, R. Castagnani and M. Petrini, J. Org. Chem., 1992, 57, 2160–2162 CrossRef CAS; (d) C. Jubert and P. Knochel, J. Org. Chem., 1992, 57, 5431–5438 CrossRef CAS.
- For related condensation-Diels–Alder reaction sequences, see: (a) H. Neumann, A. Jacobi von Wangelin, D. Gördes, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2001, 123, 8398–8399 CrossRef CAS; (b) A. Jacobi von Wangelin, H. Neumann, D. Gördes, A. Spannenberg and M. Beller, Org. Lett., 2001, 3, 2895–2898 CrossRef CAS PubMed; (c) H. Neumann, A. Jacobi von Wangelin, D. Gördes, A. Spannenberg, W. Baumann and M. Beller, Tetrahedron, 2002, 58, 2381–2387 CrossRef CAS; (d) S. Klaus, S. Hübner, H. Neumann, D. Strübing, A. Jacobi von Wangelin, D. Gördes and M. Beller, Adv. Synth. Catal., 2004, 346, 970–978 CrossRef CAS; (e) D. Strübing, A. Jacobi von Wangelin, H. Neumann, D. Gördes, S. Hübner, S. Klaus, A. Spannenberg and M. Beller, Eur. J. Org. Chem., 2005, 107–113 CrossRef.
- (a) Review: D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580–2627 CrossRef CAS. Selected applications of trans-1,2-cyclohexane diamines: (b) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed; (c) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS; (d) M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 4901–4902 CrossRef CAS; (e) Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153–156 CrossRef CAS PubMed; (f) R. Wu, X. Chang, A. Lu, Y. Wang, G. Wu, H. Song, Z. Zhou and C. Tang, Chem. Commun., 2011, 47, 5034–5036 RSC; (g) S. Luo, H. Xu, J. Li, L. Zhang and J.-P. Cheng, J. Am. Chem. Soc., 2007, 129, 3074–3075 CrossRef CAS PubMed. Selected applications of cis-1,2-cyclohexane diamines: (h) A. Berkessel, T. Günther, Q. Wang and J.-M. Neudörfl, Angew. Chem., Int. Ed., 2013, 52, 8467–8471 CrossRef CAS PubMed; (i) S. A. Moteki, J. Han, S. Arimitsu, M. Akakura, K. Nakayama and K. Maruoka, Angew. Chem., Int. Ed., 2012, 51, 1187–1190 CrossRef CAS PubMed; (j) O. Kitagawa, K. Yotsumoto, M. Kohriyama, Y. Dobashi and T. Taguchi, Org. Lett., 2004, 6, 3605–3607 CrossRef CAS PubMed; (k) L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844–848 CrossRef CAS PubMed; (l) A. Hirabayashi, H. Mukaiyama, H. Kobayashi, H. Shiohara, S. Nakayama, M. Ozawa, K. Miyazawa, K. Misawa, H. Ohnota and M. Isaji, Bioorg. Med. Chem., 2008, 16, 7347–7357 CrossRef CAS PubMed; (m) K. Yoshikawa, S. Kobayashi, Y. Nakamoto, N. Haginoya, S. Komoriya, T. Yoshino, T. Nagata, A. Mochizuki, K. Watanabe, M. Suzuki, H. Kanno and T. Ohta, Bioorg. Med. Chem., 2009, 17, 8206–8220 CrossRef CAS PubMed; (n) K. Caron, V. Lachapelle and J. W. Keillor, Org. Biomol. Chem., 2011, 9, 185–197 RSC; (o) T. Govindaraju, R. G. Gonnade, M. M. Bhadbhade, V. A. Kumar and K. N. Ganesh, Org. Lett., 2003, 5, 3013–3016 CrossRef CAS PubMed; (p) A. Trapero and A. Llebaria, ACS Med. Chem. Lett., 2011, 2, 614–619 CrossRef CAS PubMed.
- (a) K. Patora-Komisarska, M. Benohoud, H. Ishikawa, D. Seebach and Y. Hayashi, Helv. Chim. Acta, 2011, 94, 719–745 CrossRef CAS; (b) B. Potthoff and E. Breitmeier, Chem. Ber., 1987, 120, 255–257 CrossRef CAS; (c) G. Bobowski, B. West and D. Omecinsky, J. Heterocycl. Chem., 1992, 29, 33–49 CrossRef CAS; (d) J. Barluenga, F. Aznar and M. Fernández, Tetrahedron Lett., 1995, 36, 6551–6554 CrossRef CAS; (e) K. Bogdanowicz-Szwed and A. Budzowski, Monatsh. Chem., 2001, 132, 947–957 CrossRef CAS; (f) H. Sunden, R. Rios, Y. Xu, L. Eriksson and A. Cordova, Adv. Synth. Catal., 2007, 349, 2549–2555 CrossRef CAS; (g) B. Han, Y.-C. Xiao, Z.-Q. He and Y.-C. Chen, Org. Lett., 2009, 11, 4660–4663 CrossRef CAS PubMed.
- (a) For similar equilibria with N-acylamines, see: D. Gördes, A. Jacobi von Wangelin, S. Klaus, H. Neumann, D. Strübing, S. Hübner, H. Jiao, W. Baumann and M. Beller, Org. Biomol. Chem., 2004, 2, 845–851 RSC; (b) A. Jacobi von Wangelin, H. Neumann, D. Gördes, S. Klaus, D. Strübing and M. Beller, Chem. – Eur. J., 2003, 9, 4286–4294 CrossRef PubMed.
- A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887–2939 CrossRef CAS PubMed.
- For further preparative and spectroscopic details, see the ESI.†.
- (a) B.-C. Hong, M.-F. Wu, H.-C. Tseng, G.-F. Huang, C.-F. Su and J.-H. Liao, J. Org. Chem., 2007, 72, 8459–8471 CrossRef CAS PubMed; (b) P. Kotame, B.-C. Hong and J.-H. Liao, Tetrahedron Lett., 2009, 50, 704–707 CrossRef CAS PubMed.
- Sequential cycloaddition and oxidation reactions: (a) S. Mekidechea and L. Désaubry, Tetrahedron Lett., 2008, 49, 5268–5270 CrossRef PubMed; (b) J. T. Vessels, S. Z. Janicki and P. A. Petillo, Org. Lett., 2000, 2, 73–76 CrossRef CAS PubMed; (c) G. Hilt and K. I. Smolko, Angew. Chem., Int. Ed., 2003, 42, 2795–2797 CrossRef CAS PubMed; (d) G. Hilt and M. Danz, Synthesis, 2008, 2257–2263 CrossRef CAS PubMed; (e) G. Hilt, S. Lüers and K. I. Smolko, Org. Lett., 2005, 7, 251–253 CrossRef CAS PubMed; (f) H. Neumann, A. Jacobi von Wangelin, S. Klaus, D. Strübing, D. Gördes and M. Beller, Angew. Chem., Int. Ed., 2003, 42, 4503 CrossRef CAS PubMed; (g) R. Fichtler, J.-M. Neudörfl and A. Jacobi von Wangelin, Org. Biomol. Chem., 2011, 9, 7224–7236 CAS.
- (a) R. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017–1047 CrossRef CAS PubMed; (b) W. E. Noland, Chem. Rev., 1955, 55, 137–155 CrossRef CAS.
- The best enantioselectivities of amine/acid co-catalyzed sequential cyclisation/elimination reactions were obtained with (S)-diphenyl-prolinol (or the TMS-ether thereof) and 2-nitrobenzoic acid (each 20 mol%): up to 40% yield and 62% ee of nitrocyclohexadiene 36.
- Oxidative aromatisation of cyclohexadienes: (a) T. Moriuchi, K. Kikushima, T. Kajikawa and T. Hirao, Tetrahedron Lett., 2009, 50, 7385–7387 CrossRef CAS PubMed; (b) H. Tanaka, T. Ikeno and T. Yamada, Synlett, 2003, 576–578 CAS; (c) N. Nakamichi, H. Kawabata and M. Hayashi, J. Org. Chem., 2003, 68, 8272–8273 CrossRef CAS PubMed; (d) C. A. Busacca and Y. Dong, Synth. Commun., 2000, 30, 501–509 CrossRef CAS; (e) J. Cossy and D. Belotti, Org. Lett., 2002, 4, 2557–2559 CrossRef CAS PubMed; (f) M. Hayashi, K. Yamada and S. Nakayama, J. Chem. Soc., Perkin Trans. 1, 2000, 1501–1503 RSC; (g) J. C. Leffingwell and H. J. Bluhm, J. Chem. Soc., Chem. Commun., 1969, 1, 1151–1152 RSC; (h) for more examples, see also: J. March, Advanced Organic Chemistry, Wiley, New York, 4th edn, 1992, p. 1162 Search PubMed.
- (a) H. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, 2001, ch. 6.3, p. 170 CrossRef; (b) M. R. Michaelides, Y. Hong, S. DiDomenico Jr., E. K. Bayburt, K. E. Asin, D. R. Britton, C. W. Lin and K. Shiosaki, J. Med. Chem., 1997, 40, 1585–1599 CrossRef CAS PubMed.
- (a) R. Ballini and G. Bosica, Tetrahedron, 1995, 51, 4213–4222 CrossRef CAS; (b) M. V. Gil, E. Roman and J. A. Serrano, Tetrahedron Lett., 2001, 42, 4625–4628 CrossRef CAS.
- For an enantioselective synthesis of related aminocyclohexenes, see: D. Strübing, H. Neumann, S. Klaus, A. Jacobi von Wangelin, D. Gördes, M. Beller, P. Braiuca, C. Ebert, L. Gardossi and U. Kragl, Tetrahedron, 2004, 60, 683 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data of all new compounds. CCDC 973119–973122. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob00913d |
| This journal is © The Royal Society of Chemistry 2014 |