
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent switchable cycloaddition: a (one-pot) metal-free approach towards N-substituted benzo[e]- or [f]isoindolones via Csp2–H functionalization†
Pratik A.
Ambasana
ab,
Dipak D.
Vachhani
*a,
Marzia
Galli‡
a,
Jeroen
Jacobs
c,
Luc
Van Meervelt
c,
Anamik K.
Shah
b and
Erik V.
Van der Eycken
*a
aLaboratory for Organic & Microwave-Assisted Chemistry (LOMAC), Department of Chemistry, University of Leuven (KU Leuven), Celestijnenlaan 200F, B-3001, Leuven, Belgium. E-mail: erik.vandereycken@chem.kuleuven.be; ddvachhani@gmail.com
bDepartment of Chemistry, Saurashtra University, Rajkot – 360005, India
cBiomolecular Architecture, Department of Chemistry, University of Leuven (KULeuven), Celestijnenlaan 200F, B-3001, Leuven, Belgium
First published on 18th September 2014
Abstract
The tuning of selective ring closure is a nontrivial challenge in synthetic organic chemistry. Herein we report a solvent switchable metal-free [4 + 2] cycloaddition approach via Csp2–H functionalization. The protocol is highly atom economical with water being the only by-product, delivering N-substituted benzo[e]- or [f]isoindolones in high yields.
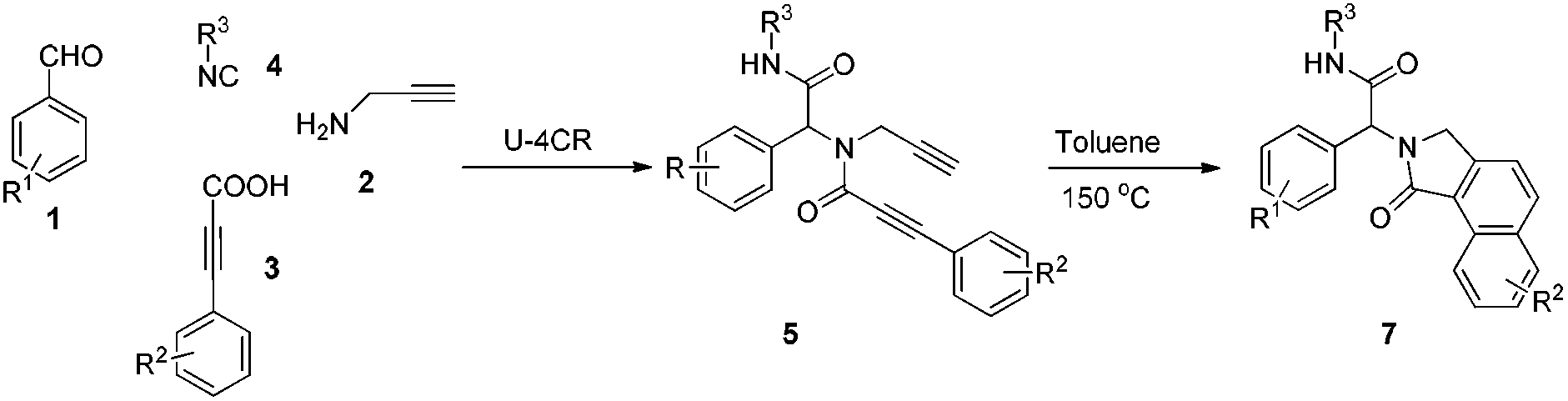
The elaboration of methodologies to selectively access heterocycles, while guaranteeing molecular diversity and eco-compatibility, represents a great challenge to organic chemists. In this regard, multi-component reactions (MCR) inherently bequeath molecular diversity and complexity in a single step.1 Moreover, post-MCR transformations could provide powerful ways to generate libraries of unprecedented molecular skeletons.2,3 Employing this strategy, merged with gold-catalyzed dual σ–π activation,4 we recently reported a post-Ugi gold-catalyzed regioselective tandem cyclization via Csp3–H functionalization (Scheme 1).5
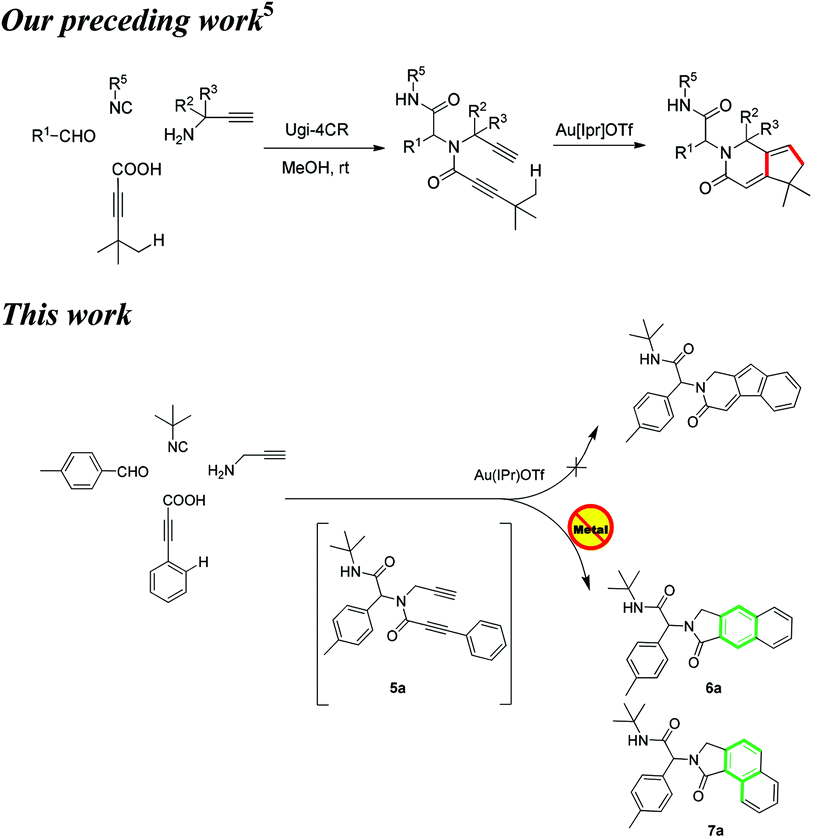 | ||
| Scheme 1 Different cyclization of N-propynyl propiolamides. | ||
Stimulated by these findings and our recent endeavours regarding post-Ugi transformations,6 we aimed to extend this methodology to the Csp2–H functionalization employing cascade cyclizations4c of N-propynyl phenylpropiolamides to access indenopyridinones (Scheme 1). These compounds are known to possess antihistaminic and antidepressant activity.7
According to the literature, cationic gold,4c,5 palladium,8–10 zinc,11 copper,12 and iron13 have been used alongside phosphorous containing ligands,9 Lewis acids and excess of phenolic additives,13–15 to catalyze similar intramolecular cyclizations. The access to the precursors for such cyclizations is often rather tedious.13,15,16
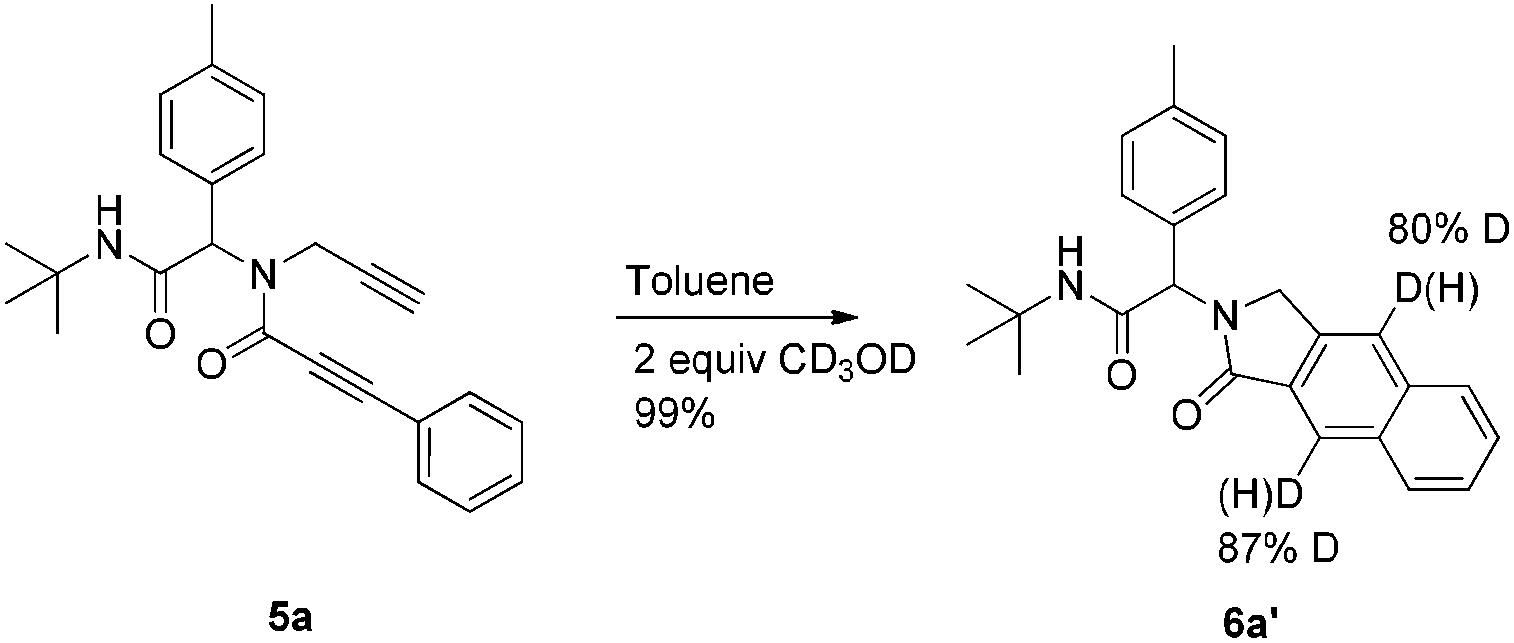
To investigate the workability of the concept, N-propynyl phenylpropiolamide (5a), synthesized via Ugi four-component reaction (U-4CR)17 of p-tolualdehyde (1a) with propargyl amine (2a), 3-phenylpropiolic acid (3a) and tert-butyl isonitrile (4a) in methanol, was subjected to our established reaction conditions using 5 mol% of in situ generated IPrAuOTf.5 To our surprise, we did not observe, by NMR spectroscopy, the desired product. Instead, as revealed by MS-analysis, we observed two other products with similar molecular weight as the starting material. Finally, 1H NMR and X-ray crystallographic analysis18 proved that 38% of the benzo[f]isoindolone 6a was furnished,4c next to 36% of the benzo[e]isoindolone 7a (Scheme 1 and Fig. 1). Interestingly, this benzo[f]isoindolones are known to act as potent human 17,20-lyase inhibitors for the treatment of castration-resistant prostate cancer (CRPC).8 Further, a control experiment ruled out the necessity of the metal-catalyst (Table 1, entry 1).
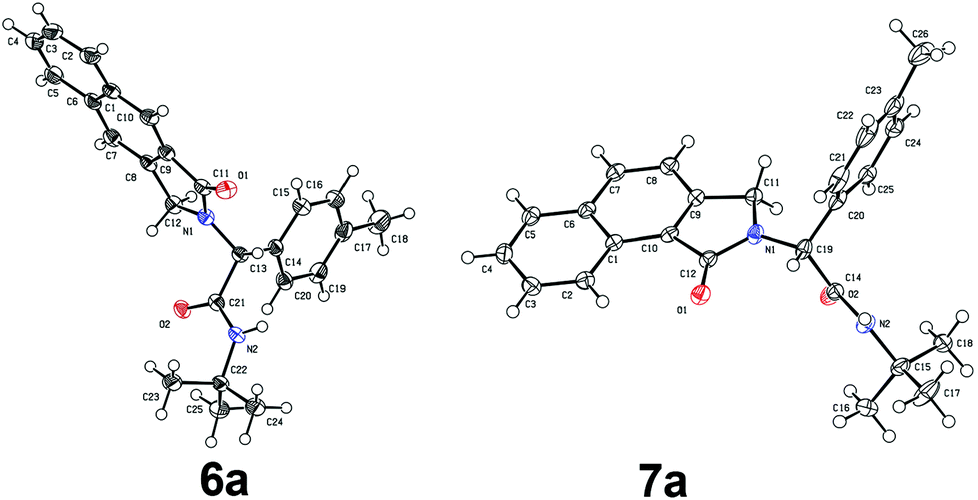 | ||
| Fig. 1 Crystal structure of compound 6a and 7a. Thermal ellipsoids set at 50% probability.17 Only one molecule is shown for 6a. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temp (°C) | Time (h) | Yieldb (%) | Ratio (6a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7a)b :7a)b |
| a Unless otherwise stated, all reactions were performed using 0.1 mmol of 5a in the indicated solvent (1 mL), under N2 atmosphere and conventional heating. b The combined yield and ratio of 6a and 7a was estimated on the basis of 1H NMR analysis using 2,4,6-trimethoxy benzaldehyde as an internal standard (0.1 mmol). c Isolated yields. d A one-pot sequence was applied using n-BuOH as common solvent; for the Ugi-4CR 8 h at 50 °C; followed by 3 h at 140 °C. | |||||
| 1 | Toluene | 120 | 16 h | 71 | 54:46 |
| 2 | Toluene | 140 | 15 h | 76 | 31:69 |
| 3 | Xylene | 140 | 15 h | 73 | 49:51 |
| 4 | DCE | 140 | 15 h | 78 | 47:53 |
| 5 | Toluene | 150 | 15 h | 95 |
19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :81 :81
|
| 6 | Toluene–MeOH (1:1) |
140 | 15 h | 99 | 99:01 |
| 7 | iPrOH | 140 | 15 h | 99 | 97:03 |
| 8 | n-BuOH | 140 | 15 h | 97c | >99:01 |
| 9 | 2,2,2-Trifluoroethanol | 140 | 15 h | 95 | >99:01 |
| 10 | n-BuOH | 140 | 8 h | 98c | >99:01 |
| 11 | n-BuOH | 140 | 3 h | 98c | >99:01 |
| 12 | n-BuOH | 50/140 | 8 h/3 h | 97 |
>99:01
|
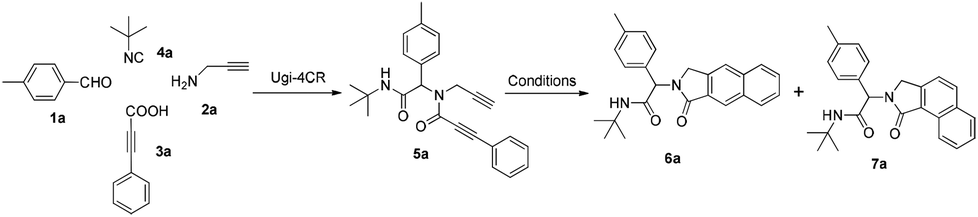
To get some mechanistic insight, a deuterium labelling experiment was performed with compound 5a (Scheme 2). Use of 2 equiv. of CD3OD in the reaction resulted in ∼87% of deuterium incorporation in the product 6a′. However, incorporation of deuterium was also observed on the terminal carbon from the propargyl amine part, probably due to facile deprotonation of the terminal acetylene. Based on these observations and previous mechanistical studies,15 a plausible mechanism with two distinct possibilities is depicted in Scheme 3.
 | ||
| Scheme 2 Deuterium labelling experiment with compound 5a. | ||
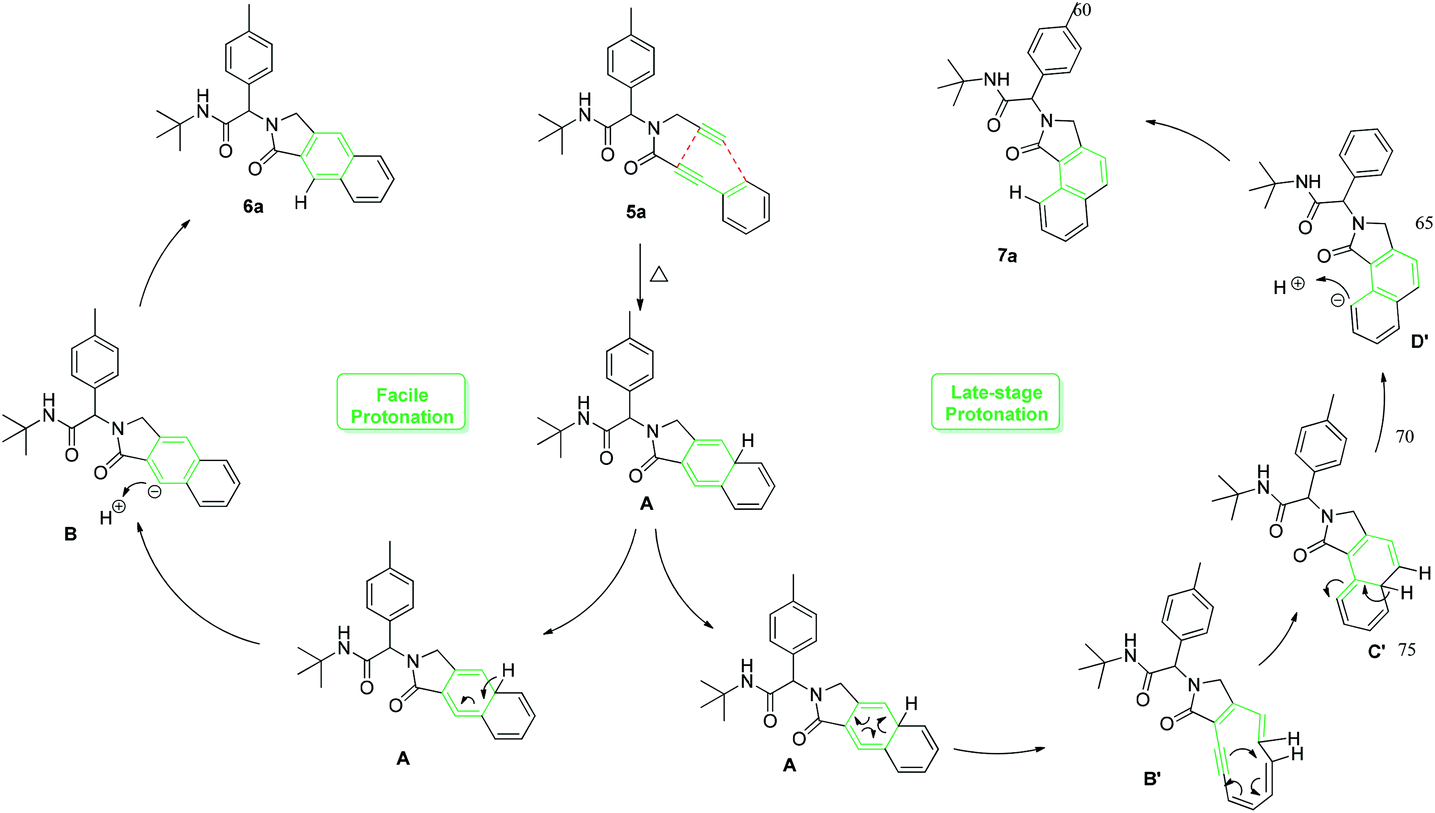 | ||
| Scheme 3 Plausible mechanism for the metal-free post-Ugi cyclization. | ||
The cyclohexatriene A, formed by initial [4 + 2] cyclization, can undergo either isomerization followed by protonation to deliver benzo[f]isoindolone 6a, or a six-electron electrocyclic ring-opening process to afford 1,2-dehydro-[10]annulene B′. [1,6]-Electrocyclization of B′ then leads to cyclic allene C′, which upon aromatization, results in the formation of the rearranged benzo[e]isoindolone 7a.
Motivated by these findings, we further investigated the conditions to optimize the selective ring closure. Among different solvents screened, toluene was found to be the best, when used at elevated temperature, for the formation of benzo[e]isoindolone 7a (Table 1, entries 2–5). The use of a polar protic solvent solely, or as co-solvent with toluene, dramatically shifts the pendulum towards the formation of benzo[f]isoindolone 6a (Table 1, entries 6–11). To our great satisfaction, an attempt to carry out the sequence in a tandem fashion resulted in an almost quantitative yield with complete selectivity for 6a (Table 1, entry 12). Besides the atom economy and the catalyst free process, the application of n-BuOH also contributes to the green aspect of this protocol.19,20
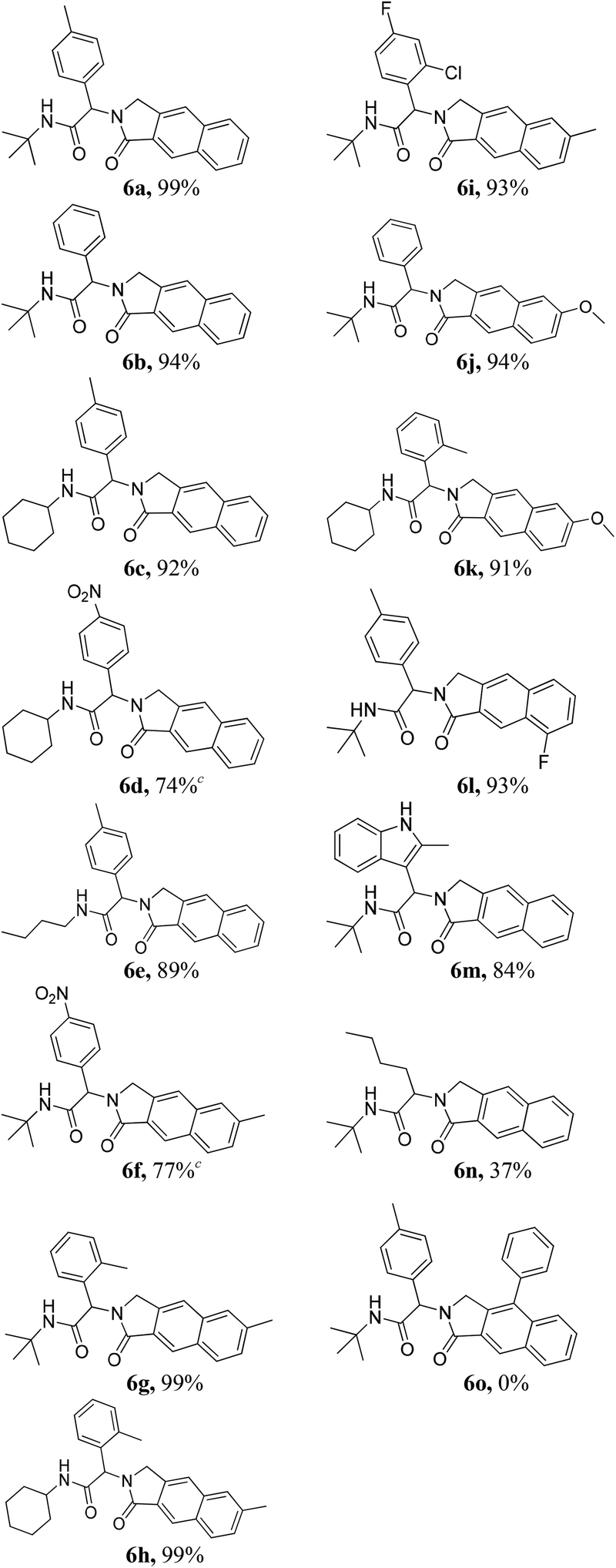
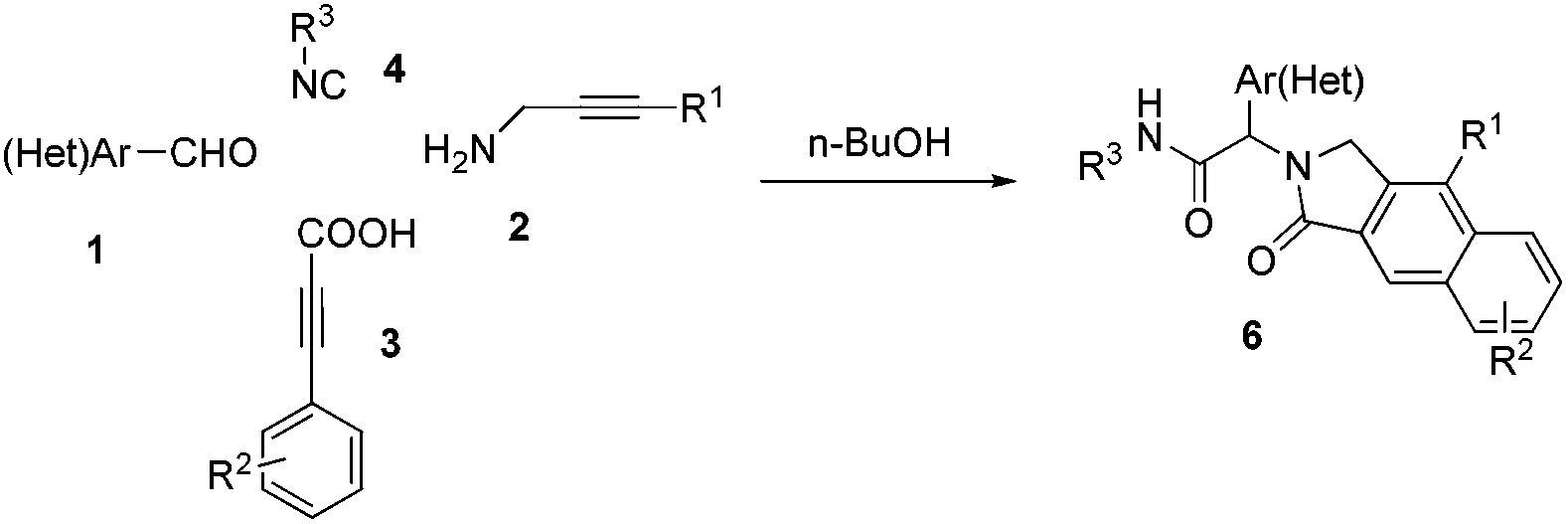
Having optimized the regioselective cycloadditions (Table 1, entries 5 and 12), we first explored the substrate scope for the one-pot synthesis of benzo[f]isoindolones 6. In most cases, compounds 6 were obtained in excellent yields (84–99%), without the need of further chromatographic purification (Table 2). Ugi-product resulting from various aromatic aldehydes, isonitriles and acids were well tolerated.
|
|
|
|---|---|
| One-pot cycloadduct | One-pot cycloadduct |
| a Unless otherwise stated, all reactions were performed using aldehyde 1 (0.1 mmol), amine 2 (1.05 equiv.), acid 3 (1.05 equiv.) and isonitrile 4 (1.05 equiv.) in n-butanol (2 mL) at 50 °C for 8–12 h followed by heating at 140 °C for 3–8 h. b Yields are isolated yields. c Isolated after chromatographic purification. | |
|
|
Notably, 2-methylindole-3-carbaldehyde afforded the indolyl-benzo[f]isoindolone 6m in high yield. However, a significant loss of the yield was observed when aliphatic valeraldehyde was used (6n, Table 2). The use of phenyl propargylamine failed to give the desired product (6o, Table 2).
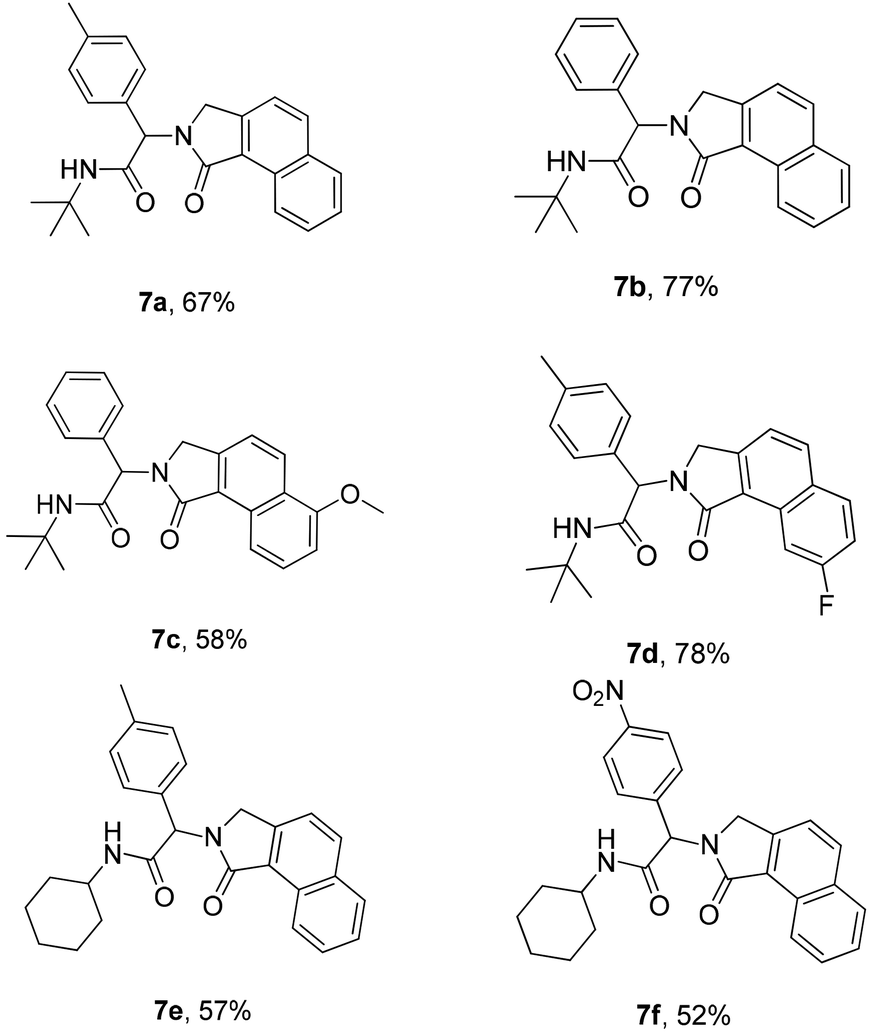
Next, we turned our attention to generate a small library of rearranged benzo[e]isoindolones 7, employing our optimized conditions (Table 1, entry 5). The reaction proved to be versatile with respect to aldehyde, acid and isonitrile used in the Ugi-reaction, giving the desired compounds 7 in 52–78% yield (Table 3). The presence of an electron withdrawing group on the acid improves the yield of desired product, while the use of an electron deficient aldehyde and electron rich acid lowered the yields (Table 3, 7c, d and f). Importantly, employment of Ugi-product 5d, derived from an o-substituted acid, under optimized condition for the formation of rearranged product 7 (Table 1, entry 5), resulted in only one isomer of benzo[e]isoindolone 7d (Table 3, 7d). The structures of compounds 7 are well characterized by NMR spectroscopy and compounds 7b and 7d were also unambiguously assigned by X-ray crystallography (Table 3, entries 3 and 4).18
|
|
|
|---|---|
| Product 7 | Product 7 |
| a For U-4CR: all reactions were performed using aldehyde 1 (0.1 mmol), amine 2 (1.05 equiv.), acid 3 (1.05 equiv.) and isonitrile 4 (1.05 equiv.) in n-butanol (2 mL) at 50 °C for 8–12 h. b For cycloaddition: reactions were run using 5 (0.25 mmol) in dry toluene (2 mL) under N2 atmosphere at 150 °C for 8–15 h. | |
|
|
Conclusions
We have developed a (one-pot) facile metal-free and atom economical protocol for the synthesis of diversely substituted benzo[e]isoindolones 6 and benzo[f]isoindolones 7 from readily available starting materials. This one of the rare examples where the selectivity of the ring-closure could be controlled by a simple switch of the solvent.Acknowledgements
Support was provided by the research fund of the University of Leuven (KU Leuven) and the FWO (Fund for Scientific Research–Flanders, Belgium). PAA is thankful to EXPERTS Asia (Second Cohort) and DDV is thankful to PDM-kort, University of Leuven (KU Leuven) for providing a postdoctoral fellowship. We thank the Hercules Foundation for supporting the purchase of a single crystal diffractometer through project AKUL/09/0035.Notes and references
- For multicomponent reactions based on isocyanide, see:
(a) A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168 CrossRef
; (b) A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed
- For gold-catalyzed post-Ugi transformations, see:
(a) S. G. Modha, D. D. Vachhani, J. Jacobs, L. Van Meervelt and E. V. Van der Eycken, Chem. Commun., 2012, 48, 6550 RSC
- For post-Ugi transformations using transition-metals other than gold see:
(a) G. Cunny, M. Bois-Choussy and J. Zhu, J. Am. Chem. Soc., 2004, 126, 14475 CrossRef PubMed
-
(a) L. Ye, Y. Wang, D. H. Aue and L. Zhang, J. Am. Chem. Soc., 2012, 134, 31 CrossRef CAS PubMed
- D. D. Vachhani, M. Galli, J. Jacobs, L. Van Meervelt and E. V. Van der Eycken, Chem. Commun., 2013, 49, 7171 RSC
-
(a) S. G. Modha, D. D. Vachhani, J. Jacobs, L. Van Meervelt and E. V. Van der Eycken, Chem. Commun., 2012, 48, 6550 RSC
-
(a) J. Augstein, A. L. Ham and P. R. Leeming, J. Med. Chem., 1972, 15, 466 CrossRef CAS
- T. Kaku, N. Matsunaga, A. Ojida, T. Tanaka, T. Hara, M. Yamaoka, M. Kusaka and A. Tasaka, Bioorg. Med. Chem., 2011, 19, 1751 CrossRef CAS PubMed
- Y. Sato, T. Tamura1, A. Kinbara1 and M. Mori, Adv. Synth. Catal., 2007, 349, 647 CrossRef CAS
-
A. C. Childs, G. M. Paul Giblin and M. P. Healy, PCT Int. Appl, WO2009019281 A1, 2009 Search PubMed
- H. Yeo, Y. Li, L. Fu, J.-L. Zhu, E. A. Gullen, G. E. Dutschman, Y. Lee, R. Chung, E. S. Huang, D. J. Austin and Y. C. Cheng, J. Med. Chem., 2005, 48, 534 CrossRef CAS PubMed
- D. Rodríguez, L. Castedo, D. Domínguez and C. Saá, Synthesis, 2004, 761 Search PubMed
- R. L. Danheiser, A. E. Gould, R. F. de la Pradilla and A. L. Helgason, J. Org. Chem., 1994, 59, 5514 CrossRef CAS
- M. V. Santander, M. B. Pastor, J. N. Nelson, C. Castro and W. L. Karney, J. Org. Chem., 2013, 78, 2033 CrossRef CAS PubMed
- D. Rodríguez, A. Navarro-Vázquez, L. Castedo, D. Domıínguez and C. Saa, J. Am. Chem. Soc., 2001, 123, 9178 CrossRef
- Y. Kita, R. Okunaka, T. Honda, M. Kondo, O. Tamura and Y. Tamura, Chem. Pharm. Bull., 1991, 39, 2106 CrossRef CAS
-
(a) I. Ugi, R. Meyr, U. Fetzer and C. Steinbrucker, Angew. Chem., 1959, 71, 386 Search PubMed
- CCDC 1009252 (6a), CCDC 1009249 (7a), CCDC 1009250 (7b), CCDC 1009251 (7d).†.
- C. R. Hargreaves and J. B. Manley, http://www.acs.org/content/dam/acsorg/greenchemistry/industriainnovation/roundtable/solvent-selection-guide.pdf, January 18, 2014.
- C. Capello, U. Fischer and K. Hungerbuhler, Green Chem., 2007, 9, 927 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, X-ray crystal and spectral data. CCDC 1009249–1009252. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob01644k |
| ‡ Present address: School of Biological and Chemical Science, Queen Mary University of London, Joseph Priestley Building, Mile End Road, London, E1 4NS, UK. |
| This journal is © The Royal Society of Chemistry 2014 |