Glutathione-triggered disassembly of isothermally responsive polymer nanoparticles obtained by nanoprecipitation of hydrophilic polymers†
Daniel J.
Phillips
,
Joseph P.
Patterson
,
Rachel K.
O'Reilly
and
Matthew I.
Gibson
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, UK CV2 7AL. E-mail: m.i.gibson@warwick.ac.uk; Fax: +44 (0)2476 524112
First published on 15th August 2013
Abstract
The encapsulation and selective delivery of therapeutic compounds within polymeric nanoparticles offers hope for the treatment of a variety of diseases. Traditional approaches to trigger selective cargo release typically rely on polymer degradation which is not always sensitive to the biological location of a material. In this report, we prepare nanoparticles from thermoresponsive polymers with a ‘solubility release catch’ at the chain-end. This release catch is exclusively activated in the presence of intracellular glutathione, triggering an ‘isothermal’ response and promoting a change in polymer solubility. This solubility switch leads to specific and rapid nanoparticle disassembly, release of encapsulated cargo and produces completely soluble polymeric side-products.
Introduction
The application of polymeric nanoparticles for the delivery of therapeutic compounds exhibiting poor pharmacological profiles (e.g. low solubility and high toxicity) holds exciting promise for the treatment of many diseases, particularly cancer.1–5 Nanomedicine approaches should enable improved patient compliance by reducing side-effects and administration frequency. To achieve this aim it is preferable that the nanoparticles can release their cargo specifically at the site of action (normally in the cytosol). Typical release mechanisms include the GFLG peptide sequence, which is sensitive to lysosomal cathepsins,6 or more commonly the degradation of main-chains or cross-links within the constituent polymers.7 Ester and amide linkages are routinely employed for this purpose as they slowly hydrolyse in biological milieu with the assistance of ester-/peptid ases. For example, Harth and co-workers have reported cross-linked polyesters with potential for intracellular delivery.8,9 These particles have been used to entrap high concentrations of hydrophobic therapeutics whilst displaying linear degradation characteristics dependent on their crystallinity. When functionalised with a targeting peptide these particles were used successfully to deliver paclitaxel to tumours exposed to ionising radiation.10 Yang et al. reported the use of polyester/DNA nanoparticles to deliver the gene for vascular endothelial growth factor into stem cells to promote limb growth in mice,11 and Palamoor and Jablonski exploited the gradual degradation of poly(orthoesters) for ocular applications, achieving delivery of epinephrine over a period of several months.12Perhaps the most appealing feature of polyesters is for sustained (slow) release applications. For example, cross-linked polyester nanoparticles required ∼10 days to degrade by 82.5% in the best case,8 whilst emulsion-derived polyesters degraded by only ∼50% after several months.13 However, the non-specific degradation mechanism of such functionality poses two challenges: unwanted, non-specific cargo leaching of targeted delivery agents, and the generation of acidic side-products due to ester hydrolysis.14,15 For instance, the generation of acid within degrading microparticles of poly(lactic-co-glycolic acid) led to regions with pH as low as 1.5 after several days.16 An alternative degradable linkage is the disulfide bond which is sensitive to the primary in vivo reducing agent glutathione (GSH). GSH is attractive given the 1000-fold concentration differential between intra- and extracellular environments.17 This gradient has been exploited to ensure nanocarriers are stable in the circulation, but rapidly disassemble in the intracellular environment.18–21 Zhong and co-workers compared the rate of doxorubicin release from poly(ethyleneglycol)-block-poly(caprolactone) micelles with and without a disulfide linker in a reducing environment. Complete drug release from the disulfide-containing micelles occurred after 10 hours whilst only 20% doxorubicin was released via ester hydrolysis from the non-disulfide containing structures, highlighting the advantage of bioreduction over ester hydrolysis when rapid release is desirable.22,23 However, introduction of disulfide linkages between two distinct polymer blocks can require macromolecular coupling reactions which are not always efficient and require extensive purification.22 We have previously incorporated a disulfide linkage into thermoresponsive polymers whereby the lower critical solution temperature (LCST) can be modulated in response to intracellular GSH. This enabled the soluble–insoluble transition associated with an LCST to be manipulated using biochemical-triggers rather than temperature changes, an “isothermal” transition.24–27 Other examples of isothermal responses include the use of salt gradients,28 redox-active spin labels29 and bacterial binding,30 the key advantage of which is that known responsive polymers can be easily modified to initiate complex responses under more biologically relevant conditions.
The incorporation of redox and thermally responsive components into nanoparticles via complex block copolymer self-assembly is particularly challenging from a synthetic perspective. Alternatively, nanoprecipitation (or interfacial deposition) has emerged as a facile alternative for the preparation of nanoparticles requiring only a hydrophobic polymer. Benefits of this methodology include fast processing times, control over particle size and the ready inclusion of cargo molecules.31,32 However, the need for hydrophobic polymers has essentially constrained this technique to poly(esters), plus some examples of poly(styrenes).32
In this manuscript we prepare nanoparticles from thermoresponsive polymers containing a disulfide-linked ‘solubility release catch’ at their chain-end by the nanoprecipitation technique. This release catch is designed to promote rapid nanoparticle disassembly and cargo release upon encountering intracellular levels of GSH. Furthermore, reduction of the end-group switches the material's LCST rendering the polymer and resulting by-products fully hydrophilic.
Results and discussion
We reasoned that thermoresponsive polymers displaying an LCST could be used to prepare particles via nanoprecipitation if they were held above their transition temperature. Furthermore, by introducing a redox-responsive chain-end ‘solubility release catch’ to generate an isothermal response, it should be possible to programme the polymer such that its LCST increases in the presence of intracellular GSH. This will allow the particle solubility to switch, triggering disassembly and cargo release using only end-group reduction events rather than random degradation of the polymer backbone. Furthermore, the released polymer will be hydrophilic and of sufficiently low molecular weight to be removed by renal elimination, without the need for polymer degradation (Fig. 1).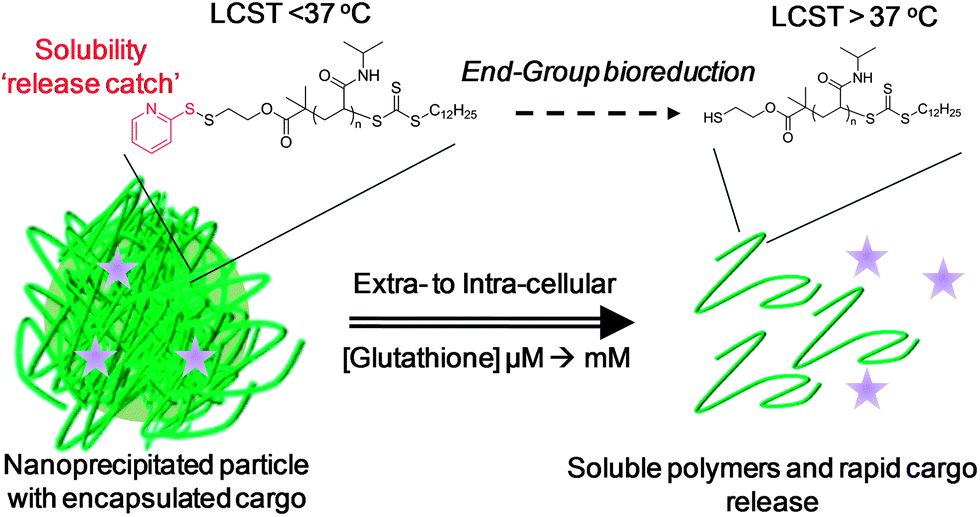 | ||
| Fig. 1 Isothermal disassembly concept: Glutathione reduction of the end-group shifts the LCST to induce a solubility switch and cargo release. | ||
In this study poly(N-isopropylacrylamide), PNIPAM, was selected as the responsive polymer given its strong shift in cloud point (measurable property of the LCST) upon polymer end-group modification provides the desired ‘solubility release catch’.24,25,30 PNIPAMs were prepared using a pyridyl disulfide (PDS)-functionalised RAFT agent and were characterised by 1H NMR, SEC and turbidimetry (ESI†). PNIPAM-1 with Mn = 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1, Mw/Mn = 1.24 and cloud point ∼32 °C was selected for use here, Scheme 1.
400 g mol−1, Mw/Mn = 1.24 and cloud point ∼32 °C was selected for use here, Scheme 1.
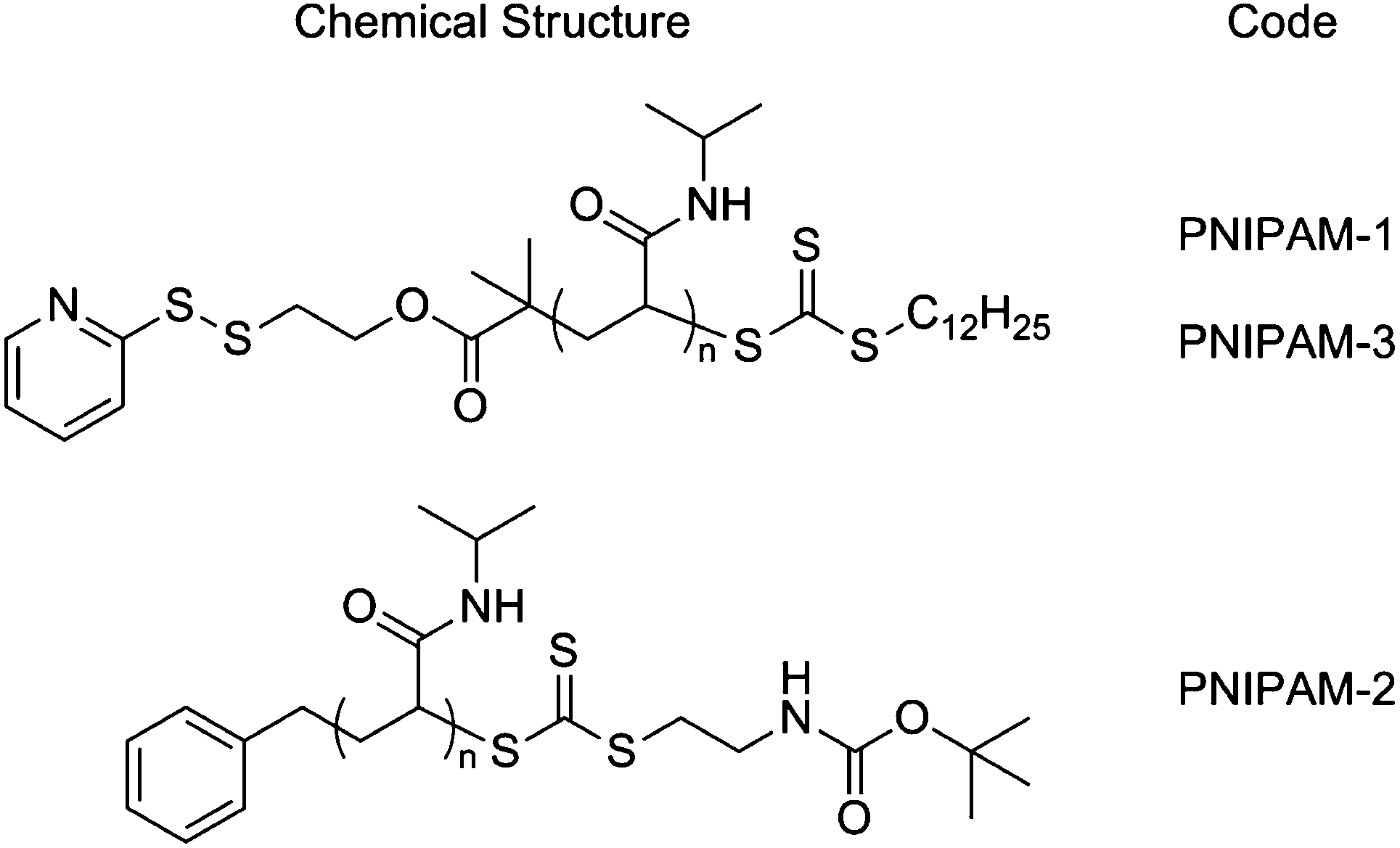 | ||
| Scheme 1 Chemical structures of polymers used in this study: PNIPAM-1 Mn (SEC) = 25400 g mol−1, Mw/Mn = 1.24; PNIPAM-2 Mn (SEC) = 18000 g mol−1, Mw/Mn = 1.13; PNIPAM-3 Mn (SEC) = 19200 g mol−1, Mw/Mn = 1.19 (full characterisation in ESI†). | ||
PNIPAM-1 was dissolved in acetone (1 mg mL−1) and added dropwise into 50 °C water to give a pale blue, opalescent suspension, indicative of nanoparticle formation. Dynamic light scattering (DLS) at 50 °C revealed nanoparticles with an average hydrodynamic diameter of 197 nm (polydispersity 0.2, Fig. 2A) and demonstrated colloidal stability for 7 days without aggregation. As the particles prepared with this particular polymer must be held above the polymer cloud point (to be hydrophobic), cryogenic transmission electron microscopy (cryo-TEM) with rapid vitrification was employed. Cryo-TEM of these soft, responsive, nanoparticles is non-trivial, but with the rapid cooling it was possible to visualize the nanostructures. As exemplified by Fig. 2A, cryo-TEM revealed nanoparticles with mean diameter of 138 nm, which is in broad agreement with the DLS data. These differences can be rationalised by considering the average particle size provided by cryo-TEM is a number average compared to the z-average provided by DLS. Additional images and image-analysis are included in the ESI.†
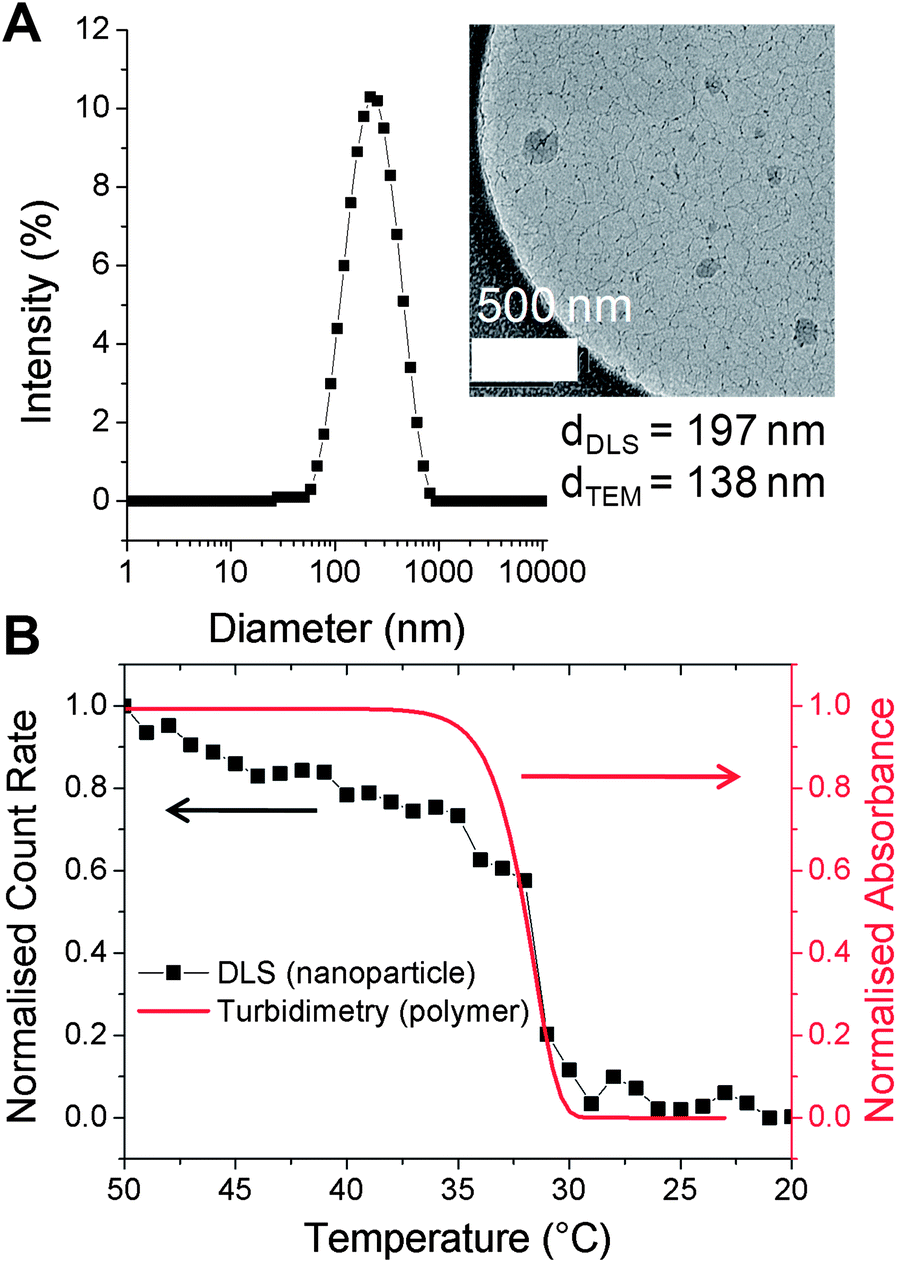 | ||
| Fig. 2 (A) DLS of nanoparticles prepared from PNIPAM-1. (inset: cryo-TEM image); (B) thermal disassembly of nanoparticles (black, DLS) and turbidity data of linear PNIPAM-1 (red). Concentration = 0.5 mg mL−1. Indicated diameters are z-average (DLS) and number-average (TEM); see ESI† for further discussion. | ||
To ensure that the resulting particles retained the intrinsic thermo-responsiveness of their constituent polymer, temperature-dependent DLS was conducted (Fig. 2B). As the nanoparticles were cooled from 50 to 20 °C a decrease in scattering intensity (count rate) was observed indicating disassembly of the particles. This was also confirmed by the visual change from an opalescent suspension to a colourless solution. An overlay of this DLS data with turbidimetric analysis of PNIPAM-1 indicated that disassembly correlated with the cloud point of the polymer.
The principal aim of this study was to evaluate whether the nanoparticles would specifically disassemble in response to intracellular levels of GSH via reduction of the PDS solubility release catch. To verify a GSH-induced physical response, the nanoparticles were held at 50 °C, GSH was added at 10 μM (extracellular) or 1 mM (intracellular) and the particle size measured. Addition of 10 μM GSH showed no changes visually, or by DLS. In contrast, addition of 1 mM GSH resulted in rapid precipitation of the nanoparticles (Fig. 3A, inset) and a significant increase in the particle size to ca. 1.5 microns. It should be noted that this experiment is not intended to mimic in vivo conditions, but to demonstrate the concept of isothermal disassembly. Hence, aggregation is observed as the experimental temperature, which is above the cloud point of both the original and GSH-modified polymer (see ESI†), means the resulting disassembled polymer chains exist in a globular conformation. Further exemplification under more physiological conditions, leading to dissolution of the polymers is described later (vide infra).
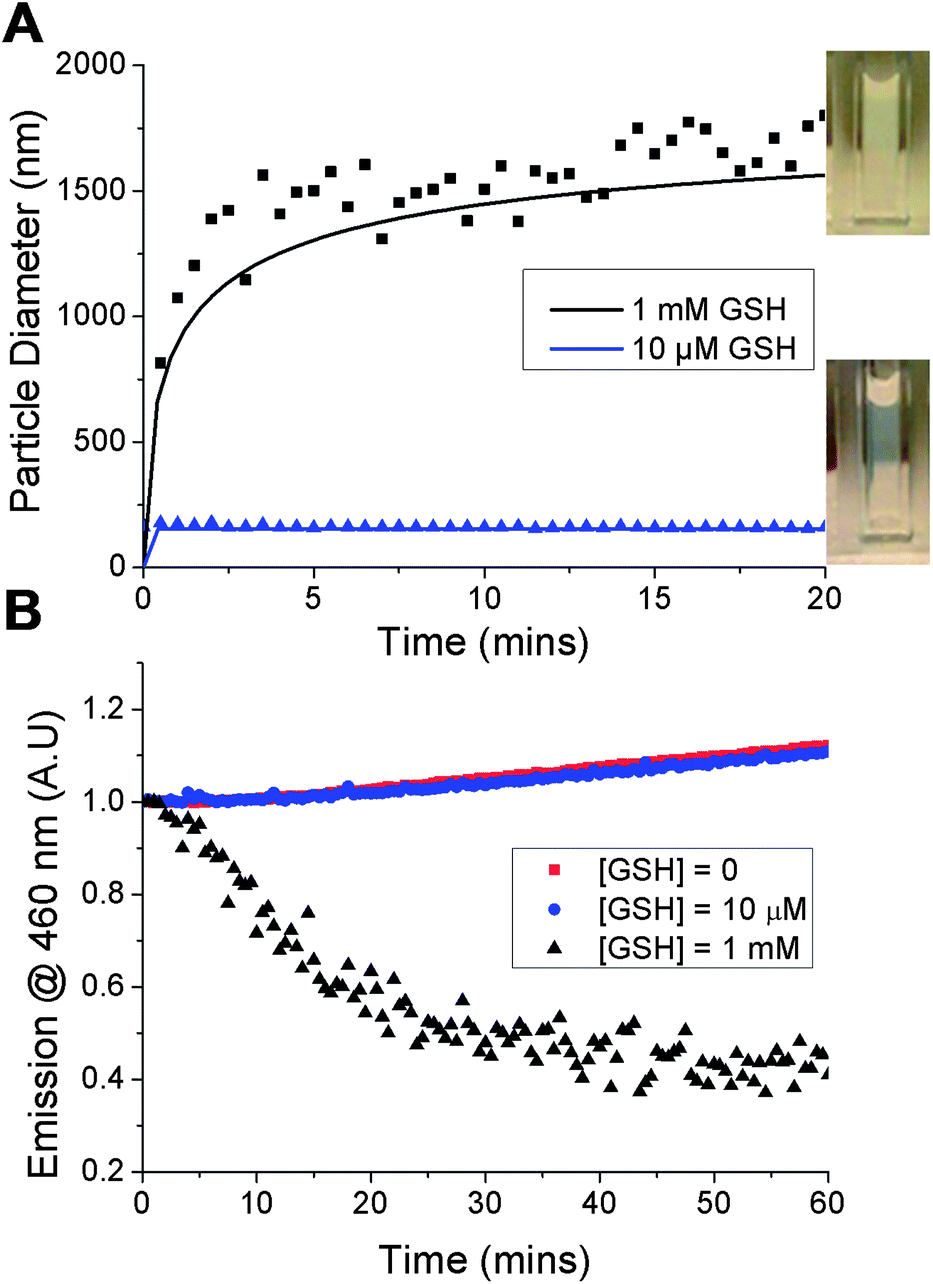 | ||
| Fig. 3 GSH-triggered disassembly of nanoparticles: (A) dynamic light scattering – line of best fit is shown as solid curve (inset: nanoparticle appearance following addition of GSH); (B) DPH fluorescence change of loaded nanoparticles incubated with varying GSH concentrations. | ||
A fluorescent probe, diphenylhexatriene (DPH), was encapsulated in the particle by co-precipitation as a model cargo. DPH only fluoresces in hydrophobic environments allowing direct observation of its release into an aqueous environment.33 Addition of up to 10 μM GSH caused no decrease in fluorescence intensity, with the slight increase observed attributable to evaporation given the elevated (50 °C) temperatures being used. Addition of 1 mM GSH led to an almost immediate decrease in fluorescence intensity demonstrating that particles assembled from thermoresponsive polymers are capable of releasing a cargo in response to a biochemical stimulus (Fig. 3B). The observed release agreed with DLS data and confirmed the polymer phase transition as the trigger. Release was both specific, yet significantly faster than that observed with disulfide-linked micelles which required over 10 hours7 or 40 hours34 to release their cargo. We believe the fast release kinetics may be due to the cooperative nature of pNIPAM's LCST; if a small fraction of the polymer chains undergo an increase in their LCST (due to bioreduction), then the LCST of those remaining is also raised, facilitating disassembly.35 This cooperative LCST behaviour is not found with all thermoresponsive polymers, such as poly[oligo (ethyleneglycol)methacrylates], highlighting the importance of the choice of PNIPAM.35 Other polymers with co-operative LCST behaviour include elastin-side chain poly(methacrylates)36 and poly(N-vinyl lactams). The observation may also be explained by considering the location of encapsulated material in different nanoparticle structures. For example, cross-linked micelles contain hydrophobic cargo within the core. In our particles however, we expect the cargo molecules to be diffusely spread throughout the structure, meaning cargo release occurs as soon as the outer polymers pass through their phase transition.
To ensure that any LCST change was due to reduction of the pyridyl disulfide α-end-group by the cysteine residue present in GSH, and not displacement of the trithiocarbonate ω-end-group by the glutamic acid residue (ESI† for mechanism), control UV-vis experiments were performed using two trithiocarbonate-containing PNIPAM samples of similar molecular weights. The samples differed by their α-end-group: PNIPAM-2 and PNIPAM-3 contained a tert-butyl carbamate and PDS group respectively (see ESI† for characterisation). The distinctive UV-vis absorption of the RAFT agent at 309 nm was monitored as a function of GSH concentration and no change in absorbance was observed with PNIPAM-2 (see ESI†). A slight increase in absorbance at 309 nm and 343 nm was observed with PNIPAM-3 and was attributed to the release of pyridine thione, a by-product of thiol-PDS exchange, whose spectrum exhibits some absorbance overlap with that of the trithiocarbonate (Fig. 4).
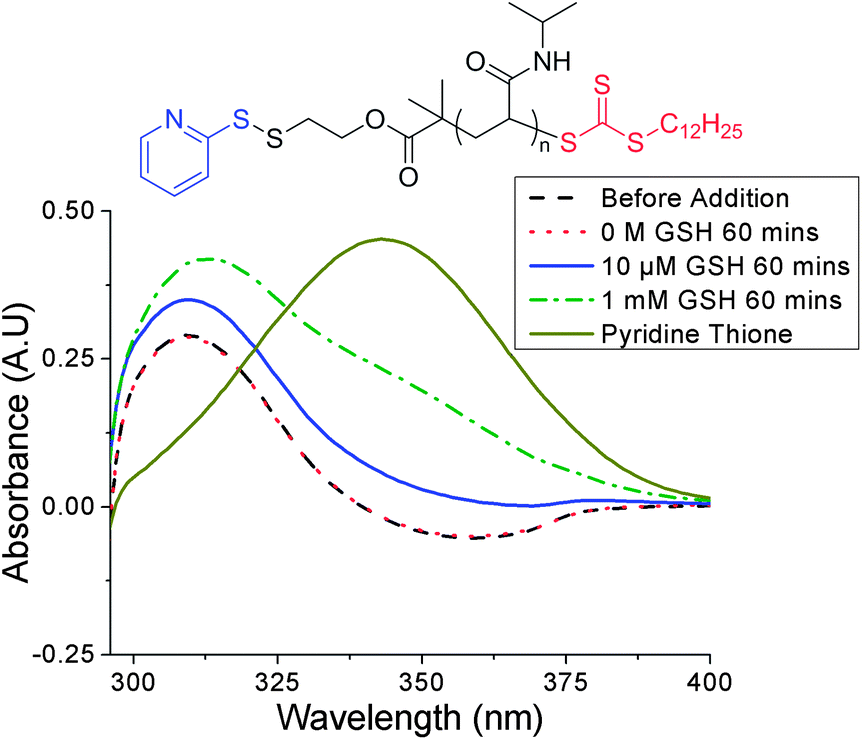 | ||
| Fig. 4 UV-vis data before and after incubation of PNIPAM-3 with varying GSH concentrations. No significant change in absorbance at 309 nm indicated no trithiocarbonate cleavage (slight increases at 309 nm and 343 nm due to release of pyridine thione). | ||
The motivation for using an isothermally responsive polymer nanoparticle, as opposed to a conventional hydrophobic polymer, lies in the solubility switch observed upon end-group bioreduction. PNIPAM-1 had an LCST ∼ 32 °C which is not suitable for an in vivo application, therefore statistical copolymers of NIPAM and N-hydroxyethyl acrylamide (HEA) were prepared and their thermoresponsivity measured (ESI†). The polymer with 5 mol% HEA was found to have a cloud point ∼35 °C at 0.5 mg mL−1 which increased to ∼37 °C upon GSH reduction (Fig. 5A). This polymer was used to form nanoparticles with diameter ∼200 nm (DLS and cryoTEM in ESI†). The nanoparticles were stable at 37 °C and in the presence of 10 μM GSH, the extracellular concentration. Increasing the GSH concentration to 1 mM (as found in many intracellular compartments) resulted in particle disassembly (blue suspension to colourless solution) and dye release experiments showed quenching of DPH fluorescence (Fig. 5B). This demonstrates that by fine-tuning the polymer structure and hence transition temperature, isothermal disassembly can be employed to trigger cargo release from nanoparticles specifically in response to biochemical triggers. We anticipate that this concept could also be translated to degradable thermoresponsive polymers such as elastin-based peptides37 or poly(phosphoesters).38
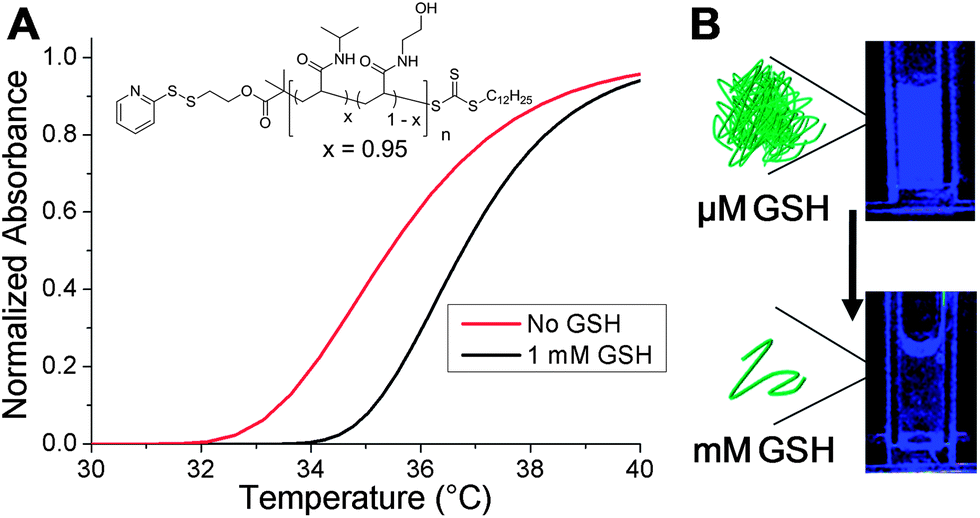 | ||
| Fig. 5 (A) Turbidimetric response of linear P(NIPAM95-co-HEA5) with and without addition of 1 mM GSH; (B) visual change in the fluorescence of nanoparticles (37 °C), before and after addition of 1 mM GSH. Fluorescence change is due to DPH release and quenching. | ||
Conclusions
To summarise, this manuscript demonstrates the use of isothermally responsive polymers which are capable of shifting their phase behaviour upon the application of the specific biochemical stimulus glutathione. Nanoparticles were formed by holding PNIPAM above its transition temperature allowing the use of nanoprecipitation; a method typically associated with hydrophobic polymers. The nanoparticles were shown to specifically disassemble and release a cargo upon exposure to intracellular levels of glutathione with complete release in around 30 minutes. The fast, but highly specific release kinetics offers a significant advantage over other reducible carriers such as micelles. Importantly the particles were stable for extended periods in the presence of circulatory-levels of glutathione which is essential for a disulfide-linked delivery system where cargo leaching is undesirable. The mechanism of disassembly was shown to be due to the isothermal phase transition of the PNIPAM, caused by rapid and selective cleavage of the disulfide-linked end-group. The ability to tune this transition temperature using simple co-polymerisation methodologies was also demonstrated to generate responses at physiological temperatures. This report represents a powerful strategy to generate nanoparticles from hydrophilic polymers, without the need for cross-linking or block copolymers. Furthermore, these structures can be tuned to trigger release in response to specific cellular stimuli without requiring a non-specific polymer degradation step. Future work will include in vitro assessment of drug delivery, the incorporation of degradable functionality into the polymer main-chain and targeting functionalities.Experimental section
Synthetic procedures
The synthesis and characterisation of the functionalised RAFT agents and polymers are described in the ESI.†General procedure for nanoparticle preparation
Polymer (5 mg) was dissolved in acetone (5 mL) and added dropwise to a round bottom flask containing a stir bar and deionised water (10 mL) held at 50 °C over a period of one hour. The flask was left stirring, open to air at 50 °C for several hours to allow acetone to evaporate. The resulting pale blue, partially turbid solution was stored at 50 °C and used within 7 days. The preparation of 1,6-diphenyl-1,3,5-hexatriene-loaded nanoparticles followed the same methodology only DPH was dissolved alongside polymer in acetone at a concentration of 0.05 mg mL−1.Analytical and physical methods
NMR spectroscopy (1H, 13C) was conducted on a Bruker DPX-400, Bruker DRX-500, a Bruker AV III-600 or a Bruker AV II-700 spectrometer using deuterated chloroform as solvent. All chemical shifts are reported in ppm (δ) relative to tetramethylsilane (TMS). High resolution mass spectra were recorded on a Bruker Electrospray Ultra-High Resolution tandem TOF mass spectrometer using electrospray ionization (ESI) in positive mode on samples prepared in methanol. Degraded sample mass spectral analysis was carried using a Bruker MaXis UHR-Q-TOF mass spectrometer using ESI in positive mode over a scan range of 500–7000 m/z. Samples were prepared in methanol, diluted 20-fold in 50:50 methanol–water and introduced by direct infusion at 90 μL h−1. Source conditions were: end plate offset at −500 V; capillary at −4500 V; nebulizer gas (N2) at 1.6 bar; dry gas (N2) at 8 L min−1; dry temperature at 180 °C. Ion transfer conditions were: ion funnel RF at 400 Vpp; multiple RF at 400 Vpp; quadruple low mass set at 455 m/z; collision energy at 5.0 eV; collision RF at 1200 Vpp; ion cooler RF at 250–600 Vpp; transfer time set at 121 μs; pre-pulse storage time set at 15 μs. Calibration was completed with sodium formate (10 mM). FTIR spectra were acquired using a Bruker Vector 22 FTIR spectrometer with a Golden Gate diamond attenuated total reflection cell. A total of 64 scans were collected on samples in their native (dry) state. UV-visible spectra were obtained using an Agilent Cary 60 UV-visible spectrophotometer at a “medium” scan-speed. SEC analysis was performed on a Varian 390-LC MDS system equipped with a PL-AS RT/MT autosampler, a PL-gel 3 μm (50 × 7.5 mm) guard column, two PL-gel 5 μm (300 × 7.5 mm) mixed-D columns using DMF with 5 mM NH3BF4 at 50 °C as the eluent at a flow rate of 1.0 mL min−1. The GPC system was equipped with ultraviolet (UV) (set at 280 nm) and differential refractive index (DRI) detectors. Narrow molecular weight PMMA standards (200–1.0 × 106 g mol−1) were used for calibration using a second order polynomial fit. The cloud points were measured using an Optimelt MPA100 system (Stanford Research Systems). The recorded turbidimetry curve was normalized between values of 0 and 1. The cloud point was defined as the temperature corresponding to a normalized absorbance of 0.5. A polymer concentration of 0.5 mg mL−1 and a constant heating rate of 1 °C min−1 were used for all experiments. Fluorescence measurements were performed using a Biotech Synergy HT and processed using the Gen5 software package. Particle size analysis was determined by dynamic light scattering using a Malvern Zetasizer Nano ZS instrument. A 4 mW He–Ne 633 nm laser module was used and scattered light was measured at 173° (back scattering). The attenuator and position was selected automatically by the instrument.
Cryogenic transmission electron microscopy samples (0.5 mg mL−1 in water) were examined using a Jeol 2010F TEM operated at 200 kV and imaged using a GatanUltrascan 4000 camera. Images were captured using Digital Micrograph software (Gatan). A 3 μL droplet of the sample solution held at 50 °C was rapidly transferred to a holey carbon-coated copper grid, and the grid was blotted to remove excess solution. Subsequently, the grid was plunged into liquid ethane to vitrify the sample. The temperature of the cryogenic stage was maintained below −170 °C, using liquid nitrogen, during imaging. Where appropriate, particle size analysis was performed using ImageJ.
Nanoparticle fluorescence assay
A generalised procedure for the fluorescence assay is as follows. Nanoparticles (190 μL) were incubated in a 96-well plate at 50 °C for 1 hour. 10 μL water or concentrated glutathione solution was added to give final glutathione concentrations of 0, 0.01 and 1 mM and the fluorescence monitored over a period of 2 hours. Excitation wavelength set at 360 nm, emission wavelength at 460 nm and the plate maintained at 50 °C throughout.Acknowledgements
Equipment used was supported by the Innovative Uses for Advanced Materials in the Modern World (AM2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). MIG is a Birmingham Science City Interdisciplinary Research Fellow funded by the Higher Education Funding Council for England (HEFCE). DJP thanks the University of Warwick for Funding.Notes and references
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–359 CrossRef CAS.
- R. Duncan and R. Gaspar, Mol. Pharmaceutics, 2011, 8, 2101–2141 CrossRef CAS.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- R. Duncan, J. B. Lloyd and J. Kopeček, Biochem. Biophys. Res. Commun., 1980, 94, 284–290 CrossRef CAS.
- S. R. Abulateefeh, S. G. Spain, K. J. Thurecht, J. W. Aylott, W. C. Chan, M. C. Garnett and C. Alexander, Biomater. Sci., 2013, 1, 434–442 RSC.
- A. E. van der Ende, E. J. Kravitz and E. Harth, J. Am. Chem. Soc., 2008, 130, 8706–8713 CrossRef CAS.
- A. E. van der Ende, V. Sathiyakumar, R. Diaz, D. E. Hallahan and E. Harth, Polym. Chem., 2010, 1, 93–96 RSC.
- R. J. Passarella, D. E. Spratt, A. E. van der Ende, J. G. Phillips, H. Wu, V. Sathiyakumar, L. Zhou, D. E. Hallahan, E. Harth and R. Diaz, Cancer Res., 2010, 70, 4550–4559 CrossRef CAS.
- F. Yang, S.-W. Cho, S. M. Son, S. R. Bogatyrev, D. Singh, J. J. Green, Y. Mei, S. Park, S. H. Bhang, B.-S. Kim, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2009, 107(8), 3317–3322 CrossRef.
- M. Palamoor and M. M. Jablonski, Mol. Pharmaceutics, 2012, 10, 701–708 Search PubMed.
- A. Musyanovych, J. Schmitz-Wienke, V. Mailänder, P. Walther and K. Landfester, Macromol. Biosci., 2008, 8, 127–139 CrossRef CAS.
- M. Vert, Biomacromolecules, 2005, 6, 538–546 CrossRef CAS.
- D. Putnam, Nat. Mater., 2008, 7, 836–837 CrossRef CAS.
- K. Fu, D. W. Pack, A. M. Klibanov and R. Langer, Pharm. Res., 2000, 17, 100–106 CrossRef CAS.
- C. Wu, C. Belenda, J.-C. Leroux and M. A. Gauthier, Chem.–Eur. J., 2011, 17, 10064–10070 CrossRef CAS.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS.
- T.-i. Kim and S. W. Kim, React. Funct. Polym., 2011, 71, 344–349 CrossRef CAS.
- B. Khorsand Sourkohi, A. Cunningham, Q. Zhang and J. K. Oh, Biomacromolecules, 2011, 12, 3819–3825 CrossRef CAS.
- S. Aleksanian, B. Khorsand, R. Schmidt and J. K. Oh, Polym. Chem., 2012, 3, 2138–2147 RSC.
- H. Sun, B. Guo, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2009, 30, 6358–6366 CrossRef CAS.
- Y.-L. Li, L. Zhu, Z. Liu, R. Cheng, F. Meng, J.-H. Cui, S.-J. Ji and Z. Zhong, Angew. Chem., Int. Ed., 2009, 48, 9914–9918 CrossRef CAS.
- D. J. Phillips and M. I. Gibson, Chem. Commun., 2012, 48, 1054–1056 RSC.
- D. J. Phillips and M. I. Gibson, Biomacromolecules, 2012, 13, 3200–3208 CrossRef CAS.
- M. J. Summers, D. J. Phillips and M. I. Gibson, Chem. Commun., 2013, 49, 4223–4225 RSC.
- M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2013, 42, 7204–7213 RSC.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS.
- H. Fu, D. M. Policarpio, J. D. Batteas and D. E. Bergbreiter, Polym. Chem., 2010, 1, 631–633 RSC.
- J. Shepherd, P. Sarker, K. Swindells, I. Douglas, S. MacNeil, L. Swanson and S. Rimmer, J. Am. Chem. Soc., 2010, 132, 1736–1737 CrossRef CAS.
- T. Govender, S. Stolnik, M. C. Garnett, L. Illum and S. S. Davis, J. Controlled Release, 1999, 57, 171–185 CrossRef CAS.
- C. Zhang, V. J. Pansare, R. K. Prud'homme and R. D. Priestley, Soft Matter, 2012, 8, 86–93 RSC.
- Y. Saaka, R. C. Deller, A. Rodger and M. I. Gibson, Macromol. Rapid Commun., 2012, 33, 779–784 CrossRef CAS.
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS.
- N. S. Ieong, M. Hassan, D. J. Phillips, Y. Saaka, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2012, 3, 794–799 RSC.
- F. Fernàndez-Trillo, J. C. M. van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Chem. Commun., 2008, 2230–2232 RSC.
- M. Shah, P.-Y. Hsueh, G. Sun, H. Y. Chang, S. M. Janib and J. A. MacKay, Protein Sci., 2012, 21, 743–750 CrossRef CAS.
- Y.-C. Wang, L.-Y. Tang, Y. Li and J. Wang, Biomacromolecules, 2008, 10, 66–73 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: This includes additional turbidimetry curves for all polymers tested. See DOI: 10.1039/c3py00991b |
| This journal is © The Royal Society of Chemistry 2014 |