Bi- and tri-dentate imino-based iron and cobalt pre-catalysts for ethylene oligo-/polymerization
Jing
Ma
a,
Chun
Feng
a,
Shaoli
Wang
b,
Ke-Qing
Zhao
a,
Wen-Hua
Sun
*b,
Carl
Redshaw
*ac and
Gregory A.
Solan
d
aCollege of Chemistry and Materials Science, Sichuan Normal University, Jing-An Road 5, Chengdu, 610066, China
bKey Laboratory of Engineering Plastics, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: whsun@iccas.ac.cn; Fax: +86 10 62618239; Tel: +86 10 62557955
cDepartment of Chemistry, University of Hull, Cottingham Rd, Hull, HU6 7RX, UK. E-mail: c.redshaw@hull.ac.uk; Fax: +44 (0)1482 466410; Tel: +44 (0)1482 465219
dDepartment of Chemistry, University of Leicester, University Road, Leicester LE1 7RH, UK. E-mail: gas8@le.ac.uk
First published on 16th December 2013
Abstract
Recent progress on the use of iron and cobalt complex pre-catalysts for ethylene reactivity is reviewed. The review is organized in terms of the denticity of the chelate ligands employed, with particular reference to the influence of the ligand frameworks and their substituents on the catalytic performance for ethylene oligomerization/polymerization catalysis. The majority of the systems bear tri-dentate ligation at the iron/cobalt centre, though it is clear that bi-dentate iron/cobalt complex pre-catalysts have also attracted significant attention. Such systems produce in most cases highly linear products ranging from oligomeric α-olefins to high molecular weight polyethylene, and as such are promising candidates for both academic and industrial considerations.
 Jing Ma | Jing Ma received her BSc from Sichuan Normal University in July 2011. She is currently a second-year graduate student with Carl Redshaw in the College of Chemistry and Materials Science, Sichuan Normal University. Her current research interests focus on olefin polymerization. |
 Chun Feng | Chun Feng received his BSc from Sichuan University of Science & Engineering and Ph.D. degree from Chengdu Institute of Biology, Chinese Academy of Sciences. He was a postdoctoral fellow with Professor Zhangjie Shi at Peking University, and is currently an associate professor at Sichuan Normal University. Research interests include synthetic methodology. |
 Shaoli Wang | Shaoli Wang received her M.S. at Beijing University of Technology in 2011, and now is a Ph.D. candidate at the Institute of Chemistry, Chinese Academic Sciences. Her current research focuses on late transition metal complexes in ethylene polymerization. |
 Ke-Qing Zhao | Ke-Qing Zhao received his Ph.D. from Sichuan University in 1997 under Professor Liang-Fu Zhang. After postdoctoral research at the National Taiwan University with Professors J.-T. Chen and S.-T. Liu on late-transition metal catalyzed olefin polymerization, he worked in Yo Shimizu's group (AIST, Japan) for a year on discotic liquid crystalline semiconductors. He is now a full professor at Sichuan Normal University. Current research interests include liquid crystalline semiconductors as well as transition metal catalyzed polymerizations. |
 Wen-Hua Sun | Wen-Hua Sun received his B.Sc. in chemistry at Lanzhou University (1986), his M.S./Ph.D. degrees in physical chemistry at Lanzhou Institute of Chemical Physics (LICP, 1989/1994). He worked in LICP as a Research Associate (1989) and Associate Professor (1993), and at Hokkaido University with fellowships from Japan Society for the Promotion of Science (1995), Center of Excellence (1997) and Japan Science and Technology Corporation (1998). Since 1999, he has served as a Professor of Chemistry and Polymer Science at the Institute of Chemistry, Chinese Academy of Sciences. He has over 250 publications and 45 granted patents; several have been developed for industrial (pilot) processes. |
 Carl Redshaw | Carl received his BSc (Hons) and Ph.D. from Newcastle University, was a Robert A. Welch Fellow at the University of Texas, Austin, and a postdoc with the late Prof. Sir G. Wilkinson at Imperial College (IC). Following further post-docs at Durham University and at IC, he was awarded a Leverhulme Special Research Fellowship. In 1999, he moved to the University of East Anglia (UEA) and was Lecturer, Senior lecturer and Reader in Chemistry. He moved to the University of Hull in 2012 to take up the Chair of Inorganic Materials. He is also currently Guest Professor at Sichuan Normal University in Chengdu, China. |
 Gregory A. Solan | Greg Solan received his B.Sc. (Hons) degree in Chemistry with European studies from the University of Sussex in 1988 before moving to the University of Cambridge to begin his doctoral work with Martin Mays. After completing his Ph.D. degree (1992), he was a postdoctoral fellow at the Université de Lausanne with the late Carlo Floriani, at the University of Edinburgh with Richard Winpenny, and at Imperial College London with Vernon Gibson. In 1999, he was appointed to a lectureship in Inorganic Chemistry at the University of Leicester and promoted to Senior Lecturer in 2007. His research interests cover coordination and organometallic chemistry and their use in catalysis and new inorganic materials synthesis. |
1. Introduction
The annual consumption of plastics continues to rise and is expected to increase by 4% year on year between now and 2016.1 Of this, almost half the production revolves around the formation of polyethylene and polypropylene products (Chart 1), and so there not only remains a drive for new competitive processes to produce known α-olefin derived plastics, but also a need to develop new polymer types. Given this, the search for new catalyst systems remains of great interest in the academic community and pivotal to the plastics industry.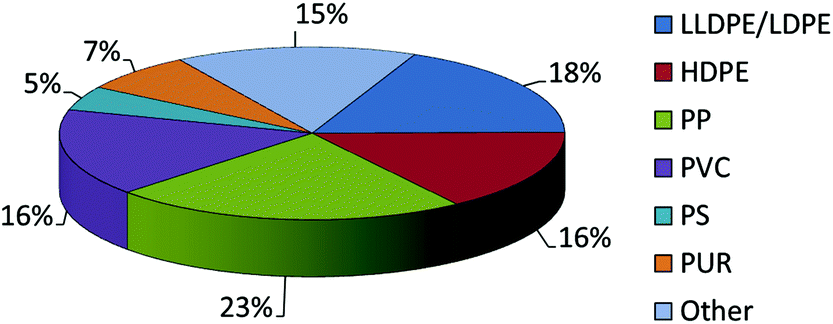 | ||
| Chart 1 2012 World polymer demand. | ||
Metallocenes,2 and a number of supported catalyst systems, for example the Phillips catalyst,3 remain central to current plastic production; however, the search for cheaper and more active catalytic systems, particularly those that will operate at high temperatures remains an area of immense interest. Of the more modern homogeneous systems developed, those based on the metals of iron or cobalt and bearing bis(imino)pyridine ligand sets have made a significant impact in the literature, particularly as they allow for controllable product formation (oligomers versus polymers).4,5 Indeed, the first reports initiated extensive investigations into the use of iron or cobalt-based complex pre-catalysts bearing bis(imino)pyridine type ligands (Chart 2) that continues apace to this day. The reader is directed to a raft of review articles that focus on early and some more recent developments.6–19 The main driving force for such studies is to access new highly branched polyethylene products, whilst getting away from the more oxophilic nature of early transition metal systems. In the past half-dozen or so years, stable complex pre-catalysts that exhibit high thermal stability have been reported for both cobalt20 and iron complexes.21 Inspired by catalytic systems which revealed high activities at high temperature, but which were inert at room temperature, our group revisited the bis(imino)pyridylmetal (Fe or Co) complexes derived from benzhydryl-substituted anilines,17,22,23 and confirmed high activity for both iron and cobalt systems during ethylene polymerization. As well as active pre-catalysts bearing N^N^N tri-dentate ligation at the metal (Fe or Co), a number of N^N bi-dentate metal (Fe or Co) complexes have also been investigated for ethylene reactivity.24–26 Subsequently, numerous complex pre-catalysts formed by fine tuning of the (imino)pyridine motifs and related ligands have been screened, and in light of this, a full review article covering recent progress on such systems is presented here. In this review, the synthesis of each family of iron and cobalt complex pre-catalysts is discussed together with characterization data and their oligo-/polymerization behaviour; complexes bearing bi-dentate ligand sets are dealt with initially, followed by tri-dentate ligand sets and finally combinations thereof.
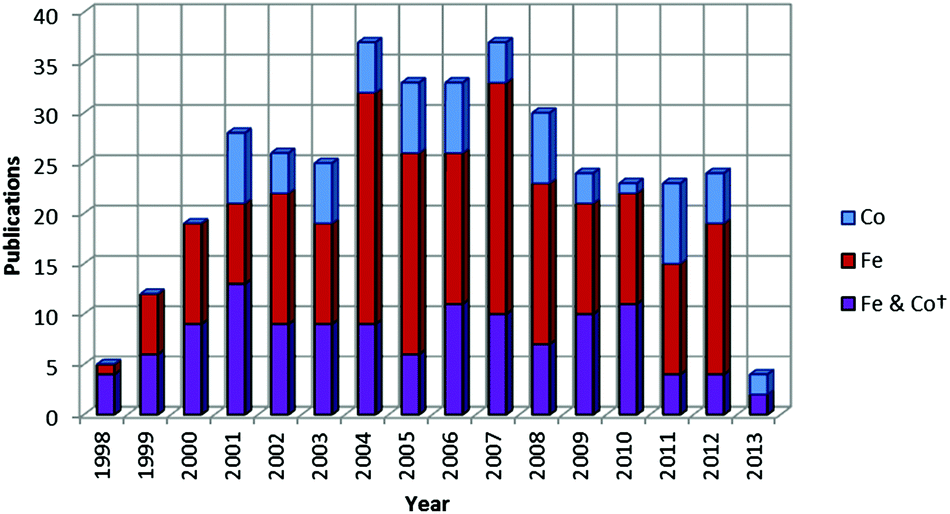 | ||
| Chart 2 Number of publications per year concerned with the study of iron and cobalt olefin polymerization/oligomerization catalysts since their discovery in 1998 (*as of 05/06/13; patent applications not included). | ||
2. Bi-dentate iron and cobalt pre-catalysts
Studies of iron/cobalt complex pre-catalysts were initiated on tri-dentate systems and good activities were generally reported,4,5 whilst by comparison, bi-dentate iron/cobalt complexes have tended to exhibit relatively low activity for ethylene reactivity. Such low activity is probably due to the formation of unstable active species, which given the greater open space available at the metal, tend to be more prone to coordinating with other species present in the system (leading to deactivation). The result of this poor performance is that less attention has been paid to bi-dentate iron/cobalt complex pre-catalysts, although readily available iminopyridine derivatives have been used to form cobalt complexes,27–30 which were found to exhibit high activity with selectivity toward dimerization. Following on from this, recent investigations involving the design of new N^N bi-dentate ligands and the metal complexes thereof, have revealed promising results for ethylene polymerization.23,24Methylene-linked imino and pyridyl N^N bi-dentate iron and cobalt complexes of the type 1–5 (Scheme 1) were found to exhibit good activities towards ethylene oligomerization in the presence of MMAO or MAO (Table 1),24 with activities as high as 8.01 × 105 g mol −1 (M) h−1; the main products were C4 or C6 (entry 8, Table 1).
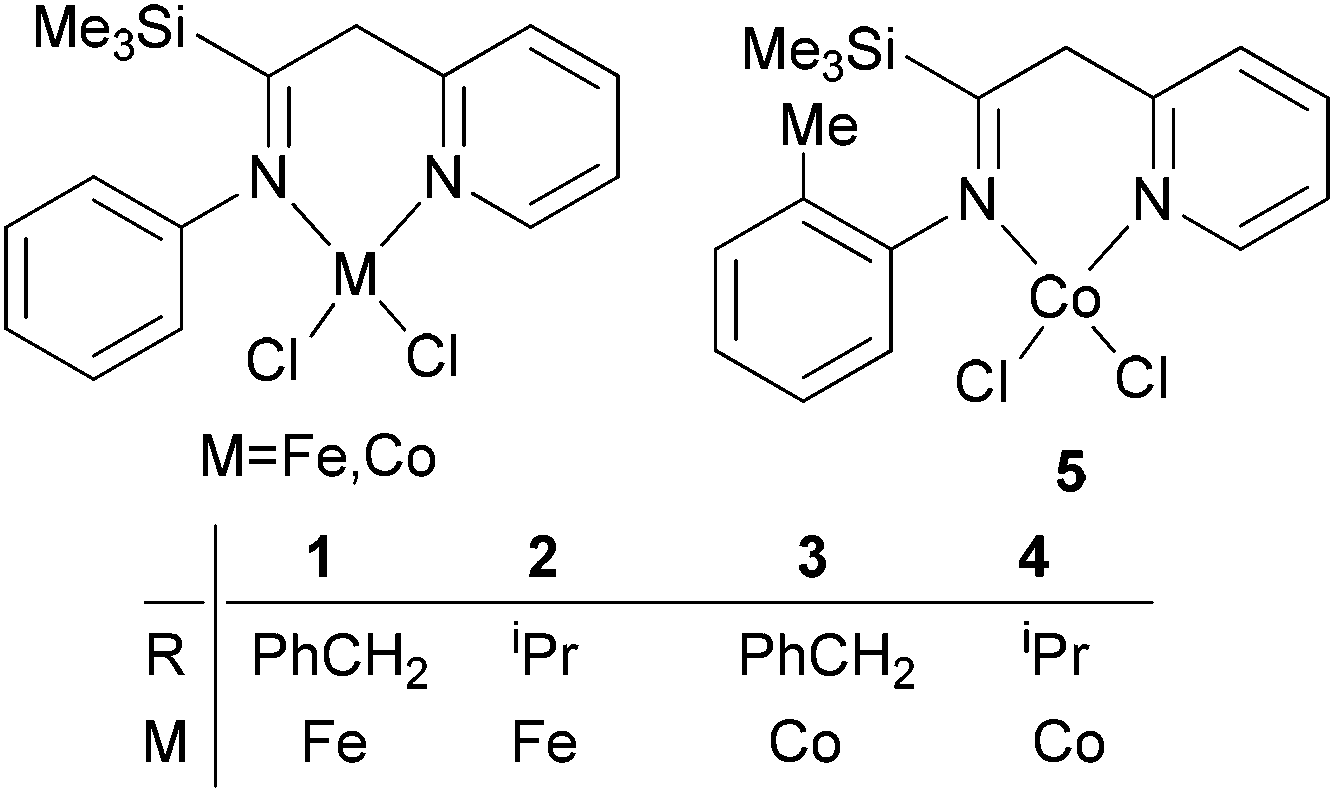 | ||
| Scheme 1 N,N-Bi-dentate iron and cobalt complexes.24 | ||
| Entry | Complex | Co-catalyst | Al/M | Activityb | C4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c [%] c [%] |
C6c [%] |
|---|---|---|---|---|---|---|
| a Conditions: 5 μmol of pre-catalyst, 30 mL of toluene, 1 atm ethylene, 20 °C, 0.5 h. b kg mol−1 (M) h−1. c Weight percent determined by GC analysis. | ||||||
| 1 | 1 | MMAO | 1000 | 233 | 98.9 | 1.1 |
| 2 | 1 | MMAO | 1500 | 550 | 92.8 | 7.2 |
| 3 | 1 | MMAO | 2000 | 518 | 99.3 | 0.7 |
| 4 | 2 | MMAO | 1500 | 430 | 99.4 | 0.6 |
| 5 | 3 | MMAO | 1000 | 426 | 99.4 | 0.6 |
| 6 | 3 | MMAO | 1500 | 470 | 99.5 | 0.5 |
| 7 | 3 | MMAO | 2000 | 623 | 99.8 | 0.2 |
| 8 | 4 | MMAO | 2000 | 801 | 99.6 | 0.4 |
| 9 | 3 | MAO | 500 | 8.5 | 3.0 | 97.0 |
| 10 | 3 | MAO | 800 | 24.6 | 19.8 | 80.2 |
| 11 | 3 | MAO | 1000 | 12.8 | 10.5 | 89.5 |
| 12 | 4 | MAO | 800 | 11 | 21.0 | 79.0 |
| 13 | 5 | MMAO | 800 | 142 | 98.4 | 1.6 |
| 14 | 5 | MMAO | 2000 | 129 | >99 | |
| 15 | 5 | MAO | 800 | 4 | 81.6 | 18.4 |
| 16 | 5 | MAO | 2000 | 1.2 | 37.8 | 62.2 |
Kempe et al. have investigated the 6-aryl-substituted N,N-bi-dentate iron and cobalt complexes 6–9 (Scheme 2) for ethylene oligomerization.31 In the case of the iron complexes 6 and 8, scant activity was observed (entries 1–4, Table 2), whereas the cobalt systems 7 and 9 exhibited high selectivity towards 1-butene (entries 5–8, Table 2). High ethylene dimerization selectivities were also observed for iminopyridine complexes bearing less sterically demanding substituents at the pyridine 6-position.31 Upon activation with triethylaluminium (TEA) rather than methylaluminoxane (MAO), reduced activity was observed and whilst the formation of polymeric by-products was suppressed, the dimerization selectivity increased to over 95% (entries 5–8, Table 2). The results obtained with cobalt complexes 7 and 9 were similar to (nearly as good as) the results observed by Bianchini and coworkers, who observed for iminopyridine cobalt complexes (activated with MAO) bearing a 6-phenyl or 6-naphthyl substituent on the pyridine ring, the formation of oligomerization products and butenes.27–30
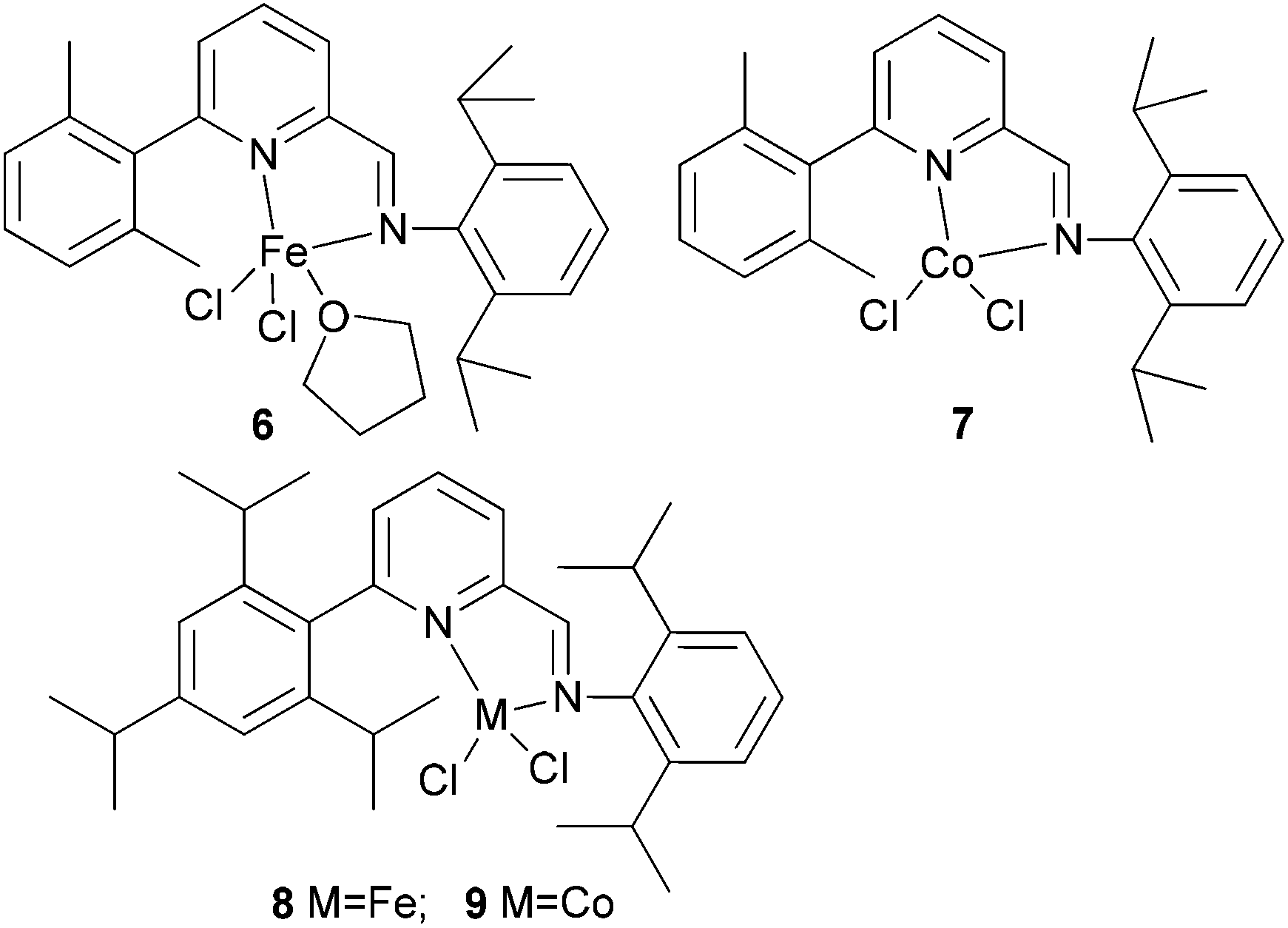 | ||
| Scheme 2 N,N-Bi-dentate iron and cobalt complexes with 6-aryl-substituted iminopyridine ligands.31 | ||
| Entry | Complex | Activator | Al/M | Conversionb | m pol/g | C4c/g |
C6/g |
|---|---|---|---|---|---|---|---|
| a Conditions: 10 μmol of pre-catalyst, 260 mL of toluene, 5 bar ethylene, 30 °C, 15 min. b kg (mol h bar)−1. c >95% 1-butene. | |||||||
| 1 | 6, Fe | MAO | 500 | 2 | 0.05 | — | — |
| 2 | 6, Fe | TEA | 200 | 0 | — | — | — |
| 3 | 8, Fe | MAO | 500 | 2 | 0.06 | — | — |
| 4 | 8, Fe | TEA | 200 | 0 | — | — | — |
| 5 | 7, Co | MAO | 500 | 358 | 0.06 | 3.95 | 0.55 |
| 6 | 7, Co | TEA | 200 | 152 | — | 1.85 | 0.05 |
| 7 | 9, Co | MAO | 500 | 472 | 0.06 | 5.00 | 0.80 |
| 8 | 9, Co | TEA | 200 | 172 | — | 2.10 | 0.05 |
Solan et al. have used the 6-aryl group to bridge two iminopyridine N,N-bi-dentate iron (10) and cobalt (11) complexes (Scheme 3).32 While the diiron species proved inactive on treatment with MAO, the dicobalt complex did exhibit low activity and afforded mixtures of oligomeric products based on short chain α-olefins and internal olefins.
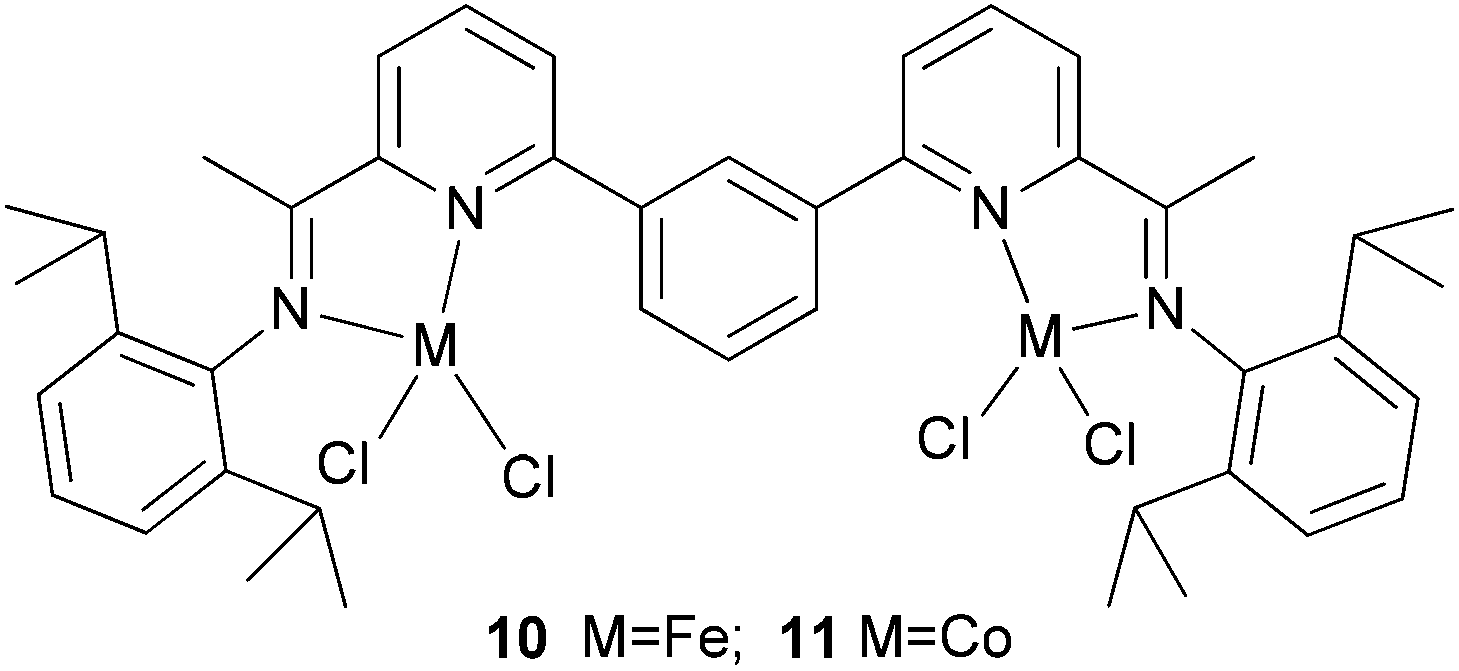 | ||
| Scheme 3 Aryl-linked bimetallic N,N-bi-dentate iron and cobalt complexes bearing iminopyridine ligands.32 | ||
Avilés et al. investigated the cobalt(II) complexes 12–18 (Scheme 4) bearing a variety of α-diimine ligands for ethylene polymerization.33 The influence of the ligand framework (DAB versus BIAN) and also the halide X was studied. The highest activities were observed at 20 °C with an Al/Co ratio at 500:1. Catalyst system 16/MAO, containing the mesityl-BIAN ligand exhibited the lowest activity, far lower than the o,o′-iPr2Ph-BIAN analogue 17/MAO (entries 5–8, Table 3). Contrastingly, the o,o′-iPr2Ph-DAB based catalyst system 15/MAO, containing the bulkier iPr substituents on the phenyl groups, exhibited lower activity in comparison to the mesityl analogue 14/MAO (entries 1–4, Table 3), the latter being the most active of all the catalysts studied. The products obtained via12–18 were mainly oily branched oligomers at elevated ethylene pressure (3–5.5 bar).
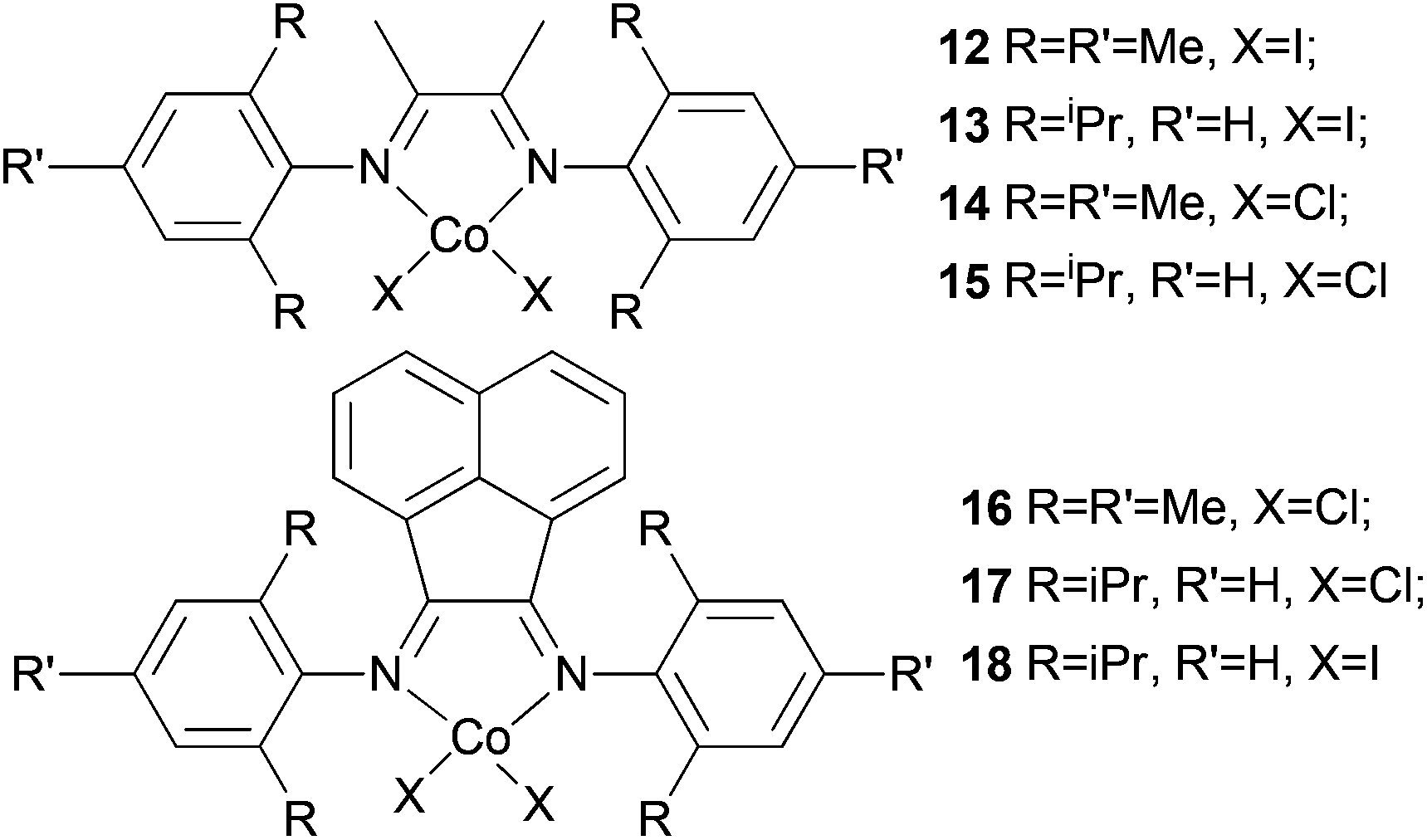 | ||
| Scheme 4 N,N-Bi-dentate cobalt complexes with α-diimine.33 | ||
| Entry | Complex | Al/Co | m PE (mg) | Activityc | No. branches/1000 Cd |
|---|---|---|---|---|---|
| a Conditions: 10 μmol of pre-catalyst, 50 mL of toluene, 2 bar ethylene, 20 °C, 2 h. b Traces. c g (mmol cat h bar)−1. d Estimated by 1H NMR. | |||||
| 1 | 14 | 500 | 43 | 1.08 | 25 |
| 2 | 14 | 1000 | 32 | 0.80 | 25 |
| 3 | 15 | 500 | 5 | 0.13 | — |
| 4 | 15 | 1000 | 9 | 0.23 | 55 |
| 5 | 16 | 500 | 20 | — | |
| 6 | 16 | 1000 | 4 | 0.10 | 25 |
| 7 | 17 | 500 | 16 | 0.41 | — |
| 8 | 18 | 1000 | 26 | 0.65 | 46 |
The catalyst systems based on 14 and 16, which possess ligands with mesityl substituents, afforded branched products with 25/1000 C (entries 1, 2, and 6, Table 3), whereas 15 and 17, containing the bulkier o,o′-iPr2Ph substituents, afforded PE with about twice the branching of the other systems (see entries 4 and 8, Table 3).33
Our group has synthesized N,N-bidentate iron and cobalt complexes bearing 2-(benzimidazolyl)pyridine ligands and have investigated their ethylene oligomerization potential.34 Upon activation with different organoaluminium co-catalysts at an ambient pressure of ethylene, the complexes 19–26 (Scheme 5) exhibited very low activities (Table 4). The results obtained showed that complexes of type 21 possessed relatively higher activities than did complexes 19 (entries 2, 3, 8, and 9, Table 4); the same tendency was also observed for 23–26 (entries 6, 7, 11, and 12, Table 4). The trends were attributed to deprotonation of the N–H group to afford anionic amide ligands, and formation of N–Al species which upon activation by the organoaluminium co-catalyst led to increased catalytic activity. The iron complexes 23 and 24, possessing a carboxylate group at the 6-position of the pyridine ring, exhibited activities somewhat higher than those of their analogues 19 and 20 (entries 1, 5–7, Table 4). This was assumed to be either the result of enhanced solubility or possibly an electron-withdrawing effect of the carboxylate group and a weak metal–oxygen interaction.27 Interestingly, the presence of an ester group in such catalysts also led to lower amounts of C6 in the products than for the other systems; this was thought to be due to faster elimination. Overall though, the activities associated with the cobalt complexes remained relatively unchanged on changing the substituents at pyridine in such benzimidazole cobalt complexes.34
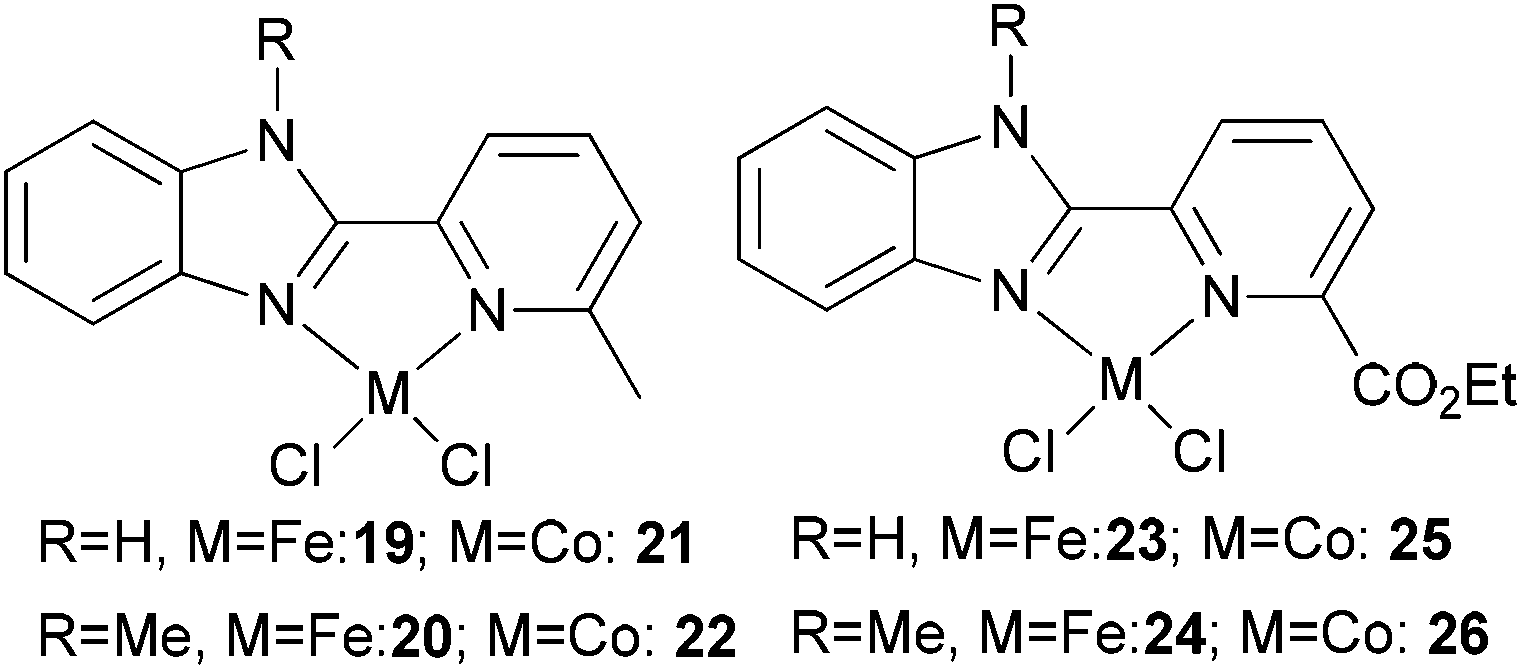 | ||
| Scheme 5 N,N-Bi-dentate iron and cobalt complexes with 2-(benzimidazolyl)pyridine derivatives.34 | ||
| Entry | Complex | Co-catalyst | Al/M | Activityb | Oligomer distributionc (%) | |
|---|---|---|---|---|---|---|
| C4/∑C | C6/∑C | |||||
| a Conditions: 5 μmol of pre-catalyst, 100 mL of toluene, 20 atm of ethylene, 20 °C, 30 min. b In units of 104 g (mol of M)−1 h−1 atm−1. c Determined by GC; ∑C signifies the total amounts of oligomers. | ||||||
| 1 | 19 | MMAO | 1000 | 0.86 | 81.4 | 18.6 |
| 2 | 19 | Et2AlCl | 500 | 0.56 | 58.7 | 41.3 |
| 3 | 19 | Et2AlCl | 200 | 0.38 | 60.2 | 39.8 |
| 4 | 19 | MAO | 1000 | 0.26 | 53.9 | 46.1 |
| 5 | 20 | MMAO | 1000 | 0.78 | 68.3 | 31.7 |
| 6 | 23 | MMAO | 1000 | 5.15 | 96.3 | 3.4 |
| 7 | 24 | MMAO | 1000 | 3.33 | 96.3 | 3.7 |
| 8 | 21 | Et2AlCl | 500 | 1.29 | 67.4 | 32.6 |
| 9 | 21 | Et2AlCl | 200 | 1.41 | 70.5 | 29.5 |
| 10 | 22 | Et2AlCl | 200 | 0.80 | 37.4 | 62.6 |
| 11 | 25 | Et2AlCl | 200 | 1.13 | 81.6 | 18.4 |
| 12 | 26 | Et2AlCl | 200 | 0.76 | 77.9 | 22.1 |
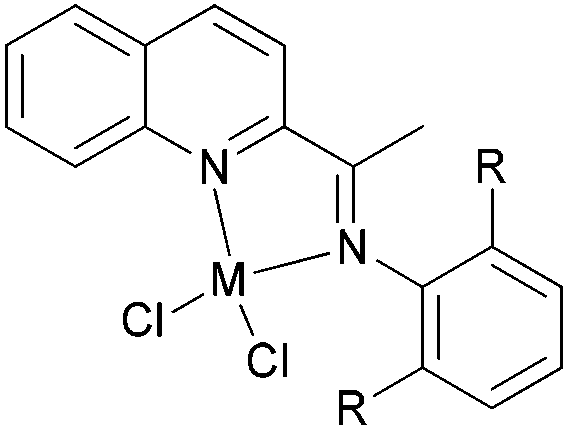
Our group has synthesized a series of N,N-bidentate iron and cobalt complexes bearing either 2-(1-aryliminopropylidene)quinolines (Scheme 6, 27–32),35 8-(1-aryliminoethylidene)quinaldines (Scheme 7, 33–42)36 or 2-(1-aryliminoethylidene)quinolines (Scheme 8, 43–52).37
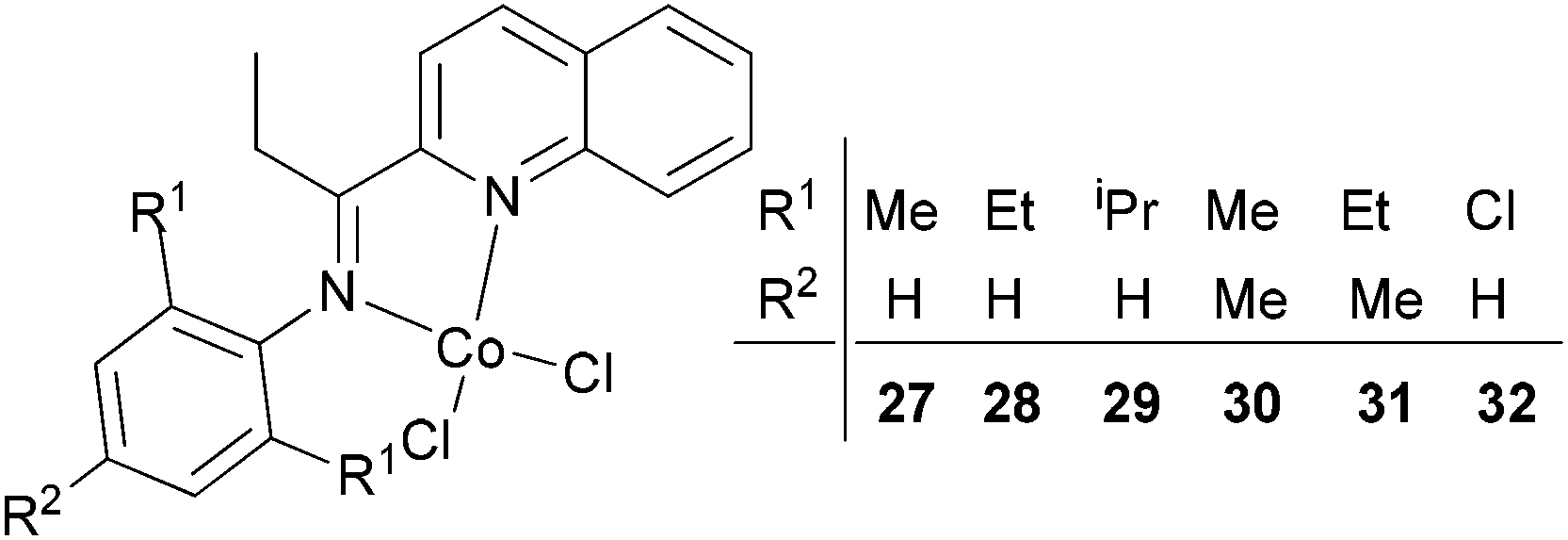 | ||
| Scheme 6 N,N-Bidentate cobalt(II) complexes bearing 2-(1-aryliminopropylidene)quinolines.35 | ||
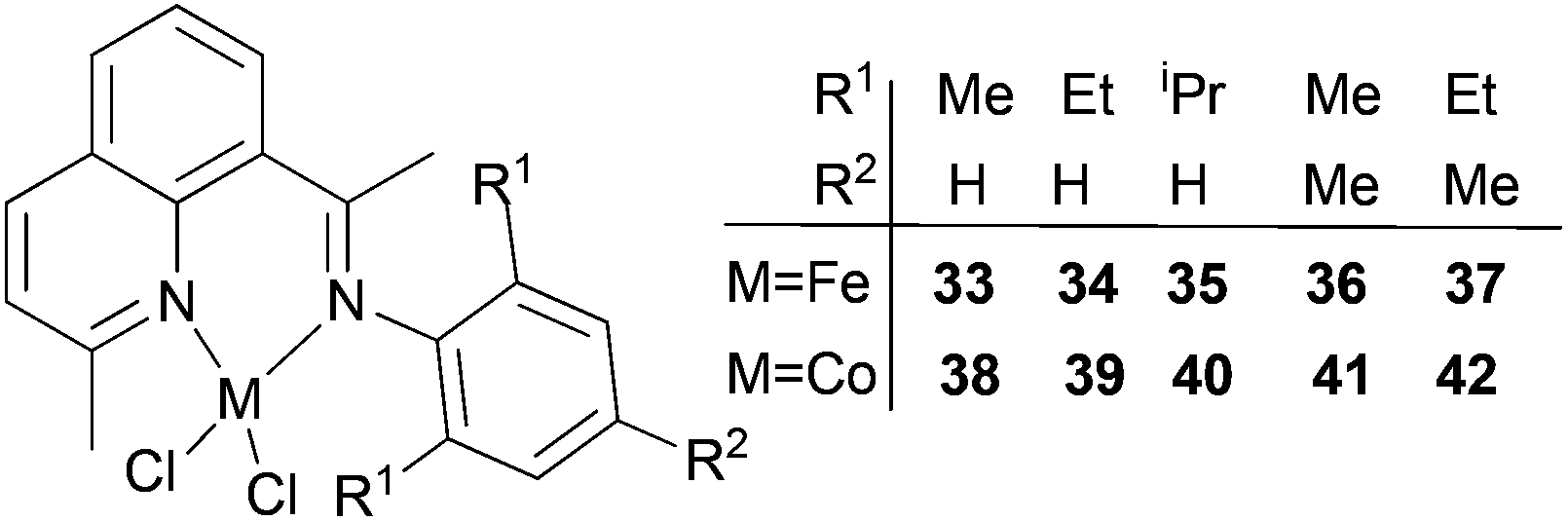 | ||
| Scheme 7 N,N-Bidentate cobalt(II) and iron(II) complexes bearing 8-(1-aryliminoethylidene)quinaldines.36 | ||
 | ||
| Scheme 8 N,N-Bidentate cobalt(II) and iron(II) complexes bearing 2-(1-aryliminoethylidene)quinolines.37 | ||
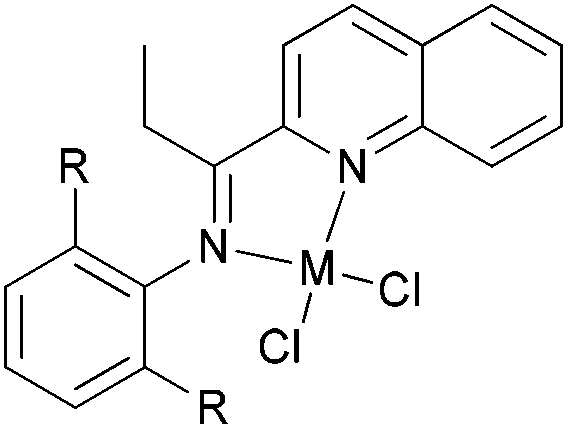
For the 2-(1-aryliminopropylidene)quinolylcobalt(II) dichlorides (Scheme 6, 27–32), low activities were observed at ambient pressure of ethylene.35 However, on increasing the pressure to 10 atm of ethylene and using a molar ratio [Al/Co] of 1000:1 at room temperature, a dimerization activity of 1.9 × 106 g mol−1 (Co) h−1 was achieved with MAO, and 9.4 × 105 g mol−1 (Co) h−1 in the presence of MMAO. Variation of the Al/Co molar ratio or an increase in the reaction temperature led to reduced catalytic activity. At 80 °C, both butenes and polyethylene were observed albeit with low activity, whilst on further increasing the temperature (90 °C), polyethylene solely was obtained. Further studies using all pre-catalysts at an Al/Co molar ratio of 1000 and at 20 °C and 10 atm revealed the observed activity order 29 > 28 > 27 and 31 > 30 (Scheme 6, 27–32), all of which were higher than 32 (entries 1–6, Table 5). The observed trend was consistent with bulkier substituents affording enhanced protection at the active site, and thereby maintaining the stability of the catalytic system.32 It should be noted that in these system, bi-dentate ligation provided less electronic donation versus the tridentate ligand sets discussed in section 3. In the case of the observations for 28vs. 31 and 27vs. 30, ligands bearing an additional methyl group were found to exhibit enhanced activity which was attributed to better solubility (entries 1, 2, 4, and 5, Table 5). At 90 °C and 10 atm ethylene over 30 min, the activity trend was 29 > 28 > 27 > 32 and 31 > 30 > 32 (entries 1–6, Table 6), consistent with the activity trends observed for ethylene dimerization at room temperature.35 The polyethylene products possessed similar molecular weights with narrow molecular weight distributions (2.82–3.98); similar cobalt active species were assumed. The use of higher temperatures and increased amounts of MAO afforded polyethylene of broader molecular weight distribution, i.e. catalysis by multi-active species.19,38–42
| Entry | Complex | Butene yieldb/g | Activityc | α-C4/∑C4 |
|---|---|---|---|---|
| a Conditions: 5 μmol of pre-catalyst, 1000 of Al/Co, 100 mL of toluene, 10 atm of ethylene, 20 °C, 30 min. b Determined by GC. c 105 g mol−1 (Co) h−1. | ||||
| 1 | 27 | 2.11 | 8.4 | 53.3% |
| 2 | 28 | 2.51 | 10 | 57.6% |
| 3 | 29 | 4.75 | 19 | 59.8% |
| 4 | 30 | 2.43 | 9.7 | 51.8% |
| 5 | 31 | 3.02 | 12 | 59.8% |
| 6 | 32 | 1.91 | 7.6 | 56.4% |
| Entry | Complex | Product yieldb/mg | Activityb |
M
wc × 10−4 |
M
w/Mnc |
|---|---|---|---|---|---|
| a Conditions: 5 μmol of pre-catalyst, 3000 of Al/Co, 100 mL of toluene, 10 atm of ethylene, 90 °C, 30 min. b 104 g mol−1 (Co) h−1. c Determined by GPC vs. polystyrene standards. | |||||
| 1 | 27 | 105 | 4.2 | 11.16 | 3.87 |
| 2 | 28 | 115 | 4.6 | 9.54 | 3.98 |
| 3 | 29 | 130 | 5.2 | 12.54 | 3.79 |
| 4 | 30 | 113 | 4.5 | 10.73 | 3.97 |
| 5 | 31 | 123 | 4.9 | 9.25 | 2.82 |
| 6 | 32 | 98 | 4.0 | 10.41 | 3.72 |
For the 2-(1-aryliminoethylidene)quinoline complexes 33–42 (Scheme 7),36 activation with MMAO led to good activity for ethylene dimerization; the iron(II) pre-catalysts exhibited higher activities and a better selectivity for α-butene.36 The activity trends (iron) 35 > 37 > 36 > 34 > 33 (entries 1, 5–8, Table 7) and (cobalt) 40 > 42 > 41 > 39 > 38 (entries 9, 13–16, Table 7) were observed. As elsewhere, such observations were consistent with bulkier substituents affording better catalytic activity (an additional substituents at the para-position of the aryl group also yields higher activity).36 For the cobalt systems, relatively lower activities were exhibited with lower selectivity for α-butene (entries 1–16, Table 7). On increasing the reaction temperature from 30 °C to 60 °C, the activity for 35 decreased rapidly (from 5.71 × 105 g mol−1 (Fe) h−1 to 0.78 × 105 g mol−1 (Fe) h−1) (entries 1–4, Table 7); similarly, for 40 increasing the reaction temperature from 20 °C to 50 °C resulted in a large decrease in the activity (from 4.89 × 105 g mol−1 (Fe) h−1 to 1.97 × 105 g mol−1 (Co) h−1) (entries 9–12, Table 7), consistent with thermally unstable active species.38
| Entry | Complex | T/°C | Al/M | Activityb | Oligomer distributionc (%) | ||
|---|---|---|---|---|---|---|---|
| α-C4 | C4/∑C | C6/∑C | |||||
| a Conditions: 5 μmol of pre-catalyst, 100 mL of toluene, 10 atm of ethylene, 30 min. b 105 g mol−1 (M) h−1. c Determined by GC; ∑C denotes the total amounts of oligomers. | |||||||
| 1 | 35 (Fe) | 30 | 1500 | 5.71 | 99.6 | 97.5 | 2.5 |
| 2 | 35 (Fe) | 40 | 1500 | 4.64 | 95.5 | 96.2 | 3.2 |
| 3 | 35 (Fe) | 50 | 1500 | 3.52 | 96.4 | 95.7 | 4.3 |
| 4 | 35 (Fe) | 60 | 1500 | 0.78 | 94.6 | 96.8 | 3.2 |
| 5 | 33 (Fe) | 30 | 1500 | 4.37 | 99.1 | 97.7 | 2.3 |
| 6 | 34 (Fe) | 30 | 1500 | 4.77 | 98.9 | 97.8 | 2.2 |
| 7 | 36 (Fe) | 30 | 1500 | 4.94 | 98.2 | 99.1 | 0.9 |
| 8 | 37 (Fe) | 30 | 1500 | 4.97 | 99.3 | 98.5 | 1.5 |
| 9 | 40 (Co) | 20 | 2500 | 4.89 | 59.9 | 98.4 | 1.6 |
| 10 | 40 (Co) | 30 | 2500 | 3.88 | 53.3 | 98.9 | 1.1 |
| 11 | 40 (Co) | 40 | 2500 | 2.20 | 68.6 | 98.7 | 1.3 |
| 12 | 40 (Co) | 50 | 2500 | 1.97 | 80.4 | 98.3 | 1.7 |
| 13 | 38 (Co) | 20 | 2500 | 3.03 | 58.4 | 98.2 | 1.8 |
| 14 | 39 (Co) | 20 | 2500 | 3.34 | 57.2 | 96.5 | 3.5 |
| 15 | 41 (Co) | 20 | 2500 | 3.07 | 57.6 | 98.2 | 1.8 |
| 16 | 42 (Co) | 20 | 2500 | 3.56 | 56.5 | 97.2 | 2.8 |
For the 2-(1-aryliminoethylidene)quinoline family (Scheme 8, 43–52),37 cobalt complexes 43–47 exhibited higher activity, a better selectivity for α-C4 and enhanced thermo-stability for ethylene dimerization over the analogous pre-catalysts bearing 2-(1-aryliminopropylidene)quinolines;35 no polyethylene was obtained at high temperatures (entries 1–11, Table 8). The use of bulkier substituents on the arylimino group led to enhanced activity as well as a high selectivity for ethylene dimerization. The presence of an additional para methyl group also led to improved activities. Surprisingly, the cobalt pre-catalysts 43–47 were more active than their iron counterparts 48–52 (entries 1, 8–16, Table 8). For example, the cobalt pre-catalyst 47 was found to possess an activity of 1.82 × 106 g mol−1 (Co) h−1versus the iron pre-catalyst 52 at 5.89 × 105 g mol−1 (Fe) h−1 (entries 11, 16, Table 8); note however that different aluminium co-catalysts were employed.37 In addition, the cobalt pre-catalyst 45/MAO maintained a reasonable activity (for ethylene dimerization – entries 1–7, Table 8) at temperatures as high as 60 °C, which was indicative of enhanced thermal stability versus the active species derived from the iron complexes.
| Entry | Complex | T/°C | Al/M | Activityb | Oligomer distributionc (%) | ||
|---|---|---|---|---|---|---|---|
| α-C4 | C4/∑C | C6/∑C | |||||
| a Conditions: 5 μmol of pre-catalyst, 100 mL of toluene, 10 atm of ethylene, 30 min. b 105 g mol−1 (M) h−1. c Determined by GC; ∑C denotes the total amounts of oligomers. d Co-catalyst: MAO. e Co-catalyst: MMAO. | |||||||
| 1d | 45 (Co) | 20 | 1000 | 16.5 | 67.1 | 99.8 | 0.2 |
| 2d | 45 (Co) | 30 | 1000 | 12.7 | 65.1 | 99.8 | 0.2 |
| 3d | 45 (Co) | 40 | 1000 | 11.2 | 62.1 | 99.6 | 0.4 |
| 4d | 45 (Co) | 60 | 1000 | 10.6 | 65.3 | 99.3 | 0.7 |
| 5d | 45 (Co) | 80 | 1000 | 8.0 | 66.7 | 98.6 | 1.4 |
| 6d | 45 (Co) | 90 | 1000 | Trace | — | — | — |
| 7d | 45 (Co) | 100 | 1000 | — | — | — | — |
| 8d | 43 (Co) | 20 | 1000 | 12.2 | 67.2 | 99.4 | 0.6 |
| 9d | 44 (Co) | 20 | 1000 | 13.0 | 65.3 | 99.6 | 0.4 |
| 10d | 46 (Co) | 20 | 1000 | 17.1 | 67.9 | 99.7 | 0.3 |
| 11d | 47 (Co) | 20 | 1000 | 18.2 | 65.5 | 99.8 | 0.2 |
| 12e | 48 (Fe) | 20 | 2500 | 3.61 | 61.5 | 100 | — |
| 13e | 49 (Fe) | 20 | 2500 | 3.91 | 62.2 | 100 | — |
| 14e | 50 (Fe) | 20 | 2500 | 3.99 | 64.1 | 100 | — |
| 15e | 51 (Fe) | 20 | 2500 | 4.49 | 65.5 | 100 | — |
| 16e | 52 (Fe) | 20 | 2500 | 5.89 | 66.3 | 100 | — |
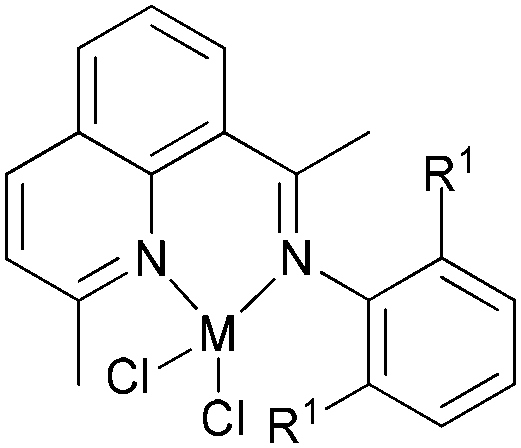
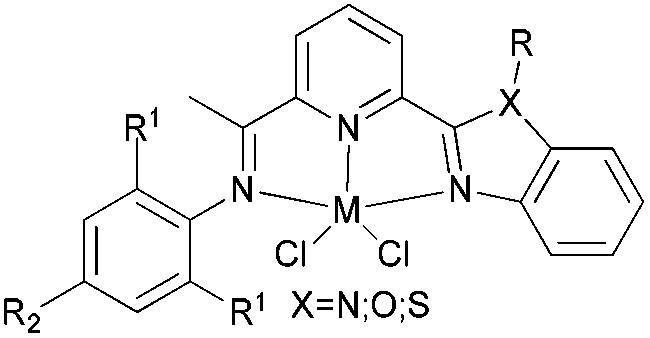
Our group has also investigated the ethylene oligo-/polymerization behaviour of iron and cobalt complexes bearing 8-(benzoimidazol-2-yl)quinolones (Scheme 9).25,26 At 1 bar ethylene, the catalytic behaviour of type 67 pre-catalyst with co-catalysts of methylaluminoxane (MAO), modified methylaluminoxane (MMAO), and triisobutylaluminium (iBu3Al) was evaluated. The various oligomers obtained ranged from C4 to C18 with high selectivity for α-olefins (>95%); a Schulz–Flory distribution was observed.26 At 10 bar ethylene, on increasing the reaction temperature from 20 to 60 °C, the catalytic activity decreased for cobalt pre-catalyst 67, and more low-molecular-weight oligomers were formed. At 80 °C however, only trace polyethylene was isolated, but no oligomers.26 The substituents R1 and R2 were found to have an influence on the catalytic performance of 67–80. On fixing R2, the oligomerization activity gradually increased on increasing the bulk of R1. By contrast, on fixing R1, the catalytic performance observed followed the order H > Me > Et > iPr.26 At 30 bar ethylene, iron(II) and cobalt complexes bearing 2-R1-8-(1-R2-benzimidazol-2-yl)quinolines afforded highly active ethylene polymerization catalysts upon activation with MAO at 100 °C.25 Under the optimized reaction conditions (30 atm ethylene, Al/Fe = 3000, 100 °C), the activity of 53 reached 6.11 × 106 g mol−1 (Fe) h−1 and that of 67 reached 1.83 × 106 g (mol of Co)−1 h−1 Cethylene−1.25,26 In the series of pre-catalysts with R1 = Et, with the exception of 55 with R2 = H, the activity order observed, namely 56 > 57 > 58, appeared to be governed by the electronic influence of the ligands (electron-donating alkyl substituents R2 = Me (56) < Et (57) < iPr (58)).25 This is consistent with slow ethylene insertion at the electron-rich active species.20 The same trend of catalytic activities was also observed in the series of iron pre-catalysts with R1 = Me, Pr, or Ph, respectively. For the series of iron pre-catalysts with R2 = Me, the activities order was R1 = Me (54) > Ph (64) > iPr (60) > Et (56).25 As well as the electronic influence used to explain the better activities of pre-catalysts 54 (Me) and 64 (Ph),20 the presence of bulkier substituents could protect the active species, i.e. Ph (64) > Pr (60) > Et (56).4,5,38,43–47 Similar (R1 and R2) substituent influences on catalytic activities were also observed by the cobalt analogs bearing related ligands.26
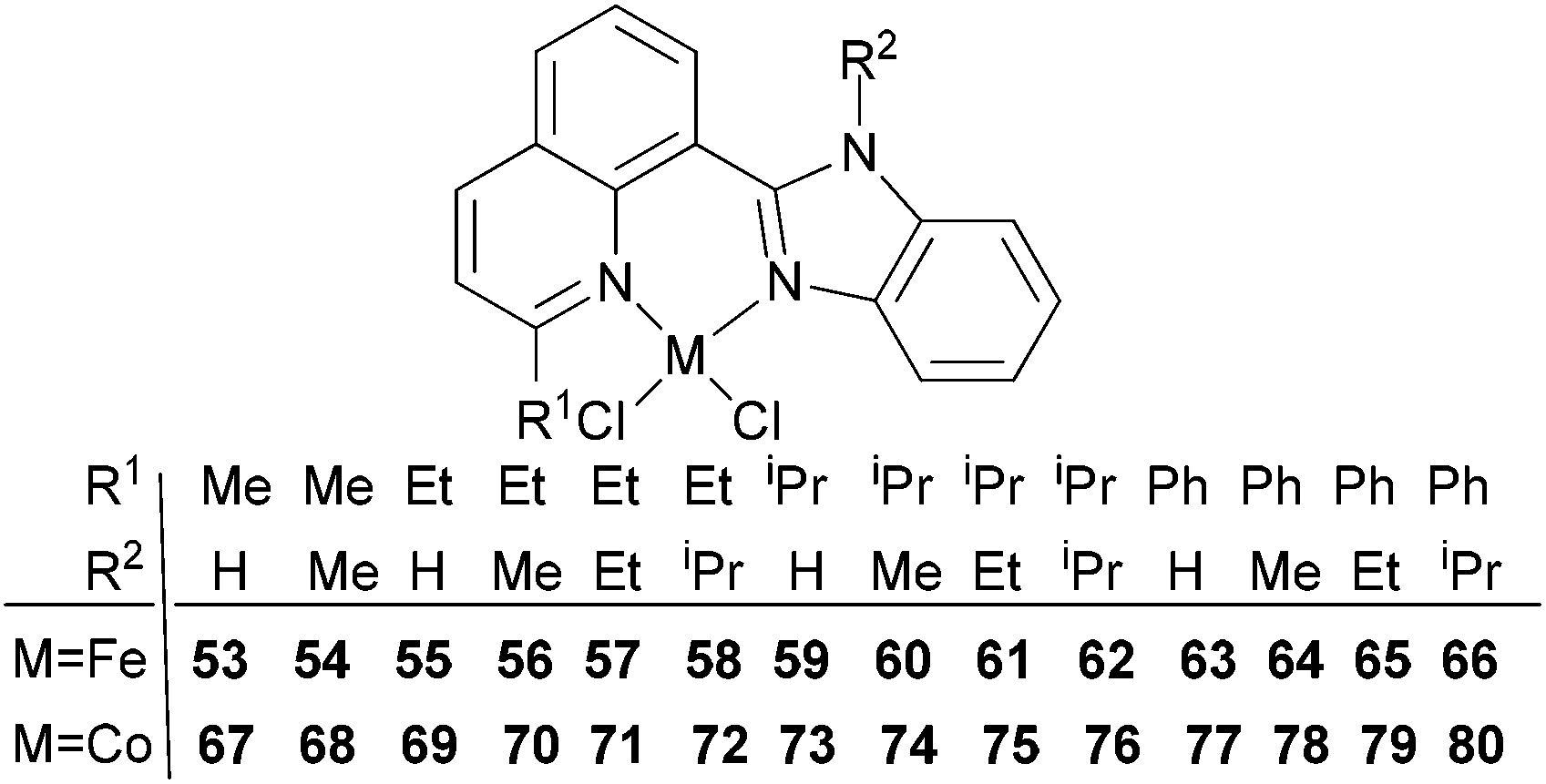 | ||
| Scheme 9 N,N-Bi-dentate iron(II) and cobalt(II) complexes bearing 8-(benzoimidazol-2-yl)quinolines.25,26 | ||
3. Tri-dentate iron and cobalt pre-catalysts
Tri-dentate iron/cobalt pre-catalysts usually exhibit higher activities compared to those exhibited by bi-dentate iron/cobalt pre-catalysts for both ethylene oligomerization and polymerization, and so there has been much progress in this area in recent years.3.1. Bis(imino)pyridine type ligation
Following the discovery of iron and cobalt 2,6-bis(imino)pyridyl catalysts, most investigations have focused on modifying the ligand framework of the parent 2,6-bis(imino)pyridine ligand set either through changing the steric and/or electronic properties of the substituents at the N-bound aryl groups present.4,5,43,48 Thus, numerous variations of the original tri-dentate 2,6-bis(imino)pyridyl ligand have been reported, with many of them maintaining the same [N,N,N]-metal core in the catalyst systems.6,7,18,19,49,50The Alt group investigated a family of bis(arylimino)pyridine iron(II) complexes bearing ω-alkenyl substituents (Scheme 10) for ethylene oligomerization and polymerization.51 Complexes bearing substituents both at the 2- and 6-positions of the iminophenyl rings only produced polyethylene, whereas if only one ortho imino group was present, the complexes produced, depending on bulk of the substituent, either oligomer–polymer mixtures or only oligomeric mixtures; the reaction conditions could also affect the products formed.52 Usually, iron(III) complexes with alkyl substituted iminophenyl rings exhibit higher polymerization activities than do their iron(II) analogues, however the α-alkenyl substituted iron(III) complexes exhibited similar activities to their iron(II) analogues.51 Longer α-alkenyl chains on these bis(arylimino)pyridine had a positive effect on the observed polymerization activities, whereas longer alkyl chains on the bis(arylimino)pyridine led to decreased activity.
 | ||
| Scheme 10 N,N,N-Tri-dentate bis(arylimino)pyridine iron(II) complexes with ω-alkenyl substituted.51 | ||
The Alt group also studied the influence of the para aryl substituent (Scheme 11) on the oligomerization and polymerization of ethylene.51 The complex bearing fluorine substituents44,53,54 exhibited by far the highest activity amongst the 4-halogen-2-methyl substituted family of complexes (Chart 3).44,53 Whilst those bearing chloro or bromo substituents at the para position afforded relatively lower activities (Scheme 11), the introduction of iodo substituents led to increased activity. Both the size and the electro-negativity of these substituents played an important role in determining activity. For small halogen substituents (F, Cl, Br), electronegativity was the decisive factor (highest activity for the fluoro complex), whereas iodo substituents again led to a higher polymerization activity along with an increased content of higher molecular weight olefins.53 When sterically demanding alkynyl substituted cyclopentadienyl/fluorenyl moieties were introduced, via Sonogashira coupling reactions, the resulting complexes produced exclusively polymers. The polymerization activities of 82 and 83 were somewhat higher than 81 (Scheme 12) (entries 1–3, Table 9). Bulky groups at the para positions also exerted an influence on the molecular weights of the resultant polymerization products. Although only one of the ortho positions of the ligand in 82 (Scheme 12) is substituted, the steric bulk of the cyclopentadienyl and fluorenyl motifs appeared high enough to significantly decrease the rate of β-H elimination. The average molecular weights Mn and Mw produced with 82/MAO (Scheme 12) were 14050 and 198900 g mol−1, respectively, and are very similar to those obtained for the polyethylene produced using 83/MAO (Mn = 16200 g mol−1, Mw = 158000 g mol−1) (entries 1–3, Table 9). Given this, it appears that occupation of both ortho positions on the iminoaryl groups is not a pre-requisite for polymer production in such catalysts, though the need to occupy the para positions with sterically demanding groups then becomes a factor.53
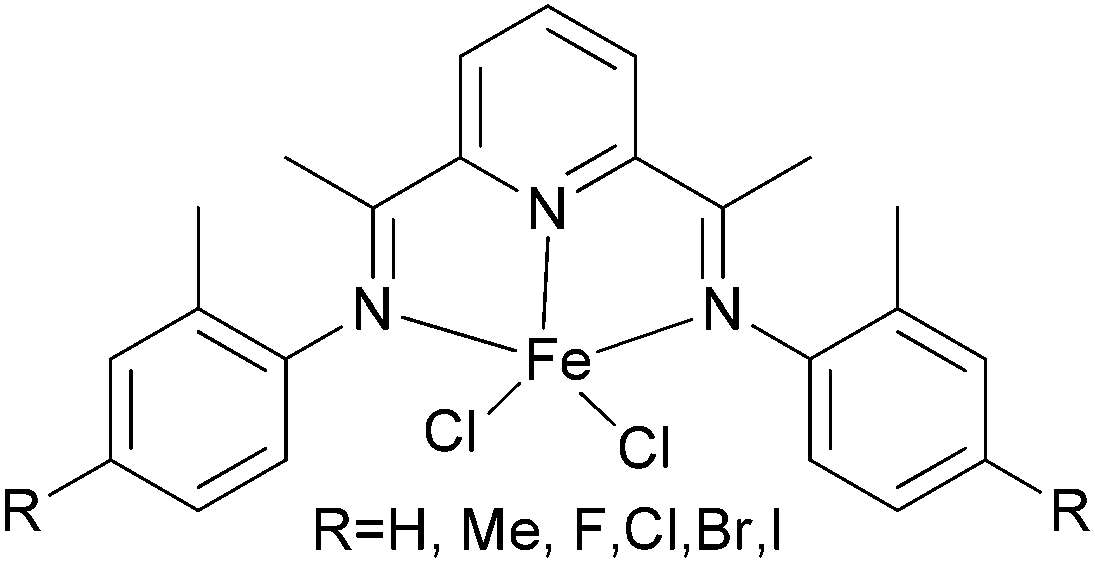 | ||
| Scheme 11 N,N,N-Tridentate bis(arylimino)pyridine iron(II) complexes.43,53 | ||
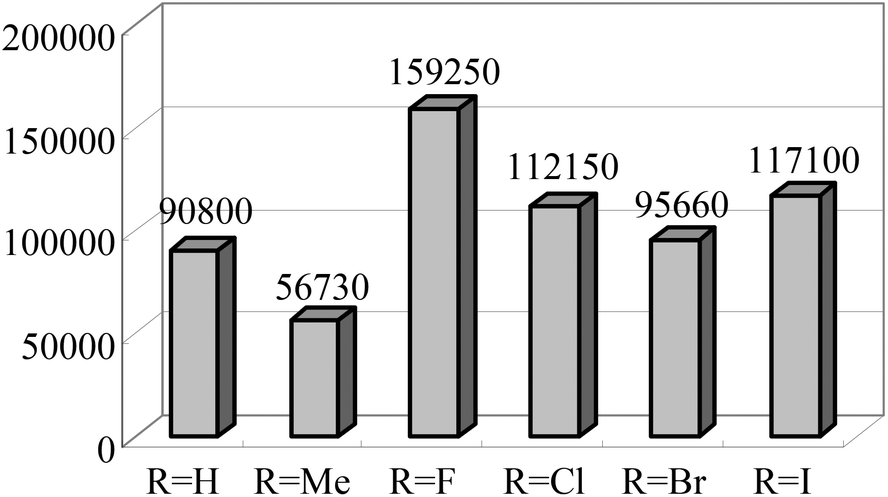 | ||
| Chart 3 Activities of 4-halogen-2-methyl substituted 2,6-bis(arylimino) pyridine iron complexes. All complexes produce 100% oligomers.44,53 | ||
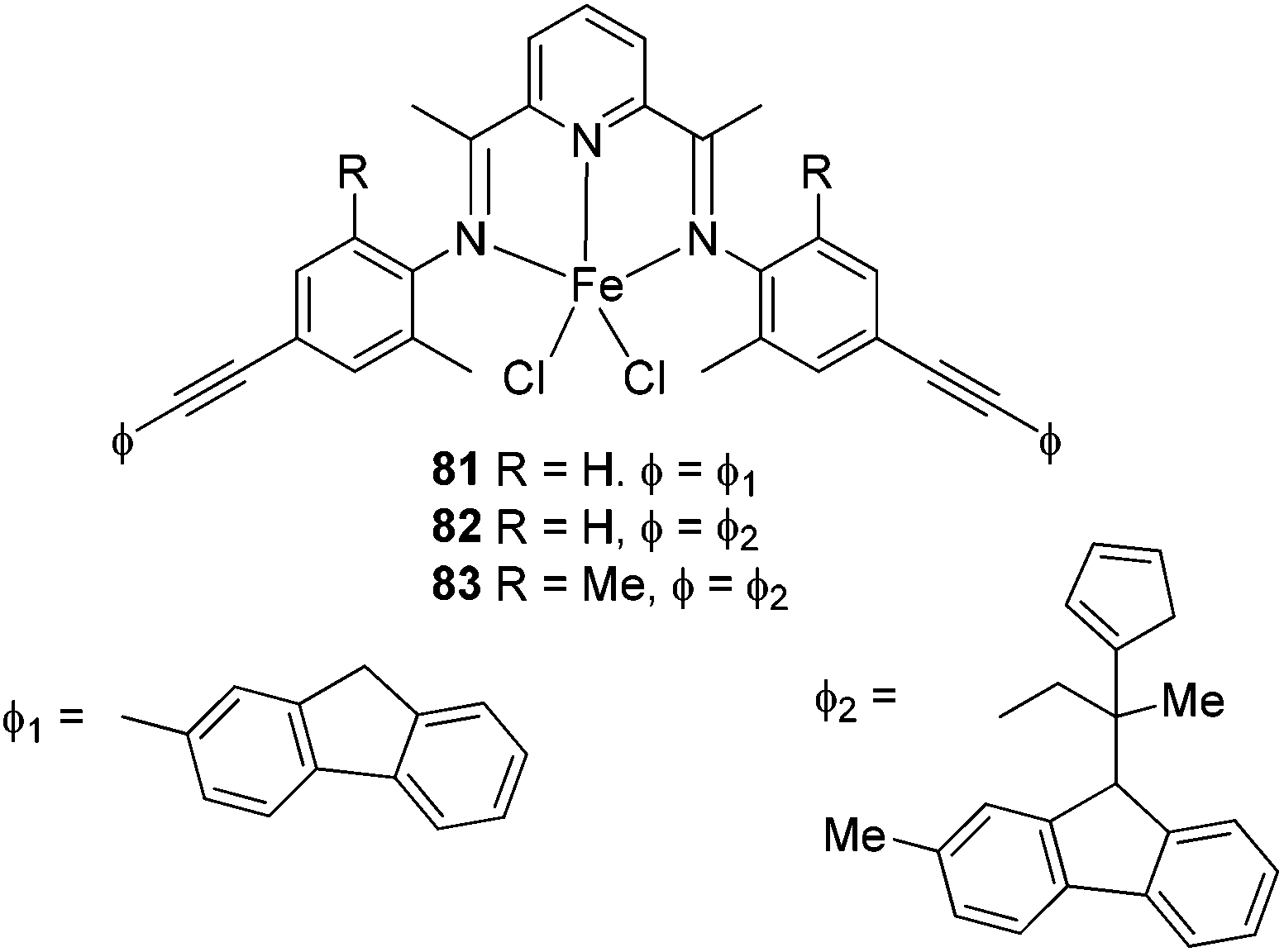 | ||
| Scheme 12 N,N,N-Tridentate bis(arylimino)pyridine iron(II) complexes.53 | ||
The Herrmann group has investigated the iron complexes 84–89 (Scheme 13) for the oligomerization and polymerization of ethylene and propylene using modified methylaluminoxane (MMAO) as activator.55 Complex 87 showed very little activity for the polymerization of either ethylene or propylene. It is tempting to attribute the low activity of 87 to the distorted geometry brought about by the introduction of bulky biphenyl groups. In contrast, 85 exhibited activities as high as 104 kg of PE ((mol of Fe) h bar)−1 for ethylene polymerization, but the activity for propylene was far lower. Complex 86 afforded, in the case of propylene polymerization, only a low activity of about 200 kg of PP ((mol of Fe) h bar)−1, affording in the process atactic oligomers with molecular weight 300 and a polydispersity of 2.2. In contrast, use of 86 for ethylene polymerization led to much higher activity, up to 7.3 × 104 kg of PE ((mol of Fe) h bar)−1, but the polymers produced were of lower molecular weight compared to those obtained from 84. This was explained in terms of the decreased steric hindrance imparted by the chelating ligand: one ortho substituent at the N-aryl ring is missing compared to 84. Complex 89 acts as a hybrid of the two symmetric complexes 84 and 86, and exhibits an average activity. The polymerization activity of the unsymmetrical catalyst 88 was found to be equivalent to the symmetric catalysts 85 and 86, viz. 8 × 104 kg of PE ((mol of Fe) h bar)−1. The molecular weights of the polymers formed were slightly higher for 88 than in the case of the symmetric catalyst 85. However, use of 88 for propylene polymerization was less successful. Indeed, at 0 °C, the activity was 1500 kg of PP ((mol of Fe) h bar)−1, which decreased rapidly on increasing the temperature (60 °C). The oligomers obtained on reaction at 0 °C had a molecular weight of around 200.55
 | ||
| Scheme 13 N,N,N-Tridentate bis(imino)pyridine-substituted iron(II) complexes.55 | ||
Ionkin et al. have investigated iron(II) and iron(III) complexes modified using a cyano group for the production of α-olefins in 120 °C.56 It was found that the both types of complexes 91–95 (Scheme 14) and 98–102 (Scheme 14) afforded productive catalysts for the synthesis of α-olefins (entries 2–7, 10–14, Table 10). The Schulz–Flory distributions of α-olefins appear more ideal and their K values higher than for the parent symmetric methyl substituted Fe(II) complex 90 (Scheme 14) (entries 1–7, 10–14, Table 10). Complexes functionalized with a para cyano (nitrilo) group 98–102 tended to afford α-olefins with higher Schulz–Flory K values and with smaller amounts of insoluble α-olefins than did the corresponding complexes minus the para cyano groups 91–95 (entries 2–7, 10–14, Table 10). However, the nitrilo complexes 98–102 were found to be less productive (entries 10–14, Table 10). It was also found that symmetrically substituted 96 with nitrilo groups at both ortho positions produced trace (if any) α-olefins even when used at high concentrations (entry 8, Table 10). Symmetric 97, with CNs in both para positions, in contrast to 90, had a very high K of 0.69 (entries 1, 14, Table 10).56
 | ||
| Scheme 14 N,N,N-Tridentate bis(imino)pyridine-substituted iron(II) and iron(III) complexes.56 | ||
| Entry | Pre-catalyst and amount (μmol) | Amount of the co-catalyst, MMAO (mmol) | “K” Value of the Schultz–Flory distributionb | Kilograms of α-olefins per gram of catalyst | % Solids in total α-olefinc | Schultz–Flory distribution R2 |
|---|---|---|---|---|---|---|
| a Conditions: solvent, o-xylene; pressure, 700 psig; temperature, 120 °C. b Determined from GC, using extrapolated values for C-10 and C-12. c Xylenes insoluble fraction of α-olefins. | ||||||
| 1 | 90; 0.06 | 1.13 | 0.59 | 458 | 3.44 | 0.9862 |
| 2 | 91; 0.08 | 2.26 | 0.62 | 263 | 3.3 | 0.9876 |
| 3 | 92; 0.18 | 2.26 | 0.62 | 115 | 8.22 | 0.9989 |
| 4 | 93; 0.64 | 2.26 | 0.61 | 102 | 2.76 | 0.9928 |
| 5 | 94; 0.07 | 2.26 | 0.61 | 165 | 3.43 | 0.9849 |
| 6 | 95; 0.19 | 2.26 | 0.62 | 210 | 1.09 | 0.9928 |
| 7 | 95; 0.06 | 2.26 | 0.61 | 208 | 1.80 | 0.997 |
| 8 | 96; 0.80 | 2.26 | Trace | |||
| 9 | 97; 0.77 | 2.26 | 0.69 | 15 | 15.36 | 0.9764 |
| 10 | 98; 0.20 | 2.26 | 0.63 | 147 | 3.45 | 0.9989 |
| 11 | 99; 0.20 | 2.26 | 0.61 | 103 | 1.84 | 0.9983 |
| 12 | 100; 0.09 | 2.26 | 0.62 | 189 | 2.20 | 0.9979 |
| 13 | 101; 0.15 | 2.26 | 0.64 | 75 | 1.95 | 0.999 |
| 14 | 102; 0.18 | 2.26 | 0.61 | 138 | 1.72 | 0.9916 |
Ionkin and co-workers have also studied iron(II) complexes modified by a boryl group for the production of α-olefins at high temperature.57 The non-symmetrical mono-borylated 103 (Scheme 15) was found to behave as a polymerization catalyst at high temperatures; the solids produced were thought to be the result of disproportionation affording a bis(mesitylimino)pyridyl iron(II) complex. The non-symmetric complexes 104 and 105 (Scheme 15) exhibited greater thermal stability, but were less productive than the parent symmetric complex 90 (Scheme 14), though more desirable product distributions were achieved.57
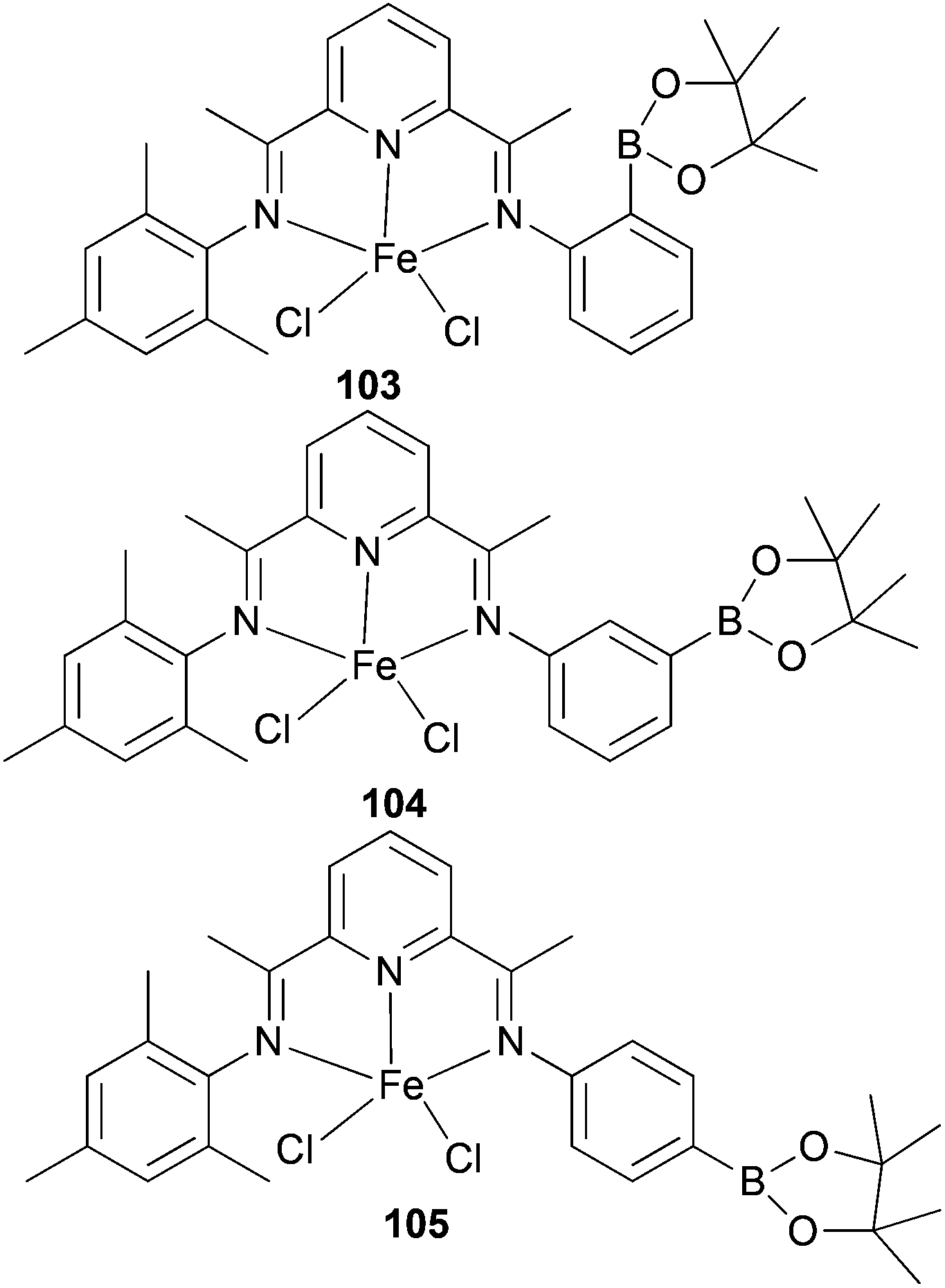 | ||
| Scheme 15 N,N,N-Tridentate bis(imino)pyridine-substituted iron(II) complexes modified by a boryl group.57 | ||
The same group also investigated ethylene oligomerization by the iron complexes 106–108 (Scheme 16) and the Co-based complex 109 (Scheme 16), each functionalized with double patterns of substitutions, i.e. o-methyl plus o-fluorine in the same iminoaryl arm.58 The Fe-based 106–108 afforded very active catalysts for the production of α-olefins with a more ideal Schulz–Flory distribution of α-olefins and with higher K values than did the parent methyl substituted Fe(II) complex 90 (Scheme 14). Complex 109 was found to have a very low activity for oligomerization, which is typical of such cobalt complexes.4 The limited observations appeared to suggest that non-symmetrical 107 and 108 possessed longer lifetimes than did the symmetrical 90 (Scheme 14) and 106 (Scheme 16) systems. The steric hindrance at the active center, particularly in terms of the number of ortho substituents present, appeared to affect the product distribution as well as catalyst productivities. It was noted that larger steric hindrance seemed to shift the distribution to longer chain α-olefins, but also led to reduced productivity.58
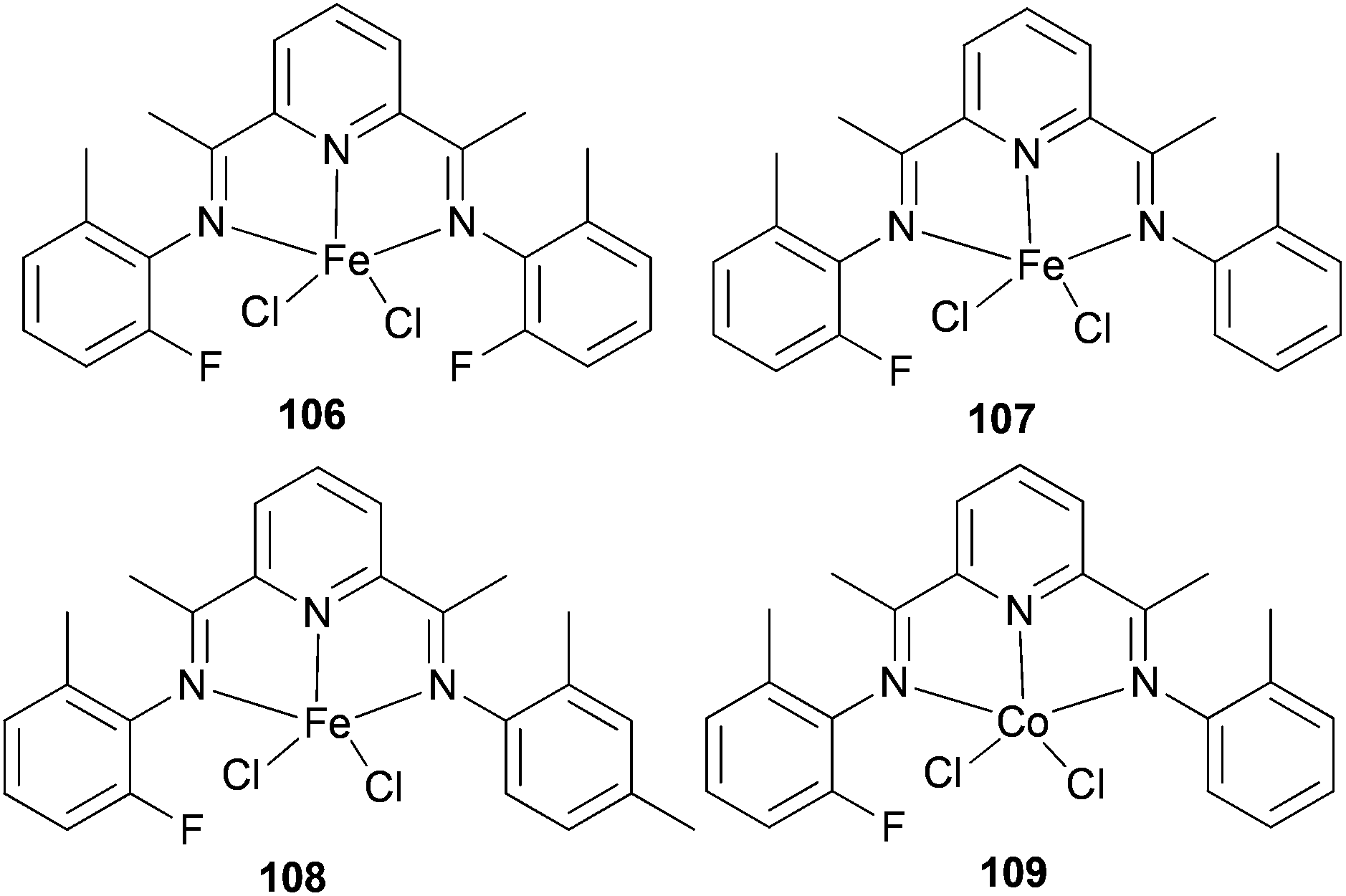 | ||
| Scheme 16 N,N,N-Tridentate bis(imino)pyridine-substituted iron(II) and cobalt(II) complexes.58 | ||
The Erker group has studied the effect of a pendant alkenyl group on the ethylene oligo-/polymerization of bis(imino)pyridine-type iron(II) and cobalt(II) complexes.59 It was found that for iron, 110a/MAO (Scheme 17) afforded the highest activity (60 g mmol−1 (Fe) h−1 bar−1) together with 114/MAO (90 g mmol−1 (Co) h−1 bar−1).60 For the cobalt systems 110/MAO or 114/MAO, linear polyethylene was obtained. Treatment of the N-allyl iron complex 111a/MAO gave a rather active catalyst system: an overall activity of 470 g mmol−1 (Fe) h−1 bar−1) was observed. Direct activation in a Büchi autoclave gave a less (albeit slightly) active catalyst (activity of 420 g mmol−1 (Fe) h−1 bar−1). The reaction produced a mixture of polyethylene and a substantial oligomeric fraction. The saturated N-(n-butyl) iron complex 111d/MAO (Scheme 17) gave a slightly less active catalyst (activity ∼130 g of PE + olig mmol−1 (Fe) h−1 bar−1), and produced mostly oligoethylenes (12.8 g isolated after 60 min of reaction time) and scant polyethylene (0.9 g) (cf. 111a/MAO: 5.5 g of oligomers and 17.8 g of polyethylene after 30 min reaction time). The remaining catalyst systems 111b–d/MAO fall between these two extremes: they all produced mixtures of C10–C15 oligoethylenes, with substantial amounts of polyethylene.59
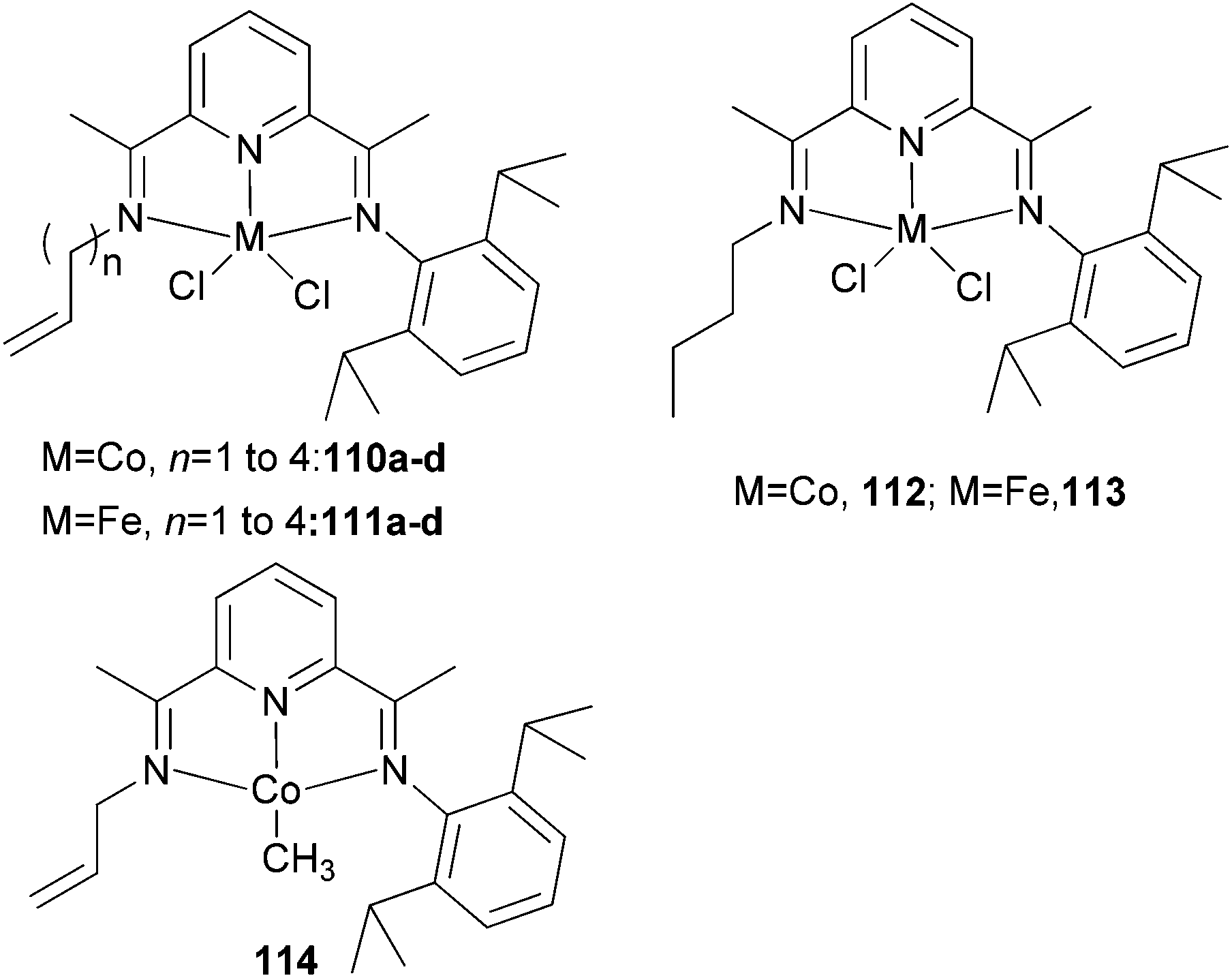 | ||
| Scheme 17 N,N,N-Tridentate bis(imino)pyridine-substituted iron(II) and cobalt(II) complexes with a pendant alkenyl group.36,59 | ||
The Cámpora group have investigated whether the presence of 4-neophyl, 4-benzyl or 4-allyl groups would affect the ethylene polymerization properties of the complexes 115–120 (Scheme 18).61 The impact of an alkyl substituent on the catalytic properties of the 4-neophyl and 4-benzyl derivatives was negligible, and basically only served to increase the solubility of the system. This suggests that the methodology could well be advantageous for the introduction of groups suitable for immobilization; other methods tend to have the linker functionality placed in a position where it may have a significant influence on the catalyst activity. As for the neophyl and benzyl groups, the introduction of a 4-allyl group on the pyridine ring did not alter the productivity of the corresponding Fe and Co catalysts (117 and 120), although the polymers generated were appreciably less soluble. GPC analyses of the polymers produced by 117 revealed much larger values of Mw (up to 180000) and a larger polydispersity index (Mw/Mn = 21). This might perhaps be due to a self-immobilization occurring via co-polymerization of the pendant allyl group into the growing polyethylene chain.61
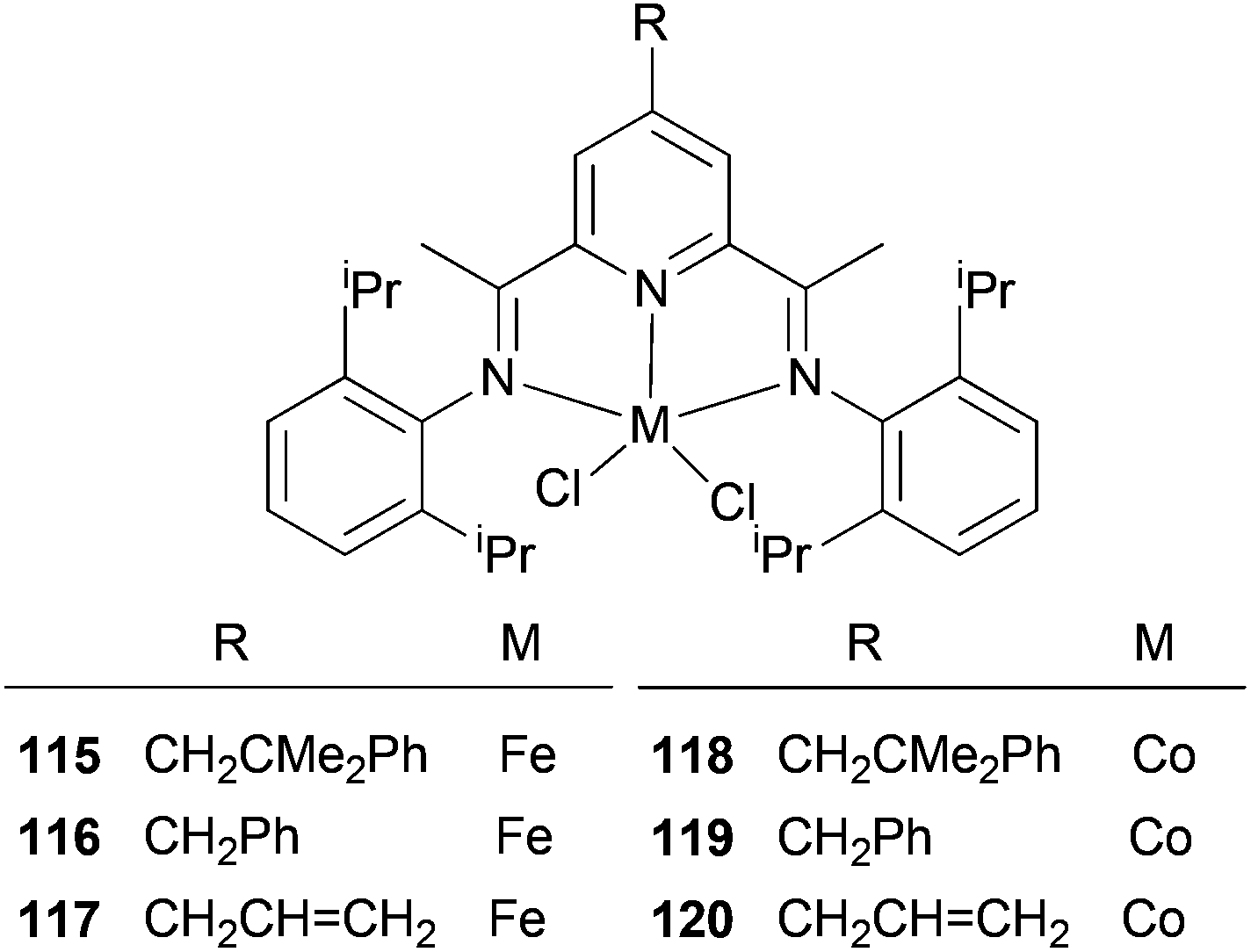 | ||
| Scheme 18 N,N,N-Tridentate iron(II) and cobalt(II) complexes bearing 4-alkyl-2,6-diiminopyridine.61 | ||
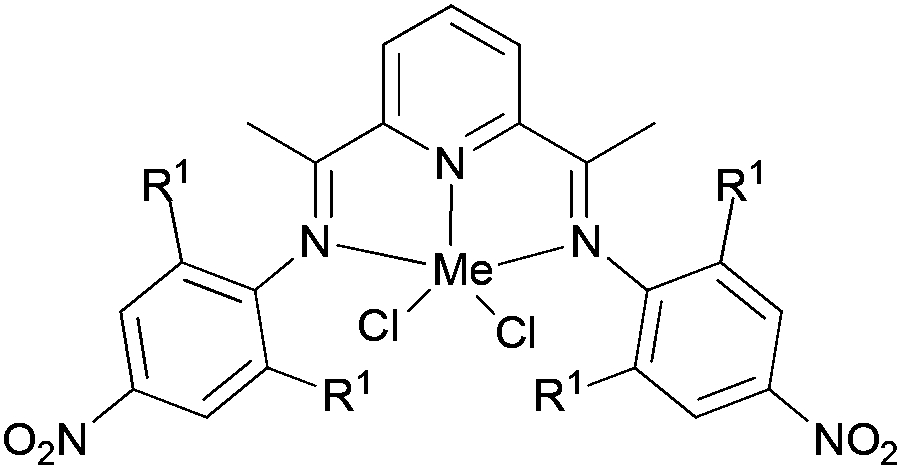
Wu and co-workers focused on the ethylene polymerization properties of para nitro substituted 2,6-bis(phenylimino)pyridyl Fe(II) and Co(II) complexes.62 The iron pre-catalyst 122 (Scheme 19) displayed moderately increased catalytic activities relative to the non-nitro substituted analogue 125, which was ascribed to the strong electron-withdrawing properties of the para nitro groups leading to increased Lewis acidic character at the cationic iron center. The ortho steric effect in such systems also played a significant role in controlling the activity and polymer properties, for example 121 possessing ortho methyl substituents on the aryl rings revealed much lower polymerization activities than did the ortho isopropyl analogue 122. Pre-catalysts 121/MAO and 122/MAO produced linear, high molecular weight polymers with low branching, while the cobalt pre-catalysts 123 and 124 possessed low activities and formed low Mw products on treatment with MAO.62
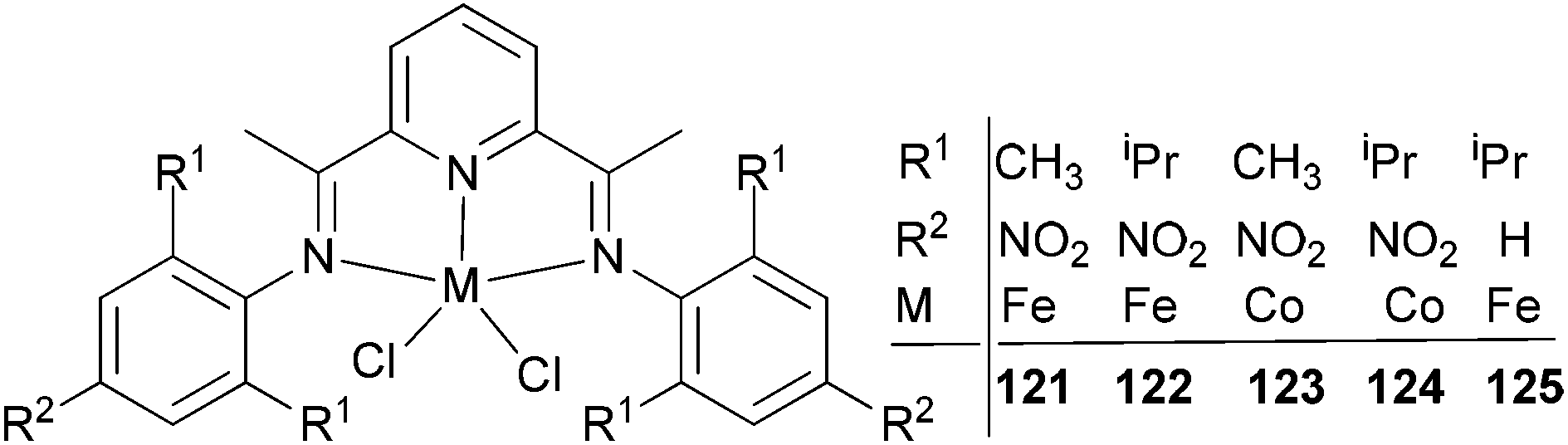 | ||
| Scheme 19 N,N,N-Tridentate bis(imino)pyridine-substituted iron(II) and cobalt(II) complexes.62 | ||
The Yang group investigated the ethylene polymerization characteristics of a series of iron and cobalt acetylacetonate complexes bearing different bis(imino)pyridyl ligands.63 The use of solely Fe(acac)3 did not produce an active system when MAO was used as the co-catalyst, whilst the strategy of adding Fe(acac)3 and the ligands sequentially also led to no activity. To activate such systems, the bis(imino)-pyridyl ligands and the Fe(acac)3 must be pre-mixed. In addition, the use of equal molar amounts of Fe(acac)3 and ligand are a suitable ratio for achieving the highest activity.63 The active species was formed by coordination between equal amounts of the iron acetylacetonate complex and the ligands. Broad bimodal polyethylene and oligomers could be obtained with these catalyst systems. The bis(imino)pyridyl ligands were capable of influencing these catalyst systems. High Mw polyethylene was obtained with the Fe(acac)3/L1, Fe(acac)3/L2 and Fe(acac)3/L3 (Scheme 20) systems. The main products were oligomers and polymers for the Fe(acac)3/L4 and Fe(acac)3/L5 systems. However, polymer was the predominant product with the Fe(acac)3/L6 and Fe(acac)3/L7 systems; only a comparatively small amount of oligomer was formed. The main products were 1-butene and 1-hexene, whilst no polymer was obtained with the Co(acac)3/L4 and Co(acac)3/L5 systems. The polymerization results revealed that on increasing the reaction temperature, the Mw was lowered. With decreased steric bulk, the number of active centers for preparing high Mw polyethylene decreased, and the effect of temperature on the MWD of the products became much more notable. The effect of the Al/Fe ratio on the polymer properties was also sensitive to the steric bulk. When AlMe3 present in commercial MAO was removed and the Al/Fe ratio was increased, higher Mw polyethylene was obtained, and the Mw value of the low Mw part was increased. It was proposed that two kinds of active centers were present in the system. One used for preparing the low molar mass part was sensitive to AlMe3, whilst the other used for preparing the high molar mass part was less dependent on AlMe3; the latter needed more activation energy to form in comparison to the former active center. As a result, the combination of the multi-active centers and chain transfer could explain the results of the polymerization observed when using such Fe(acac)3/Ln (n = 1–7) (Scheme 20) catalytic systems.63
 | ||
| Scheme 20 Structure of the bis(imino)pyridyl ligands.63 | ||
The Xie group has looked into the factors effecting ethylene oligomerization when using unsymmetric bis(imino)pyridine iron(II) complexes bearing alkyl and halogen substituents.64 Such Fe(II) complexes were highly active for ethylene oligomerization with a high selectivity for linear α-olefins. The catalytic activities of 126a, 126b and 127b (Scheme 21) were in excess of 106 g mmol−1 (Fe) h−1 bar−1, which is higher than that of the methyl or fluoro substituted symmetric 2,6-bis(arylimino)pyridyl iron(II) complexes. The products were mainly linear α-olefins, with the highest yield recorded at over 98%. The distribution of α-olefins was between C4 and C24; no polymers were observed. The catalytic performance, especially the oligomer distribution, could be tuned by synergistic steric and electronic effects. The selectivity for C6–C16 was more than 80% in oligomers when catalyzed by 126a, 126b and 126d, which is 15–30% higher than that catalyzed by methyl or fluoro substituted symmetric 2,6-bis(imino)pyridyl iron(II) complexes and demonstrates potential for industrial application.64
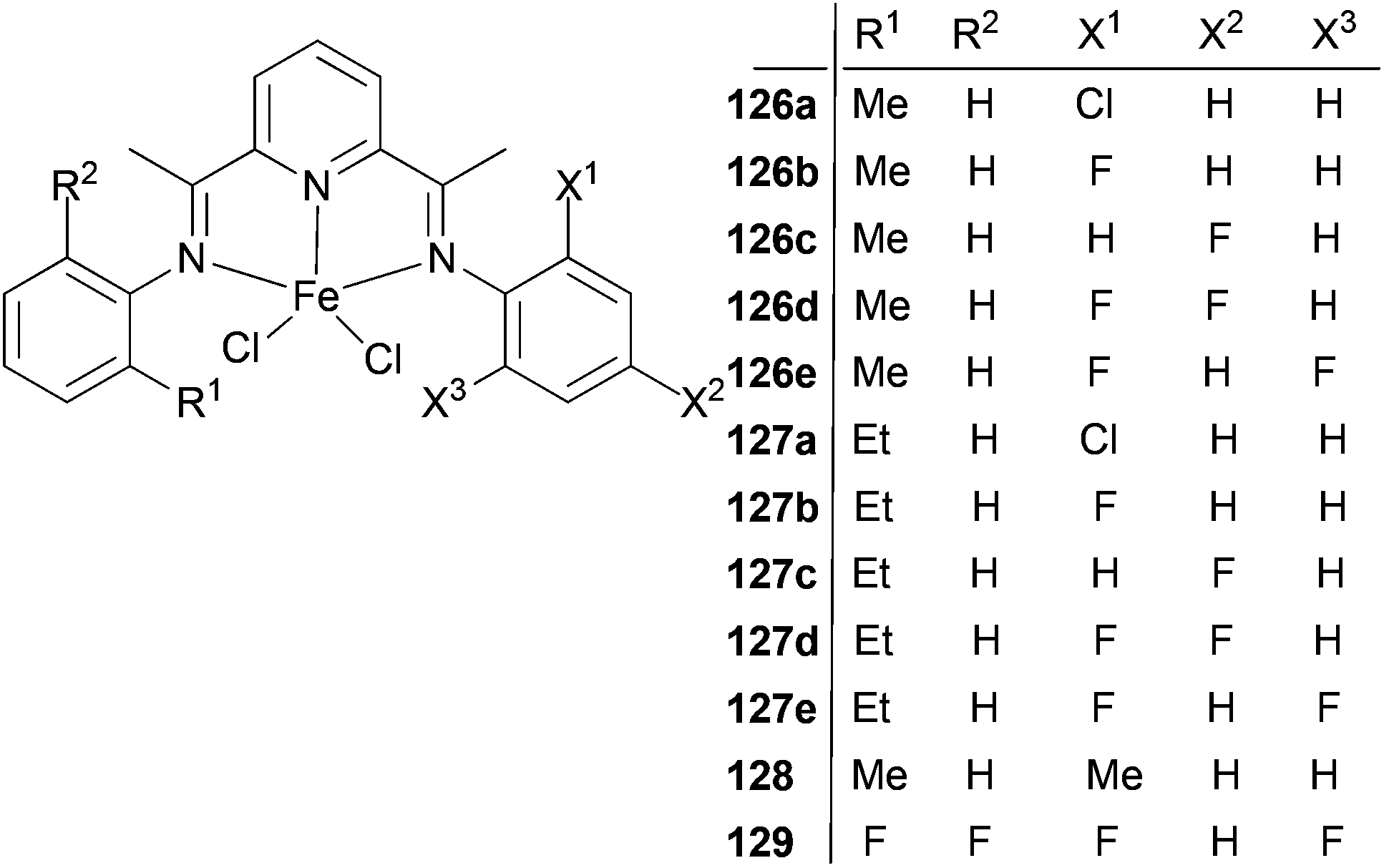 | ||
| Scheme 21 N,N,N-Tridentate asymmetric bis(imino)pyridine iron(II) complexes with alkyl and halogen substituents.64 | ||
The sterics associated with the alkyl substituents had a profound effect on the oligomer distribution. When the halogen substituents were the same but the alkyl substituents were different as in 126b (methyl) and 127b (ethyl), then 126b produced significantly more C4 than 127b, which was attributed to the smaller alkyl-steric effect and subsequently enhanced rates of chain transfer or β-hydrogen elimination. Moreover, similar trends were also found for 126c/127c and 126d/127d, which is in accordance with the catalytic behavior of symmetric alkyl-substituted 2,6-bis(arylimino)pyridyl iron(II) complexes. The steric effects apparently also worked in unsymmetric 2,6-bis(imino)pyridyl iron(II) complexes, and could be used to inhibit the production of low molar mass oligomers (C4).
Electron withdrawing halogen groups could also exert an influence on the catalytic behavior of unsymmetric 2,6-bis(imino)pyridyl iron(II) complexes, such that ligands containing halogen substituents enhanced the catalytic performance.45,54 Complexes with only one fluoro substituent at the imino-N aryl ring ortho position had the highest oligomerization activity. The introduction of an electron withdrawing group could increase the electrophilicity of the central metal Fe and the coordination of ethylene, favoring the catalytic activity.44,54 However, the catalytic activity decreased as more fluoro substituents were introduced into the phenyl ring, similar to the situation observed for the symmetric 2,6-bis(imino)pyridyl iron(II) complexes. A possible explanation is that the stronger electron withdrawing fluoro atoms could weaken the electron donation ability of the ligand, making the active species more prone to decomposition during the polymerization process.65,66 The electronic effect of the halogen groups was also demonstrated in the oligomer distributions. When one of the methyl groups on the aryl rings of complex 128 was substituted by a fluoro substituent (126b), more C4 and less C18 were produced. Furthermore, when comparing the complexes 126a, 126b, 126d and 129, where the electron-withdrawing effect is gradually enhanced, a similar trend was seen. Similar results were also observed for 127a, 127b, 127d and 129, i.e. the electronic effect of a halogen can be utilized to inhibit the production of high molecular oligomers.64
Three new unsymmetrical bis(imino)pyridyl iron(II) complexes (130–132, Scheme 22) were explored for their potential better thermo-stability.67 Complex 131, containing 2-methyl-6-sec-phenethyl substituents at the aniline moiety, exhibited a better activity and produced much higher molecular weight polyethylene as compared to the singly ortho substituted analogues 130 and 132 and the more established symmetrical 2,6-diisopropylphenyl-substituted complex 84 (Scheme 13). Furthermore, at 70 °C, the catalyst 131 maintained a high activity and relatively stable kinetics. The polymers obtained by the unsymmetrical and bulky alkyl-substituted catalysts possessed a bimodal molecular weight distribution due to the co-existence of two chain transfer pathways. The content of the low molecular weight fraction increased on increasing the Al/Fe ratio.67
 | ||
| Scheme 22 N,N,N-Tridentate bis(imino)pyridine iron(II) complexes bearing bulky and unsymmetrical substituted aniline groups.67 | ||
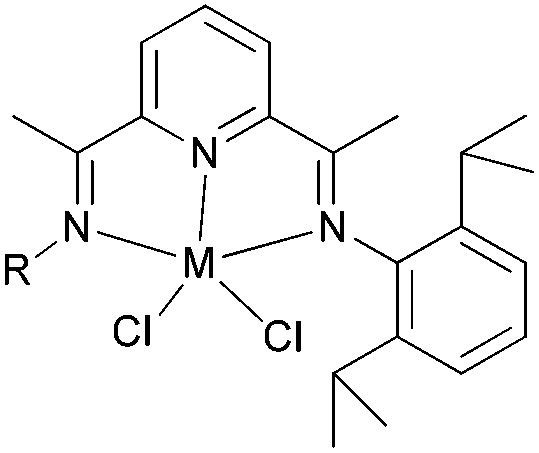
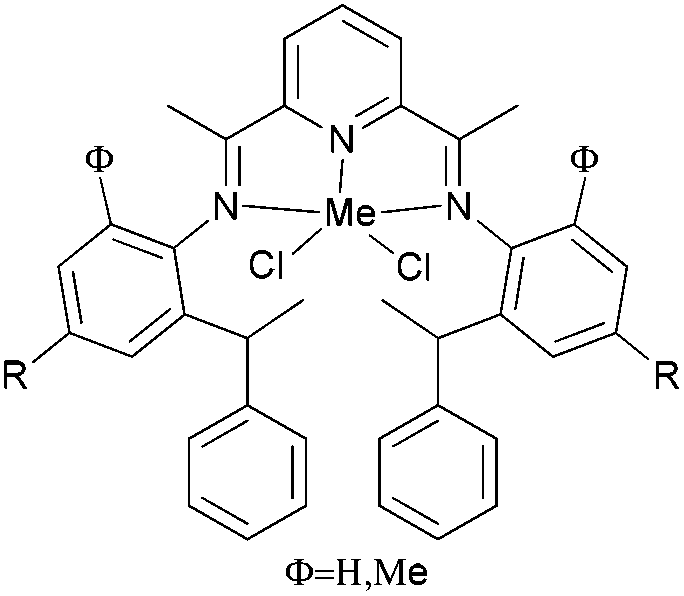
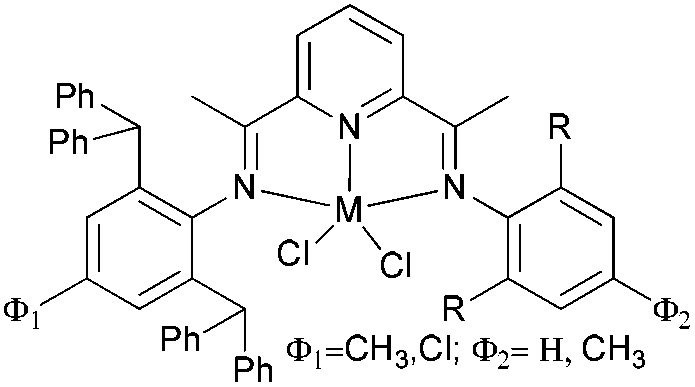
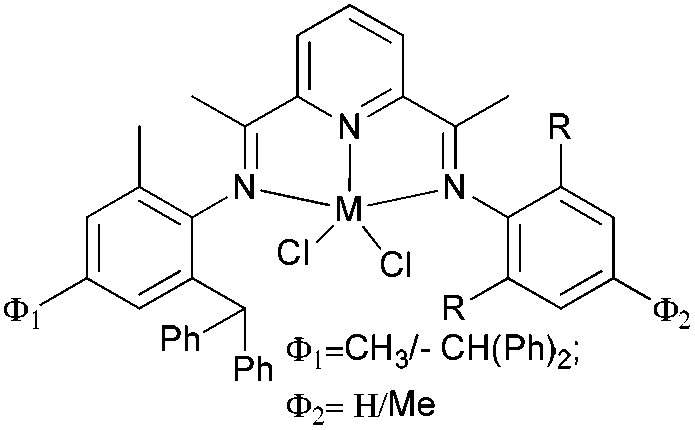
Our group has systematically studied the ethylene polymerization characteristics of bis(imino)pyridine iron(II) complexes bearing different benzhydryl-substituents.22,23,68–74 The complexes 133, 137, 140, 142, 144 (Scheme 23 – R1 = Me, Et, iPr; R2 = H, Me) exhibited high activity during ethylene polymerization, producing linear polyethylene without any trace of oligomers. This was particularly the case upon activated with MMAO, and the observed activity was the highest reported for an iron-based pre-catalysts of this type. Typically, the activity of 133 was 2.15 × 107 g mol−1 (Fe) h−1 at 80 °C, 137 was 2.27 × 107 g mol−1 (Fe) h−1 at 70 °C, 140 was 2.69 × 107 g mol−1 (Fe) h−1 at 60 °C and 142 was 3.15 × 107 g mol−1 (Fe) h−1 at 60 °C, 144 was 1.53 × 107 g mol−1 (Fe) h−1 at 60 °C, respectively. These results not only showed the highest activity for non-symmetric 2,6-bis(imino)pyridine iron(II) complexes bearing benzhydryl-substituted (133, 137, 140, 142, 144), but they also illustrated improved thermal stability, potentially making such systems of interest for industrial consideration.22,68,70,72,74 However, 135 and 146 displayed only moderate activity and 139 exhibited low activity due to the steric bulk.20,65,71 By incorporating a para chloro substituent, 137 was found to exhibit extremely high activity and produced highly linear polyethylene of low molecular weight and with narrow PDI, indicative of single-site catalytic behaviour.68 Modification of the ligand by changing the relative positions of the benzhydryl substituents resulted in 140, which revealed both high activity and good thermal stability.70 In addition, when a para fluorophenyl was employed instead of the phenyl moiety within the benzhydryl substituents, the resultant iron pre-catalyst 142 exhibited even higher activities (in the 108 g mol−1 h−1 range), producing polyethylene of relatively high molecular weight.72 When less benzhydryl substituents were present, pre-catalyst 144 exhibited a relatively lower activity, revealing that the presence of the bulky dibenzhydryl substituted N-aryls enhanced the catalytic performance of their metal complexes. When using cobalt instead of iron, the activity for ethylene polymerization of the pre-catalysts 134, 138, 141, 143, and 145 was one order of magnitude lower than the corresponding iron pre-catalysts.22,32,68–71 In addition, polymers obtained via the cobalt pre-catalysts 134, 138, 141, 143, and 145 possessed narrower molecular weight distributions, indicative of the formation of single-site active species.23,69,71,73,74
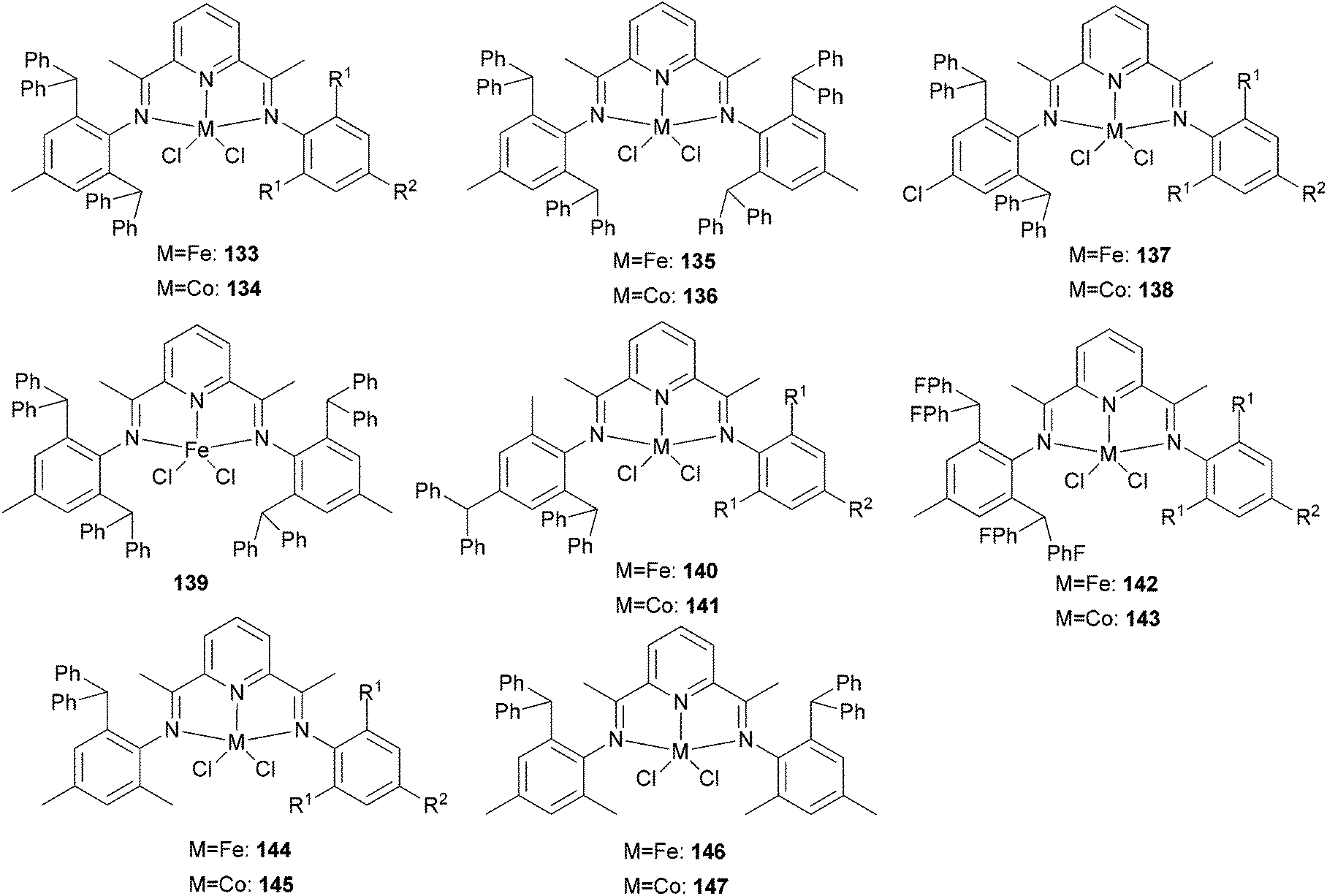 | ||
| Scheme 23 N,N,N-Tridentate bis(imino)pyridine iron(II) complexes bearing benzhydryl-substituted.22,23,68–74 | ||
The 1,8-diimino-2,3,4,5,6,7-hexahydroacridine iron and cobalt complexes 148 and 149 (Scheme 24, left) were prepared via a one-pot synthesis and exhibited high activity for ethylene polymerization when treated with methylaluminoxane.75 The ortho substituents of the aryl rings and the type of metal employed played a significant role on ethylene activation and specifically on product distribution. The Fe complex bearing a 2,6-diisopropyl-substituted bis(imino)tetrahydroacridyl ligand produced polyethylene of moderate molecular weight (18000). On changing the ortho substitution from 2,6-diisopropyl to 2,6-dimethyl, the Co complexes produced polyethylene waxes and oligomers of predominantly α-olefins simultaneously, and in the process obeying the Schulz–Flory distribution. The Co complexes bearing ligands with less sterically hindered substituents at the ortho position behaved exclusively as dimerization catalysts.75 A series of 2,8-bis(arylimino)-5,6,7-trihydroquinoline iron and cobalt complexes 150 and 151 (Scheme 24, right) were also prepared and were screened for ethylene polymerization.76,77 Upon treatment with either MAO or MMAO, complexes 150 and 151 exhibited very high activities for ethylene polymerization, for example, the activity of 150 reached 2.4 × 107 g mol−1 (Fe) h−1 at 50 °C when activated with MMAO83 and the highest activity of 151 reached 1.1 × 107 g mol−1 (Fe) h−1 at 60 °C when activated with MAO under 10 bar ethylene.76 The obtained polyethylene via150 was a low molecular weight polymer,76 and the polyethylene from 151 under the optimized reaction parameters, possessed low molecular weight (waxes) and narrow polydispersity.76
 | ||
| Scheme 24 1,8-Diimino-2,3,4,5,6,7-hexahydroacridine and 2,8-bis(arylimino)-5,6,7-trihydroquinoline iron and cobalt complexes.75–77 | ||
3.2. Pyridine type ligation
During the development of new ligand systems for enhancing both activity and control of the microstructure of the resulting polymer, a number of changes to the basic bis(imino)pyridine ligand motif have been targeted. As well as the modifications to the N-imine substituents, there are many other strategies which can be used to modify the skeleton of the bis(imino)pyridine ligand.Our group has synthesized two five-coordinate iron(II) chloride complexes (152 and 153, Scheme 25) bearing 2-imine-6-(methyl alcohol)pyridine chelates via an aluminium-mediated methyl migration route from the corresponding 2-acetyl-6-iminopyridine and studied the ethylene polymerization properties of the complexes 152 and 153.10 Both iron complexes displayed moderate activity (170 g mmol−1 (Fe) h−1 bar−1 of 152 and 276 g mmol−1 (Fe) h−1 bar−1 of 153) for ethylene polymerization on treatment with excess methylaluminoxane, significantly lower than for related bis(arylimino)pyridine iron systems. The products were highly linear polymers and some oligomeric products. Furthermore, analysis of the vinylic region of the spectra revealed the presence of greater than 99% α-olefins. It was apparent that these systems, as with bis(imino)pyridine iron catalysts,43 could facilitate chain-transfer reactions by both β-H elimination and chain transfer to aluminium.10
 | ||
| Scheme 25 N,N,O-Chelates for five-coordinate iron(II) chloride complexes.10 | ||
The Karam group investigated iron and cobalt complexes bearing tris(2-pyridyl)phosphine and tris(2-pyridyl)amine ligands (Scheme 26).78 Such catalysts exhibited relatively high activities (range = 32–271 g mmol−1 (M) h−1 bar−1) for ethylene polymerization.79 The polymerization temperature had an impact on the activity of the systems bearing either ligand set. In the case of 154 and 155, a five times increase was observed by increasing the temperature from 60 °C (50 g mmol−1 (Fe) h−1 bar−1 for 154 and 57 g mmol−1 (Fe) h−1 bar−1 for 155) to 80 °C (243 g mmol−1 (Fe) h−1 bar−1 for 154 and 271 g mmol−1 (Fe) h−1 bar−1 for 155). The analogous cobalt complexes 156 and 157 exhibited similar behaviour, revealing a 6-fold activity increase. The iron complexes possessed higher activities than did their cobalt counterparts, for example, the activity of 154 was 243 g mmol−1 (Fe) h−1 bar−1, whilst that for 156 was 213 g mmol−1 (Co) h−1 bar−1. In addition, it was found that the bridgehead atom of the ligand TpX (X = P, N) did not affect significantly the activity of the active species nor the molecular weight of the polymers. The polyethylene obtained was linear HDPE with a broad mono-modal distribution.78
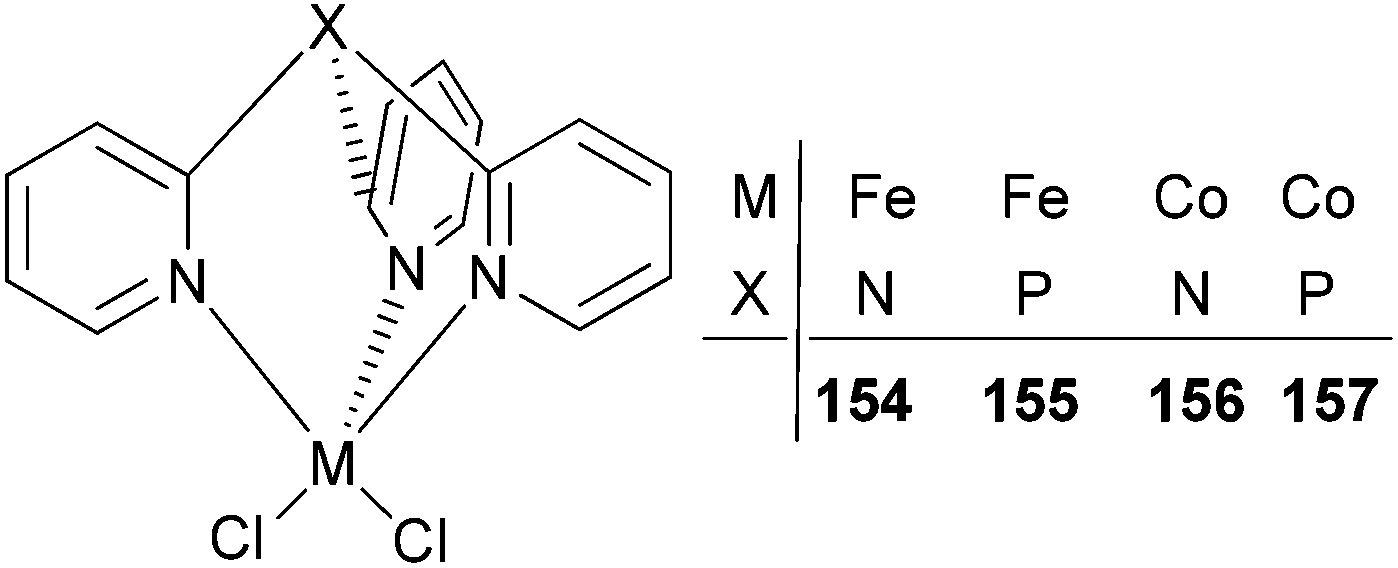 | ||
| Scheme 26 Tris(2-pyridyl)phosphine and tris(2-pyridyl)amine iron(II) and cobalt(II) catalysts.78 | ||
Our group has systematically studied the ethylene polymerization and oligomerization characteristic of a number of N,N,N-tridentate pyridyl iron(II) and cobalt(II) catalysts with different ligand frameworks (Scheme 27).20,50,80–85 The iron pre-catalyst 158 exhibited high activity for ethylene oligomerization in the presence of MAO with butene as the major product at atmospheric pressure.50 Similarly, the cobalt pre-catalyst 159 also exhibited high activity for ethylene oligomerization in the presence of MMAO with butene as the major product at atmospheric pressure, for example, the typical activity of 158 was 1.67 × 105 g (mol Co)−1 h−1 for oligomerization and the C4 percent was 96.4%. On increasing the ethylene pressure, both the activity and the content of longer-chain oligomers increased.50 In some cases, polyethylene waxes were collected to afford overall good polymerization activity. A typical example exhibited an activity of up to 2.24 × 106 g (mol Fe)−1 h−1 for oligomerization, and 8.56 × 105 g (mol Fe)−1 h−1 for polymerization using 159.50 The analogs 160 and 161 revealed high activity towards ethylene oligomerization; some polyethylene waxes were also formed.80–82 The activity order R = H78 > R = Me79 > R = iPr77 was observed due to the electronic influence exerted by the substituents. When the benzimidazole contained different substituents such as Me or Cl, the activity of 162 and 163versus160a exhibited slight differences.50,83 When activated with MMAO, 162 afforded high activities (1.86 × 106 g mmol−1 (M) h−1) for ethylene oligomerization, lower than the activity of 160a, whilst 163 revealed high activities (2.82 × 106 g mmol−1(Fe) h−1) upon treatment with MAO, higher than the activity of 160a.83 All the oligomers produced were in the range C4–C28 with a very high selectivity for linear α-olefins and high K values. The complex 164a showed moderate to good activities of up to 106 g (product) (mol Fe)−1 h−1 bar−1 for the oligomerization and polymerization of ethylene, with high selectivity for vinyl-terminated oligomers or polyethylene waxes.84 In contrast, the 2-(benzothiazolyl)-6-(1-(arylimino)ethyl)pyridine complex 164b exhibited activities of up to 107 g mol−1 (Fe) h−1 for oligomers and 7.01 × 105 g mol−1 (Fe) h−1 for waxes in the presence of MMAO. An increase in the temperature resulted in deactivation and a lower selectivity for α-olefins. In most cases, the wax-like products obtained were confirmed to be vinyl-type olefins.85
 | ||
| Scheme 27 N,N,N-Tridentate pyridyl iron(II) and cobalt(II) catalysts.20,50,80–85 | ||
3.3. Other tri-dentate ligands
There are also many other examples beyond the iminopyridyl-type ligand set. For example, quinoline and phenanthrolyl derivatives have potential for the preparation of active iron and cobalt pre-catalyst, and our group has made some progress in this area.We developed a synthetic template method to prepare the complexes 166 and 167 (Scheme 28) bearing N-((pyridin-2-yl)methylene)-quinolin-8-amine ligands. Upon activation with MMAO, complexes 166 and 167 exhibited high catalytic activities (up to 106 g mol−1 (Fe) h−1) for ethylene oligomerization, with dimers and trimers as products, and moderate to high selectivity for 1-C4. For ligands bearing bulky substituents, enhanced activity was observed for the iron(II) system, however lower catalytic activity was obtained when using Co(II) complexes.86 Interestingly, complexes 168 and 169 ligated by 2,8-bis-(1-aryliminoethyl)quinolines showed unique properties toward ethylene polymerization: no activity was observed at low temperature, but high activity was achieved at temperatures higher than 80 °C (up to 7.61 × 106 g mmol−1 (M) h−1 at 100 °C) upon activation with methylaluminoxane (MAO).21 Moreover, the polyethylene obtained was of high molecular weight and narrow distribution. This was the first example of iron or cobalt pre-catalysts with such high activity for ethylene polymerization at high temperatures.21 Similarly, 2-(1-(arylimino)methyl)-8-(1H-benzimidazol-2-yl)-quinolyl iron(II) and cobalt chloride complexes 170 and 171 were also prepared and upon activation with MAO, they exhibited much lower activity than 168 and 169; the highest activity for the iron pre-catalyst was 6.5 × 105 g mmol−1 (Fe) h−1 and that of the cobalt pre-catalyst was 4.0 × 105 g mmol−1 (Co) h−1 for ethylene polymerization, affording highly linear polyethylene. However, such systems possessed good thermal stability as evidenced by the high activities at high temperatures (up to 100 °C).87
 | ||
| Scheme 28 N,N,N-Tri-dentate iron(II) and cobalt(II) complexes bearing tridentate quinoline derivatives.21,86–90 | ||
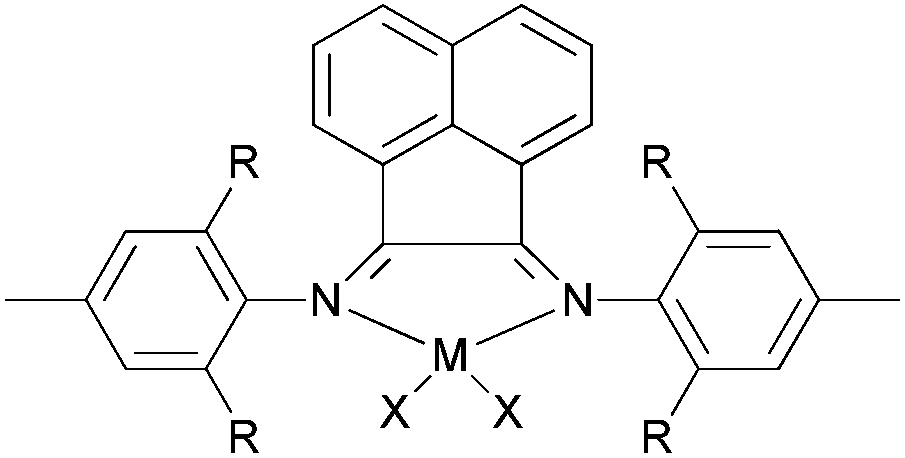
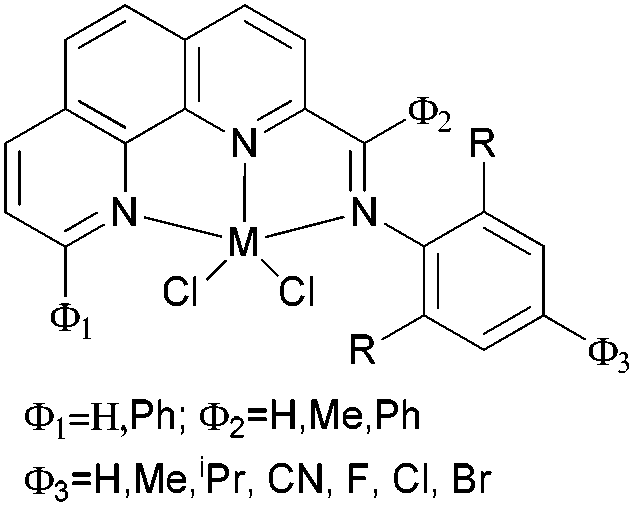
Our group has also synthesized a family of 2-imino-1,10-phenanthroline ligands and the iron and cobalt complexes thereof.91,93–95 The preparative route was somewhat complicated starting from phenanthroline, 2-acetyl-1,10-phenanthrolines and their substituted analogues were prepared.91,93 Imino-phenanthrolines were then prepared by the reaction of the phenanthroline ketones or aldehydes with anilines, from which the corresponding metal complexes were obtained.91,93–95 The iron complexes ligated by 2-imino-1,10-phenanthrolines 172b (Scheme 29) exhibited very high activity (up to 4.91 × 107 g mmol−1 (Fe) h−1) for ethylene oligomerization with high selectivity for α-olefins (>94%), with Schulz–Flory distribution.91 Notably, N-aryl linked bimetallic iron complexes have been reported, which afforded higher molecular weight polymeric materials than did their monometallic analogues.92 The cobalt complexes ligated by 2-imino-1,10-phenanthrolines 173, reported by Solan et al., exhibited lower activity than the iron complexes.93 In addition, it was found that the substituent on the arylimino moiety affected the product formed, for example, for the phenyl bearing an Et substituent, i.e.172b, the best activity was observed, whilst stronger electron withdrawing groups decreased the ethylene reactivity (when the phenyl substituent R1 was a halogen, the activity order decreased as follows: Br > Cl > F).91 Variation of the R substituent on the imino-C of ligands of the type, 2-(ArN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR)-1,10-phenanthroline, also resulted in changes to the catalytic performance. The aldimine (R = H, 172a), ethyl-ketimine (R = Et, 172c) and phenyl-ketimine (R = Ph, 172d) complexes showed relatively lower catalytic activities than did the corresponding methyl-ketimine (R = Me, 172b) complex. All the obtained oligomers exhibited Schulz–Flory distribution. However, upon treatment with MAO or MMAO, 172c, which bears an ethyl substituent on the imine-C, revealed a better thermal stability (10 °C higher compared to 172b) and possessed a higher content of α-olefins (C6–C16).91,94,95 When a phenyl substituent was introduced at the 9-position of the 1,10-phenanthroline by reaction of PhLi with 2-acetyl-1,10-phenanthroline, the resultant 2-imino-9-phenyl-1,10-phenanthrolines complexes 174a, 174b, 174d exhibited much lower activities (2.33 × 106 g mmol−1 (Fe) h−1) in comparison to their analogues 172a–d, and the products included butene (major product, >90%) and hexene; there was no polymer formation.96 Replacement of the imine group with benzimidazoles resulted in iron complexes of type 176, bearing 2-(benzimidazol-2-yl)-1,10-phenanthrolines. On treatment with MMAO, these complexes oligomerized ethylene to dimers and trimers with high activities and with good selectivity for α-olefins.97 The introduction of a methyl group at the 9-position of the phenanthroline ring led to a decrease in the oligomerization activity as well as a slight increase in α-C4 selectivity, as evidenced by higher activities and lower α-C4 selectivity obtained by 176a (R = H) versus176b (R = Me). The incorporation of an alkyl group (R2) on the N atom of the benzimidazole led to a decrease in oligomerization activity and selectivity for 1-butene, whereas other alkyl groups such as methyl, ethyl, isopropyl and benzyl, revealed no obvious influence on the α-C4 selectivity.97 When the imidazole group was replaced by 2-benzoxazole or oxazoline, the resultant complexes 178, 180 exhibited much lower activity and the products were comprised of oligomers of butene and hexane, with lower α-C4 selectivity.98 It should be emphasised that most of the previous research on bis(imino)pyridyl iron pre-catalysts has recorded ethylene polymerization and oligomerization, with much of the focus being on the polymerization products. However, iron catalytic systems bearing 2-iminophenanthrolines can readily deliver oligomerization products91 and have recently been successfully scaled up for use in a 500 tonne pilot plant.
CR)-1,10-phenanthroline, also resulted in changes to the catalytic performance. The aldimine (R = H, 172a), ethyl-ketimine (R = Et, 172c) and phenyl-ketimine (R = Ph, 172d) complexes showed relatively lower catalytic activities than did the corresponding methyl-ketimine (R = Me, 172b) complex. All the obtained oligomers exhibited Schulz–Flory distribution. However, upon treatment with MAO or MMAO, 172c, which bears an ethyl substituent on the imine-C, revealed a better thermal stability (10 °C higher compared to 172b) and possessed a higher content of α-olefins (C6–C16).91,94,95 When a phenyl substituent was introduced at the 9-position of the 1,10-phenanthroline by reaction of PhLi with 2-acetyl-1,10-phenanthroline, the resultant 2-imino-9-phenyl-1,10-phenanthrolines complexes 174a, 174b, 174d exhibited much lower activities (2.33 × 106 g mmol−1 (Fe) h−1) in comparison to their analogues 172a–d, and the products included butene (major product, >90%) and hexene; there was no polymer formation.96 Replacement of the imine group with benzimidazoles resulted in iron complexes of type 176, bearing 2-(benzimidazol-2-yl)-1,10-phenanthrolines. On treatment with MMAO, these complexes oligomerized ethylene to dimers and trimers with high activities and with good selectivity for α-olefins.97 The introduction of a methyl group at the 9-position of the phenanthroline ring led to a decrease in the oligomerization activity as well as a slight increase in α-C4 selectivity, as evidenced by higher activities and lower α-C4 selectivity obtained by 176a (R = H) versus176b (R = Me). The incorporation of an alkyl group (R2) on the N atom of the benzimidazole led to a decrease in oligomerization activity and selectivity for 1-butene, whereas other alkyl groups such as methyl, ethyl, isopropyl and benzyl, revealed no obvious influence on the α-C4 selectivity.97 When the imidazole group was replaced by 2-benzoxazole or oxazoline, the resultant complexes 178, 180 exhibited much lower activity and the products were comprised of oligomers of butene and hexane, with lower α-C4 selectivity.98 It should be emphasised that most of the previous research on bis(imino)pyridyl iron pre-catalysts has recorded ethylene polymerization and oligomerization, with much of the focus being on the polymerization products. However, iron catalytic systems bearing 2-iminophenanthrolines can readily deliver oligomerization products91 and have recently been successfully scaled up for use in a 500 tonne pilot plant.
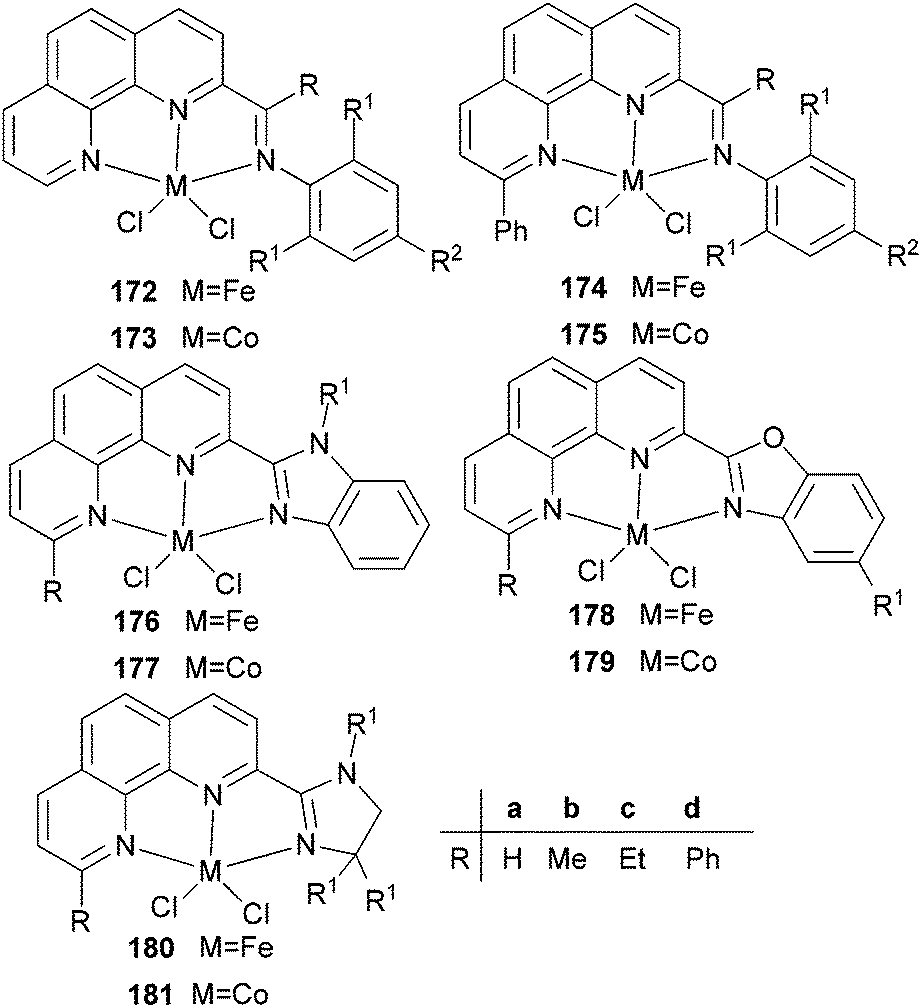 | ||
| Scheme 29 N,N,N-Tridentate iron(II) and cobalt(II) complexes bearing 1,10-phenanthroline derivatives.91–98 | ||
To highlight the catalytic performances of the different complex pre-catalysts, a summary of promising model complexes is collected in Table 11.
| The type | M | Co-catalyst | R | Oligomerization | Polymerization | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| Activ.a | Distri. | Activ.a |
M
wb |
|||||
| a 105 g mol−1 h−1. b 104 g mol−1. | ||||||||
|
Fe/Co | MMAO | PhCH2 | 2.3–8.0 | C4 >92.8% | — | — | 24 |
| iPr | >4.3 | C4 >99.4% | — | — | 24 | |||
|
Co | MAO | Me/iPr | 0.01–0.02/trace | — | — | — | 33 |
|
Co | MAO | Me | Trace | — | — | — | 33 |

|
Fe/Co | MAO/MMAO/Et2AlCl | H/Me | 0.5–2.8 | C4 (37.4–81.4%) | — | — | 34 |
| C6 (18.6–62.6%) | ||||||||

|
Fe/Co | MMAO/Et2AlCl | H/Me | 1.5–10.3 | C4 77.9–96. 3% | — | — | 34 |
|
Co | MAO | Me/Et/iPr/Cl | 7.6–19 | C4 (56.4–59.8%) | 0.4–0.5 | 9.5–12.5 | 35 |
|
Fe/Co | MMAO | Me/Et/iPr | 0.78–5.71 | C4 (95.7–98.9%) | — | — | 36 |
|
Fe/Co | MAO | Me/Et/iPr | 8.0–16.5 | C4 (99.3–100%) | — | — | 37 |

|
Fe | MAO | H/Me/F/Cl/Br/I | 567.3–1592.5 | 100% oligomer | — | — | 44,53,54 |
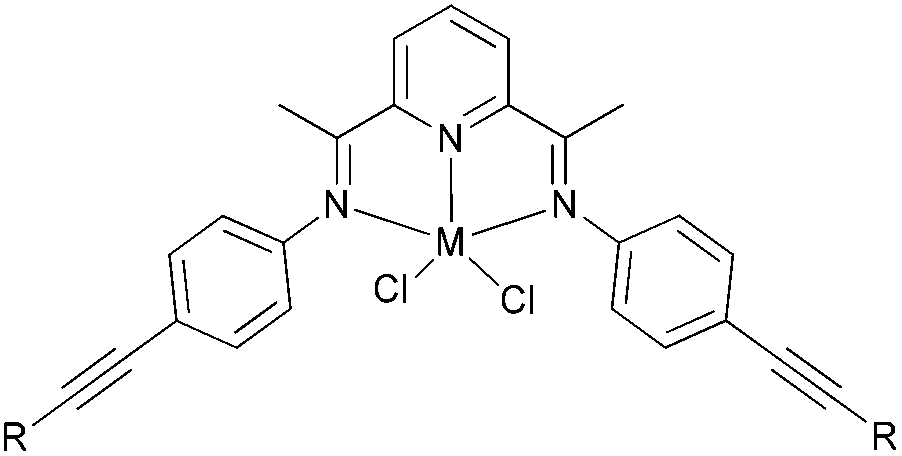
|
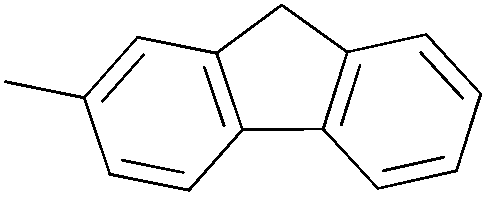
Fe | MAO |
|
245.9 | 100% oligomer | — | — | 53 |
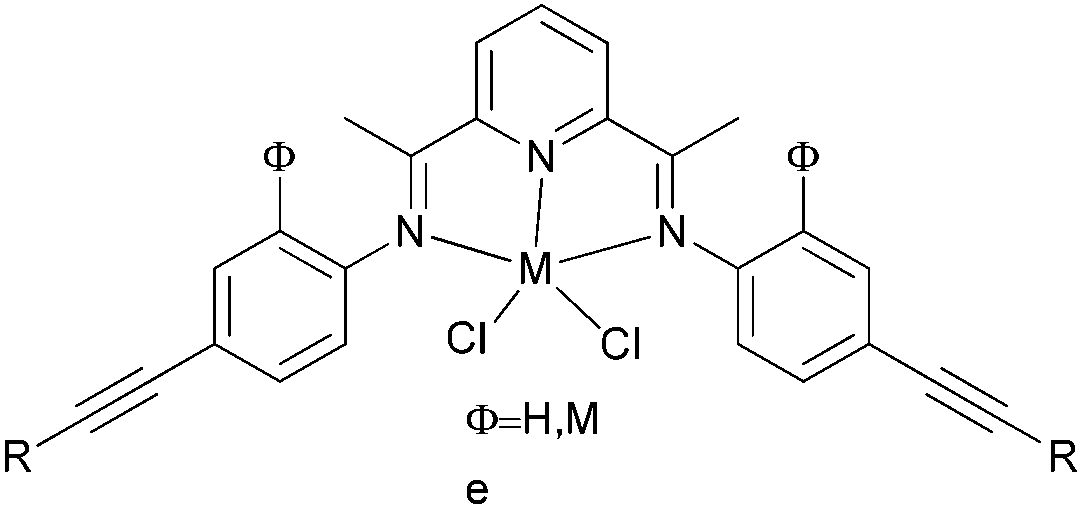
|
Fe | MAO |
|
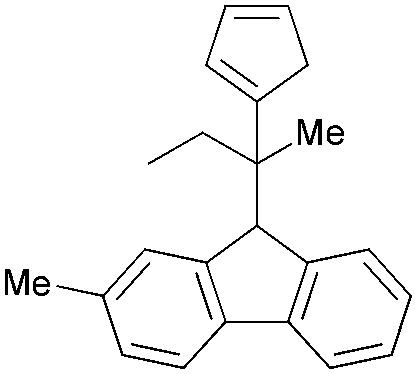
— | — | 742.7–898.2 | 15.8–19.9 | 53 |

|
Fe | MMAO | 2,6-Diisopro-pylphenyl/1-anthracenyl/2-isopropyl-phenyl/2-biphenyl | 14.0–81.8 | — | 13.4–2000 | 1–10 | 55 |
|
Fe | MMAO | 1-Anthracenyl/2-isopropyl-phenyl | — | — | 20–2000 | 0.1–1 | 55 |

|
Fe/Co | MAO | CH2CMe2Ph/CH2Ph/CH2CHCH2 |
— | — | 7.7–20.9 | 2.57–18.0 | 61 |
|
Fe/Co | MAO | Me/iPr | — | — | 4.0–60 | 20.7–64.6 | 62 |
|
Fe | MAO | Me/OCH3 | — | — | 3.1–12.0 | 0.3–149.9 | 67 |
|
Fe/Co | MAO/MMAO | Me/Et/iPr/–CH(Ph)2 | — | — | Trace–269 | 0.23–120 | 22, 23,70, 71 |
|
Fe/Co | MAO/MMAO | Me/Et/iPr/–CH(Ph)2 | — | — | 9.3–352 | 1.26–9449.9 | 72–74 |
|
Fe/Co | MAO/MMAO/Et2AlCl | H/Me | 0.01–41.1 | α-Olefin >70% | Trace | — | 77–82 |
|
Fe/Co | MAO/MMAO | Me/Et/iPr/F/Cl | 0.76–491 | α-Olefin >79% | Trace–304 | — | 91–98 |
4. Outlook
The discovery of highly active 2,6-bis(arylimino)pyridyl metal (metal = iron or cobalt) complexes proved to be a milestone in the development of late transition metal olefin polymerization catalysts. Much effort has been devoted to catalyst modification with a view to enhancing not only the catalytic activity and control over the microstructure of the resulting polymer, but also the thermal stability of such systems. Some encouraging results has been achieved, for example, the phenanthroline-based iron catalysts, which were found to exhibit very high activity for ethylene oligomerization (comparable to the bis(imino)pyridine iron system), have been successfully employed in a 500 tonne pilot plant managed by Sinopec in China. However, if late transition metal catalysts of this type are to be utilized in industry, then many problems need still to be solved, including rapid deactivation at temperatures akin to those required for industrial operation, as well as the problem that polymeric products are not solely formed. On-going research continues to try to address these issues, and the use of bulky groups such as the benzhydryl motif has already gone some way to addressing such problems.Acknowledgements
CR thanks the EPSRC for an overseas travel award, the RSC for an international author grant and Sichuan Normal University for financial support.Notes and references
- Source: The Plastics Portal by PlasticsEurope, see http://www.plasticseurope.org/information-centre/press-room-1351/press-releases-2012/first-estimates-suggest-around-4-increase-in-plastics-global-production-from-2010.aspx.
- See for example, (a) A. L. McKnight and R. M. Waymouth, Chem. Rev., 1998, 98, 2587–2598 CrossRef CAS PubMed; (b) H. H. Brintzinger, D. Fischer, R. Mulhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143–1170 CrossRef CAS; (c) G. W. Coates, Chem. Rev., 2000, 100, 1223–1252 CrossRef CAS PubMed; (d) M. Bochmann, Top. Catal., 1999, 7, 9–22 CrossRef CAS; (e) M. Bochmann, Dalton Trans., 1996, 255–279 RSC.
- M. P. Daniel, Adv. Catal., 2010, 53, 128–584 Search PubMed and references therein.
- G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 849–850 RSC.
- B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049–4050 CrossRef CAS.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1203 CrossRef CAS PubMed.
- V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–315 CrossRef CAS PubMed.
- C. Bianchini, G. Giambastiani, I. G. Rios, G. Mantovani, A. Meli and A. M. Segarra, Coord. Chem. Rev., 2006, 250, 1391–1418 CrossRef CAS PubMed.
- V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745–1776 CrossRef CAS PubMed.
- V. C. Gibson, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, Organometallics, 2007, 26, 5119–5123 CrossRef CAS.
- K. P. Bryliakov, Russ. Chem. Rev., 2007, 76, 253–277 CrossRef CAS PubMed.
- W.-H. Sun, S. Zhang and W. Zuo, C. R. Chim., 2008, 11, 307–316 CrossRef CAS PubMed.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS PubMed.
- S. Jie, W.-H. Sun and T. Xiao, Chin. J. Polym. Sci., 2010, 28, 299–304 CrossRef CAS PubMed.
- C. Bianchini, G. Giambastiani, L. Luconi and A. Meli, Coord. Chem. Rev., 2010, 254, 431–455 CrossRef CAS PubMed.
- T. Xiao, W. Zhang, J. Lai and W.-H. Sun, C. R. Chim., 2011, 14, 851–855 CrossRef CAS PubMed.
- W. Zhang, W.-H. Sun and C. Redshaw, Dalton Trans., 2013, 42, 8988–8997 RSC.
- V. C. Gibson and G. A. Solan, in Catalysis without Precious Metals, ed. M. Bullock, Wiley-VCH, Weinheim, 2010, pp. 111–141 Search PubMed.
- V. C. Gibson and G. A. Solan, Top. Organomet. Chem., 2009, 26, 107–158 CrossRef CAS.
- R. Gao, K. Wang, Y. Li, F. Wang, W.-H. Sun, C. Redshaw and M. Bochmann, J. Mol. Catal. A: Chem., 2009, 309, 166–171 CrossRef CAS PubMed.
- S. Zhang, W.-H. Sun, T. Xiao and X. Hao, Organometallics, 2010, 29, 1168–1173 CrossRef CAS . Additional correction S. Zhang, W.-H. Sun, T. Xiao and X. Hao, Organometallics, 2010, 29, 6837–6840 CrossRef.
- J. Yu, H. Liu, W. Zhang, X. Hao and W.-H. Sun, Chem. Commun., 2011, 47, 3257–3259 RSC.
- J. Yu, W. Huang, L. Wang, C. Redshaw and W.-H. Sun, Dalton Trans., 2011, 40, 10209–10214 RSC.
- L. Wang, C. Zhang and Z.-X. Wang, Eur. J. Inorg. Chem., 2007, 2477–2487 CrossRef CAS.
- T. Xiao, S. Zhang, G. Kehr, X. Hao, G. Erker and W.-H. Sun, Organometallics, 2011, 30, 3658–3665 CrossRef CAS.
- T. Xiao, P. Hao, G. Kehr, X. Hao and W.-H. Sun, Organometallics, 2011, 30, 4847–4853 CrossRef CAS.
- C. Bianchini, G. Mantovani, A. Meli, F. Migliacci and F. Laschi, Organometallics, 2003, 22, 2545–2547 CrossRef CAS.
- C. Bianchini, G. Giambastiani, G. Mantovani, A. Meli and D. Mimeau, J. Organomet. Chem., 2004, 689, 1356–1361 CrossRef CAS PubMed.
- C. Bianchini, G. Giambastiani, A. Meli and A. Toti, Organometallics, 2007, 26, 1303–1305 CrossRef CAS.
- C. Bianchini, D. Gatteschi, G. Giambastiani, R. Guerrero Rios, A. Lenco, F. Laschi, C. Mealli, A. Meli, L. Sorace, A. Toti and F. Vizza, Organometallics, 2007, 26, 726–739 CrossRef CAS.
- T. Irrgang, S. Keller, H. Maisel, W. Kretschmer and R. Kempe, Eur. J. Inorg. Chem., 2007, 4221–4228 CrossRef CAS.
- Y. D. M. Champouret, J. Fawcett, W. J. Nodes, K. Singh and G. A. Solan, Inorg. Chem., 2006, 45, 9890–9900 CrossRef CAS PubMed.
- V. Rosa, S. Carabineiro, T. Avilés, P. T. Gomes, R. Welter, J. M. Campos and M. R. Ribeiro, J. Organomet. Chem., 2008, 693, 769–775 CrossRef CAS PubMed.
- W.-H. Sun, P. Hao, S. Zhang, Q. Shi, W. Zuo, X. Tang and X. Lu, Organometallics, 2007, 26, 2720–2734 CrossRef CAS.
- T. Xiao, J. Lai, S. Zhang, X. Hao and W.-H. Sun, Catal. Sci. Technol., 2011, 1, 462–469 CAS.
- S. Song, T. Xiao, C. Redshaw, X. Hao, F. Wang and W.-H. Sun, J. Organomet. Chem., 2011, 696, 2594–2599 CrossRef CAS PubMed.
- S. Song, W. Zhao, L. Wang, C. Redshaw, F. Wang and W.-H. Sun, J. Organomet. Chem., 2011, 696, 3029–3035 CrossRef CAS PubMed.
- G. J. P. Britovsek, S. Mastroianni, G. A. Solan, S. P. D. Baugh, C. Redshaw, V. C. Gibson, A. J. P. White, D. J. Williams and M. R. J. Elsegood, Chem.–Eur. J., 2000, 6, 2221–2231 CrossRef CAS.
- E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed.
- X. Tang, D. Zhang, S. Jie, W.-H. Sun and J. Chen, J. Organomet. Chem., 2005, 690, 3918–3928 CrossRef CAS PubMed.
- W. Zhang, W.-H. Sun, S. Zhang, J. Hou, K. Wedeking, S. Schultz, R. Fröhlich and H. Song, Organometallics, 2006, 25, 1961–1969 CrossRef CAS.
- W. Zhang, W.-H. Sun, X. Tang, T. Gao, S. Zhang, P. Hao and J. Chen, J. Mol. Catal. A: Chem., 2007, 265, 159–166 CrossRef CAS PubMed.
- G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Strömberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728–8740 CrossRef CAS.
- Y. Chen, C. Qian and J. Sun, Organometallics, 2003, 22, 1231–1236 CrossRef CAS.
- I. S. Paulino and U. Schuchardt, J. Mol. Catal. A: Chem., 2004, 211, 55–58 CrossRef CAS PubMed.
- Z. Zhang, S. Chen, X. Zhang, H. Li, Y. Ke, Y. Lu and Y. Hu, J. Mol. Catal. A: Chem., 2005, 230, 1–8 CrossRef CAS PubMed.
- J.-Y. Liu, Y. Zheng, Y.-G. Li, L. Pan, Y.-S. Li and N.-H. Hu, J. Organomet. Chem., 2005, 690, 1233–1239 CrossRef CAS PubMed.
- B. L. Small and M. Brookhart, Macromolecules, 1999, 32, 2120–2130 CrossRef CAS.
- W.-H. Sun, W. Zhao, J. Yu, W. Zhang, X. Hao and C. Redshaw, Macromol. Chem. Phys., 2012, 213, 1266–1273 CrossRef CAS.
- W.-H. Sun, P. Hao, G. Li, S. Zhang, W. Wang, J. Yi, M. Asma and N. Tang, J. Organomet. Chem., 2007, 692, 4506–4518 CrossRef CAS PubMed.
- C. Görl and H. G. Alt, J. Mol. Catal. A: Chem., 2007, 273, 118–132 CrossRef PubMed.
- B. L. Small and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 7143–7144 CrossRef CAS.
- C. Görl and H. G. Alt, J. Organomet. Chem., 2007, 692, 4580–4592 CrossRef PubMed.
- Y. Chen, R. Chen, C. Qian, X. Dong and J. Sun, Organometallics, 2003, 22, 4312–4321 CrossRef CAS.
- F. Kaul, K. Puchta, G. Frey, E. Herdtweck and W. Herrmann, Organometallics, 2007, 26, 988–999 CrossRef CAS.
- A. S. Ionkin, W. J. Marshall, A. J. Adelman, B. B. Fones, B. M. Fish, M. F. Schiffhauer, P. D. Soper, R. L. Waterland, R. E. Spence and T. Xie, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 585–611 CrossRef CAS.
- A. S. Ionkin, W. J. Marshall, A. J. Adelman, B. B. Fones, B. M. Fish and M. F. Schiffhauer, Organometallics, 2008, 27, 1902–1911 CrossRef CAS.
- A. S. Ionkin, W. J. Marshall, A. J. Adelman, B. B. Fones, B. M. Fish, M. F. Schiffhauer, R. E. Spence and T. Xie, Organometallics, 2008, 27, 1147–1156 CrossRef CAS.
- G. Wallenhorst, G. Kehr, H. Luftmann, R. Fröhlich and G. Erker, Organometallics, 2008, 27, 6547–6556 CrossRef.
- J. Jin, D. R. Wilson and E. Y.-X. Chen, Chem. Commun., 2002, 708–709 RSC.
- J. Cámpora, A. Marcos Naz, P. Palma, A. Rodriguez-Delgado, E. Alvarez, I. Tritto and L. Boggioni, Eur. J. Inorg. Chem., 2008, 1871–1879 CrossRef.
- Z. Long, B. Wu, P. Y. Yang, G. Li, Y. Liu and X.-J. Yang, J. Organomet. Chem., 2009, 694, 3793–3799 CrossRef CAS PubMed.
- J. Wang, W. Li, B. Jiang and Y. Yang, J. Appl. Polym. Sci., 2009, 113, 2378–2391 CrossRef CAS.
- G. Xie, T. Li and A. Zhang, Inorg. Chem. Commun., 2010, 13, 1199–1202 CrossRef CAS PubMed.
- S. Matsui and T. Fujita, Catal. Today, 2001, 66, 63–73 CrossRef CAS.
- T. Zhang, W.-H. Sun, T. Li and X. Yang, J. Mol. Catal. A: Chem., 2004, 218, 119–124 CrossRef CAS PubMed.
- L. Guo, H. Gao, L. Zhang, F. Zhu and Q. Wu, Organometallics, 2010, 29, 2118–2125 CrossRef CAS.
- W. Zhao, J. Yu, S. Song, W. Yang, H. Liu, X. Hao, C. Redshaw and W.-H. Sun, Polymer, 2012, 53, 130–137 CrossRef CAS PubMed.
- J. Lai, W. Zhao, W. Yang, C. Redshaw, T. Liang, Y. Liu and W.-H. Sun, Polym. Chem., 2012, 3, 787–793 RSC.
- X. Cao, F. He, W. Zhao, Z. Cai, X. Hao, T. Shiono, C. Redshaw and W.-H. Sun, Polymer, 2012, 53, 1870–1880 CrossRef CAS PubMed.
- F. He, W. Zhao, X. Cao, T. Liang, C. Redshaw and W.-H. Sun, J. Organomet. Chem., 2012, 713, 209–216 CrossRef CAS PubMed.
- W.-H. Sun, W. Zhao, J. Yu, W. Zhang, X. Hao and C. Redshaw, Macromol. Chem. Phys., 2012, 213, 1266–1273 CrossRef CAS.
- S. Wang, W. Zhao, X. Hao, B. Li, C. Redshaw, Y. Li and W.-H. Sun, J. Organomet. Chem., 2013, 731, 78–84 CrossRef CAS PubMed.
- S. Wang, B. Li, T. Liang, C. Redshaw, Y. Li and W.-H. Sun, Dalton Trans., 2013, 42, 8988–8997 RSC.
- V. K. Appukuttan, Y. Liu, B. C. Son, C.-S. Ha, H. Suh and I. Kim, Organometallics, 2011, 30, 2285–2294 CrossRef CAS.
- W. Zhang, W. Chai, W.-H. Sun, X. Hu, C. Redshaw and X. Hao, Organometallics, 2012, 31, 5039–5048 CrossRef CAS.
- W.-H. Sun, S. Kong, W. Chai, T. Shiono, C. Redshaw, X. Hu, C. Guo and X. Hao, Appl. Catal., A, 2012, 447–448, 67–73 CrossRef CAS PubMed.
- A. Karam, R. Tenia, M. Martínez, F. López-Linares, C. Albano, A. Diaz-Barrios, Y. Sánchez, E. Catarí, E. Casas, S. Pekerar and A. Albornoz, J. Mol. Catal. A: Chem., 2007, 265, 127–132 CrossRef CAS PubMed.
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428–447 CrossRef CAS.
- Y. Chen, P. Hao, W. Zuo, K. Gao and W.-H. Sun, J. Organomet. Chem., 2008, 693, 1829–1840 CrossRef CAS PubMed.
- L. Xiao, R. Gao, M. Zhang, Y. Li, X. Cao and W.-H. Sun, Organometallics, 2009, 28, 2225–2233 CrossRef CAS.
- P. Hao, Y. Chen, T. Xiao and W.-H. Sun, J. Organomet. Chem., 2010, 695, 90–95 CrossRef CAS PubMed.
- L. Zhang, X. Hou, J. Yu, X. Chen, X. Hao and W.-H. Sun, Inorg. Chim. Acta, 2011, 379, 70–75 CrossRef CAS PubMed.
- R. Gao, Y. Li, F. Wang, W.-H. Sun and M. Bochmann, Eur. J. Inorg. Chem., 2009, 4149–4156 CrossRef CAS.
- S. Song, R. Gao, M. Zhang, Y. Li, F. Wang and W.-H. Sun, Inorg. Chim. Acta, 2011, 376, 373–380 CrossRef CAS PubMed.
- K. Wang, K. Wedeking, W. Zuo, D. Zhang and W.-H. Sun, J. Organomet. Chem., 2008, 693, 1073–1080 CrossRef CAS PubMed.
- T. Xiao, S. Zhang, B. Li, X. Hao, C. Redshaw, Y.-S. Li and W.-H. Sun, Polymer, 2011, 52, 5803–5810 CrossRef CAS PubMed.
- V. K. Appukuttan, Y. Liu, B. C. Son, C.-S. Ha, H. Suh and I. Kim, Organometallics, 2011, 30, 2285–2294 CrossRef CAS.
- W. Zhang, W. Chai, W.-H. Sun, X. Hu, C. Redshaw and X. Hao, Organometallics, 2012, 31, 5039–5048 CrossRef CAS.
- W.-H. Sun, S. Kong, W. Chai, T. Shiono, C. Redshaw, X. Hu, C. Guo and X. Hao, Appl. Catal., A, 2012, 447–448, 67–73 CrossRef CAS PubMed.
- W.-H. Sun, S. Jie, S. Zhang, W. Zhang, Y. Song, H. Ma, J. Chen, K. Wedeking and R. Fröhlich, Organometallics, 2006, 25, 666–677 CrossRef CAS.
- G. A. Solan and J. D. A. Pelletier, PCT Int. Appl, WO 2005118605 (ExxonMobil Chemical), 2005 Search PubMed.
- J. D. A. Pelletier, Y. D. M. Champouret, J. C. Cadarso, L. Clowes, M. Gañete, K. Singh, V. Thanarajasingham and G. A. Solan, J. Organomet. Chem., 2006, 691, 4114–4123 CrossRef CAS PubMed.
- S. Jie, S. Zhang, W.-H. Sun, X. Kuang, T. Liu and J. Guo, J. Mol. Catal. A: Chem., 2007, 269, 85–96 CrossRef CAS PubMed.
- M. Zhang, W. Zhang, T. Xiao, J.-F. Xiang, X. Hao and W.-H. Sun, J. Mol. Catal. A: Chem., 2010, 320, 92–96 CrossRef CAS PubMed.
- S. Jie, S. Zhang and W.-H. Sun, Eur. J. Inorg. Chem., 2007, 5584–5598 CrossRef CAS.
- M. Zhang, P. Hao, W. Zuo, S. Jie and W.-H. Sun, J. Organomet. Chem., 2008, 693, 483–491 CrossRef CAS PubMed.
- M. Zhang, R. Gao, X. Hao and W.-H. Sun, J. Organomet. Chem., 2008, 693, 3867–3877 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2014 |