Metal-ion controlled solid-state reactivity and photoluminescence in two isomorphous coordination polymers†
Received
30th September 2013
, Accepted 18th December 2013
First published on 13th January 2014
Abstract
Reactions of [M(NH3)2]OH (M = Ag and Cu) and 4-(1H-pyrazol-4-yl)pyridine (Hpypz), using p-xylene (C8H10) as the template, gave isomorphous two-dimensional porous metal azolate frameworks [Ag2(pypz)2]·0.5C8H10 (MAF-36(Ag), 1·g) and [Cu2(pypz)2]·0.5C8H10 (MAF-36(Cu), 2·g). Powder and single-crystal X-ray diffraction studies showed that, upon guest removal, 1·g transformed into a three-dimensional nonporous framework [Ag6(pypz)6] (1′), while 2·g can retain its porous two-dimensional structure. Depending on the metal ion, framework flexibility/rigidity, and/or porosity, 1·g and 2·g, as well as their guest-free derivatives, showed distinctly different photoluminescence and guest-responsive behaviors.
Introduction
Coordination polymers (CPs) have attracted much attention due to their diverse structures and interesting properties.1 Because CPs are constructed by non-covalent bonding interactions and configuration changeable organic ligands, they are more flexible than carbon- or oxide-based solid materials.2 For example, many porous CPs could expand or contract upon guest desorption/adsorption.3 In addition, the crystalline nature of CPs makes it possible to directly visualize the structure transformations through diffraction techniques.4 The structural changes of CPs commonly originate from the rotation of building blocks around covalent or coordination single bonds, or sliding between different supramolecular networks. More interestingly, some structural transformations may involve the cleavage and generation of coordination and/or covalent bonds.5
Understanding the structure–property relationship of CPs is an important yet distant goal because the property of a CP is the combined result of various structural parameters. Comparison of the structure–property differences between isomorphous or polymorphic compounds may be an efficient solution to understand how a particular structure parameter influences the overall properties.4e,6 This strategy has been successfully applied in evaluating the effects of molecular building blocks on adsorption affinity,4e,6a,b photoswitchable ability,6c polarization capability,6d the direction of magnetic anisotropy,6e and other physical properties. For instance, the univalent coinage metal bipyrazolate [M2(bpz)] (M = Ag or Cu, H2bpz = 3,3′,5,5′-tetramethyl-4,4′-bipyrazole) can crystallize as four isomeric/polymorphic structures. Systematic comparison of their adsorption properties demonstrated that coordinatively unsaturated AgI/CuI clusters can bind unsaturated hydrocarbon molecules via weak M⋯π interactions, and the Ag⋯π interaction is stronger than that of Cu⋯π.4e However, to the best of our knowledge, there is still no related research on the structural flexibility and solid-state reactivity of isomorphous CPs. Herein, we report the syntheses, structures, and guest-responsive behaviors of two isomorphous coordination polymers, which illustrate the important roles of metal ions in controlling structural transformations, porosity, and photoluminescence.
Experimental
Materials and methods
Commercially available reagents were used as received without further purification. 4-(1H-Pyrazol-4-yl)pyridine (Hpypz) was synthesized by a reported method.7 Elemental analyses (C, H, N) were performed on an Elementar Vario EL elemental analyzer. Thermogravimetry (TG) analyses were performed using a TA-Q50 instrument. Powder X-ray diffraction (PXRD) patterns were recorded on a Bruker D8 Advance diffractometer (Cu Kα).
Synthesis of [Ag2(pypz)2]·0.5C8H10 (1·g)
Single crystals: p-Xylene (1.0 mL) and an ethanol solution of Hpypz (0.025 mol L−1, 2.0 mL) were successively, slowly and carefully layered onto an aqueous ammonia (25%, 2.0 mL) solution of a mixture of Ag2O (0.01 mmol, 2.3 mg) and AgCl (0.01 mmol, 1.4 mg). After two weeks, colorless needle-like crystals were collected, washed with ethanol, and dried in air (yield ca. 35%). Bulk microcrystalline powder: To an aqueous ammonia (25%, 12 mL) solution of Ag2O (0.3 mmol, 69.6 mg), p-xylene (6.0 mL) and an ethanol (12 mL) solution of Hpypz (0.6 mmol, 87.0 mg) were successively added under rigorous stirring at room temperature. The reaction mixture was continued to stir at room temperature for 12 h, filtered, and dried at reduced pressure to give a white powder (yield 117 mg, 70%). EA calcd (%) for guest-free [Ag2(pypz)2] (C16H12N6Ag2): C 38.13, H 2.40, N 16.67; found: C 38.29, H 2.45, N 16.53.
Synthesis of [Cu2(pypz)2]·0.5C8H10 (2·g)
Single crystals of 2·g can be obtained by sealing a solution of [Cu(NH3)2]OH (1.0 mmol) in aqueous ammonia–methanol (2/4 mL) and p-xylene (1.0 mL) in a 15 mL Teflon-lined vessel, which was heated in an oven to 140 °C for 90 h, and then cooled to room temperature at a rate of 5 °C h−1. The resulting mixture was washed with ethanol, and pale yellow needle-like crystals were collected and dried in air (yield ca. 50%). Bulk microcrystalline powder was obtained (yield 130 mg, 75%) using the similar method used for 1·g, except that Ag2O was replaced by [Cu(NH3)2]OH (0.75 mmol) and the whole process was carried out under a N2 atmosphere. EA calcd (%) for guest-free [Cu2(pypz)2] (C16H12N6Cu2) calcd: C 46.26, H 2.91, N 20.23; Found: C 45.99, H 2.97, N 19.86.
Crystallography
Single-crystal X-ray diffraction data were recorded on a Rigaku R-AXIS SPIDER IP diffractometer with Mo Kα radiation. Integration of the raw data frames and empirical absorption corrections were applied using the Rapid AUTO package. The structures were solved with the direct method and refined with the full-matrix least-squares technique using the SHELXTL package. Anisotropic thermal parameters were assigned to all non-hydrogen atoms. The hydrogen atoms were generated by the riding mode. Data collection and structural refinement parameters are given in Table 1. CCDC 963314 and 963315 contain the supplementary crystallographic data for 1·g and 2·g.
Table 1 Crystallographic data and structural refinement details
| Compound |
1·g
|
2·g
|
1′
|
|
R
1 = ∑||Fo| − |Fc||/∑|Fo|.
wR2 = [∑wc(Fo2 − Fc2)2/∑wc(Fo2)2]1/2, and wc = (σ2(Fo2)2 + {0.1[max(0,Fo2) + 2Fc2]/3}2)−1.
R
p = Σ|cYsim(2θi) − Iexp(2θi) + Yback(2θi)|/Σ|Iexp (2θi)|.
R
wp = {wp[cYsim(2θi) − Iexp(2θi) + Yback(2θi)]2/Σwp [Iexp(2θi)]2}1/2, and wp = 1/Iexp(2θi).
|
| Method |
Single crystal |
Single crystal |
Powder |
| Formula |
C20H17Ag2N6 |
C20H17Cu2N6 |
C48H36Ag6N18 |
| F.W. |
557.14 |
468.50 |
1512.17 |
|
T (K) |
150(2) |
150(2) |
298(2) |
| Space group |
P21/c |
P21/c |
P21/c |
|
a (Å) |
5.7782(2) |
5.7359(5) |
12.941(2) |
|
b (Å) |
20.4081(8) |
20.449(2) |
23.463(2) |
|
c (Å) |
16.1009(8) |
15.430(1) |
15.836(2) |
|
β (°) |
94.674(1) |
94.154(1) |
98.94(2) |
|
V (Å3) |
1892.34(14) |
1805.1(3) |
4750(1) |
|
Z
|
4 |
4 |
4 |
|
D
c (g cm−3) |
1.956 |
1.724 |
2.129 |
|
μ (mm−1) |
2.088 |
2.375 |
2.500 |
R
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (>2σ) a (>2σ) |
0.0304 |
0.0334 |
R
pc = 3.18% |
| wR2b (all data) |
0.1096 |
0.1117 |
R
wp
= 4.31% |
| GOF |
1.005 |
1.037 |
/ |
The crystal structure of 1′ was determined by single-crystal X-ray diffraction and refined by PXRD. A single crystal of 1′ was obtained by heating 1·g from room temperature to 473 K at a rate of 0.2 °C min−1. The structural model obtained from single-crystal analysis was used as the initial structural model for the Rietveld refinement. For comparison with 1·g, the six independent Ag atoms in 1′ were named Ag11/Ag12/Ag13 and Ag21/Ag22/Ag23, indicating that they originated from Ag1 and Ag2 in 1′, respectively. The pseudo-Voigt profile parameters, background parameters, the cell parameters, the zero point of the diffraction pattern, the global isotropic atom displacement parameters, the Berar–Baldinozzi asymmetry correction parameters, and the March–Dollase preferred orientation correction parameters were optimized step by step to improve the agreement between the calculated and the experimental powder diffraction patterns for 1′, which was carried on the Reflex Powder Refinement module of the Material Studio package.
Results and discussion
Syntheses and structures
Liquid diffusion and solvothermal reactions of [M(NH3)2]OH (M = Ag and Cu) and 4-(1H-pyrazol-4-yl)pyridine (Hpypz) in alcohol–aqueous ammonia solution with p-xylene (C8H10) as the template gave single crystals of metal azolate frameworks [Ag2(pypz)2]·0.5C8H10 (MAF-36(Ag), 1·g) and [Cu2(pypz)2]·0.5C8H10 (MAF-36(Cu), 2·g). Microcrystalline powders of 1·g and 2·g were synthesized in high yield by the rapid solution mixing method. Similar to other MAFs, aqueous ammonia is beneficial for the crystal growth of 1·g and 2·g.8
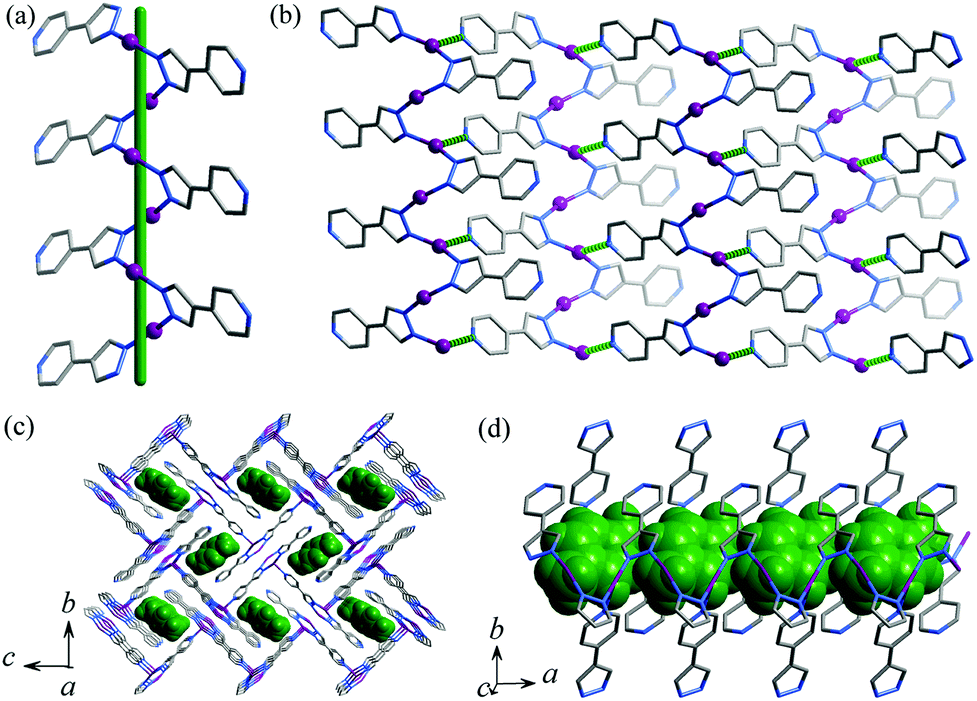
Single-crystal X-ray diffraction study demonstrated that 1·g and 2·g are isomorphous and crystallize in the space group of P21/c, with two metal ions, two deprotonated pypz− ligands and half of a p-xylene molecule in the asymmetric unit (Fig. S1, ESI†). One of the Ag/Cu ions is coordinated by two pyrazolate nitrogen donors from two pypz− ligands in a bent geometry (Ag–N 2.089(3)–2.094(3) Å, N–Ag–N 161.15(12)°, Cu–N 1.875(3)–1.877(3) Å, N–Cu–N 165.33(13)°), and another Ag/Cu ion is three-coordinated by two pyrazolates (Ag–N 2.127(3)–2.130(3) Å, N–Ag–N 160.80(11)°, Cu–N 1.907(3)–1.910(3) Å, N–Cu–N 156.99(12)°) and one pyridine (Ag–N 2.459(3) Å, N–Ag–N 99.99(11)°, Cu–N 2.187(3) Å, N–Cu–N 102.86(12)°) from three pypz− ligands in a T-shaped coordination geometry. Obviously, pyrazolate has stronger coordination bonds than pyridine. The Ag/Cu ions are firstly connected by the pyrazolate moiety of pypz− into a 21 helix (Fig. 1a). The intrachain M⋯M contacts (Ag⋯Ag 3.1413(4)–3.2075(4) Å, Cu⋯Cu 3.0788(6)–3.1609(6) Å) are consistent with literature values of other Ag/Cu pyrazolates.9 Adjacent 21 helices interconnect with each other into a two-dimensional (2D) zigzag layer by the pyridyl ends of pypz− ligands on one of the two sides of the chain and half of the metal ions (Fig. 1b). The pyridines of the other side remain uncoordinated and point outward from the layer. Regarding the 3-coordinated components as 3-connected nodes and 2-coordinated ones as linkers, the 2D network can be simplified as a honeycomb (hcb) topology. These zigzag layers stack in an interdigitation fashion, showing interlayer M⋯M contacts (Ag⋯Ag 3.1353(4)–3.1535(4) Å, Cu⋯Cu 3.3034(6)–3.3483(6) Å, see Fig. S1, ESI†) and weak hydrogen bonds (C9–H⋯N3 3.310(5) Å). The ligand-unsupported Cu⋯Cu contacts are longer than the Ag⋯Ag contacts, even though CuI is much smaller than AgI (sum of the van der Waals radii are 2.80 Å for Cu⋯Cu and 3.44 Å for Ag⋯Ag),10 which demonstrates clearly that argentophilicity is stronger than cuprophilicity.11 Because the dangling pyridyl groups are far smaller than the gaps of the layers, 1D small channels (void = 18.5%) are formed between two adjacent interdigitated layers, which are occupied by p-xylene molecules (Fig. 1c and 1d). There are some weak C–H⋯π interactions between the guest and the host (the distances are 3.551(4)–3.732(4) Å for 1·g and 3.498(3)–3.646(4) Å for 2·g).
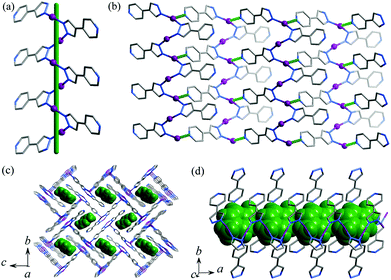 |
| | Fig. 1 Perspective views of (a) a helical Ag-pyrazolate fragment (a green stick represents the 21 screw axis), (b) a 2D zigzag layer (coordination bonds between helical chains are highlighted in green dotted lines), (c) the packing structure along the channel direction and (d) perpendicular to the channel direction in 1·g (p-xylene molecules are shown in space-filling mode and highlighted in green). | |
Framework stability and structural transformation
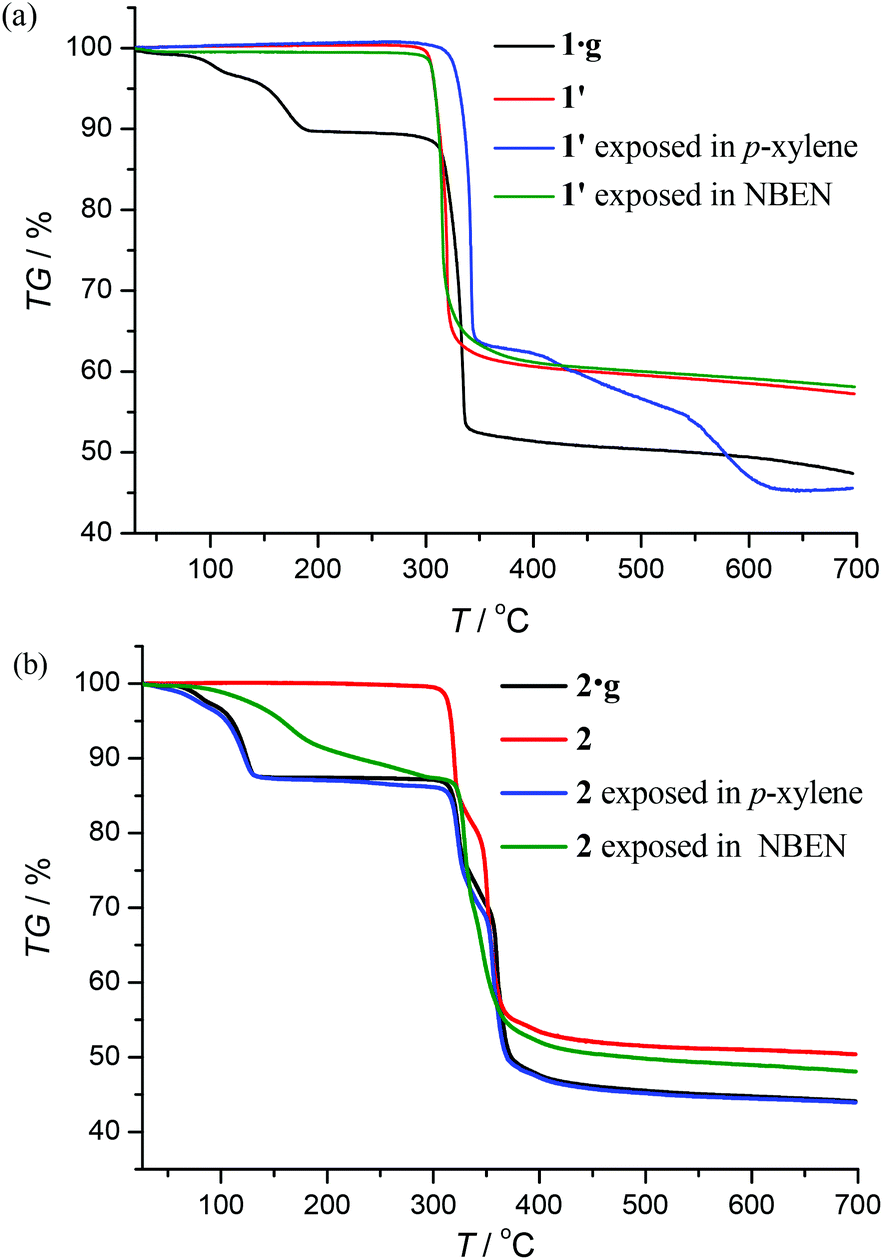
Thermogravimetry (TG) analyses revealed that 1·g and 2·g could lose all p-xylene molecules above 463 K and 407 K, and decomposed above 563 K and 600 K, respectively (Fig. 2). Powder X-ray diffraction (PXRD) revealed that the guest-free product obtained from 1·g was a new phase (hereafter denoted as 1′). 1′ could not be changed back to 1·g even after the sample was immersed in various solvents for a few weeks (Fig. S2, ESI†). However, the PXRD pattern of 2 is only slightly different from that of 2·g. Moreover 2 can convert back to 2·g upon adsorption of solvent vapors (Fig. S3, ESI†). These phenomena indicate that there should be drastic structural change from 1·g to 1′, whereas only a slight framework distortion is likely to occur between 2·g and 2.
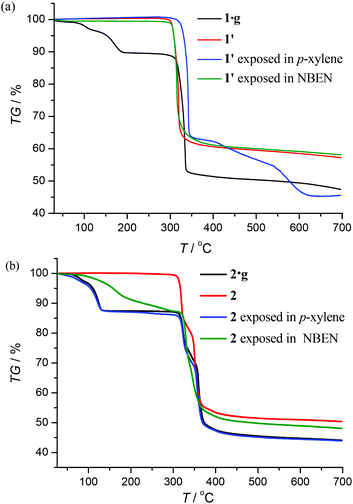 |
| | Fig. 2 TG curves and sorption properties. | |
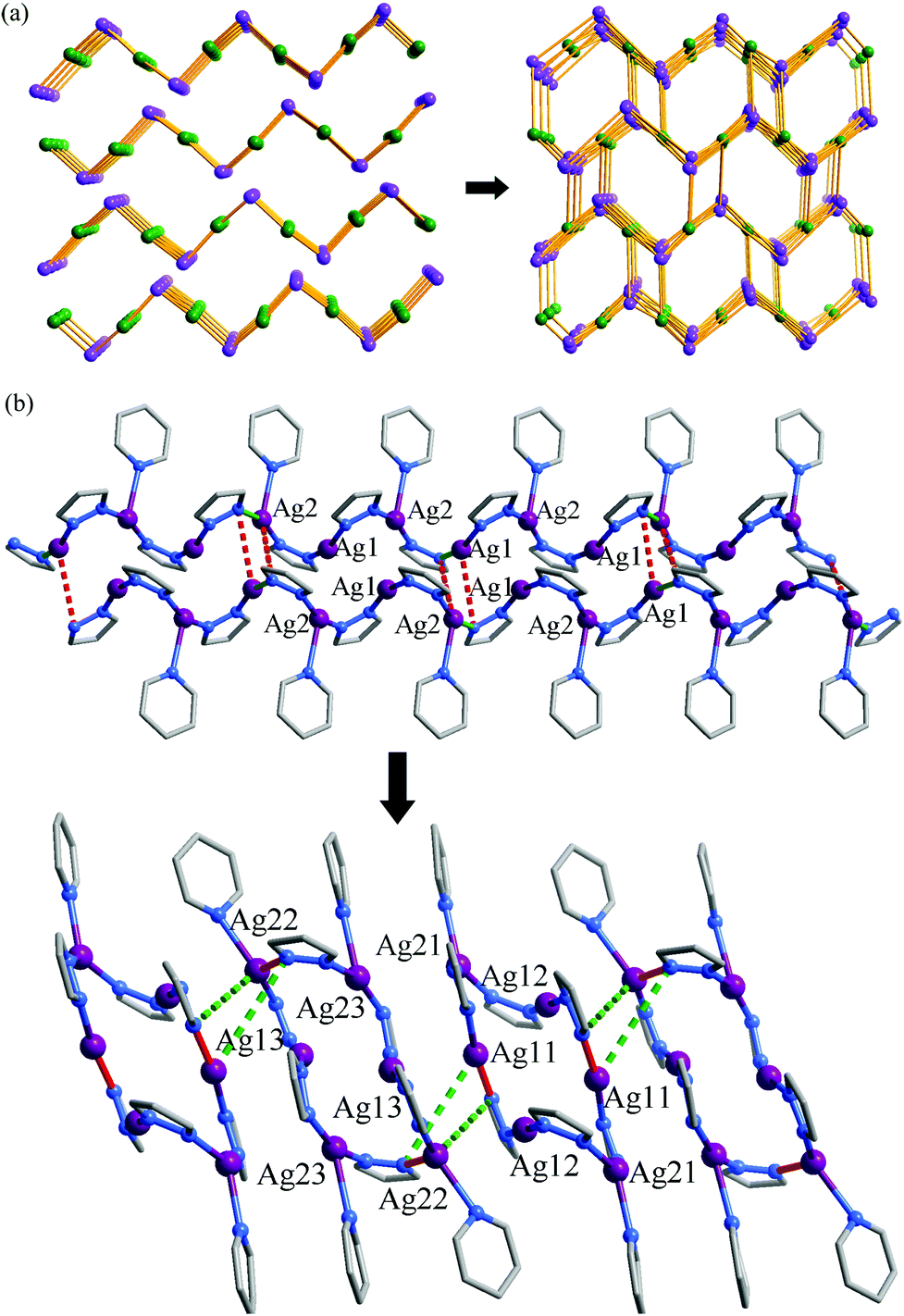
In spite of the poor single crystallinity and complexed crystal structure of 1′, we fortunately determined its single-crystal structure, which was formulated as [Ag6(pypz)6]. Nevertheless, the single-crystal diffraction data was poor. Therefore, the final crystal structure of 1′ was obtained from the Rietveld refinement of its PXRD pattern. The results revealed that 1′ crystallizes in a space group of P21/c with a unit-cell volume ca. 2.5 times of 1·g (Table 1). In the asymmetric unit of 1′, there are six Ag ions and six deprotonated pypz− ligands (Fig. S4, ESI†). The above data illustrate that the structural complexity of 1′ is three times that of 1·g, while losing the p-xylene molecules makes a 17% contraction in volume. In 1′, metal ions and pypz− ligands show similar coordination modes as those in 1·g. That is, half Ag ions/organic ligands are two coordinated, and the others are three coordinated. However, 1′ has a new network connectivity, in which the Ag ions are connected by the pyrazolate moiety of pypz− into Ag6(pz)6 rings rather than infinite Ag(pz) helices in 1·g (Fig. 3). Regarding the 3-coordinated metal ions and ligands as 3-connected nodes, and the coordination bonds as linkers, the 3D network can be simplified to be a binodal 3-connected net with a point symbol (4·8·10)(8·102) (Fig. 4a). There are very rare examples of structural transformations from 2D to 3D coordination polymers.12 Obviously, the transformation from 1·g to 1′ requires not only distortion of the nets but also cleavages and formations of coordination bonds. Based on the structural relationship between 1·g and 1′, a possible network rearrangement mechanism can be suggested (Fig. 4b).
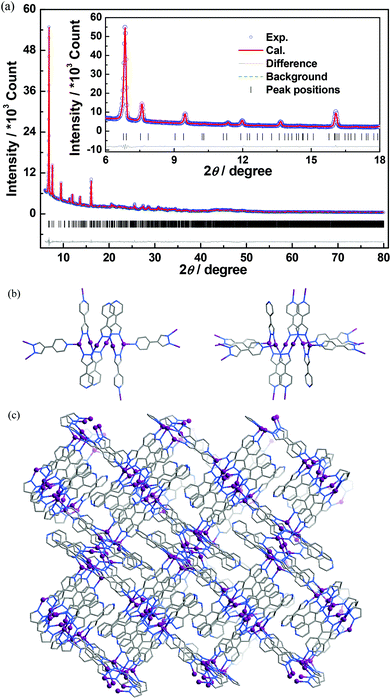 |
| | Fig. 3 (a) The final Rietveld refinement plots, (b) two kinds of Ag6(pz)6 rings, and (c) the 3D coordinated structure of 1′. | |
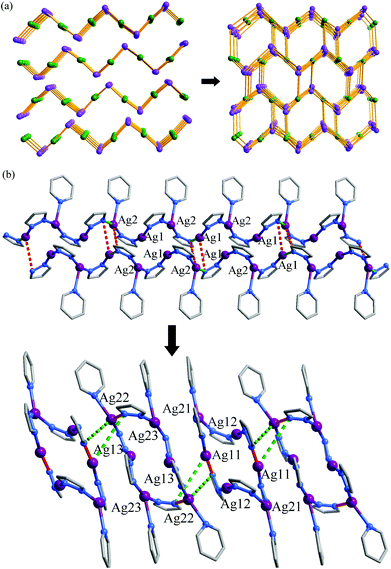 |
| | Fig. 4 (a) 2D to 3D network topology transformation (3-coordinated Ag ions and pypz− are shown as purple and green spheres, respectively) and (b) a possible structural transformation mechanism (red and green sticks denote bonds involved in cleavage/formation, solid for cleavage and dashed for formation) from 2D 1·g to 3D 1′. | |
Photoluminescence property
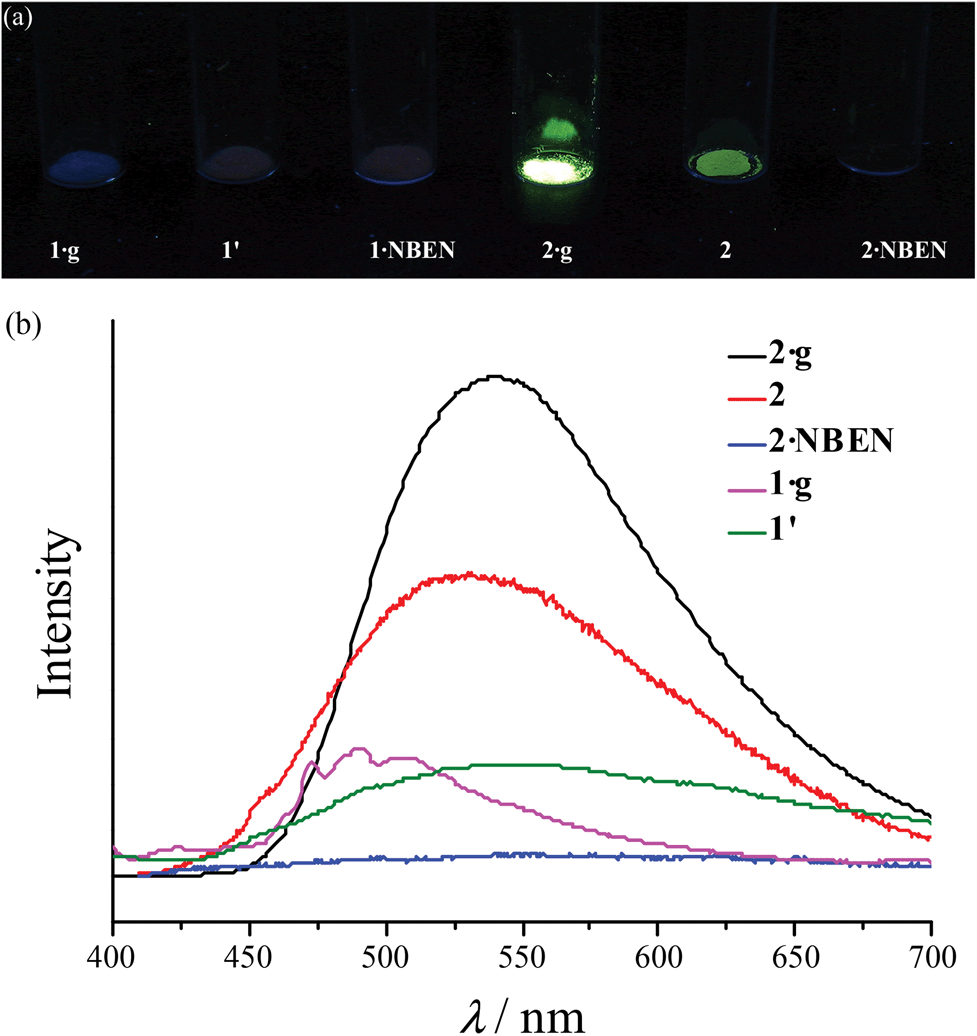
Under 365 nm ultraviolet irradiation, 2·g displays bright yellow-green photoluminescence, while that of 1·g is much weaker and blue (Fig. 5a). As shown in Fig. 5b and Fig. S5,† the broad and structureless emission spectra (λmax = 538 nm), as well as the microsecond emission lifetimes of 2·g and 2, is consistent with the 3[MLCT] phosphorescence of similar CuI complexes.13 The emission peak of 1·g possesses a vibronic structure with the maximum located at 492 nm, which is similar to the ligand-based triplet phosphorescence emission of [Ag(tz)] (Htz = 1,2,4-triazole) affected by heavy metal effect.13 After guest removal, the photoluminescence of these d10 coinage metal complexes exhibit a distinctly different response. Compared with 1·g, the emission peak of 1′ shows a large red shift (61 nm). For 2·g and 2, desolvation gives rise to a notable decrease of the photoluminescence intensity and a blue shift of the emission peak (Δλ = 26 nm). Moreover, p-xylene vapour can restore the luminescence color from 2 to 2·g, but cannot affect 1′ (Fig. S6†), which is consistent with their reversible and irreversible structural transformations, respectively. It is well known that nitrobenzene is a strong electron-withdrawing molecule that can effectively quench photoluminescence based on the donor–acceptor electron-transfer mechanism.14 Interestingly, the luminescence of 2 can be effectively quenched with nitrobenzene (NBEN). However, the luminescence of 1·g, 1′ and 2·g are almost unchanged upon exposing them in saturated NBEN vapor, indicating that porosity is crucial for luminescence detection of guest molecules.1d As shown in Fig. 2, 2 can readily adsorb about 13% solvent (p-xylene, nitrobenzene, etc.) inside its microporous channels, but 1′ can only adsorb trace solvent vapours on its outer surface. These results indicate the porosity of 2 and the nonporous nature of 1′.
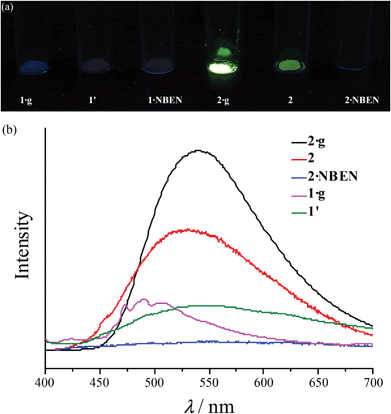 |
| | Fig. 5 (a) Photographs (under 365 nm ultraviolet) and (b) photoluminescence spectra. | |
Conclusions
In summary, two isomorphous univalent Ag/Cu coordination polymers with a porous layer structure were obtained via liquid diffusion and solvothermal reactions. Upon guest removal, the Ag-based compound transforms into a 3D nonporous framework, which requires cleavage and reformation of coordination bonds. On the other hand, the Cu-based compound can retain its porous 2D structure for adsorption of other types of guests. Moreover, these compounds show distinctly different photoluminescence properties and guest-responsive behaviors, which can be attributed to the variation of metal ion and porosity. These results should be insightful for understanding the structure–property relationship of coordination polymers.
Acknowledgements
This work was supported by the “973 Project” (2012CB821706) and NSFC (21121061 and 21225105).
Notes and references
-
(a) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724–781 CrossRef PubMed;
(b) C. Janiak, Dalton Trans., 2003, 2781–2804 RSC;
(c) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed;
(d) Y.-J. Cui, Y.-F. Yue, G.-D. Qian and B.-L. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed;
(e) J.-P. Zhang, X.-C. Huang and X.-M. Chen, Chem. Soc. Rev., 2009, 38, 2385–2396 RSC;
(f) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
-
(a) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed;
(b) K. C. Stylianou, J. Rabone, S. Y. Chong, R. Heck, J. Armstrong, P. V. Wiper, K. E. Jelfs, S. Zlatogorsky, J. Bacsa, A. G. McLennan, C. P. Ireland, Y. Z. Khimyak, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2012, 134, 20466–20478 CrossRef CAS PubMed;
(c) G. Ferey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
-
(a) B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390–1393 CrossRef CAS PubMed;
(b) S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC;
(c) J.-H. Wang, M. Li and D. Li, Chem. Sci., 2013, 4, 1793–1801 RSC;
(d) Y.-S. Wei, K.-J. Chen, P.-Q. Liao, B.-Y. Zhu, R.-B. Lin, H.-L. Zhou, B.-Y. Wang, W. Xue, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2013, 4, 1539–1546 RSC;
(e) H.-L. Zhou, R.-B. Lin, C.-T. He, Y.-B. Zhang, N. Feng, Q. Wang, F. Deng, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2013, 4, 2534–2541 Search PubMed.
-
(a) J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781–1795 CrossRef CAS PubMed;
(b) C. D. Wu and W. B. Lin, Angew. Chem., Int. Ed., 2005, 44, 1958–1961 CrossRef CAS PubMed;
(c) M. S. Chen, M. Chen, S. Takamizawa, T. Okamura, J. A. Fan and W. Y. Sun, Chem. Commun., 2011, 47, 3787–3789 RSC;
(d) N. Li, F. L. Jiang, L. A. Chen, X. J. Li, Q. H. Chen and M. C. Hong, Chem. Commun., 2011, 47, 2327–2329 RSC;
(e) J.-P. Zhang and S. Kitagawa, J. Am. Chem. Soc., 2008, 130, 907–917 CrossRef CAS PubMed;
(f) Y.-Q. Chen, G.-R. Li, Z. Chang, Y.-K. Qu, Y.-H. Zhang and X.-H. Bu, Chem. Sci., 2013, 4, 3678–3682 RSC.
-
(a) G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755–1775 RSC;
(b) S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 CrossRef CAS PubMed;
(c) L. Wen, P. Cheng and W. B. Lin, Chem. Commun., 2012, 48, 2846–2848 RSC;
(d) J. Sun, F. N. Dai, W. B. Yuan, W. H. Bi, X. L. Zhao, W. M. Sun and D. F. Sun, Angew. Chem., Int. Ed., 2011, 50, 7061–7064 CrossRef CAS PubMed.
-
(a) J.-B. Lin, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2010, 132, 6654–6656 CrossRef CAS PubMed;
(b) J.-P. Zhang, A.-X. Zhu, R.-B. Lin, X.-L. Qi and X.-M. Chen, Adv. Mater., 2011, 23, 1268–1271 CrossRef CAS PubMed;
(c) J. W. Brown, B. L. Henderson, M. D. Kiesz, A. C. Whalley, W. Morris, S. Grunder, H. Deng, H. Furukawa, J. I. Zink, J. F. Stoddart and O. M. Yaghi, Chem. Sci., 2013, 4, 2858–2864 RSC;
(d) W. Zhang and R.-G. Xiong, Chem. Rev., 2012, 112, 1163–1195 CrossRef CAS PubMed;
(e) V. Mereacre, A. Baniodeh, C. E. Anson and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 15335–15337 CrossRef CAS PubMed;
(f) D.-F. Weng, Z.-M. Wang and S. Gao, Chem. Soc. Rev., 2011, 40, 3157–3181 RSC.
- H. Adams, S. R. Batten, G. M. Davies, M. B. Duriska, J. C. Jeffery, P. Jensen, J. Lu, G. R. Motson, S. J. Coles, M. B. Hursthouse and M. D. Ward, Dalton Trans., 2005, 1910–1923 RSC.
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed.
-
(a) N. Masciocchi, M. Moret, P. Cairati, A. Sironi, G. A. Ardizzoia and G. La Monica, J. Am. Chem. Soc., 1994, 116, 7668–7676 CrossRef CAS;
(b) F. Gong, Q. Wang, J. Chen, Z. Yang, M. Liu, S. Li, G. Yang, L. Bai, J. Liu and Y. Dong, Inorg. Chem., 2010, 49, 1658–1666 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- J.-P. Zhang, Y.-B. Wang, X.-C. Huang, Y.-Y. Lin and X.-M. Chen, Chem. – Eur. J., 2005, 11, 552–561 CrossRef CAS PubMed.
-
(a) Y. C. He, J. Yang, G. C. Yang, W. Q. Kan and J. F. Ma, Chem. Commun., 2012, 48, 7859–7861 RSC;
(b) S. K. Ghosh, W. Kaneko, D. Kiriya, M. Ohba and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 8843–8847 CrossRef CAS PubMed;
(c) R. Medishetty, L. L. Koh, G. K. Kole and J. J. Vittal, Angew. Chem., Int. Ed., 2011, 50, 10949–10952 CrossRef CAS PubMed;
(d) W. M. Bloch, R. Babarao, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441–10448 CrossRef CAS PubMed.
- J.-P. Zhang, Y.-Y. Lin, X.-C. Huang and X.-M. Chen, J. Am. Chem. Soc., 2005, 127, 5495–5506 CrossRef CAS PubMed.
- S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization. CCDC 963314 and 963315. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qi00061c |
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.